

Abstract
Hypercapnia, the elevation of CO2 in blood and tissues, commonly occurs in severe acute and chronic respiratory diseases, and is associated with increased risk of mortality. Recent studies have shown that hypercapnia adversely affects innate immunity, host defense, lung edema clearance and cell proliferation. Airway epithelial dysfunction is a feature of advanced lung disease, but the effect of hypercapnia on airway epithelium is unknown. Thus, in the current study we examined the effect of normoxic hypercapnia (20% CO2 for 24 h) vs normocapnia (5% CO2), on global gene expression in differentiated normal human airway epithelial cells. Gene expression was assessed on Affymetrix microarrays, and subjected to gene ontology analysis for biological process and cluster-network representation. We found that hypercapnia downregulated the expression of 183 genes and upregulated 126. Among these, major gene clusters linked to immune responses and nucleosome assembly were largely downregulated, while lipid metabolism genes were largely upregulated. The overwhelming majority of these genes were not previously known to be regulated by CO2. These changes in gene expression indicate the potential for hypercapnia to impact bronchial epithelial cell function in ways that may contribute to poor clinical outcomes in patients with severe acute or advanced chronic lung diseases.
Introduction
Hypercapnia, the elevation of CO2 in blood and tissue, commonly occurs in patients with acute respiratory failure and chronic pulmonary disorders1–3. It has long been recognized that hypercapnia is associated with an increased risk of death in chronic obstructive pulmonary disease (COPD)4–8. Other studies have shown that hypercapnia is an independent risk factor for mortality in adults with community-acquired pneumonia9, children with adenoviral pneumonia10, cystic fibrosis patients awaiting lung transplantation11, and patients receiving mechanical ventilation for acute respiratory distress syndrome (ARDS)12.
Recent research has shed light on a variety of mechanisms by which hypercapnia may adversely affect clinical outcomes in patients with lung disease. We and others have shown that elevated CO2 inhibits LPS-induced expression of IL-6 and TNF in macrophages, fibroblasts and alveolar epithelial cells13,14. We also found that hypercapnia impairs bacterial clearance through inhibition of both phagocytosis and autophagy in macrophages13,15. Moreover, hypercapnia worsened lung injury and decreased bacterial clearance in mechanically ventilated rats with E. coli pneumonia16 and increased the mortality of Pseudomonas pneumonia in mice17. In the latter study, elevated CO2 decreased the early release of TNF, IL-6 and multiple chemokines into the lung, inhibited bacterial phagocytosis and NADPH-oxidase-mediated reactive oxygen species generation by lung neutrophils, and increased bacterial loads in the lungs and other organs17. Hypercapnia was also shown to inhibit alveolar fluid clearance in the rat lung, which was due to downregulation of Na,K-ATPase activity caused by endocytosis of the enzyme due to activation of AMP-activated kinase and PKC-zeta18,19. Additionally, elevated CO2 inhibited proliferation of alveolar epithelial cells and lung fibroblasts, which resulted from mitochondrial dysfunction triggered by mirR-183-dependent downregulation of isocitrate mitochondrial dehydrogenase 2 (IDH2)20. Thus, hypercapnia adversely affects innate immune responses, host defense, lung edema clearance, and proliferation of cells required for lung repair. Notably, in almost all of the foregoing studies, elevated CO2 produced these effects independently of acidosis.
While the above studies have focused on the impact of hypercapnia on macrophages, neutrophils, fibroblasts, and alveolar epithelial cells, how elevated CO2 affects bronchial epithelial cells has not previously been investigated. The airway epithelium is the first line of defense against inhaled pathogens and other noxious agents, and its integrity is critical for host defense and maintenance of lung homeostasis21,22. Also, diseases associated with hypercapnia including COPD, asthma, cystic fibrosis and ARDS are all characterized by airway epithelial dysfunction23–26. Thus, in the current study we examined the effect of hypercapnia on global gene expression in normal human bronchial epithelial (NHBE) cells that were differentiated at the air-liquid interface (ALI). We show that exposure for 24 h to normoxic hypercapnia (20% CO2), as opposed to normocapnia (5% CO2) downregulates genes linked to innate immunity, host defense, and nucleosome assembly and upregulates genes required for cholesterol biosynthesis and lipid metabolism. These changes in gene expression indicate the potential for hypercapnia to alter bronchial epithelial cell function in ways that may contribute to poor clinical outcomes in patients with severe acute or advanced chronic lung diseases.
Results
Differential gene expression induced by hypercapnia
To determine whether elevated CO2 induces transcriptional changes in airway epithelium, NHBE cells differentiated at ALI were exposed to hypercapnia (20% CO2) for 24 h or maintained in normocapnia (5% CO2) as a control, and global gene expression was analyzed using Affymetrix GeneChip Hybridization. Microarray global gene expression analysis revealed that hypercapnia significantly modified the expression of 309 genes ≥±1.4 fold (expressed as log2 [fold change]) with an adjusted P value ≤ 0.05. This represents only 1.5% of the 20,390 transcripts represented on the Affymetrix chip, indicating that the impact of elevated CO2 on gene expression is highly selective. The proportion of genes whose expression was significantly upregulated (126) or downregulated (183) in response to high CO2 is depicted in Fig. 1a. Differential gene expression is also represented as a volcano plot of log2 (fold change ratio) vs. −log10 (P values) (Fig. 1b) and as a heat map with hierarchical clustering (Supplementary Fig. 1a). The names and fold-changes for all genes downregulated or upregulated by ≥1.4 fold are listed in Supplementary Tables 1,2, respectively. These transcriptional changes cannot be attributed to cytotoxicity since exposure to 20% CO2 for 24 h caused no increase in LDH release from the cells (Supplementary Fig. 2).
Figure 1.

Hypercapnia induces transcriptional changes in NHBE cells. Global gene expression was assessed in ALI-differentiated NHBE cells after exposure to 5% CO2 (normocapnia) or 20% CO2 (hypercapnia) for 24 h. (a) Pie chart indicating proportion of genes downregulated or upregulated by hypercapnia. (b) Volcano plot showing statistical significance (−log10 [P value]) plotted against log2 fold change for hypercapnia vs normocapnia. Plot indicates significantly upregulated genes (log2 [fold change]≥+0.5, adjusted P value ≤ 0.05) in red and downregulated genes (log2 [fold change]≤−0.5, adjusted P value ≤ 0.05) in blue. (c) Bars represent the top 10 GO biological processes downregulated (blue) and upregulated (red) by high CO2.
Biological processes targeted by hypercapnia
Gene Ontology (GO) analysis showed that for hypercapnia-downregulated genes, the most enriched process was nucleosome assembly (Fig. 1c and Supplementary Fig. 1b), which includes multiple histone genes (Supplementary Fig. 1b). Other hypercapnia-downregulated processes include chemotaxis, cell surface receptor signaling, response to LPS, defense responses to bacteria, and cell adhesion (Fig. 1c). Specific CO2-downregulated genes associated with these processes are listed in Supplementary Fig. 1b. Among hypercapnia-upregulated genes, the most enriched processes involved lipid metabolism including cholesterol, isoprenoid, fatty acid and steroid biosynthesis, as well as oxidation-reduction, and negative regulation of translation initiation (Fig. 1c). Specific CO2-upregulated genes associated with some of these processes are listed in Supplementary Fig. 1b.
GO biological process-associated gene clusters targeted by hypercapnia
Major clusters from hypercapnia-downregulated genes are linked to immune response, nucleosome assembly, cell differentiation, oxidation reduction, and ion and lipid transport (Fig. 2). Clusters from upregulated genes induced by high CO2 (Fig. 3) involve biological processes related to lipid metabolism, cholesterol biosynthesis, signal transduction, and transport. A number of these important clusters, labelled A-E in Figs 2 and 3, are analyzed in more detail in the following sections. Their corresponding gene lists are depicted in Figs 4,5, and Supplementary Fig. 3.
Figure 2.

Networks of GO biological processes downregulated by hypercapnia. Gene clusters associated with GO biological processes containing five or more hypercapnia-downregulated genes and their intra- and inter-cluster connections, as determined by unbiased analysis using Mathematica® v11.2.
Figure 3.

Networks of GO biological processes upregulated by hypercapnia. Gene clusters associated with GO biological processes containing five or more hypercapnia-upregulated genes and their intra- and inter-cluster connections, as determined by unbiased analysis using Mathematica® v11.2.
Figure 4.

Hypercapnia alters expression of genes involved in innate immunity and host defense. ALI-differentiated NHBE cells were exposed to normocapnia (NC) or hypercapnia (HC) for 24 h prior to analysis. (a) Cluster A genes altered by hypercapnia and their associated GO biological processes. (b) Heatmap and hierarchical clustering of gene expression profiles in normocapnia and hypercapnia. (c) CXCL1, CXCL14, CCL28, IL6R, and TLR4 mRNA expression levels were assessed by qPCR and expression in hypercapnia was expressed as fold change relative to normocapnia. Results shown are means ± SE; n = 3. (d) Representative fluorescence micrographs of NHBE cells double-stained for TLR4 (red) and the cilia marker acetylated tubulin (green), and counter stained with Hoechst (blue). Non immune control (NIC) cells stained without primary antibodies. (e) Immunoblotting of whole cell lysates for TLR4. Histogram shows densitometry of TLR4 normalized to β-actin (loading control). Results shown are means ± SE; n = 3.
Figure 5.
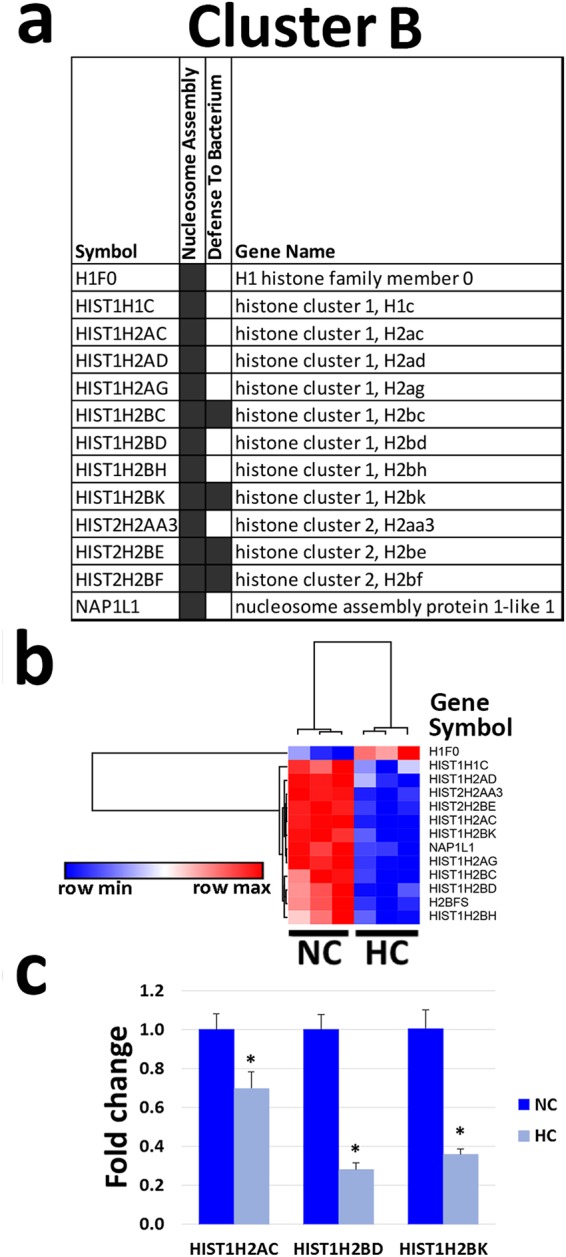
Hypercapnia alters expression of genes involved in nucleosome assembly. ALI-differentiated NHBE cells were exposed to normocapnia (NC) or hypercapnia (HC) for 24 h prior to analysis. (a) Cluster B genes altered by hypercapnia and their associated GO biologic processes. (b) Hierarchical clustering of the gene expression profiles in normocapnia and hypercapnia. (c) HIST1H2AC, HIST1H2BD and HIST1H2BK mRNA expression assessed by qPCR and hypercapnia was expressed as fold change relative to normocapnia. Results shown are means ± SE; n = 3.
Hypercapnia differentially regulates genes associated with innate immunity and nucleosome assembly
Cluster A, represented in Fig. 4a, includes hypercapnia-regulated genes involved in signal transduction, immune and inflammatory responses, and leukocyte chemotaxis. Notably, TLR4, multiple chemokines (CCL28, CXCL1, CXCL2, CXCL6, and CXCL14) and the IL-6 receptor gene (IL6R) were all downregulated by elevated CO2. On the other hand, the IL-1 receptor like 1 gene (IL1RL1) was upregulated by hypercapnia. To validate the microarray results related to CO2-induced changes in key immunoregulatory genes, expression of CXCL1, CXCL14, CCL28, ILR6 and TLR4 was also assessed by qPCR. We found that these genes were all downregulated at levels similar to those in the microarray analysis (Fig. 4c). Indeed, the degree of CO2-induced downregulation of these transcripts assessed by qPCR and microarray was highly correlated (r2 = 0.7981).
In addition, to determine whether downregulation of a key immunoregulatory transcript by hypercapnia was accompanied by a similar change in protein expression, we assessed expression of TLR4 protein in differentiated NHBE cells. Immunofluorescence microscopy (Fig. 4d) and immunoblotting (Fig. 4e) both showed that exposure to 20% CO2 for 24 h decreased NHBE cell expression of TLR4 protein. Full-length blots are included in Supplementary Fig. 4. Taken together, these results suggest that hypercapnia would suppress airway epithelial innate immune response to microbial pathogens and other inflammatory stimuli.
Next, we analyzed cluster B, which includes hypercapnia-regulated genes that codify proteins involved in nucleosome assembly (Fig. 5a). The heat map in Fig. 5b shows that hypercapnia downregulates genes encoding multiple family members of the core histones H2A and H2B27, as well as the nucleosome assembly protein 1-like 1 (NAP1L1), which regulates protein complex assembly, chromosome organization and DNA metabolism. The only upregulated gene in cluster B is H1F0, encoding histone H1, which is normally expressed in terminally differentiated and slowly dividing cells. To validate the microarray data from cluster B, we performed qPCR for selected transcripts whose expression was significantly altered in the microarray analysis. Figure 5c shows that expression of the histone genes HIST1H2AC, HIST1H2BD, and HIST1H2BK was downregulated by hypercapnia as assessed by qPCR, again similar to the microarray results.
Other gene clusters impacted by hypercapnia
Cluster C (Supplementary Fig. 3) includes CO2-regulated genes associated with cholesterol biosynthesis (DHCR7, FDFT1, HMGCS1, IDI1) and transport (ABCA1, ABCC3, ABCC5), other lipid metabolism (ALDH3B1, ALDH3B2, ALOX15B, FADS1–2), and oxidation reduction (several members of the cytochrome P450 family). Cluster D (Supplementary Fig. 3) includes hypercapnia-regulated genes involved in transcription (EGR3, JUN, RBM14) and DNA repair (BCCIP, CUL4A, RMB14). Genes in cluster E (Supplementary Fig. 3) regulate cell differentiation (CADM1, NRP2, NOTCH2NL and others), cell surface receptor signaling (EGFR, IFNAR1, IL6R and others) and apoptosis (BCL2L15, DAPL1, SEMA6A and others). The impact of elevated CO2 on expression of these genes would be expected to alter epithelial metabolism and barrier function, as well as innate immune and inflammatory responses.
Discussion
To our knowledge, the present study is the first to investigate the impact of hypercapnia on global gene expression in airway epithelial cells. Of importance, we utilized primary NHBE cells cultured at ALI to achieve a differentiated state closely resembling normal human bronchial epithelium. Our principal finding was that hypercapnia altered expression of a small number of specific genes (309 out of 20,390 transcripts assayed, or 1.5%) in differentiated NHBE cells. Of these, 183 genes (59%) were downregulated, while 126 (41%) were upregulated. Thus, the effects of elevated CO2 are highly selective, involving both differential repression and differential activation of specific gene subsets. The overwhelming majority of these genes were not previously known to be regulated by CO2. Furthermore, gene ontogeny analysis showed enrichment of hypercapnia-regulated genes involved in a variety of fundamentally important cellular processes. Altering expression of genes related to these processes would be expected to impart functional changes in bronchial epithelial cells, which could in turn influence the pathophysiology and outcomes of many respiratory diseases.
Our data show that hypercapnia alters expression of multiple components of the innate immune system, including downregulation of the IL-6 receptor (IL6R); the neutrophil chemokines CXCL1, CXCL2 and CXCL628; the mucosal-associated chemokines CCL28 and CXCL1429–31 and importantly TLR4. Hypercapnia also upregulated CD55 and CD86, which bind virus at the cell surface32,33. While hypercapnia downregulated TLR4, it increased the expression of IL1RL1, which has been shown to inhibit TLR4 activation34. Of note, Schneberger et al. previously reported that hypercapnia reduced LPS-induced secretion of IL-6 and IL-8 in the airway epithelial cell line BEAS-2B35. These observations are relevant because of the well-documented role of TLR4 in host defense against multiple respiratory pathogens36–40. Interestingly, airway epithelial TLR4 expression was reduced in patients with severe COPD as compared to those with less severe COPD41, possibly due to hypercapnia in patients with advanced disease. Reduced expression of immune response genes was also seen in the lungs of newborn mice exposed to moderate hypercapnia (8% CO2) for the first two weeks of life42. While the immune genes downregulated by hypercapnia in the newborn mice differed from those we found in NHBE cells, the mucosal immunity chemokine CXCL1443 was commonly downregulated in both systems. Taken together, these observations indicate that the airway epithelium is an important target for hypercapnic suppression of innate immune gene expression. This, along with the suppressive effects of elevated CO2 on macrophage, neutrophil, alveolar epithelial cell functions13–15,17–19 likely contributes to the deleterious impact of elevated CO2 on lung injury and host defense.
Another cluster impacted by hypercapnia includes genes related to nucleosome assembly, which also have antibacterial properties. The nucleosome consists of 145–147 base-pair-segments of DNA wrapped around a histone octamer containing one (H3–H4)2 tetramer, two H2A–H2B dimers, and histone chaperones or linkers that facilitate nucleosome assembly44. Regulation of nucleosome assembly following DNA replication, DNA repair and gene transcription is critical for the maintenance of genome stability and epigenetic information44. Within this gene cluster, hypercapnia downregulated transcripts for the core histones H2A and H2B27, the histone chaperone NAP1L145, and the linker histone H127. Downregulation of histone gene expression can be triggered by DNA-damage or indirect inhibition of DNA synthesis46 and might lead to alterations of chromatin structure that would influence transcriptional regulation of many genes and even genome stability47. Exchange of core histones with histone variants might also alter the chemical nature and physical properties of the nucleosome, thereby affecting distinct cellular processes48. In addition, histones H2A and H2B also can inactivate endotoxin and function as antimicrobial proteins49,50.
We also found that elevated CO2 upregulated NHBE cell expression of cholesterol and fatty acid biosynthesis genes, while downregulating ATP-binding cassette (ABC) transporters, which promote the efflux of cholesterol and phospholipids from the cell51. Interestingly, enveloped viruses subvert preexisting lipids for viral entry and trafficking and also reprogram lipid synthesis and lipid distribution in lipid rafts to establish an optimal environment for their replication, assembly and egress52. Furthermore, host defense against viral infection involves interferon-mediated downregulation of sterol biosynthesis53. Thus, hypercapnia-induced cholesterol accumulation might contribute to the entry, replication, and shedding of respiratory viruses in the airways.
As noted above, in a previous study, we showed that hypercapnia downregulates the TCA cycle enzyme IDH2, resulting in mitochondrial dysfunction and impaired proliferation of fibroblasts and A549 lung epithelial cells20. However, in the current study, hypercapnia did not alter IDH2 expression in NHBE cells, indicating that CO2-mediated regulation of gene expression is cell-type-specific. On the other hand, a number of genes involved in mitochondrial function were regulated by hypercapnia in NHBE cells. Among these, upregulated genes included acyl-CoA dehydrogenase short/branched chain (ACADSB) and acyl-CoA synthetase short chain family member 2 (ACSS2), which encode enzymes involved in fatty acid synthesis and oxidation54. Genes downregulated by elevated CO2 included gamma-butyretaine hydroxlase 1 (BBOX 1), which catalyzes synthesis of L-carnitine, an essential co-factor in beta-oxidation55; kynurenine 3-monooxygenase (KMO), an outer mitochondrial membrane protein that hydroxylates tryptophan to form kynurenine56; BCL2 interacting protein 3 (BNIP3), a BH3 domain protein with pro-apoptotic activity57; and mitochondrial assembly of ribosomal large subunit 1 (MALSU1), an inhibitor of translation at the mitochondrial ribosome58. The diverse activities of these genes indicate the potential for hypercapnia to disrupt multiple mitochondrial functions in NHBE cells.
While the current study does not reveal the molecular mechanism(s) underlying hypercapnia-induced changes in gene transcription, other recent work suggests a path to elucidating components of a putative CO2-induced signaling pathway leading to inhibition of innate immune gene expression and impaired host defense. We previously reported that elevated CO2 inhibits expression of antimicrobial peptide genes and suppresses antibacterial host defense in Drosophila59, suggesting that the immunosuppressive effect of hypercapnia is evolutionarily conserved. Using a genome-wide RNAi screen, we identified a small number of genes whose expression is required for CO2-induced immunosuppression in Drosophila cells, and which are conserved in mammalian systems60. Flies deficient in of one these genes, a zinc finger homeodomain transcription factor known as zfh2, were protected from CO2-induced mortality associated with bacterial infection60. This opens up the opportunity to test whether orthologues of zfh2 and other genes identified in the Drosophila screen mediate hypercapnic immunosuppression in mice and ultimately in humans.
Alterations in expression of innate immune and other genes in airway epithelial cells may be of central importance in the CO2-induced increase in mortality of Pseudomonas pneumonia we previously observed in mice17. In addition, the suppressive effect of elevated CO2 on immune gene expression in the airway epithelium, along with similar effects on immune cells, suggest a reason why severe COPD and other lung disease associated with hypercapnia all carry a high risk of pulmonary infection. Bacterial and viral infections are a principal cause of acute COPD exacerbations61–64, which are linked to the need for hospitalization and to mortality65,66. Thus, CO2-induced alterations in airway epithelial gene expression may underlie the increase in mortality associated with hypercapnia in advanced COPD, as well as community-acquired pneumonia9, adenoviral lung infections10 and cystic fibrosis11. It is notable in this regard that reducing hypercapnia with noninvasive ventilatory support has been shown to decrease hospital readmissions and mortality in patients with severe COPD67,68. Further investigation of the molecular mechanisms and mediators of CO2 effects on gene expression may reveal targets for pharmacologic intervention to prevent hypercapnic immune suppression in patients with advanced respiratory disease.
Methods
Primary Normal Human Bronchial Epithelial Cells
Primary NHBE cells isolated from airways of humans without known lung disease were obtained from a commercial source (Lonza). The cells were plated on collagen-coated plastic dishes, grown to confluence in BEGMTM Bronchial Epithelial Cell Growth Medium (Lonza), and passaged after enzyme dissociation with trypsin69. Cells from passage-3 were seeded onto 24-mm, 0.4 μm pore size, polyester, transwell inserts (Corning) at 0.5 × 106 cells per insert (4.67 cm2) and cultured in a serum-free medium70, comprised of 1:1 mixture of BEBM (Lonza): DMEM (Mediatech), supplemented with hydrocortisone (0.5 μg/ml), insulin (5 μg/ml), transferrin (10 μg/ml), epinephrine (0.5 μg/ml), triiodothyronine (6.5 ng/ml), epidermal growth factor (0.5 ng/ml), retinoic acid (50 nM), bovine pituitary extract (0.4%), gentamycin (50 μg/ml), and amphotericin B (50 ng/ml). After the cells reached confluence in submersion culture, the medium above the inserts was removed and the cells were maintained in ALI culture for two more weeks, at which point differentiation to a pseudostratified mucociliary epithelium with characteristics of airway epithelium in vivo was established69,71. Differentiation after ∼2 wk on ALI culture was confirmed by the presence of beating cilia and mucus production, as previously described72. Culture of NHBE cells up to the point of full differentiation was carried out in an atmosphere of humidified 5% CO2/95% air at 37 °C.
Hypercapnia Exposure
After differentiation, NHBE cells were cultured in ALI for an additional 24 h in humidified 20% CO2/21% O2/59% N2 (hypercapnia) or maintained in humidified 5% CO2/95% air (5% CO2/20% O2/75% N2; normocapnia), as control. The growth medium was pre-saturated with appropriate CO2 concentration for 4 h prior to the addition to the cells. The PCO2 and pH of the pre-saturated media were measured using a pHOx Plus Blood Gas Analyzer (Nova Biomedical Corp). For the normocapnia- and hypercapnia-equilibrated media, the PCO2s were 44 and 112 mmHg, and the corresponding pH values were 7.4 and 7.1 respectively.
Cytotoxicity Assay
To determine whether hypercapnia induces cytotoxicity, lactate dehydrogenase (LDH) release to the apical and basolateral compartments was assessed using a colorimetric Cytotoxicity Detection Kit (Roche) according to the manufacturer’s instructions. Absorbance at 490 nm was measured using a VersaMax Tunable Microplate Reader (Molecular Devices). Percent LDH release was calculated as the amount of LDH measured in the basolateral supernatant or apical wash divided by the total amount of LDH in the culture (LDH in cell lysates plus that measured in apical and basolateral compartments) times 100.
RNA Isolation and Affymetrix GeneChip Hybridization
Total RNA was isolated using the RNeasy Mini kit (Qiagen). Quality and quantity of each RNA sample were assessed using a 2100 BioAnalyzer (Agilent). RNA was hybridized to GeneChip® Human Genome U133 2.0 Plus Array (Affymetrix). A total of 6 chips, each hybridized to a cRNA from different normocapnic (n = 3) or hypercapnic (n = 3) NHBE cell cultures were used in this study. The U133 2.0 Plus Arrays contain probes for approximately 56,921 transcripts and variants, including over 45,000 well characterized human genes. Fluorescent images were detected in a GeneChip® Scanner 3000 and expression data were extracted using the GeneChip Operating System v 1.2 (Affymetrix).
Microarray Data Analysis
Differential gene expression between normocapnia and hypercapnia was assessed by a statistical linear model analysis using the BioConductor package limma73,74 (https://www.bioconductor.org/help/faq/), in which an empirical Bayes method is used to moderate the standard errors of the estimated log-fold changes of gene expression. The moderated t-statistic p-values derived from the limma analysis were further adjusted for multiple testing by Benjamini and Hochberg’s method75 to control false discovery rate (FDR). Many genes whose expression signals were below background were defined as “absent”. Transcripts absent in all samples were filtered out, leaving 54,675 probes corresponding to 20,390 genes in the downstream analysis. The lists of differentially expressed genes were obtained by the FDR criteria of <0.05 and fold-change cutoff of >1.4. Differential gene expression in hypercapnia versus normocapnia was depicted in a pie chart, volcano plot of statistical significance (−log10 P value) plotted against log2 fold change, and hierarchical clustering by Pearson correlation represented as heat maps generated using Heatmapper76 and Gene-E (https://software.broadinstitute.org/GENE-E/).
Over representation analysis (ORA) of gene ontology (GO) terms from biological processes of all genes downregulated or upregulated by hypercapnia were separately analyzed using the Gene Ontology Analysis InnateDB tool77 which utilizes a manually-curated knowledgebase of genes, proteins, interactions and signaling pathways involved in mammalian innate immune responses. Results from the Innate DB analysis were confirmed using GeneGo Metacore (Thomson Reuter), a separately curated database and pathway analysis tool. Microarray data have been deposited to the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/projects/geo) complied with MIAME standards (accession number GSE110362).
Network Ontology Analysis
Subsequent analysis of global expression changes and ontology network assessment on the differentially selected genes was performed using Mathematica® v11.2 (Wolfram Research, Inc., Mathematica, Version 11.2, Champaign, IL (2017)). Ontology groups were generated using inbuilt GenomeData, matching the annotated genes with pre-defined processes and intracellular functions. Two approaches were used for analysis of genome wide expression changes: unbiased measurements of intra-network gene expression and fold-change ranked segmentation. Unbiased intra-network changes were assessed for cellular processes that contained at least five genes in the post-screen data. Mean-fold change, the variance of the fold-change, and Pearson correlation of expression were measured for each process. Intra-network heterogeneity of relative expression was measured by calculating the standard deviation of the relative expression for genes within any given ontological process. For instance, if a gene was classified as belonging to both “Nucleosome Assembly” and “Signal Transduction”, it was assigned to both groups and a connection between these processes was indicated. To further understand the impact of hypercapnia-induced differential gene expression, cluster domains of GO biological processes containing 5 or more genes and with at least 4 connections were also generated using Mathematica® v11.2. These processes were broadly grouped based on gene function and by their connections.
Quantitative TaqMan Real-time RT-PCR
Total RNA was isolated from NHBE cells and first-strand cDNA was generated using MultiScribe™ MuLV reverse transcriptase (Applied Biosystems). The first-strand cDNA was used to quantitate the mRNA levels by TaqMan real-time PCR system (Applied Biosystems). The level of expression of eukaryotic translation elongation factor 1 alpha 1 (EEF1A1) was used as reference, and fold change of target genes was calculated by the ∆∆CT method78.
Immunofluorescence Staining for TLR4
After exposure to normocapnia (5% CO2) or hypercapnia (20% CO2) for 24 h, differentiated NHBE cells were fixed with ice-cold 50% acetone/50% methanol for 5 min. Cells were blocked in PBS containing 2% BSA and 0.1% triton X-100 then double-stained with 1:200 polyclonal rabbit anti-human TLR4 antibody (H-80, Santa Cruz Biotechnology) followed by 1:200 Alexa Fluor 555-conjugated goat-anti-rabbit IgG (red) (Invitrogen), and 1:500 monoclonal mouse anti-human acetylated tubulin antibody (Clone 6–11B-1, Sigma) followed by 1:200 Alexa fluor 488-conjugated goat anti-mouse IgG (green) (Invitrogen). Nuclei were identified by staining with 1 µg/ml Hoescht (blue) (Sigma). Images were obtained using a Nikon TE200 inverted fluorescence microscope (Nikon) equipped with a SPOT RT Monochrome Digital Camera (Diagnostic Instruments). All images were captured with the same gain and exposure time using Metamorph software.
Immunoblotting for TLR4
After exposure to normocapnia (5% CO2) or hypercapnia (20% CO2) for 24 h, differentiated NHBE cells were lysed in RIPA buffer (Santa Cruz Biotechnology) supplemented with PMSF, sodium orthovanadate and protease inhibitor cocktail. Lysate proteins (30 μg/well) were resolved by SDS/PAGE 4–20% gradient gels and transferred to nitrocellulose (Bio-Rad Laboratories). Membranes were probed with polyclonal rabbit anti-human TLR4 (H-80) antibody followed by HRP-conjugated anti-rabbit secondary antibody (Pierce). Blots were stripped and re-probed with monoclonal mouse anti-human β-actin (Abcam) followed by HRP-conjugated anti-mouse secondary antibody (Pierce) to confirm the equal loading. The signals were detected using enhanced chemiluminescence SuperSignal West Dura Substrate kit (Pierce). TLR4/βactin ratios were assessed using ImageJ79.
Statistical analysis
Data are presented as means ± SE. Differences between two groups were assessed using Student’s t test. Levene’s test was used to analyze the homogeneity of variances. Significance was accepted at p < 0.05.
Electronic supplementary material
Acknowledgements
This work was supported by National Institutes of Health grants R01 HL107629, R56 HL131745, R01 HL131745 and R01 HL085534; and by a Merit Review from the Department of Veterans Affairs.
Author Contributions
N.W. and P.H.S.S. designed the experiments. N.W., M.C.N. and A.N. performed the experiments. S.M.C.M., P.T.R. and H.M. performed the bioinformatics analysis. S.M.C.M., N.W., P.T.R., H.M. and P.H.S.S. analyzed and interpreted the data. I.S. G.J.B. and J.I.S. contributed reagents or analytic tools, S.M.C.M. and P.H.S.S. wrote the manuscript. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
S. Marina Casalino-Matsuda and Naizhen Wang contributed equally.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-32008-x.
References
- 1.Miniño, A. M., Xu, J. & Kochanek, K. D. Deaths: Preliminary Data for 2008. National Vital Statistics Reports59 1–72 http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_02.pdf (2010). [PubMed]
- 2.Sheikh HS, Tiangco ND, Harrell C, Vender RL. Severe hypercapnia in critically ill adult cystic fibrosis patients. J Clin Med Res. 2011;3:209–212. doi: 10.4021/jocmr612w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laffey JG, Kavanagh BP. Carbon dioxide and the critically ill–too little of a good thing? Lancet. 1999;354:1283–1286. doi: 10.1016/S0140-6736(99)02388-0. [DOI] [PubMed] [Google Scholar]
- 4.Moser KM, Shibel EM, Beamon AJ. Acute respiratory failure in obstructive lung disease. Long-term survival after treatment in an intensive care unit. JAMA. 1973;225:705–707. [PubMed] [Google Scholar]
- 5.Martin TR, Lewis SW, Albert RK. The prognosis of patients with chronic obstructive pulmonary disease after hospitalization for acute respiratory failure. Chest. 1982;82:310–314. doi: 10.1378/chest.82.3.310. [DOI] [PubMed] [Google Scholar]
- 6.Goel A, Pinckney RG, Littenberg B. APACHE II predicts long-term survival in COPD patients admitted to a general medical ward. Journal of general internal medicine. 2003;18:824–830. doi: 10.1046/j.1525-1497.2003.20615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groenewegen KH, Schols AM, Wouters EF. Mortality and mortality-related factors after hospitalization for acute exacerbation of COPD. Chest. 2003;124:459–467. doi: 10.1378/chest.124.2.459. [DOI] [PubMed] [Google Scholar]
- 8.Mohan A, et al. Clinical presentation and predictors of outcome in patients with severe acute exacerbation of chronic obstructive pulmonary disease requiring admission to intensive care unit. BMC pulmonary medicine. 2006;6:27. doi: 10.1186/1471-2466-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sin DD, Man SF, Marrie TJ. Arterial carbon dioxide tension on admission as a marker of in-hospital mortality in community-acquired pneumonia. Am J Med. 2005;118:145–150. doi: 10.1016/j.amjmed.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 10.Murtagh P, Giubergia V, Viale D, Bauer G, Pena HG. Lower respiratory infections by adenovirus in children. Clinical features and risk factors for bronchiolitis obliterans and mortality. Pediatric Pulmonology. 2009;44:450–456. doi: 10.1002/ppul.20984. [DOI] [PubMed] [Google Scholar]
- 11.Belkin RA, et al. Risk factors for death of patients with cystic fibrosis awaiting lung transplantation. American journal of respiratory and critical care medicine. 2006;173:659–666. doi: 10.1164/rccm.200410-1369OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nin N, et al. Severe hypercapnia and outcome of mechanically ventilated patients with moderate or severe acute respiratory distress syndrome. Intensive care medicine. 2017;43:200–208. doi: 10.1007/s00134-016-4611-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang N, et al. Elevated CO2 selectively inhibits interleukin-6 and tumor necrosis factor expression and decreases phagocytosis in the macrophage. FASEB J. 2010;24:2178–2190. doi: 10.1096/fj.09-136895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cummins EP, et al. NF-κB Links CO2 Sensing to Innate Immunity and Inflammation in Mammalian Cells. J. Immunol. 2010;185:4439–4445. doi: 10.4049/jimmunol.1000701. [DOI] [PubMed] [Google Scholar]
- 15.Casalino-Matsuda SM, Nair A, Beitel GJ, Gates KL, Sporn PH. Hypercapnia Inhibits Autophagy and Bacterial Killing in Human Macrophages by Increasing Expression of Bcl-2 and Bcl-xL. J Immunol. 2015;194:5388–5396. doi: 10.4049/jimmunol.1500150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Croinin DF, et al. Sustained hypercapnic acidosis during pulmonary infection increases bacterial load and worsens lung injury. Critical care medicine. 2008;36:2128–2135. doi: 10.1097/CCM.0b013e31817d1b59. [DOI] [PubMed] [Google Scholar]
- 17.Gates KL, et al. Hypercapnia impairs lung neutrophil function and increases mortality in murine pseudomonas pneumonia. Am J Respir Cell Mol Biol. 2013;49:821–828. doi: 10.1165/rcmb.2012-0487OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Briva A, et al. High CO2 levels impair alveolar epithelial function independently of pH. PLoS One. 2007;2:e1238. doi: 10.1371/journal.pone.0001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vadasz I, et al. AMP-activated protein kinase regulates CO2-induced alveolar epithelial dysfunction in rats and human cells by promoting Na,K-ATPase endocytosis. J Clin Invest. 2008;118:752–762. doi: 10.1172/JCI29723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vohwinkel CU, et al. Elevated CO(2) levels cause mitochondrial dysfunction and impair cell proliferation. J Biol Chem. 2011;286:37067–37076. doi: 10.1074/jbc.M111.290056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Proud D, Leigh R. Epithelial cells and airway diseases. Immunol Rev. 2011;242:186–204. doi: 10.1111/j.1600-065X.2011.01033.x. [DOI] [PubMed] [Google Scholar]
- 22.Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol. 2011;45:189–201. doi: 10.1165/rcmb.2011-0011RT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puchelle E, Zahm JM, Tournier JM, Coraux C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:726–733. doi: 10.1513/pats.200605-126SF. [DOI] [PubMed] [Google Scholar]
- 24.Moskwa, P. et al. A Novel Host Defense System of Airways is Defective in Cystic Fibrosis. American journal of respiratory and critical care medicine (2006). [DOI] [PMC free article] [PubMed]
- 25.Lang JD, McArdle PJ, O’Reilly PJ, Matalon S. Oxidant-antioxidant balance in acute lung injury. Chest. 2002;122:314S–320S. doi: 10.1378/chest.122.6_suppl.314S. [DOI] [PubMed] [Google Scholar]
- 26.Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nature immunology. 2015;16:27–35. doi: 10.1038/ni.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol. 2015;16:178–189. doi: 10.1038/nrm3941. [DOI] [PubMed] [Google Scholar]
- 28.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 29.Liu B, Wilson E. The antimicrobial activity of CCL28 is dependent on C-terminal positively-charged amino acids. Eur J Immunol. 2010;40:186–196. doi: 10.1002/eji.200939819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu J, Chatterjee M, Schmid H, Beck S, Gawaz M. CXCL14 as an emerging immune and inflammatory modulator. J Inflamm (Lond) 2016;13:1. doi: 10.1186/s12950-015-0109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dai C, et al. CXCL14 displays antimicrobial activity against respiratory tract bacteria and contributes to clearance of Streptococcus pneumoniae pulmonary infection. J Immunol. 2015;194:5980–5989. doi: 10.4049/jimmunol.1402634. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Bergelson JM. Adenovirus receptors. Journal of virology. 2005;79:12125–12131. doi: 10.1128/JVI.79.19.12125-12131.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Johnson JB, Parks GD. Parainfluenza virus 5 upregulates CD55 expression to produce virions with enhanced resistance to complement-mediated neutralization. Virology. 2016;497:305–313. doi: 10.1016/j.virol.2016.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brint EK, et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nature immunology. 2004;5:373–379. doi: 10.1038/ni1050. [DOI] [PubMed] [Google Scholar]
- 35.Schneberger D, et al. Effect of elevated carbon dioxide on bronchial epithelial innate immune receptor response to organic dust from swine confinement barns. Int Immunopharmacol. 2015;27:76–84. doi: 10.1016/j.intimp.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abel B, et al. Toll-like receptor 4 expression is required to control chronic Mycobacterium tuberculosis infection in mice. J Immunol. 2002;169:3155–3162. doi: 10.4049/jimmunol.169.6.3155. [DOI] [PubMed] [Google Scholar]
- 37.Branger J, et al. Toll-like receptor 4 plays a protective role in pulmonary tuberculosis in mice. International immunology. 2004;16:509–516. doi: 10.1093/intimm/dxh052. [DOI] [PubMed] [Google Scholar]
- 38.Wang X, et al. Toll-like receptor 4 mediates innate immune responses to Haemophilus influenzae infection in mouse lung. J Immunol. 2002;168:810–815. doi: 10.4049/jimmunol.168.2.810. [DOI] [PubMed] [Google Scholar]
- 39.Branger J, et al. Role of Toll-like receptor 4 in gram-positive and gram-negative pneumonia in mice. Infect Immun. 2004;72:788–794. doi: 10.1128/IAI.72.2.788-794.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurt-Jones EA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nature immunology. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 41.MacRedmond RE, Greene CM, Dorscheid DR, McElvaney NG, O’Neill SJ. Epithelial expression of TLR4 is modulated in COPD and by steroids, salmeterol and cigarette smoke. Respir Res. 2007;8:84. doi: 10.1186/1465-9921-8-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li G, et al. Effect of carbon dioxide on neonatal mouse lung: a genomic approach. J Appl Physiol. 2006;101:1556–1564. doi: 10.1152/japplphysiol.01031.2005. [DOI] [PubMed] [Google Scholar]
- 43.Hernandez-Ruiz M, Zlotnik A. Mucosal Chemokines. J Interferon Cytokine Res. 2017;37:62–70. doi: 10.1089/jir.2016.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol. 2013;20:14–22. doi: 10.1038/nsmb.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okuwaki M, Kato K, Nagata K. Functional characterization of human nucleosome assembly protein 1-like proteins as histone chaperones. Genes Cells. 2010;15:13–27. doi: 10.1111/j.1365-2443.2009.01361.x. [DOI] [PubMed] [Google Scholar]
- 46.Su C, et al. DNA damage induces downregulation of histone gene expression through the G1 checkpoint pathway. The EMBO journal. 2004;23:1133–1143. doi: 10.1038/sj.emboj.7600120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonisch C, Hake SB. Histone H2A variants in nucleosomes and chromatin: more or less stable? Nucleic Acids Res. 2012;40:10719–10741. doi: 10.1093/nar/gks865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Talbert PB, Henikoff S. Histone variants–ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010;11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 49.Kawasaki H, Koyama T, Conlon JM, Yamakura F, Iwamuro S. Antimicrobial action of histone H2B in Escherichia coli: evidence for membrane translocation and DNA-binding of a histone H2B fragment after proteolytic cleavage by outer membrane proteinase T. Biochimie. 2008;90:1693–1702. doi: 10.1016/j.biochi.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 50.Kim HS, et al. Endotoxin-neutralizing antimicrobial proteins of the human placenta. J Immunol. 2002;168:2356–2364. doi: 10.4049/jimmunol.168.5.2356. [DOI] [PubMed] [Google Scholar]
- 51.Tarr PT, Tarling EJ, Bojanic DD, Edwards PA, Baldan A. Emerging new paradigms for ABCG transporters. Biochim Biophys Acta. 2009;1791:584–593. doi: 10.1016/j.bbalip.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heaton NS, Randall G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011;19:368–375. doi: 10.1016/j.tim.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blanc M, et al. Host defense against viral infection involves interferon mediated down-regulation of sterol biosynthesis. PLoS Biol. 2011;9:e1000598. doi: 10.1371/journal.pbio.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanchez-Lazo L, et al. Fatty acid synthesis and oxidation in cumulus cells support oocyte maturation in bovine. Mol Endocrinol. 2014;28:1502–1521. doi: 10.1210/me.2014-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tarhonskaya H, et al. Non-enzymatic chemistry enables 2-hydroxyglutarate-mediated activation of 2-oxoglutarate oxygenases. Nat Commun. 2014;5:3423. doi: 10.1038/ncomms4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith JR, Jamie JF, Guillemin GJ. Kynurenine-3-monooxygenase: a review of structure, mechanism, and inhibitors. Drug Discov Today. 2016;21:315–324. doi: 10.1016/j.drudis.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 57.Choe SC, Hamacher-Brady A, Brady NR. Autophagy capacity and sub-mitochondrial heterogeneity shape Bnip3-induced mitophagy regulation of apoptosis. Cell Commun Signal. 2015;13:37. doi: 10.1186/s12964-015-0115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rorbach J, Gammage PA, Minczuk M. C7orf30 is necessary for biogenesis of the large subunit of the mitochondrial ribosome. Nucleic Acids Res. 2012;40:4097–4109. doi: 10.1093/nar/gkr1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Helenius IT, et al. Elevated CO2 suppresses specific Drosophila innate immune responses and resistance to bacterial infection. Proc Natl Acad Sci USA. 2009;106:18710–18715. doi: 10.1073/pnas.0905925106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Helenius IT, et al. Identification of Drosophila Zfh2 as a Mediator of Hypercapnic Immune Regulation by a Genome-Wide RNA Interference Screen. J Immunol. 2016;196:655–667. doi: 10.4049/jimmunol.1501708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mallia P, Johnston SL. Influenza infection and COPD. Int J Chron Obstruct Pulmon Dis. 2007;2:55–64. doi: 10.2147/copd.2007.2.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Serres G, et al. Importance of viral and bacterial infections in chronic obstructive pulmonary disease exacerbations. J Clin Virol. 2009;46:129–133. doi: 10.1016/j.jcv.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerke AK, et al. Predicting chronic obstructive pulmonary disease hospitalizations based on concurrent influenza activity. COPD. 2013;10:573–580. doi: 10.3109/15412555.2013.777400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359:2355–2365. doi: 10.1056/NEJMra0800353. [DOI] [PubMed] [Google Scholar]
- 65.Celli BR, MacNee W, Force AET. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932–946. doi: 10.1183/09031936.04.00014304. [DOI] [PubMed] [Google Scholar]
- 66.Global-Initiative-for-Chronic-Obstructive-Lung-Disease. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease, Updated 2010. http://www.goldcopd.org/Guidelineitem.asp?l1=2&l2=1&intId=989 (2010).
- 67.Kohnlein T, et al. Non-invasive positive pressure ventilation for the treatment of severe stable chronic obstructive pulmonary disease: a prospective, multicentre, randomised, controlled clinical trial. The Lancet. Respiratory medicine. 2014;2:698–705. doi: 10.1016/S2213-2600(14)70153-5. [DOI] [PubMed] [Google Scholar]
- 68.Murphy PB, et al. Effect of Home Noninvasive Ventilation With Oxygen Therapy vs Oxygen Therapy Alone on Hospital Readmission or Death After an Acute COPD Exacerbation: A Randomized Clinical Trial. JAMA. 2017;317:2177–2186. doi: 10.1001/jama.2017.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bernacki SH, et al. Mucin gene expression during differentiation of human airway epithelia in vitro. Muc4 and muc5b are strongly induced. Am J Respir Cell Mol Biol. 1999;20:595–604. doi: 10.1165/ajrcmb.20.4.3442. [DOI] [PubMed] [Google Scholar]
- 70.Gray TE, Guzman K, Davis CW, Abdullah LH, Nettesheim P. Mucociliary differentiation of serially passaged normal human tracheobronchial epithelial cells. Am J Respir Cell Mol Biol. 1996;14:104–112. doi: 10.1165/ajrcmb.14.1.8534481. [DOI] [PubMed] [Google Scholar]
- 71.Thornton DJ, et al. Characterization of mucins from cultured normal human tracheobronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1118–1128. doi: 10.1152/ajplung.2000.278.6.L1118. [DOI] [PubMed] [Google Scholar]
- 72.Casalino-Matsuda SM, Monzon ME, Forteza RM. Epidermal growth factor receptor activation by epidermal growth factor mediates oxidant-induced goblet cell metaplasia in human airway epithelium. Am J Respir Cell Mol Biol. 2006;34:581–591. doi: 10.1165/rcmb.2005-0386OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Benjamini YaHY. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society B. 1995;57:289–300. [Google Scholar]
- 76.Babicki S, et al. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 2016;44:W147–153. doi: 10.1093/nar/gkw419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Breuer K, et al. InnateDB: systems biology of innate immunity and beyond–recent updates and continuing curation. Nucleic Acids Res. 2013;41:D1228–1233. doi: 10.1093/nar/gks1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 79.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.