

Abstract
The threat of antibiotic resistant bacterial infections continues to underscore the need for new treatment options. Historically, small molecule metabolites from microbes have provided a rich source of antibiotic compounds, and as a result, significant effort has been invested in engineering the responsible biosynthetic pathways to generate novel analogs with attractive pharmacological properties. Unfortunately, biosynthetic stringency has limited the capacity of non-ribosomal peptide synthetases and polyketide synthases from producing substantially different analogs in large numbers. Another class of natural products, the ribosomally synthesized and post-translationally modified peptides (RiPPs), have rapidly expanded in recent years with many natively displaying potent antibiotic activity. RiPP biosynthetic pathways are modular and intrinsically tolerant to alternative substrates. Several prominent RiPPs with antibiotic activity will be covered in this review with a focus on their biosynthetic plasticity. While only a few RiPP enzymes have been thoroughly investigated mechanistically, this knowledge has already been harnessed to generate new-to-nature compounds. Through the use of synthetic biology approaches, on-going efforts in RiPP engineering hold great promise in unlocking the potential of this natural product class.
Introduction
Despite remarkable medical advances, including improved sanitation, effective vaccines, and antibiotics, bacterial infections remain a serious threat to human health [1]. Annually, over 17 million people succumb to bacterial infections, with an increasing proportion due to antibiotic resistance [1,2]. Therefore, there is urgent and continuous need for new antibiotics. Small molecule metabolites from microbes have been a highly productive source of chemical matter that ultimately led to most of today’s clinically used antibiotics [3–5]. Many of these natural products are derived from polyketide synthase (PKS) or non-ribosomal peptide synthetase (NRPS) families, including well-known antibiotics such as the beta-lactams, tetracyclines, macrolides, and glycopeptides. With the rising resistance to these proven antibiotic classes, alternative sources of antibiotics must be discovered. The ribosomally synthesized and post-translationally modified peptides (RiPPs) have been attracting interest as one such source of untapped potential. Unfortunately, the clinical deployment of RiPPs has been hindered due to issues such as poor solubility and bioavailability despite many displaying potent antibiotic activity and modes of action distinct from currently used drugs. However, the single property that sets RiPPs apart from polyketides and non-ribosomal peptides is their extraordinary biosynthetic malleability. Owing to this, on-going RiPP engineering efforts appear to be poised to mitigate the abovementioned issues [6,7].
RiPP biosynthesis begins with the ribosomal synthesis of a precursor peptide. Most commonly, RiPP precursors are comprised of an N-terminal leader region and a C-terminal core region. The former contains a recognition sequence that is bound directly by a particular biosynthetic protein while the latter receives post-translational modifications to ultimately become the mature RiPP (Figure 1). Through the physical separation of the sites responsible for substrate binding and residues that are modified, RiPP biosynthetic pathways can be highly specific yet promiscuously process highly variable core sequences. Many RiPP classes have been characterized using in vivo and in vitro methods to better understand their substrate tolerance and promiscuity. These studies have provided a starting point for utilizing RiPP biosynthetic enzymes to generate novel compounds to address the rise of antibiotic resistance.
Figure 1.
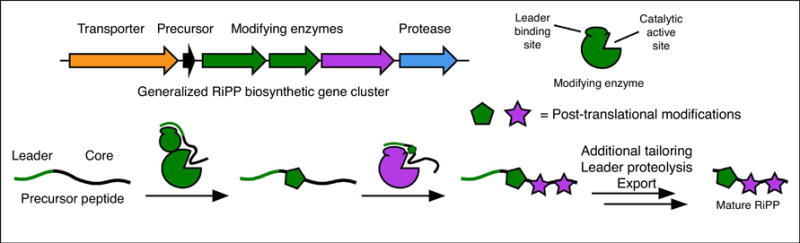
A generalized RiPP biosynthetic gene cluster complete with a precursor peptide, modifying enzymes, and proteolysis/export enzymes. RiPP precursor peptides feature leader and core regions that separate substrate recognition residues from residues that undergo modification.
In this review, several RiPPs with antibiotic activity will be examined. We detail their defining structural features, mode of action, as well as their biosynthetic malleability with an emphasis on how each class might be exploited to create analogs of future biotechnological or medicinal value.
Lasso Peptides
Lasso peptides are topologically interesting RiPPs that feature an isopeptide-linked macrocycle from the N-terminus of the core region to the side chain of a recipient Asp/Glu residue. Lasso peptide uniquely display a catenane-like structure where the C-terminal tail is threaded through the macrocycle and locked into place by disulfide bonds and/or steric interactions (Figure 2) [*8]. Despite their relative simplicity, lasso peptides display a striking array of bioactivities with several notable examples functioning as antibiotics. These include microcin J25, produced by certain strains of Escherichia coli to kill other strains of E. coli and closely related Salmonella strains. Microcin J25 gains entry to the cell by hijacking FhuA, an outer membrane siderophore receptor. Once inside, microcin J25 potently inhibits RNA polymerase. Capistruin, produced by Burkholderia thailandensis, also targets RNA polymerase. Streptomonomicin, isolated from Streptomonospora alba, causes cell lysis in Bacillus sp., especially B. anthracis, and is thought to operate by modulating the activity of the WalR response regular [9]. LP2006, lariatin, and lassomycin all harbor activity towards various Mycobacterium sp., with the latter reported to target the ClpC1 ATPase of Mycobacterium tuberculosis [*8,10–13]. RODEO-enabled genome mining has revealed many more lasso peptides await characterization [13].
Figure 2.
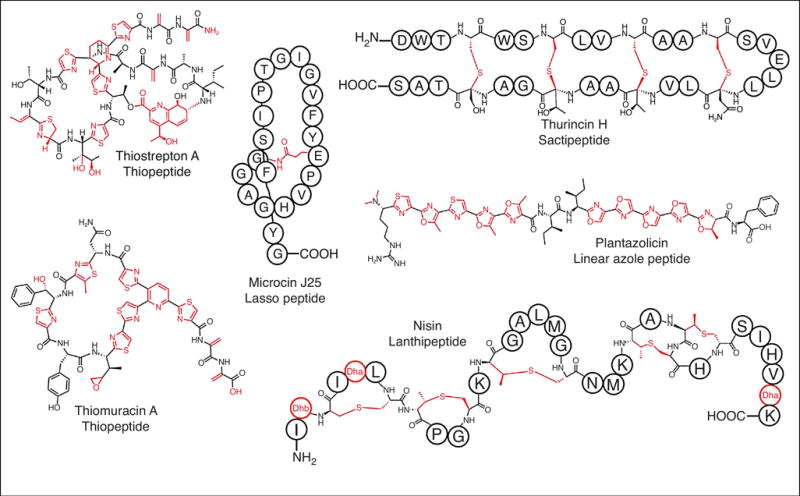
Representative members of several RiPP classes. RiPPs display an array of structural diversity and antimicrobial activity. Sites of post-translational modifications are indicated in red.
Lasso peptide biosynthesis requires a minimum of two enzymes. The first is a transglutaminase homolog that functions as a leader peptidase. The second enzyme is homologous to asparagine synthetase and responsible for forming the threaded macrocycle. [*8,14]. While a detailed mechanism for lasso peptide biosynthesis has yet to be reported, numerous heterologous expression studies suggest that the pathways are extraordinarily tolerant towards alternative substrates. Several studies have modified the precursor peptide and determined that the lasso peptide biosynthetic enzymes tolerate most substitutions, except for residues involved directly in isopeptide bond formation or leader peptide cleavage [15–18]. Lasso peptides may prove to be a useful scaffold for specific chemical biology or biomedical applications.
Plantazolicin
Plantazolicin is a member of the linear azol(in)e-containing peptide class of RiPPs. After extensive spectrum of activity testing, plantazolicin only displayed significant activity towards B. anthracis (Figure 2). Remarkably, plantazolicin lacked activity against other members of the Bacillus cereus group, despite these organisms sharing considerable genetic identity with B. anthracis [19]. Based on a series of detailed genetic, transcriptomic, and fluorescent labeling studies, plantazolicin localizes to cardiolipin-rich lipid microdomains of the plasma membrane. Unusually, B. anthracis appear to accumulate plantazolin:cardiolipin complexes in a discontinuous helix around the longitudinal axis of the cell, rather than at the poles where cardiolipin is usually found in rod-shaped bacteria [20]. Ultimately, plantazolicin causes cell lysis, a feature that is enhanced by addition of exogenous cardiolipin, which contrasts with daptomycin, another cardiolipin-dependent antibiotic [19,21].
As a linear azol(in)e-containing peptide, plantazolicin biosynthesis shares features with one of the best-studied RiPPs, the DNA gyrase inhibitor microcin B17. Knowledge into how peptidic azol(in)e heterocycles are formed was greatly aided from studies on microcin B17 biosynthesis [22–24]. The biosynthesis of plantazolicin involves the cyclodehydration of Cys, Ser, and Thr to yield ten azoline heterocycles, nine of which undergo dehydrogenation to the azole heterocycle. Next, a type II CAAX protease family removes the leader peptide and the newly created N-terminus is dimethylated [22]. Unlike the highly permissive lasso peptide synthases, the plantazolicin heterocycle synthetase features significant stringency. Substrate tolerance was initially investigated using an E. coli heterologous expression system and precursor peptide variants. While this led to detectable generation of some analogs, these were largely limited to amino acid substitutions at sites not naturally cyclized [25]. In a separate study that reconstituted the activity of the purified azol(in) synthetase, it was demonstrated that the enzymes were significantly more promiscuous in vitro, which enabled production of analogs that were not detected through the earlier heterologous expression study [25,26]. Conserved motifs in the leader region, verified by binding studies, and the distance between this recognition sequence and the core peptide played determinant roles in recognition and processing by the cyclodehydratase [26]. The plantazolicin biosynthetic enzymes are somewhat atypical in their stringency for particular positions of the core peptide, which translates to aborted processing when a cyclizable residue is missing. Instead, a more tolerant azol(in) synthetases would likely be more suitable for library generation or targeted modification of custom precursor peptides [**27].
Lanthipeptides
Lanthipeptides are distinguished from other RiPPs by their namesake lanthionine residues, which are thioether crosslinks between a donor Cys and Cβ of a recipient residue (Figure 2) [28,29]. Characterized lanthipeptides with antibiotic activity (lantibiotics) kill Gram-positive bacteria by three possible mechanisms: sequestration of lipid II, pore formation, and binding of phosphatidylethanolamine. Some lantibiotics employ more than one of these mechanisms resulting in synergistic killing and correspondingly low rates of resistance. This is exemplified in nisin, which sequesters lipid II and induces pore formation. Despite wide usage as a food preservative for over 40 years, appreciable resistance to nisin has not been reported (Figure 2) [28,29].
Lanthipeptides are biosynthesized over two steps. First, a lanthipeptide dehydratase will convert certain Ser and Thr residues to dehydroalanine and dehydrobutyrine (Dha/Dhb), respectively. This is accomplished by activating Ser/Thr for dehydration by either a tRNA-dependent glutamylation or ATP-dependent phosphorylation. The activated esters then undergo elimination to yield the Dha/Dhb residues. The second step of lanthionine formation involves a cyclase that carries a 1,4-nucleophilic addition of a Cys thiols onto specific Dha/Dhb residues to form [28,30]. This process generates the lanthionine or methyllanthionine, respectively [29].
Lanthipeptide dehydratases rank among the most substrate tolerant RiPP enzymes characterized to date. This feature is exemplified by the prochlorosins, which have on the order of 30 precursor peptides with hypervariable core regions, each giving rise to a distinct product. Additional, uncharacterized lanthipeptide biosynthetic gene clusters have as many as 80 predicted precursors [31–34]. The promiscuity of lanthipeptide dehydratases has been harnessed in numerous ways by synthetic biologists. One example involves fusing the core regions of lanthipeptide precursors from cryptic gene clusters to the nisin leader peptide to produce novel lantibiotics [35].
Lanthipeptides libraries have been produced to diversify a naturally occurring scaffold with the aim of identifying a novel lanthipeptide with a desired activity. One such study design included the heterologous expression of ProcM (from procholorosin biosynthesis) in E. coli along with a library of ProcA precursor peptide variants. This effort yielded a library of 106 distinct lanthipeptides. Detected among these variants was a new protein-protein interaction inhibitor for the HIV p6 protein and the human tumor susceptibility gene 101, a lynchpin interaction for viral budding [*36]. Phage display has also been employed to rapidly generate lanthipeptide analogs which can be screened for desired activity. Two independent studies have demonstrated that upon fusion of a lanthipeptide precursor library to the M13 phage coat protein pIII, along with co-expression of a substrate-tolerant lanthipeptide synthetase, new lanthipeptide activities can be identified [37,**38]. This led to the discovery of new ligands for urokinase plasminogen activator and streptavidin [37]. In addition to phage display, yeast display has been used in a similar manner to diversify lacticin 481, resulting in a αvβ3 integrin-binding variant [*38]. While phage and yeast display methods have not yet been employed to generate lanthipeptides with new antibiotic modes of action, we anticipate that future efforts will be aimed squarely on this objective.
Thiopeptides
Thiopeptides are macrocyclic RiPPs that feature azol(in)e heterocycles, Dha/Dhb residues, and a class-defining six-membered N-heterocycle [39]. Many thiopeptides also feature either a quinaldic- or indolic acid-containing secondary macrocycle, C-terminal amidation, methylation, oxidation, and glycosylation (Figure 2) [39]. The unique confluence of functional groups found in other RiPPs grants the thiopeptides a special designation of being a natural “hybrid” RiPP.
Thiopeptides primarily exert antibiotic activity by inhibiting protein synthesis with few exceptions: the cyclothiazomycins are reported to inhibit RNA polymerase while lactazole purportedly lacks antibiotic activity [40,41]. Of the majority that interrupt protein synthesis, the size of the primary macrocycle correlates with the physical target of the thiopeptide. Those with 29-membered macrocycles target elongation factor Tu while those with 26- and 32-membered macrocycles bind to the protein L11 and 23S rRNA interface within the 50S ribosomal subunit [39]. Thiopeptides typically display nanomolar activity against pathogenic Firmicutes, such as Staphylococcus aureus, Enterococcus faecalis, and Clostridium difficile. Semi-synthetic derivatization has been successful in fine-tuning thiopeptide spectrum of activity. For example, a derivative of GE2270A has been reported to be highly selective towards Propionibacterium acnes [*42]. Despite their potent activity, thiopeptides suffer from poor aqueous solubility which has hampered their clinical development. Separate semi-synthetic efforts have converted GE2270A into a significantly more soluble derivative, LFF571, with efficacy comparable to vancomycin in treating C. difficile infections [43]. Despite these successes, very little is known regarding the structure-activity relationships within the thiopeptide class. Therefore, intense efforts have been undertaken to understand their biosynthesis and to leverage this knowledge towards analog generation.
The first step of thiopeptide biosynthesis is identical to the linear azol(in)e-containing peptide class. For thiopeptide substrates, the cyclodehydratase primarily acts upon Cys residues but to a lesser extent, Ser/Thr residues are found in the form of (methyl)oxazoline heterocycles. Azolines are typically dehydrogenated to the corresponding azole although thiazolines are observed in mature thiopeptides [14,**27,44]. Secondly, the azole-bearing intermediate is acted upon by a tRNA-dependent lanthipeptide-like dehydratase which installs Dha/Dhb from Ser/Thr via a glutamylation and elimination reaction sequence. At this point, two Dha residues, one presumably present as the iminol tautomer, undergo a formal [4+2] cycloaddition to generate the six-membered nitrogenous heterocycle. In the case of pyridine synthases, the leader peptide is ejected as a C-terminal carboxamide while in dehydropiperidine synthases, the leader region is proteolytically removed during a later maturation step [**27,44]. While these core biosynthetic steps are common to all thiopeptides, additional tailoring distinguishes the thiopeptides into a diverse set of subclasses [45].
The biosynthetic promiscuity of thiopeptides has primarily been investigated through in vivo expression of reprogrammed precursor peptides with thiocillin and thiostrepton being the most studied [46–49]. Thiopeptide precursor variants tolerated by the full array of maturation enzymes are observable in culture extracts by mass spectrometry. To summarize a multitude of efforts in this area, thiopeptide biosynthetic pathways display varying permissiveness on a per-residue basis with only pyridine/dehydropiperidine-incorporated Ser being universally indispensable for successful analog production. Thiopeptide precursor peptides with substitutions, insertions, or deleted amino acids have produced variants with altered peripheral properties and macrocycle size to investigate structure-activity relationships [47–49,*50]. For example, Ala-substituted and contracted/expanded variants of thiocillin have revealed that certain residues of the core are crucial for bioactivity while others are readily substituted. Further, it has been noted that the rigidity imparted by azole heterocycles is crucial for adopting an active conformation [48,49].
In vitro reconstitution has enabled a more intimate study of each transformation during thiopeptide biosynthesis while overcoming the greater biosynthetic stringency encountered in vivo [**27,44,45,*51,52]. Most notably, the thiomuracin thiazole synthetase appears to only require an ~8 residue recognition sequence to facilitate heterocyclization of virtually any core Cys residue [**27]. Additionally, it has been demonstrated that the thiocillin and thiomuracin [4+2] cycloaddition enzymes are remarkably permissive toward variable core sequences and only require the two Dha residues involved in cycloaddition and a small portion of the leader peptide [**27,*51,53]. Owing to their promiscuity, thiopeptide biosynthetic enzymes harbor the potential to generate analogs with enhanced properties or unnatural functional groups for the de novo generation of new RiPPs.
Sactipeptides
The hallmark modification of a sactipeptide is a sactionine modification, whose name derives from a thioether that is sulfur-to-alpha carbon linked (Figure 2). Only five sactipeptides have been characterized of which all demonstrate a narrow spectrum of activity towards various human pathogens such as C. difficile, Listeria monocytogenes, and B. cereus [54–58]. While there have been limited studies regarding the mode of action of sactipeptides, thuricin CD and subtilosin A disrupt the cell membrane [54]. Sporulation killing factor, a sactipeptide produced by B. subtilis, induces lysis in neighboring B. subtilis as a scavenging strategy under starvation conditions [59].
Sactionines are installed by a radical S-adenosylmethionine enzyme which mediates hydrogen atom abstraction from an acceptor residue which is then covalently linked to the sulfur of a Cys donor [55]. Sactionine linkages in known sactipeptides are exclusively formed between the most N-terminal Cys and the most C-terminal acceptor and radiate inward, resulting in rigid hairpin-like structures (Figure 2) [55]. While there is limited data regarding the biosynthetic promiscuity of these enzymes, the subtilosin synthase exhibits relaxed specificity in both the position of installed sactionines as well as the identity of acceptor residues when heterologously expressed in E. coli [**60]. These results suggest that sactionine synthases may be sufficiently substrate tolerant to enable sactionine formation in unnatural contexts to facilitate broader scale production of custom sactipeptides, although further enzyme characterization is required to identify the limits of substrate tolerance.
New-to-Nature Hybrid RiPPs
One motivation for studying RiPP biosynthesis is that such knowledge could be leveraged for specific applications in synthetic biology. The past decade has greatly enhanced our knowledge of substrate recognition and tolerance in various RiPP classes (Figure 3) and it has become apparent that many RiPP enzymes require only a nominal recognition sequence (4-10 residues) and sufficient length between this sequence and the core region to facilitate recruitment and catalysis [**27,**61,62]. This concept has been harnessed to generate new-to-nature “hybrid RiPPs” that employ enzymes from disparate RiPP classes to introduce sets of modifications that are not known to naturally coexist. By rationally designing precursor peptides, bearing in mind recognition sequences, substrate tolerances, and enzyme kinetics, this approach generated hybrid RiPPs decorated with thiazolines with (methyl)lanthionines, sactionines, and D-amino acids (Figure 4) [**61]. To rationally and more broadly apply this approach to other RiPP classes will require a significant investment in the characterization of RiPP biosynthetic pathways that remain understudied. Considerations such as substrate recognition, specificity, and tolerance must all be taken into account not only the context of a particular modification, but also how each successive modification impacts the competence for subsequent modification. Layers of complexity are also added when choosing a means of production. For instance, in vivo expression offers several conveniences, such as circumventing the need to purify proteins and also a potentially robust access to library generation. However, in vivo expression requires a suitable host and creates a more complex environment for detecting the molecules that were successfully produced. Conversely, in vitro reconstitution requires each biosynthetic protein to be soluble, stable, and active after purification. There is little doubt that library generation using an in vitro reconstitution strategy would be considerably more labor intensive; however, the ability to control the concentration of reactants and other reaction parameters typically translates to greater substrate tolerance in vitro as opposed to in vivo. We expect both approaches will be fruitful, with the choice being dependent on one’s experimental objectives.
Figure 3.
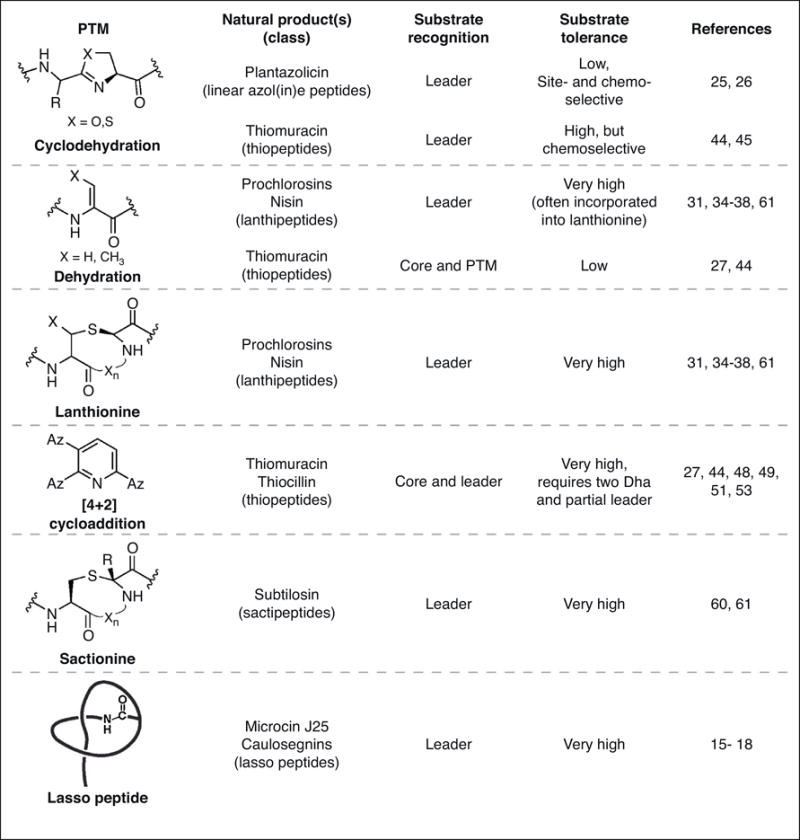
Post-translational modifications (PTMs), mode of substrate recognition, and substrate tolerance of various RiPP enzymes covered in this review. RiPP biosynthetic enzymes display a spectrum of substrate tolerances with the most tolerant enzymes likely being the best-suited for applications in synthetic biology. Az = azole.
Figure 4.
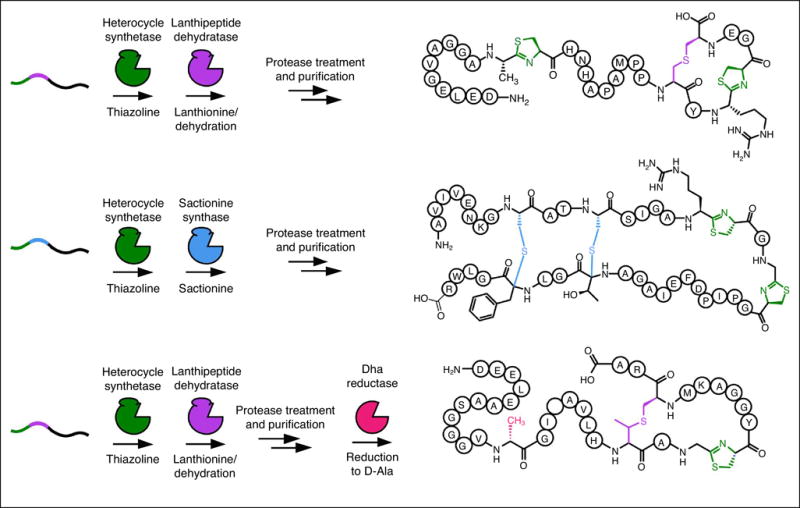
New-to-nature hybrid RiPPs generated through RiPP engineering. By leveraging knowledge of substrate recognition from various RiPP classes, chimeric precursor peptides have been designed to recruit biosynthetic enzymes not naturally found in the same context. The resulting hybrid RiPPs have functional groups not currently found together in any natural product such as thiazolines/lanthionines, thiazolines/sactionines, and thiazolines/lanthionines/D-Ala; the latter is generated by enzymatic reduction of Dha.
Summary and Outlook
RiPP natural products are structurally diverse molecules often displaying potent antimicrobial properties. If properly developed, RiPPs could contribute new scaffolds to drug discovery programs with the future potential of offsetting the rise of clinically problematic antibiotic-resistant infections. Unlike other biosynthetic paradigms (e.g., NRPS and PKS), RiPP biosynthetic enzymes typically display high levels of substrate tolerance. This biosynthetic promiscuity is attributed to a short recognition sequence being localized in a leader region of the precursor peptide which is later removed during maturation. This specific-yet-tolerant substrate processing ability endows RiPPs with unprecedented engineering potential. The community of RiPP researchers is just beginning to produce novel molecules with desired properties. We envision a future where preexisting knowledge of substrate recognition and tolerance are brought to bear on the custom design and diversification of RiPPs. Such efforts could be guided by a desire to rationally design a particular RiPP scaffold or to bind a specific target, perhaps one that is critical for a pathogenic bacterium. Encouraged by the past several years of successful RiPP research, we remain confident this class of natural products will continue to provide useful molecules and enzymes that serve the greater scientific community.
Acknowledgments
We would like to thank Christopher J. Schwalen and Xiaorui Guo for critical reading of the manuscript. We also acknowledge the U.S. National Institutes of Health (GM097142) and the David and Lucile Packard Fellowship for Science and Engineering for funding our work on RiPP biosynthesis and engineering.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
The authors declare no competing financial interests.
References
- 1.Martens E, Demain AL. The antibiotic resistance crisis, with a focus on the United States. J Antibiot. 2017;70:520. doi: 10.1038/ja.2017.30. [DOI] [PubMed] [Google Scholar]
- 2.Laxminarayan R, Amábile-Cuevas CF, Cars O, Evans T, Heymann DL, Hoffman S, Holmes A, Mendelson M, Sridhar D, Woolhouse M, et al. UN High-Level Meeting on antimicrobials—what do we need? Lancet. 2016;8:218–220. doi: 10.1016/S0140-6736(16)31079-0. [DOI] [PubMed] [Google Scholar]
- 3.Katz L, Baltz RH. Natural product discovery: past, present, and future. J Ind Microbiol Biotechnol. 2016;43:155–176. doi: 10.1007/s10295-015-1723-5. [DOI] [PubMed] [Google Scholar]
- 4.Newman DJ, Cragg GM. Natural Products as Sources of New Drugs from 1981 to 2014. J Nat Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 5.Ganesan A. The impact of natural products upon modern drug discovery. Curr Opin Chem Biol. 2008;12:306–317. doi: 10.1016/j.cbpa.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 6.Weissman KJ. Genetic engineering of modular PKSs: from combinatorial biosynthesis to synthetic biology. Nat Prod Rep. 2016;33:203–230. doi: 10.1039/c5np00109a. [DOI] [PubMed] [Google Scholar]
- 7.Winn M, Fyans JK, Zhuo Y, Micklefield J. Recent advances in engineering nonribosomal peptide assembly lines. Nat Prod Rep. 2016;33:317–347. doi: 10.1039/c5np00099h. [DOI] [PubMed] [Google Scholar]
- 8*.Hegemann JD, Zimmermann M, Xie X, Marahiel MA. Lasso peptides: an intriguing class of bacterial natural products. Acc Chem Res. 2015;48:1909–1919. doi: 10.1021/acs.accounts.5b00156. A comprehensive review of lasso peptides with a focus on their biosynthesis and physicochemical properties. Prominenantly discussed is the antibiotic lasso peptide microcin J25; the biosynthetic enzymes from this natural product are a paragon for the promiscuity of lasso peptide synthases. [DOI] [PubMed] [Google Scholar]
- 9.Metelev M, Tietz JI, Melby JO, Blair PM, Zhu L, Livnat I, Severinov K, Mitchell DA. Structure, bioactivity, and resistance mechanism of streptomonomicin, an unusual lasso Peptide from an understudied halophilic actinomycete. Chem Biol. 2015;22:241–250. doi: 10.1016/j.chembiol.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delgado MA, Rintoul MaR, Farías RN, Salomón RA. Escherichia coli RNA Polymerase Is the Target of the Cyclopeptide Antibiotic Microcin J25. J Bacteriol. 2001;183:4543–4550. doi: 10.1128/JB.183.15.4543-4550.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vincent PA, Delgado MA, Farías RN, Salomón RA. Inhibition of Salmonella enterica serovars by microcin J25. FEMS Microbiol Lett. 2004;236:103–107. doi: 10.1016/j.femsle.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 12.Semenova E, Yuzenkova Y, Peduzzi J, Rebuffat S, Severinov K. Structure-Activity Analysis of Microcin J25: Distinct Parts of the Threaded Lasso Molecule Are Responsible for Interaction with Bacterial RNA Polymerase. J Bacteriol. 2005;187:3859–3863. doi: 10.1128/JB.187.11.3859-3863.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tietz JI, Schwalen CJ, Patel PS, Maxson T, Blair PM, Tai H-C, Zakai UI, Mitchell DA. A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat Chem Biol. 2017;13:470. doi: 10.1038/nchembio.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA. A prevalent peptide-binding domain guides ribosomal natural product biosynthesis. Nat Chem Biol. 2015;11:564–570. doi: 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zimmermann M, Hegemann Julian D, Xie X, Marahiel Mohamed A. The Astexin-1 Lasso Peptides: Biosynthesis, Stability, and Structural Studies. Chem Biol. 2013;20:558–569. doi: 10.1016/j.chembiol.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 16.Hegemann JD, Zimmermann M, Xie X, Marahiel MA. Caulosegnins I–III: A Highly Diverse Group of Lasso Peptides Derived from a Single Biosynthetic Gene Cluster. J Am Chem Soc. 2013;135:210–222. doi: 10.1021/ja308173b. [DOI] [PubMed] [Google Scholar]
- 17.Pavlova O, Mukhopadhyay J, Sineva E, Ebright RH, Severinov K. Systematic Structure-Activity Analysis of Microcin J25. J Biol Chem. 2008;283:25589–25595. doi: 10.1074/jbc.M803995200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan SJ, Rajniak J, Maksimov MO, Link AJ. The role of a conserved threonine residue in the leader peptide of lasso peptide precursors. ChemComm. 2012;48:1880–1882. doi: 10.1039/c2cc17211a. [DOI] [PubMed] [Google Scholar]
- 19.Molohon KJ, Blair PM, Park S, Doroghazi JR, Maxson T, Hershfield JR, Flatt KM, Schroeder NE, Ha T, Mitchell DA. Plantazolicin Is an Ultranarrow-Spectrum Antibiotic That Targets the Bacillus anthracis Membrane. ACS Infect Dis. 2016;2:207–220. doi: 10.1021/acsinfecdis.5b00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mileykovskaya E, Dowhan W. Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim Biophys Acta. 2009;1788:2084–2091. doi: 10.1016/j.bbamem.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang T, Muraih JK, Tishbi N, Herskowitz J, Victor RL, Silverman J, Uwumarenogie S, Taylor SD, Palmer M, Mintzer E. Cardiolipin prevents membrane translocation and permeabilization by daptomycin. J Biol Chem. 2014;289:11584–11591. doi: 10.1074/jbc.M114.554444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burkhart BJ, Schwalen CJ, Mann G, Naismith JH, Mitchell DA. YcaO-Dependent Posttranslational Amide Activation: Biosynthesis, Structure, and Function. Chem Rev. 2017;117:5389–5456. doi: 10.1021/acs.chemrev.6b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li YM, Milne JC, Madison LL, Kolter R, Walsh CT. From peptide precursors to oxazole and thiazole-containing peptide antibiotics: microcin B17 synthase. Science. 1996;274:1188–1193. doi: 10.1126/science.274.5290.1188. [DOI] [PubMed] [Google Scholar]
- 24.Yorgey P, Lee J, Kordel J, Vivas E, Warner P, Jebaratnam D, Kolter R. Posttranslational modifications in microcin B17 define an additional class of DNA gyrase inhibitor. Proc Natl Acad Sci U S A. 1994;91:4519–4523. doi: 10.1073/pnas.91.10.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deane CD, Melby JO, Molohon KJ, Susarrey AR, Mitchell DA. Engineering unnatural variants of plantazolicin through codon reprogramming. ACS Chem Biol. 2013;8:1998–2008. doi: 10.1021/cb4003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deane CD, Burkhart BJ, Blair PM, Tietz JI, Lin A, Mitchell DA. In vitro biosynthesis and substrate tolerance of the plantazolicin family of natural products. ACS Chem Biol. 2016;11:2232–2243. doi: 10.1021/acschembio.6b00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27**.Zhang Z, Hudson GA, Mahanta N, Tietz JI, van der Donk WA, Mitchell DA. Biosynthetic Timing and Substrate Specificity for the Thiopeptide Thiomuracin. J Am Chem Soc. 2016;138:15511–15514. doi: 10.1021/jacs.6b08987. This study revealed previously unknown properties regarding substrate recognition, biosynthetic timing, and catalytic promiscuity within the thiopeptide class. This paper lays the groundwork for harnessing this system to generate thiopeptide analogs in an informed manner. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Repka LM, Chekan JR, Nair SK, van der Donk WA. Mechanistic Understanding of Lanthipeptide Biosynthetic Enzymes. Chem Rev. 2017;117:5457–5520. doi: 10.1021/acs.chemrev.6b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q, Yang X, Wang H, van der Donk WA. High Divergence of the Precursor Peptides in Combinatorial Lanthipeptide Biosynthesis. ACS Chem Biol. 2014;9:2686–2694. doi: 10.1021/cb500622c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li B, Sher D, Kelly L, Shi Y, Huang K, Knerr PJ, Joewono I, Rusch D, Chisholm SW, van der Donk WA. Catalytic promiscuity in the biosynthesis of cyclic peptide secondary metabolites in planktonic marine cyanobacteria. Proc Natl Acad Sci U S A. 2010;107:10430–10435. doi: 10.1073/pnas.0913677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oman TJ, van der Donk WA. Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nat Chem Biol. 2010;6:9–18. doi: 10.1038/nchembio.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y, Zhang Q, van der Donk WA. Insights into the evolution of lanthipeptide biosynthesis. Protein Sci. 2013;22:1478–1489. doi: 10.1002/pro.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cubillos-Ruiz A, Berta-Thompson JW, Becker JW, van der Donk WA, Chisholm SW. Evolutionary radiation of lanthipeptides in marine cyanobacteria. Proc Natl Acad Sci U S A. 2017;114:E5424–E5433. doi: 10.1073/pnas.1700990114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Heel AJ, Kloosterman TG, Montalban-Lopez M, Deng J, Plat A, Baudu B, Hendriks D, Moll GN, Kuipers OP. Discovery, Production and Modification of Five Novel Lantibiotics Using the Promiscuous Nisin Modification Machinery. ACS Synthetic Biology. 2016;5:1146–1154. doi: 10.1021/acssynbio.6b00033. [DOI] [PubMed] [Google Scholar]
- 36*.Yang X, Lennard KR, He C, Walker MC, Ball AT, Doigneaux C, Tavassoli A, van der Donk WA. A lanthipeptide library used to identify a protein-protein interaction inhibitor. Nat Chem Biol. doi: 10.1038/s41589-018-0008-5. accepted for publication. The wide substrate tolerance of a lanthipeptide synthase was harnessed to generate a library of variants. This yielded a custom lanthipeptide that could block the interaction between HIV p6 protein and human TSG101, a crucial interaction for viral budding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urban JH, Moosmeier MA, Aumüller T, Thein M, Bosma T, Rink R, Groth K, Zulley M, Siegers K, Tissot K, et al. Phage display and selection of lanthipeptides on the carboxy-terminus of the gene-3 minor coat protein. Nat Commun. 2017;8:1500. doi: 10.1038/s41467-017-01413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38**.Hetrick KJ, Walker MC, van der Donk WA. Development and Application of Yeast and Phage Display of Diverse Lanthipeptides. ACS Cent Sci. accepted for publication. This paper demonstrated the ability to rapidly diversify lanthipeptide scaffolds using both phage display and yeast display with the latter being previously unused for RiPPs. These platforms were leveraged to generate novel lanthipeptide compounds, some with bioactivity not seen in naturally occuring lanthipeptides. [Google Scholar]
- 39.Just-Baringo X, Albericio F, Álvarez M. Thiopeptide Antibiotics: Retrospective and Recent Advances. Mar Drugs. 2014;12:317. doi: 10.3390/md12010317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayashi S, Ozaki T, Asamizu S, Ikeda H, Omura S, Oku N, Igarashi Y, Tomoda H, Onaka H. Genome mining reveals a minimum gene set for the biosynthesis of 32-membered macrocyclic thiopeptides lactazoles. Chem Biol. 2014;21:679–688. doi: 10.1016/j.chembiol.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 41.Hashimoto M, Murakami T, Funahashi K, Tokunaga T, Nihei K, Okuno T, Kimura T, Naoki H, Himeno H. An RNA polymerase inhibitor, cyclothiazomycin B1, and its isomer. Bioorg Med Chem. 2006;14:8259–8270. doi: 10.1016/j.bmc.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 42*.Fabbretti A, He C-G, Gaspari E, Maffioli S, Brandi L, Spurio R, Sosio M, Jabes D, Donadio S. A Derivative of the Thiopeptide GE2270A Highly Selective against Propionibacterium acnes. Antimicrob Agents Chemother. 2015;59:4560–4568. doi: 10.1128/AAC.05155-14. Semi-synthetic derivatization is used to tune the spectrum of activity of potent thiopeptide antibiotic GE2270A to be selectively active against P. acnes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mullane K, Lee C, Bressler A, Buitrago M, Weiss K, Dabovic K, Praestgaard J, Leeds JA, Blais J, Pertel P. Multicenter, randomized clinical trial to compare the safety and efficacy of LFF571 and vancomycin for Clostridium difficile infections. Antimicrob Agents Chemother. 2015;59:1435–1440. doi: 10.1128/AAC.04251-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hudson GA, Zhang Z, Tietz JI, Mitchell DA, van der Donk WA. In Vitro Biosynthesis of the Core Scaffold of the Thiopeptide Thiomuracin. J Am Chem Soc. 2015;137:16012–16015. doi: 10.1021/jacs.5b10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahanta N, Zhang Z, Hudson GA, van der Donk WA, Mitchell DA. Reconstitution and Substrate Specificity of the Radical S-Adenosyl-methionine Thiazole C-Methyltransferase in Thiomuracin Biosynthesis. J Am Chem Soc. 2017;139:4310–4313. doi: 10.1021/jacs.7b00693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li C, Zhang F, Kelly WL. Heterologous production of thiostrepton A and biosynthetic engineering of thiostrepton analogs. Mol BioSyst. 2011;7:82–90. doi: 10.1039/c0mb00129e. [DOI] [PubMed] [Google Scholar]
- 47.Zhang F, Kelly WL. Saturation Mutagenesis of TsrA Ala4 Unveils a Highly Mutable Residue of Thiostrepton A. ACS Chem Biol. 2015;10:998–1009. doi: 10.1021/cb5007745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bowers AA, Acker MG, Koglin A, Walsh CT. Manipulation of Thiocillin Variants by Prepeptide Gene Replacement: Structure, Conformation, and Activity of Heterocycle Substitution Mutants. J Am Chem Soc. 2010;132:7519–7527. doi: 10.1021/ja102339q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bowers AA, Acker MG, Young TS, Walsh CT. Generation of Thiocillin Ring Size Variants by Prepeptide Gene Replacement and In Vivo Processing by Bacillus cereus. J Am Chem Soc. 2012;134:10313–10316. doi: 10.1021/ja302820x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50*.Tran HL, Lexa KW, Julien O, Young TS, Walsh CT, Jacobson MP, Wells JA. Structure– Activity Relationship and Molecular Mechanics Reveal the Importance of Ring Entropy in the Biosynthesis and Activity of a Natural Product. J Am Chem Soc. 2017;139:2541–2544. doi: 10.1021/jacs.6b10792. The promiscuity of thiocillin biosynthetic enzymes were used to generate a library of variants with some exhibiting increased potency. This study also revealed that post-translational modifications in thiopeptides have a powerful effect on reducing conformational space and that this entropic effect strongly influences bioactivity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wever WJ, Bogart JW, Baccile JA, Chan AN, Schroeder FC, Bowers AA. Chemoenzymatic Synthesis of Thiazolyl Peptide Natural Products Featuring an Enzyme-Catalyzed Formal [4 + 2] Cycloaddition. J Am Chem Soc. 2015;137:3494–3497. doi: 10.1021/jacs.5b00940. This paper presents a platform for diversifying thiopeptide scaffolds that exploits the promiscuity and efficiency of thiopeptide [4+2] cycloaddition enzymes. By harnessing solid phase peptide synthesis to generate substrate that is otherwise inaccessible and subsequently employing [4+2] cycloaddition enzymes to complete the complex transformation, this study developed a facile route for diversifying thiopeptide scaffolds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Z, Mahanta N, Hudson GA, Mitchell DA, van der Donk WA. Mechanism of a Class C Radical S-Adenosyl-l-methionine Thiazole Methyl Transferase. J Am Chem Soc. 2017;139:18623–18631. doi: 10.1021/jacs.7b10203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wever WJ, Bogart JW, Bowers AA. Identification of Pyridine Synthase Recognition Sequences Allows a Modular Solid-Phase Route to Thiopeptide Variants. J Am Chem Soc. 2016;138:13461–13464. doi: 10.1021/jacs.6b05389. [DOI] [PubMed] [Google Scholar]
- 54.Mathur H, Fallico V, O’Connor PM, Rea MC, Cotter PD, Hill C, Ross RP. Insights into the Mode of Action of the Sactibiotic Thuricin CD. Front Microbiol. 2017;8:696. doi: 10.3389/fmicb.2017.00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mathur H, Rea MC, Cotter PD, Hill C, Ross RP. The sactibiotic subclass of bacteriocins: an update. Curr Protein Pept Sci. 2015;16:549–558. doi: 10.2174/1389203716666150515124831. [DOI] [PubMed] [Google Scholar]
- 56.Wang G, Manns DC, Guron GK, Churey JJ, Worobo RW. Large-Scale Purification, Characterization, and Spore Outgrowth Inhibitory Effect of Thurincin H, a Bacteriocin Produced by Bacillus thuringiensis SF361. Probiotics Antimicrob Proteins. 2014;6:105–113. doi: 10.1007/s12602-014-9159-1. [DOI] [PubMed] [Google Scholar]
- 57.Rea MC, Sit CS, Clayton E, O’Connor PM, Whittal RM, Zheng J, Vederas JC, Ross RP, Hill C. Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc Natl Acad Sci U S A. 2010;107:9352–9357. doi: 10.1073/pnas.0913554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shelburne CE, An FY, Dholpe V, Ramamoorthy A, Lopatin DE, Lantz MS. The spectrum of antimicrobial activity of the bacteriocin subtilosin A. J Antimicrob Chemother. 2007;59:297–300. doi: 10.1093/jac/dkl495. [DOI] [PubMed] [Google Scholar]
- 59.González-Pastor JE, Hobbs EC, Losick R. Cannibalism by Sporulating Bacteria. Science. 2003;301:510–513. doi: 10.1126/science.1086462. [DOI] [PubMed] [Google Scholar]
- 60**.Himes PM, Allen SE, Hwang S, Bowers AA. Production of Sactipeptides in Escherichia coli: Probing the Substrate Promiscuity of Subtilosin A Biosynthesis. ACS Chem Biol. 2016;11:1737–1744. doi: 10.1021/acschembio.6b00042. The first paper to use heterologous expression of a sactipeptide biosynthetic pathway in E. coli to efficiently explore a library of subtilosin A variants. This revealed that sactisynthases are remarkably tolerant of changes to both location of crosslinks and identity of acceptor residue, including unnatural amino acids, and thus paved the way for engineering this burgeoning class of RiPP. [DOI] [PubMed] [Google Scholar]
- 61**.Burkhart BJ, Kakkar N, Hudson GA, van der Donk WA, Mitchell DA. Chimeric Leader Peptides for the Generation of Non-Natural Hybrid RiPP Products. ACS Cent Sci. 2017;3:629–638. doi: 10.1021/acscentsci.7b00141. A first-in-class study that leveraged over a decade of RiPP investigations to rationally design chimeric precursor peptides and exploit the biosynthetic promisuity inherent in RiPP enzymes. This unprecedented paradigm united RiPP funcitonal groups in the same molecule that naturally never co-occur and lays a groundwork for rapidly generating synthetic, new-to-nature hybrid RiPPs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khusainov R, Moll GN, Kuipers OP. Identification of distinct nisin leader peptide regions that determine interactions with the modification enzymes NisB and NisC. FEBS Open Bio. 2013;3:237–242. doi: 10.1016/j.fob.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]