

Abstract
Chronic inflammation caused by HIV infection may lead to deficient glucocorticoid (GC) signaling predisposing people living with HIV to depression and other psychiatric disorders linked to GC resistance. We hypothesized that comorbid HIV and depressive symptoms in women would synergistically associate with deficits in GC signaling. This cross-sectional study used samples obtained from the Women’s Interagency HIV Study (WIHS). The Centers for Epidemiological Studies (CES-D) was used to define depression in four groups of women from the Women’s Interagency HIV Study (WIHS): 1) HIV-negative, non-depressed (n=37); 2) HIV-negative, depressed (n=34); 3) HIV-positive, non-depressed (n=38); and 4) HIV-positive, depressed (n=38). To assess changes in GC signaling from peripheral blood mononuclear cells (PBMCs), we examined baseline and dexamethasone (Dex)-stimulated changes in the expression of the GC receptor (GR, gene: Nr3c1) and its negative regulator Fkbp5 via quantitative RT-PCR. GR sensitivity was evaluated in vitro by assessing the Dex inhibition of lipopolysaccharide (LPS)-stimulated IL-6 and TNF-α levels. Depressive symptoms and HIV serostatus were independently associated with elevated baseline expression of Fkbp5 and Nr3c1. Depressive symptoms, but not HIV status, was independently associated with reduced LPS-induced release of IL-6 and TNF-α. Counter to predictions, there was no interactive association of depressive symptoms and HIV on any outcome. Comorbid depressive symptoms with HIV infection were associated with a gene expression and cytokine profile similar to that of healthy control women, a finding that may indicate further disruptions in disease adaptation.
Keywords: HIV, depression, glucocorticoid, FKBP5, women, inflammation
1. Introduction
Advances in antiretroviral therapy (ART) have dramatically increased the life expectancy of people living with HIV (PLWH); however, PLWH still suffer from inflammation-related diseases such as cardiovascular disease, type II diabetes, cancer, and dementia at higher rates and at younger ages than uninfected individuals (Nemeth et al 2015, Valdez et al 2016). Although it has been established that persistent immune activation and inflammation contribute to non-AIDS pathologies in PLWH who are on suppressive antiretroviral therapy (ART) (Hunt 2012), the factors contributing to sustained inflammation in otherwise healthy PLWH have not been identified.
Depression co-morbid with HIV infection has emerged as a predictor of HIV-related comorbidities (Kelso-Chichetto et al 2017, Rivera-Rivera et al 2014, Rivera-Rivera et al 2016) and increased mortality (Ickovics et al 2001). Although the precise mechanistic relationship between depression and HIV-related comorbidities has not been elucidated, the biological relationship between inflammation and depression may be a critical factor (Valdez et al 2016). In seronegative individuals, a bidirectional relationship between depression and inflammation has been established such that inflammation increases the risk of depression (Miller et al 2009) and psychosocial stress and negative mood increase inflammation (Glaser & Kiecolt-Glaser 2005). Although not as well characterized in the context of HIV infection, depression and HIV-related comorbidities are more prevalent in women than in men (Kelso-Chichetto et al 2017), and middle-aged women living with HIV (WLWH) are at increased risk for systemic immune activation/inflammation compared to men living with HIV (MWLH) (Raghavan et al 2017).
A potential mechanism that may lead to pervasive inflammation and has been implicated in the pathophysiology of depression, is dysfunction of the hypothalamic-pituitary-adrenal (HPA) axis (Bekhbat et al 2017, Neigh & Nemeroff 2006). Glucocorticoids (GC) such as cortisol (CORT), are main effectors of the HPA axis. Although previous studies have demonstrated dysfunction of the HPA axis in PLWH (Patterson et al 2013), these studies have focused only on the ligand, CORT, and not the GC receptor (GR) specifically. Through direct action as a transcription factor and via transrepression of pro-inflammatory transcription factors, the GR suppresses inflammatory signaling. Conversely, reduced function of the GR can precipitate elevated inflammation (Bekhbat et al 2017).
We sought to test the central hypothesis that depression and HIV infection interact to alter the function of the GR and precipitate an enhanced inflammatory response. Hyperactivity of the HPA axis observed in depression is commonly accompanied by decreased sensitivity of GRs as evidenced by reduced translocation of GR to the nucleus and reduced transcriptional activity (Pariante & Miller 2001). Chronic inflammation caused by HIV infection may lead to deficient GC signaling, thus predisposing PLWH to psychiatric disorders linked to GC resistance, such as depression (Pace et al 2007). We hypothesized that comorbid symptoms of depression in PLWH would synergistically associate with deficits in GC signaling, demonstrated to be a sensitive marker of GC resistance in depressed patients (Menke et al 2012), both in the absence of GC stimulation and when stimulated with a synthetic GC, dexamethasone (Dex). In addition to direct assessment of GR, we also assessed a negative regulator of GR, FK506-binding protein 51 (FKBP5), and a postivie regulator, FK506-binding protein 52 (FKBP4) (Davies et al 2002). Importantly, expression of FKBP5 and its genetic variations have been linked to a number of psychiatric conditions involving HPA axis dysfunction including depression (Menke et al 2013), post-traumatic stress disorder (Klengel et al 2013), and chronic stress (Hartmann et al 2012). In addition to the established influence of FKBP5 on the GR, FKBP5 has been shown to play a role in HIV-associated cognitive deficits in PLWH (Soontornniyomkij et al 2012, Tatro et al 2009).
2. Methods
2.1. Participants
This cross-sectional study used PBMCs obtained from participants in the Women’s Interagency HIV Study (WIHS). WIHS began in 1993, and is a comprehensive multisite prospective cohort study designed to investigate the progression of HIV disease in women. WIHS is funded to the National Institutes of Health. Study visits occur every six months and include detailed interviews, physical examinations, and laboratory testing. Participants were selected as members in one of four groups: 1) HIV-negative, no depressive symptoms, based on CES-D < 16) (n=37, “HIV−/No-Dep”); 2) HIV-negative, depressive symptoms (n=34, “HIV−/Dep”); 3) HIV-positive, no depressive symptoms (n=38, “HIV+/No-Dep”); and 4) HIV-positive, depressive symptoms (n=38, “HIV+/Dep”). All participants were younger than 45 years old and African-American. Samples were excluded if the contributing subject had an AIDS diagnosis or active AIDS-defining opportunistic conditions secondary to HIV infection, current use of hormone-mediated contraceptives, was pregnant or nursing, met DSM-IV criteria for current substance/alcohol abuse or dependence (drank ≥12 glasses of alcohol/week or non-marijuana drug use [cocaine, crack, heroine, methadone, intravenous drug use]), seroconverted from HIV-negative to HIV-positive during WIHS follow-up, or had used antipsychotic, antidepressant, or antipsychotic medication or mood stabilizers within the past 4 weeks. A score of 16 or higher on the Center for Epidemiologic Studies Depression Scale (CES-D) was used to indicate a clinically relevant depressive symptom burden. Due to the high prevalence of marijuana use among HIV−/No-Dep group (approximately 35%), the four study groups were frequency-matched on marijuana use to ensure equal distribution of this potential confounder. Groups were similar on HIV RNA status (detectable/not detectable), HCV status, age, race, and CD4+ count.
2.2. Cell culture
PBMCs were cryopreserved in liquid nitrogen until plated. Cells were thawed in an RPMI medium supplemented with 5% FBS, 2mM L-glutamine, 10/10 pen/strep, and plated in duplicate at a density of 106/mL per well of a 12-well cell culture plate (Corning Inc, Corning, NY). Following overnight incubation in a humidified atmosphere at 37°C in 5% CO2, the cells were stimulated with vehicle (baseline), dexamethasone (Dex, Sigma Aldrich, product D-4902), lipopolysaccharide (LPS, Sigma Aldrich, product L2880), or concurrent Dex and LPS. Dex was given at 0 hr at a concentration of 10−8 M, and LPS was given at 6 hrs at a concentration of 100 ng/mL. Cells were harvested at 12 hrs, centrifuged at 1000rcf for 5 minutes at 4°C, and following a wash with ice-cold PBS, the cell pellets were lysed for RNA extraction. Cell lysates and cell culture supernatant were stored at −80°C until used for RNA extraction and ELISA.
2.3. Quantitative PCR
RNA was extracted from cell pellets using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. RNA integrity was assessed by a Nano-Drop 2000 spectrophotometer (ThermoScientific, Wilmington, DE, USA) and RNA samples were reverse transcribed using the High Capacity RNA to cDNA Kit (Applied Biosystems, Foster City, CA, USA). To ensure uniform amounts of total cDNA across groups, cDNA was quantified via the PicoGreen Assay (Invitrogen, Carlsbad, CA), then standardized, so that all samples started quantitative RT-PCR with 0.1 μg cDNA. The human gene Rpl13a was determined to be the optimal endogenous control based on an inter-group variance of less than 10% across groups. Primers for Fkbp5 (forward: CTTGCTGCCTTTCTGAACCT, reverse: CCCTTGGCTGACTCAAACTC), Nr3c1 (forward: CGAGCATGAGACCAGATGTA, reverse: CGACTGCTCTTTTGAAGAAA), Fkbp4 (forward: AAGCTGGAACAGAGCACCAT, reverse: GCAGCAGAGAAGGCCTGTAG), Rpl13a (forward: ATGCTGCCTCACAAGACCA, reverse: TAGGCTTCAGACGCACGAC) were designed and purchased from Applied Biosystems (Foster City, CA). The universal two-step RT-PCR cycling conditions used on the 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA) were: 50°C (2 min), 95°C (10 min), 40 cycles of 95°C (15 s) and 60°C (1 min). Samples were run in triplicate, and the coefficient of variation within the triplicates was no more than 4%. Fold changes in gene expression were calculated by the comparative 2−ΔΔCt quantification method relative to the HIV−/No-Dep group.
2.4. ELISA
Based on PBMC sample availability, a subset of the participants were also evaluated for LPS-induced cytokine expression (LPS) and its suppression by Dex (LPS+Dex). Human IL-6 and TNF-α ELISA kits were purchased from R&D Systems, and assays were performed according to the manufacturer’s instructions. The sensitivities of the assays were 0.70 pg/mL (IL-6) and 0.5-5.5 pg/mL (TNF-α). Samples were run in duplicates, and the coefficient of variance among the duplicates was less than 15%. Dex-induced suppression of LPS-induced cytokine levels were expressed as percent suppression as follows:
2.5. Statistical analysis
Too incorporate up to 4 measurements reflecting different treatment (Baseline/Dex) and gene (target/reference) combinations from each sample, repeated measures analysis was performed. All quantitative RT-PCR data were modeled with Generalized Estimating Equation (GEE) models assuming a normal distribution using robust variance estimation and an exchangeable covariance matrix. These models contained HIV status (positive vs negative), a clinically relevant depressive symptom burden (greater than 16 on CES-D), treatment (Baseline/Dex), gene (target/reference), all possible interaction terms of these 4 variables, and marijuana use as independent variables and cycle threshold (CT) values as the dependent variable. Outliers were determined using Hoaglin and Iglewicz’s method (Hoaglin & Iglewicz 1987) within each treatment level (Baseline or Dex) of dCt values and overall for ddCt values. Since ddCt values for FKBP5 were more variable – reflecting the robust increase in FKBP5 following Dex stimulation – only ddCt values > 5 IQR above the third quartile were considered outliers. If an outlier was detected, all observations for that woman were removed for that analysis.
IL-6 and TNF-α concentrations measured by ELISA were log-transformed for analysis, and the GEE models contained HIV status (positive vs negative), depressive symptoms (yes/no), treatment (LPS or LPS+Dex), all possible interaction terms, and marijuana use. GEE model selection for ELISA data was made based on QIC and clinical relevance. Whereas ELISA data from concurrent LPS+Dex treatment were modeled assuming a normal distribution using robust variance estimation and an exchangeable covariance matrix, data from LPS-only treatment were modeled assuming a gamma distribution with a log link using robust variance estimation in order to account for the robust increase in LPS-induced cytokine levels. Additionally, models for LPS-induced IL-6 concentration adjusted for CD4% while LPS-induced TNF-α models adjusted for age. All other models were unadjusted for CD4% and age. Model fit was evaluated by residual plots. Outliers were determined using Hoaglin and Iglewicz’s method (Hoaglin & Iglewicz 1987) separately within each treatment level (LPS or LPS+Dex).
3. Results
3.1. HIV infection and Depressive Symptoms Independently Predict Elevated Expression of GR and FKBP5 in PBMCs and Reduced Dex-Induced Changes in Expression
Participant demographics and biological variables are provided in Table I and II. CES-D and Quality of Life Index scores are provided in Table III. In order to assess GC signaling, we examined baseline and Dex-stimulated changes in the expression of the GC receptor (GR, gene: Nr3c1) and its negative regulator FKBP5 and positive regulator FKBP4.
Table I.
Demographic Metrics.
| HIV −/No-Dep (n=37) | HIV −/Dep (n=34) | HIV +/No-Dep (n=38) | HIV +/Dep (n=38) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| % | n | % | n | % | n | % | n | ||
| Race/Ethnicity | Other non-Hispanic | 8.1 | 3 | 5.9 | 2 | 10.5 | 4 | 13.2 | 5 |
| African-American non-Hispanic | 67.6 | 25 | 73.5 | 25 | 68.4 | 26 | 57.9 | 22 | |
| Hispanic | 24.3 | 9 | 20.6 | 7 | 21.1 | 8 | 29.0 | 11 | |
| Annual income | ≤$6,000 | 18.9 | 7 | 33.3 | 11 | 18.4 | 7 | 11.4 | 4 |
| $6,001-$12,000 | 27.0 | 10 | 30.3 | 10 | 21.1 | 8 | 37.1 | 13 | |
| $12,001-$24,000 | 21.6 | 8 | 18.2 | 6 | 34.2 | 13 | 31.4 | 11 | |
| >$24,000 | 32.4 | 12 | 18.2 | 6 | 26.3 | 10 | 20.0 | 7 | |
| Marijuana use | No | 62.2 | 23 | 67.7 | 23 | 68.4 | 26 | 63.2 | 24 |
| Yes | 37.8 | 14 | 32.4 | 11 | 31.6 | 12 | 36.8 | 14 | |
| Detectable viral load | No | 47.4 | 18 | 29.0 | 11 | ||||
| Yes | 52.6 | 20 | 71.1 | 27 | |||||
| Combination therapy/HAART use | No | 34.2 | 13 | 50.0 | 19 | ||||
| Yes | 65.8 | 25 | 50.0 | 19 | |||||
| Adherence | <95% | 37.0 | 10 | 39.1 | 9 | ||||
| ≥95% | 63.0 | 17 | 60.9 | 14 | |||||
| HCV-positive | No | 94.6 | 35 | 88.2 | 30 | 84.2 | 32 | 89.5 | 34 |
| Yes | 5.4 | 2 | 11.8 | 4 | 15.8 | 6 | 10.5 | 4 | |
| Ever sexual abuse | No | 75.7 | 28 | 88.2 | 30 | 84.2 | 32 | 79.0 | 30 |
| Yes | 24.3 | 9 | 11.8 | 4 | 15.8 | 6 | 21.1 | 8 | |
| Ever physical abuse | No | 73.0 | 27 | 76.5 | 26 | 71.1 | 27 | 57.9 | 22 |
| Yes | 27.0 | 10 | 23.5 | 8 | 29.0 | 11 | 42.1 | 16 | |
| Ever sexual abuse < 18 yrs | No | 83.8 | 31 | 94.1 | 32 | 84.2 | 32 | 89.5 | 34 |
| Yes | 16.2 | 6 | 5.9 | 2 | 15.8 | 6 | 10.5 | 4 | |
| Ever physical abuse < 18 yrs | No | 91.9 | 34 | 94.1 | 32 | 89.5 | 34 | 86.8 | 33 |
| Yes | 8.1 | 3 | 5.9 | 2 | 10.5 | 4 | 13.2 | 5 | |
Table II.
Biologic Metrics
| HIV −/No-Dep (n=37) | HIV −/Dep (n=34) | HIV +/No-Dep (n=38) | HIV +/Dep (n=38) | |||||
|---|---|---|---|---|---|---|---|---|
| Median | Q1, Q3 | Median | Q1, Q3 | Median | Q1, Q3 | Median | Q1, Q3 | |
| Age (years) | 35.0 | 26.5, 39.6 | 35.2 | 32.4, 40.1 | 36.5 | 31.8, 40.6 | 36.4 | 31.4, 40.9 |
| Viral load (copies/mL) | 11500 | 3850, 32000 | 5500 | 340, 33000 | ||||
| CD4 nadir | 294.5 | 109.0, 365.5 | 327.0 | 157.0, 431.0 | ||||
| Years on HAART | 5.1 | 3.9, 6.4 | 5.1 | 3.7, 6.7 | ||||
| Years on ART | 6.4 | 4.1, 8.4 | 6.9 | 3.9, 8.3 | ||||
| CD4 Percentage | 48.0 | 44.0, 52.0 | 48.8 | 44.5, 56.1 | 24.5 | 17.2, 33.0 | 29.2 | 19.0, 34.3 |
| CD4 count | 1151.5 | 915.0, 1509.0 | 1066.5 | 831.0, 1349.0 | 436.0 | 309.0, 606.0 | 499.5 | 313.0, 631.0 |
| Estradiol (pg/mL) | 44.0 | 34.0, 59.0 | 44.0 | 34.0, 80.0 | 43.0 | 33.5, 57.0 | 37.5 | 27.0, 46.0 |
| FSH (pg/mL) | 5.1 | 4.0, 6.5 | 4.8 | 3.7, 6.1 | 5.6 | 4.8, 7.2 | 6.0 | 4.8, 8.8 |
| Inhibin B (pg/mL) | 86.0 | 37.0, 106.0 | 49.0 | 26.0, 112.0 | 70.0 | 29.0, 103.0 | 68.0 | 36.0, 104.0 |
Table III.
Psychosocial Metrics
| HIV −/No-Dep (n=37) | HIV −/Dep (n=34) | HIV +/No-Dep (n=38) | HIV +/Dep (n=38) | |||||
|---|---|---|---|---|---|---|---|---|
| Median | Q1, Q3 | Median | Q1, Q3 | Median | Q1, Q3 | Median | Q1, Q3 | |
| CESD continuous | 7 | 3, 11 | 22 | 17, 30 | 6 | 3, 8 | 25 | 21, 37 |
| CESD Positive Affect subscale | 12 | 11, 12 | 7 | 6, 9 | 12 | 9, 12 | 4.5 | 3, 7 |
| CESD Somatic subscale | 3 | 2, 6 | 10 | 7, 12 | 3.5 | 1, 5 | 9 | 7, 13 |
| CESD Negative Affect subscale | 1 | 0, 3 | 6 | 5 12 | 0.5 | 0, 2 | 9 | 7, 14 |
| CESD Interpersonal | 0 | 0, 0 | 2 | 0, 3 | 0 | 0, 0 | 2 | 0, 3 |
| Quality of Life Index | 82.9 | 73.0, 86.6 | 62.5 | 54.1, 74.6 | 80.1 | 75.4, 91.1 | 63.4 | 52.2, 72.8 |
3.1.1. FKBP5
Baseline FKBP5 expression was elevated in HIV−/Dep (p=0.0304) as well as HIV+/No-Dep (p=0.0346) groups compared to the HIV−/No-Dep control group, as demonstrated by the reduction in Ct values (Fig 1A). Following Dex stimulation, all groups showed a significant induction of FKBP5; however, the Dex-induced increase in FKBP5 expression occurred to a lesser extent in HIV−/Dep (p=0.0039) relative to the HIV−/No-Dep control group (Fig 1B). The HIV+/Dep group displayed greater Dex-induced FKBP5 compared to the HIV−/Dep group (p=0.0153).
Figure 1.
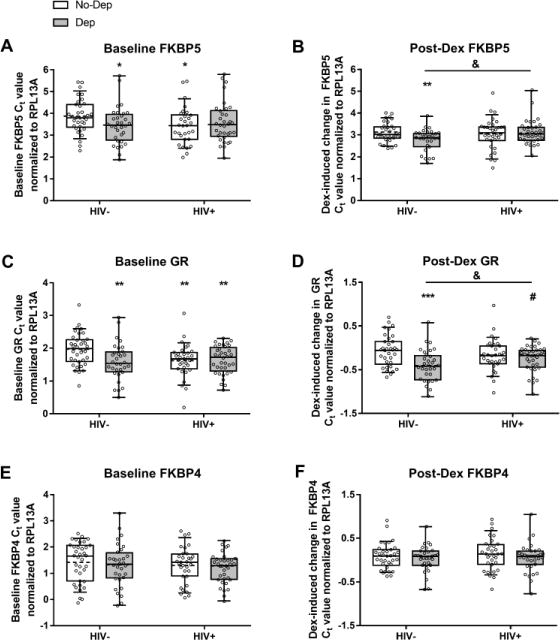
HIV and depressive symptoms are independently associated with changes to GR signaling. Baseline gene expression (dCt) and changes following stimulation with Dex (ddCt) are shown for FKBP5 (A and B), GR (C and D), and FKBP5 (E and F). All individual data points are overlaid on top of the boxplots with the box extending from 25th to 75th percentiles and whiskers representing 10th to 90th percentile. Dashed lines indicate the group mean while solid lines indicate the group median. Refer to Table 3 for representation as fold changes. *, **, *** p<0.05, p<0.01, and p<0.001 compared to the HIV−/No-Dep group. & p<0.05 across the indicated comparison. # p=0.0505 compared to the HIV−/No-Dep group.
3.1.2. GR
Baseline GR expression was higher in HIV−/Dep (p=0.0031), HIV+/No-Dep (p=0.0057), and HIV+/Dep (p=0.0027), groups compared to the HIV−/No-Dep control group, as demonstrated by the lower Ct values (Fig 1C). Following Dex, GR expression was reduced to a greater extent in HIV−/Dep (p=0.0004), and to a marginally greater extent in HIV+/Dep (p=0.0505) relative to the HIV−/No-Dep control group (Fig 1D). The HIV−/Dep group displayed greater Dex-induced reduction in GR compared to the HIV+/Dep group (p=0.0433).
3.1.3. FKBP4
Baseline FKBP4 expression did not differ among groups (Fig 1E). In addition, Dex-induced changes to FKBP4 were also similar among groups (Fig 1F).
3.2. HIV and Depressive Symptoms Independently Predict Reduced Cytokine Responsivity of PBMCs in Response to LPS Challenge
GR sensitivity was measured by the ability of Dex to suppress LPS-stimulated cytokine release. A subset of the total sample were included in these ELISA assessments of IL-6 and TNF-α based on additional sample availability (n=22, HIV−/No-Dep; n=14, HIV−/Dep; n=17, HIV+/No-Dep; and n=21, HIV+/Dep).
3.2.1. IL-6
Following LPS stimulation, IL-6 concentration in cell culture supernatant was lower in HIV−/Dep (p=0.0022) compared to the HIV−/No-Dep control group. The HIV+/Dep group displayed greater LPS-induced IL-6 compared to the HIV−/Dep group (p=0.0121) (Fig 2A). Concurrent stimulation with Dex led to suppression of LPS-stimulated IL-6 levels; however, the extent of IL-6 suppression by Dex was similar among groups (Fig 2B).
Figure 2.
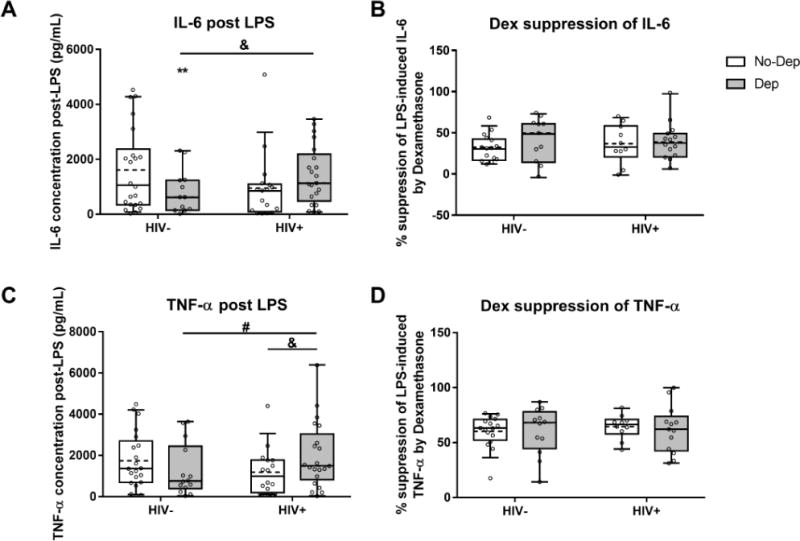
Depressive symptoms are associated with reduced LPS-induced cytokine levels, but not Dex suppression of cytokines. Cytokine concentrations following in vitro LPS treatment and percent suppression of LPS-induced cytokine by co-treatment with Dex are shown for IL-6 (A and B) and TNF-α (C and D). All individual data points are overlaid on top of the boxplots with the box extending from 25th to 75th percentiles and whiskers representing 10th to 90th percentile. Dashed lines indicate the group mean while solid lines indicate the group median. *, ** p<0.05 and p<0.01 compared to the HIV−/No-Dep group. & p<0.05 across the indicated comparison. # p=0.065 across the indicated comparison.
3.2.2. TNF-α
Following LPS stimulation, the HIV+/Dep group displayed significantly greater TNF-α concentration in cell culture supernatant compared to the HIV+/No-Dep group (p=0.0225) and marginally greater TNF-α concentration compared to the HIV−/Dep group (p=0.0650) (Fig 2C). While concurrent stimulation with Dex led to suppression of LPS-stimulated TNF-α levels in all four groups, the extent of TNF- α suppression by Dex was similar among groups (Fig 2D).
4. Discussion
The current study tested the hypothesis that depressive symptoms and HIV infection interact to alter the function of the GR and precipitate an enhanced inflammatory response in PBMCs from women. Our results indicate that while both HIV infection and depressive symptoms are associated with signs of GC resistance, these alterations are not enhanced in women with comorbid HIV infection and depressive symptoms.
The GC signaling profile associated with depressive symptoms closely mimicked that associated with HIV-infection. In the current sample, symptoms of depression were associated with increased baseline FKBP5 expression and impaired FKBP5 induction following stimulation of PBMCs with Dex, consistent with past findings in research participants meeting study criteria for depression (Menke et al 2012). We found that HIV-infection was also associated with increased baseline FKBP5, consistent with past findings in the prefrontal cortex of HIV-infected patients (Tatro et al 2009). In our study, HIV infection was not associated with Dex-induced changes in FKBP5 expression. Consistently, in translational models, HIV-1 transgenic rats do not differ in their FKBP5 response to either acute or repeated stressors compared to wildtype rats (Panagiotakopoulos et al 2015). As previously mentioned, FKBP5 plays a dual role in GC signaling, first as a negative regulator of GR’s nuclear translocation, and second as a transcriptional readout of GR’s activity. An increase in baseline FKBP5 can thus be interpreted as decreased nuclear translocation of GR (Lukic et al 2015), and the downstream consequences of such reduction in GR translocation would be lower Dex-induced transcription of FKBP5, consistent with the notion of impaired GC signaling.
Symptoms of depression and HIV infection each increased baseline GR expression, potentially suggesting a compensatory mechanism aimed at increasing GC signaling. Our finding of increased baseline GR mRNA expression in HIV-infected individuals is consistent with the findings of Norbiato et al (1994) who reported an increase in the number of GR along with decreased affinity of the receptor for GCs in patients with HIV infection who developed acquired GC resistance. However, GR expression on CD14+ monocytes has been demonstrated to be lower in individuals with HIV infection who were or were not undergoing HAART (Renga et al 2012). Therefore, it is likely that GR expression is differentially altered in distinct cellular subsets within PBMCs from PLWH. The available evidence on changes to GR expression in patients diagnosed with depression is less consistent. Changes to GR sensitivity appear to occur in people living with depression in the absence of changes in expression of the receptor itself (Pariante & Miller 2001). In response to GC stimulation, GR downregulates its expression to a small degree (Burnstein et al 1991). Dex-induced downregulation of GR occurred to a greater extent in the HIV−/Dep group compared to the HIV−/No-Dep controls. The HIV+/Dep group also showed a similar trend toward greater Dex-induced downregulation of GR, potentially indicating a mechanism aimed at compensating for the baseline GC resistance (Burnstein et al 1991). A limitation of the current study is the lack of genotype investigation into FKBP5. Known single nucleotide polymorphisms (SNPs) within the FKBP5 gene are associated with vulnerability to and recovery from major depressive disorder (Binder et al 2004, Menke et al 2013). Distinguishing between genetic variants of FKBP5 in future studies may help explain the “restored” FKBP5 profile seen in women with comorbid depressive symptoms and HIV infection.
We found that depressive symptoms were independently associated with a reduction in LPS-stimulated release of the pro-inflammatory cytokine IL-6 compared to the HIV−/No-Dep controls. A recent meta-analysis of studies measuring spontaneous concentrations of cytokines in people living with depression concluded that IL-6 and TNF-α levels were significantly higher in depressed patients compared to non-depressed controls (Dowlati et al 2010). It may therefore appear paradoxical that in the current study patients with depressive symptoms displayed lower LPS-induced cytokines. However, differences between spontaneous versus stimulated inflammatory responses, which reflect the concurrent immune activation and suppression present in depression, may reconcile the current findings with the literature. One study found that while spontaneous IL-6 release by PBMCs was higher in depressed patients, LPS-induced release of IL-6 was significantly lower in patients compared to healthy controls (Humphreys et al 2006). In contrast, HIV infection was not significantly associated with alterations in LPS-induced cytokine release, for which a number of explanations are possible. First, it is worth noting that in the current sample 47% and 29% of the HIV+/No-Dep and HIV+/Dep groups had detectable viral loads. Within these two groups, 65% and 50% percent were on ART, and adherence was around 60%, which creates a spectrum of HIV progression and potentially heterogeneous immune profiles. Previous studies showed that blood mononuclear cells from viremic patients mounted a larger TNF-α induction following stimulation by LPS than cells from virologically suppressed patients (Dutertre et al 2012), a result that was mostly attributable to the subset of CD14+/CD16+ monocytes expressing M-DC8. In addition, ART has been shown to reduce the LPS-stimulated release of IL-6 and TNF-α by monocytes compared to the cytokine response seen in unmedicated HIV+ patients (Amirayan-Chevillard et al 2000). Therefore, some of the variability in the cytokine responses in the current study may be explained by either cellular composition of the patient specimens (e.g. mixed PBMCs versus monocyte subsets) or viral load and disease progression of each patient. Secondly, it is possible that similar to the finding of Humphreys et al (2006) in depressed patients, the immune backdrop caused by HIV infection partially dampens the LPS-stimulated response of cytokines. In fact, endotoxin tolerance upon repeated exposure to LPS was found to occur in HIV-1 transgenic rats, but did not take place in the control F344 strain (Homji et al 2012), supporting the notion of heightened spontaneous, but dampened stimulated inflammation in HIV disease.
We did not find evidence of altered GC sensitivity as measured by Dex suppression of LPS-stimulated cytokine levels. A dose of 10−8 M for Dex, was selected in the current study based on its common utilization for stimulating PBMCs. One possibility is that subtle group differences such as the added impact of a comorbid condition on GC signaling could be elucidated at a lower dose of Dex. In fact, blunted anti-inflammatory function of GR was evidenced in PBMCs from patients with comorbid coronary heart disease and depression when LPS-stimulated response was suppressed by low, but not higher doses of Dex (Nikkheslat et al 2015). Similar to the gene expression experiments, we found that the impact of comorbid HIV infection and depressive symptoms on LPS-stimulated cytokines was non-additive despite similar trends of outcome due to each condition alone. Again, comorbid HIV infection and depressive symptoms were associated with a “normalization” of Dex-induced GR responsivity toward that of the HIV−/No-Dep control group. We propose that a profile similar to that of the healthy controls does not necessarily indicate restoration of healthy biological function in HIV+/Dep women. Corroborating this proposal is the finding that participants with comorbid HIV infection and depressive symptoms displayed lower scores on the Quality of Life index than healthy controls or those with either condition alone (Table III).
One possible explanation for the lack of synergistic effects due to comorbid depression and HIV infection is that immune suppression and activation are present in both depression and HIV infection (Blume et al 2011) may be driving what appears to be normal GC and immune responses that ultimately belies further dysregulation of the HPA-immune system than either condition alone would cause. It is likely that at least a subset of the HPA axis alterations seen independently in either HIV infection or depression signifies shifts in the system to adapt to the disease biology. It then follows that any disease pathology that prevents this potentially “adaptive” shift may in itself be dysfunctional. For example, consequences of repeated immune stimulation such as immune tolerance as evidenced by reduced LPS-stimulated cytokine levels, are generally assumed to be adaptive and beneficial (West & Heagy 2002). However, the comorbid presence of these two conditions may abolish the protective aspects of immune tolerance as the cytokine data presented here suggest.
It has been suggested that GC resistance reflects the disease state rather than the patient’s trait in people with chronic inflammatory conditions such as multiple sclerosis such that the severity and occurrence of GC resistance shift with remission and recurrence of the inflammatory disease (Gold et al 2012). One limitation of the current study is the use of CES-D as a measure of depressive symptoms as it does not distinguish between trait versus state depressive symptoms, which may differentially be associated with fluctuations in inflammatory markers. Depressed patients represent a heterogeneous population in terms of symptoms and inflammatory profile. The CES-D scale can be further divided into the following four subscales: Somatic, Positive affect, Depressed affect, and Interpersonal (Radloff 1977). Importantly, scores on the somatic subscale of the CES-D did not differ among our experimental groups.
Given the established role of the GR in the modulation of inflammation, an understanding of the effects of HIV infection comorbid with depression on GR function will provide insight regarding the degree to which GR dysfunction could contribute to elevated inflammation, and even viral replication, in WLWH. Importantly, future work will be necessary to examine the influence of HIV and depression on GR function in men living with HIV. Studies have attempted to treat HIV replication via targeting the GC system, but results are mixed with some reporting reduced viral replication and others reporting increased viral replication and GC-mediated increases in T cell death (Hapgood & Tomasicchio 2010). The results provided here may provide a means to explain these mixed results in the effect of GCs in HIV-infected patients and a basis for more targeted therapeutic approaches.
Table IV.
Fold Change in Gene Expression
| HIV−/Dep | HIV+/No-Dep | HIV+/Dep | ||
|---|---|---|---|---|
| Baseline expression | FKBP5 | 1.35 (103, 1.78) * | 1.33 (1.02, 1.74) * | 1.22 (0.93, 1.61) |
| GR | 1.29 (1.09, 1.54) ** | 1.26 (1.07, 1.49) ** | 1.25 (1.08, 1.44) ** | |
| FKBP4 | 1.08 (0.83, 1.39) | 1.07 (0.85, 1.36) | 1.18 (0.95, 1.46) | |
| Dex-induced expression | FKBP5 | 0.77 (0.64, 0.92) ** | 0.77 (0.54, 1.08) | 0.97 (0.82, 1.15) & |
| GR | 0.81 (0.72, 0.91) *** | 0.96 (0.85, 1.09) | 0.90 (0.81, 1.00) &# | |
| FKBP4 | 0.99 (0.87, 1.12) | 1.12 (0.98, 1.28) | 1.00 (0.90, 1.11) | |
Gene expression data were normalized to the endogenous control RPL13A, and fold changes were calculated using the 2−ΔΔCt method. Data are presented as estimated mean fold change and 95% CI.
p<0.05,
p<0.01,
p<0.001 compared to the HIV−/No-Dep referent group.
p=0.0505 compared to the HIV−/No-Dep group.
p<0.05 compared to HIV−/Dep.
Highlights.
Depressive symptoms associated with elevated baseline expression of Fkbp5 and Nr3c1.
HIV serostatus associated with elevated baseline expression of Fkbp5 and Nr3c1.
Depressive symptoms associated with reduced LPS-induced release of IL-6 and TNF-α.
Acknowledgments
Funding: Data in this manuscript were collected by the Women’s Interagency HIV Study (WIHS). The WIHS is primarily funded by NIAID, with co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Cancer Institute, and National Institute on Drug Abuse. The contents of this publication are solely the responsibility of the authors and do not represent the official views of the National Institutes of Health (NIH). WIHS (Principal Investigators): UAB-MS WIHS (Mirjam-Colette Kempf and Deborah Konkle-Parker), U01-AI-103401; Atlanta WIHS (Ighovwerha Ofotokun and Gina Wingood), U01-AI-103408; Bronx WIHS (Kathryn Anastos), U01-AI-035004; Brooklyn WIHS (Howard Minkoff and Deborah Gustafson), U01-AI-031834; Chicago WIHS (Mardge Cohen and Audrey French), U01-AI-034993; Metropolitan Washington WIHS (Seble Kassaye), U01-AI-034994; Miami WIHS (Margaret Fischl and Lisa Metsch), U01-AI-103397; UNC WIHS (Adaora Adimora), U01-AI-103390; Connie Wofsy Women’s HIV Study, Northern California (Ruth Greenblatt, Bradley Aouizerat, and Phyllis Tien), U01-AI-034989; WIHS Data Management and Analysis Center (Stephen Gange and Elizabeth Golub), U01-AI-042590; Southern California WIHS (Joel Milam), U01-HD-032632 (WIHS I – WIHS IV). The WIHS is funded primarily by the National Institute of Allergy and Infectious Diseases (NIAID), with additional co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), the National Cancer Institute (NCI), the National Institute on Drug Abuse (NIDA), and the National Institute on Mental Health (NIMH). Targeted supplemental funding for specific projects is also provided by the National Institute of Dental and Craniofacial Research (NIDCR), the National Institute on Alcohol Abuse and Alcoholism (NIAAA), the National Institute on Deafness and other Communication Disorders (NIDCD), and the NIH Office of Research on Women’s Health. WIHS data collection is also supported by UL1-TR000004 (UCSF CTSA), UL1-TR000454 (Atlanta CTSA), and P30-AI-050410 (UNC CFAR). In addition, GNN was funded by K18 MH105098 and P30AI27767.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethical Approval: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- Amirayan-Chevillard N, Tissot-Dupont H, Capo C, Brunet C, Dignat-George F, et al. Impact of highly active anti-retroviral therapy (HAART) on cytokine production and monocyte subsets in HIV-infected patients. Clin Exp Immunol. 2000;120:107–12. doi: 10.1046/j.1365-2249.2000.01201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekhbat M, Rowson SA, Neigh GN. Checks and Balances: the Glucocorticoid Receptor and NFkB in Good Times and Bad. Frontiers in neuroendocrinology. 2017 doi: 10.1016/j.yfrne.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet. 2004;36:1319–25. doi: 10.1038/ng1479. [DOI] [PubMed] [Google Scholar]
- Blume J, Douglas SD, Evans DL. Immune suppression and immune activation in depression. Brain Behav Immun. 2011;25:221–9. doi: 10.1016/j.bbi.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstein KL, Bellingham DL, Jewell CM, Powell-Oliver FE, Cidlowski JA. Autoregulation of glucocorticoid receptor gene expression. Steroids. 1991;56:52–58. doi: 10.1016/0039-128x(91)90124-e. [DOI] [PubMed] [Google Scholar]
- Davies TH, Ning YM, Sanchez ER. A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. The Journal of biological chemistry. 2002;277:4597–600. doi: 10.1074/jbc.C100531200. [DOI] [PubMed] [Google Scholar]
- Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67 doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- Dutertre CA, Amraoui S, DeRosa A, Jourdain JP, Vimeux L, et al. Pivotal role of M-DC8(+) monocytes from viremic HIV-infected patients in TNFalpha overproduction in response to microbial products. Blood. 2012;120:2259–68. doi: 10.1182/blood-2012-03-418681. [DOI] [PubMed] [Google Scholar]
- Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. 2005;5:243–51. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- Gold SM, Sasidhar MV, Lagishetty V, Spence RD, Umeda E, et al. Dynamic development of glucocorticoid resistance during autoimmune neuroinflammation. J Clin Endocrinol Metab. 2012;97:E1402–10. doi: 10.1210/jc.2012-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hapgood JP, Tomasicchio M. Modulation of HIV-1 virulence via the host glucocorticoid receptor: towards further understanding the molecular mechanisms of HIV-1 pathogenesis. Archives of virology. 2010;155:1009–19. doi: 10.1007/s00705-010-0678-0. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Wagner KV, Liebl C, Scharf SH, Wang XD, et al. The involvement of FK506-binding protein 51 (FKBP5) in the behavioral and neuroendocrine effects of chronic social defeat stress. Neuropharmacology. 2012;62:332–9. doi: 10.1016/j.neuropharm.2011.07.041. [DOI] [PubMed] [Google Scholar]
- Hoaglin DC, Iglewicz B. Fine-Tuning Some Resistant Rules for Outlier Labeling. Journal of the American Statistical Association. 1987;82:1147–49. [Google Scholar]
- Homji NF, Mao X, Langsdorf EF, Chang SL. Endotoxin-induced cytokine and chemokine expression in the HIV-1 transgenic rat. J Neuroinflammation. 2012;9:3. doi: 10.1186/1742-2094-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys D, Schlesinger L, Lopez M, Araya AV. Interleukin-6 production and deregulation of the hypothalamic-pituitary-adrenal axis in patients with major depressive disorders. Endocrine. 2006;30:371–6. doi: 10.1007/s12020-006-0016-1. [DOI] [PubMed] [Google Scholar]
- Hunt PW. HIV and inflammation: mechanisms and consequences. Curr HIV/AIDS Rep. 2012;9:139–47. doi: 10.1007/s11904-012-0118-8. [DOI] [PubMed] [Google Scholar]
- Ickovics JR, Hamburger ME, Vlahov D, Schoenbaum EE, Schuman P, et al. Mortality, CD4 cell count decline, and depressive symptoms among HIV-seropositive women: longitudinal analysis from the HIV Epidemiology Research Study. JAMA : the journal of the American Medical Association. 2001;285:1466–74. doi: 10.1001/jama.285.11.1466. [DOI] [PubMed] [Google Scholar]
- Kelso-Chichetto NE, Okafor CN, Cook RL, Abraham AG, Bolan R, Plankey M. Association Between Depressive Symptom Patterns and Clinical Profiles Among Persons Living with HIV. AIDS and behavior. 2017 doi: 10.1007/s10461-017-1822-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci. 2013;16:33–41. doi: 10.1038/nn.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukic I, Mitic M, Soldatovic I, Jovicic M, Maric N, et al. Accumulation of cytoplasmic glucocorticoid receptor is related to elevation of FKBP5 in lymphocytes of depressed patients. J Mol Neurosci. 2015;55:951–8. doi: 10.1007/s12031-014-0451-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke A, Arloth J, Putz B, Weber P, Klengel T, et al. Dexamethasone stimulated gene expression in peripheral blood is a sensitive marker for glucocorticoid receptor resistance in depressed patients. Neuropsychopharmacology. 2012;37:1455–64. doi: 10.1038/npp.2011.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke A, Klengel T, Rubel J, Bruckl T, Pfister H, et al. Genetic variation in FKBP5 associated with the extent of stress hormone dysregulation in major depression. Genes Brain Behav. 2013;12:289–96. doi: 10.1111/gbb.12026. [DOI] [PubMed] [Google Scholar]
- Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65:732–41. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neigh GN, Nemeroff CB. Reduced glucocorticoid receptors: consequence or cause of depression? Trends in endocrinology and metabolism: TEM. 2006;17:124–5. doi: 10.1016/j.tem.2006.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth CL, Bekhbat M, Neigh GN. Neural effects of inflammation, cardiovascular disease, and HIV: Parallel, perpendicular, or progressive? Neuroscience. 2015;302:165–73. doi: 10.1016/j.neuroscience.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikkheslat N, Zunszain PA, Horowitz MA, Barbosa IG, Parker JA, et al. Insufficient glucocorticoid signaling and elevated inflammation in coronary heart disease patients with comorbid depression. Brain Behav Immun. 2015;48:8–18. doi: 10.1016/j.bbi.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Norbiato G, Galli M, Righini V, Moroni M. The syndrome of acquired glucocorticoid resistance in HIV infection. Baillieres Clin Endocrinol Metab. 1994;8:777–87. doi: 10.1016/s0950-351x(05)80300-3. [DOI] [PubMed] [Google Scholar]
- Pace TW, Hu F, Miller AH. Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun. 2007;21:9–19. doi: 10.1016/j.bbi.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagiotakopoulos L, Kelly S, Neigh GN. HIV-1 proteins accelerate HPA axis habituation in female rats. Physiol Behav. 2015;150:8–15. doi: 10.1016/j.physbeh.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- Patterson S, Moran P, Epel E, Sinclair E, Kemeny ME, et al. Cortisol patterns are associated with T cell activation in HIV. PLoS One. 2013;8:e63429. doi: 10.1371/journal.pone.0063429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radloff LS. The CES-D Scale. Applied Psychological Measurement. 1977;1:385–401. [Google Scholar]
- Raghavan A, Rimmelin DE, Fitch KV, Zanni MV. Sex Differences in Select Non-communicable HIV-Associated Comorbidities: Exploring the Role of Systemic Immune Activation/Inflammation. Curr HIV/AIDS Rep. 2017 doi: 10.1007/s11904-017-0366-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renga B, Francisci D, D’Amore C, Schiaroli E, Carino A, et al. HIV-1 infection is associated with changes in nuclear receptor transcriptome, pro-inflammatory and lipid profile of monocytes. BMC Infect Dis. 2012;12:274. doi: 10.1186/1471-2334-12-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Rivera Y, Garcia Y, Toro V, Cappas N, Lopez P, et al. Depression Correlates with Increased Plasma Levels of Inflammatory Cytokines and a Dysregulated Oxidant/Antioxidant Balance in HIV-1-Infected Subjects Undergoing Antiretroviral Therapy. J Clin Cell Immunol. 2014;5 doi: 10.4172/2155-9899.1000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Rivera Y, Vazquez-Santiago FJ, Albino E, Sanchez MD, Rivera-Amill V. Impact of Depression and Inflammation on the Progression of HIV Disease. J Clin Cell Immunol. 2016;7 doi: 10.4172/2155-9899.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soontornniyomkij V, Everall IP, Moore DJ, Gouaux B, Tatro ET, et al. Increased cortical expression of FK506 binding protein-51 in HIV-associated neurocognitive disorders. Journal of neurovirology. 2012;18:313–22. doi: 10.1007/s13365-011-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatro ET, Everall IP, Masliah E, Hult BJ, Lucero G, et al. Differential expression of immunophilins FKBP51 and FKBP52 in the frontal cortex of HIV-infected patients with major depressive disorder. J Neuroimmune Pharmacol. 2009;4:218–26. doi: 10.1007/s11481-009-9146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez AN, Rubin LH, Neigh GN. Untangling the Gordian knot of HIV, stress, and cognitive impairment. Neurobiol Stress. 2016;4:44–54. doi: 10.1016/j.ynstr.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MA, Heagy W. Endotoxin tolerance: a review. Crit Care Med. 2002;30:S64–73. [PubMed] [Google Scholar]