

Abstract
The operability and substrate scope of a redesigned vinylphenol hydratase as a single biocatalyst or as part of multienzyme cascades using either substituted coumaric acids or phenols as stable, cheap, and readily available substrates are reported.
Various strategies are pursued in order to fulfill the requirement for a greener and more sustainable chemical production.1 The ability of enzymes to work under ambient conditions in a highly selective manner evidences biocatalysis as a powerful concept for eco-friendly synthetic applications of valuable compounds, which enormously benefit from the rapid progress in molecular biology and biotechnology. The use of two or even more enzymes in a cascadewise fashion can considerably improve the efficiency of a multistage synthesis by circumventing the isolation of (unstable) intermediates, thus saving time, resources, and reagents while simultaneously diminishing the consumption of energy and the production of waste.2−4 Further aspects such as the overall cascade should run energetically downhill, and the introduction of an appropriate (internal) cofactor recycling in the case of cofactor-dependent enzymes and a preferably irreversible last step in order to increase the overall yield need to be taken into account. Limitations faced in one-pot setups, such as substrate/product/reagent inhibition or the incompatibility of reaction conditions required by one catalyst to the other(s), might be circumvented by a sequential (chronological separation) order.2
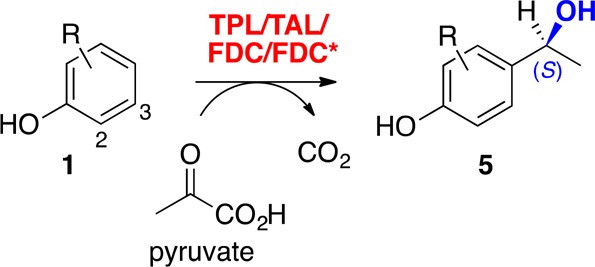
Herein, we present a redox-neutral, atom-efficient multienzyme system for the production of valuable (S)-1-(4-hydroxyphenyl)ethanols as an alternative to biocatalytic redox processes (Scheme 1).5−9 A promiscuous para-vinylphenol hydratase activity of ferulic acid decarboxylase from Enterobacter sp.,10−12 which was significantly improved by a rational redesign approach (FDC* mutant),13 was merged with a prefixed decarboxylation step catalyzed by the wild-type enzyme (FDC).14 The cascade enables the utilization of cinnamic acid derivatives from renewable feedstocks15−18 as substrates or can be prolonged by two further enzymatic steps, which have been described in the literature,19 to exploit simple phenols as starting materials.
Scheme 1. Elements of the Envisioned Multienzyme Decarboxylation/Hydration (A) and Hydroxyethylation (B) Cascade.

TPL: Tyrosine phenol lyase; TAL: tyrosine ammonia lyase; FDC: ferulic acid decarboxylase; FDC*: FDC – hydratase variant.
The replacement of a neutral valine by a carboxylate (Glu/Asp) residue as a catalytic base for the activation of water in the active site of FDC completely waived the requirement for bicarbonate as a proton relay cofactor and significantly enhanced the promiscuous hydration of 4-vinylphenols.13 In order to evaluate their biocatalytic potential, the substrate scope of the improved hydratase variants FDC_Es V46E and V46D was probed with a set of para-vinylphenols (4) bearing different substituents on the aromatic core or the vinyl side chain under previously optimized conditions13 (Scheme 2, Table 1).
Scheme 2. Substrate Scope of FDC_Es V46E and V46D Hydratase Variants.
Table 1. Substrate Scope of FDC_Es V46E and V46D Hydratase Variantsa.

Reaction conditions: purified FDC_Es variant (100 μM), substrates 4a–n (10 mM, as 10% w/w stock in propylene glycol) in potassium phosphate buffer (50 mM, pH 6.0), incubation for 24 h at 25 °C, and 700 rpm shaking (orbital shaker).
n.d. = not determined due to low conversion.
GC-MS conversion of the olefin 4k to a product with m/z of the alcohol 5k.
The E-configured olefin was used.
The Z-configured olefin was used.
The (S)-configured product was formed throughout unless otherwise stated.
Absolute configuration was not determined.
n.i. = not investigated in the literature.
A comparison of substrates 4a–e with previously reported results using wild-type decarboxylase FDC_Es in the presence of bicarbonate (3 M)10,11 reveals a superior performance of the redesigned hydratase variants (Table 1). In all cases, conversion and stereoselectivity could be significantly increased compared to the wild-type enzyme, except for the stereoselectivity with 4d, which however was low in general. In addition to chlorinated vinylphenol (4b), also fluorine (4f) and bromine (4g) substituents were well tolerated in position 2. A strong electron-withdrawing nitro group in the same position (4h) was also accepted with high stereoselectivity, albeit in low conversion. It seems that electronic effects of the substituents ortho to the phenolic OH group play only a minor role, but their influence is mainly of steric nature; i.e., the results improve in the order of R1 = NO2 < OEt < OMe < halogen < Me < H. Furthermore, the tolerance toward alkyl substituents on the styrene double bond was investigated since the hydration of such substrates would give rise to a quaternary stereocenter. However, only a methyl group on Cα (4k) was accepted, yielding the meso tertiary benzylic alcohol with moderate success, whereas substitution on Cβ (4l–n) was not tolerated at all. Overall, both variants performed fairly similar in terms of selectivity and reaction rates, with slightly better results using the glutamate variant (V46E) especially with halogens in position 3.
A major limitation for the preparation of chiral benzylic alcohols via the biocatalytic hydration of 4-vinylphenols is the preparation and storage of these compounds. A conventional protocol for the synthesis of hydroxystyrene derivatives exploits an atom-inefficient Wittig olefination of the corresponding hydroxybenzaldehydes with methyltriphenylphosphonium halides under basic conditions.20−22 The obtained hydroxystyrenes are prone to spontaneous cationic polymerization under neat conditions23,24 and require stabilization in polar solvents for storage.25 Alternatively, a preceding decarboxylation step catalyzed by phenolic and ferulic acid decarboxylases26,27 would allow the design of an enzymatic cascade that converts stable coumaric acid derivatives into chiral benzylic alcohols avoiding the isolation of troublesome vinylphenols (Scheme 1A).
A time study with 3-chlorocoumaric acid (3b) as a representative substrate showed rapid reaction progress with full consumption of the coumaric acid within 17 min and 94% conversion to the enantiomerically pure benzylic alcohol product 5b after 2 h (97% ee) (Figure 1a). Continued incubation under the reaction conditions is however accompanied by a linear decrease of the product ee, reaching 66% after 24 h. This phenomenon resembles previous observations11 and can be explained by a nonselective background hydration of 4 occurring either spontaneously13 or with FDC_Es wt in the absence of bicarbonate.11,12 Hence, careful monitoring of the reaction progress and to stop the biotransformation after an appropriate reaction time are required for optimal results.
Figure 1.
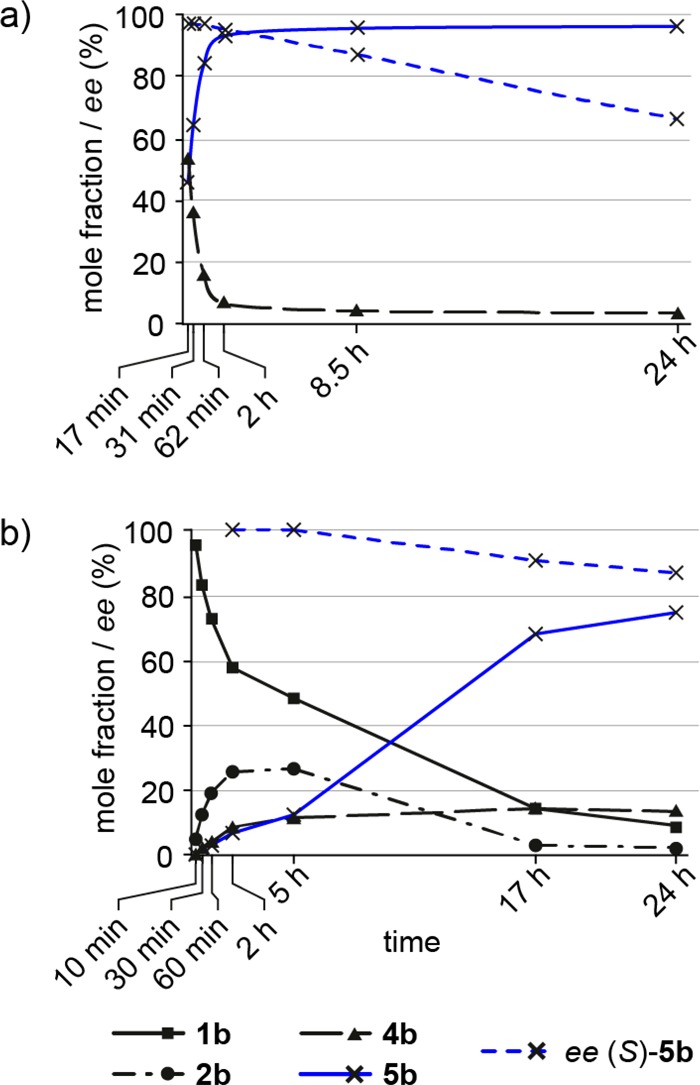
(a) Time study of the decarboxylation/hydration cascade with 3-chlorocoumaric acid (3b) as substrate at pH 6.0 (full conversion within 17 min). (b) Time profile of the four-enzyme vinylation/hydration cascade at pH 8 with 2-chlorophenol (1b) as substrate and FDC_Es V46E as hydration catalyst. The amount of coumaric acid 3b was ≤1% in all samples (not shown).
The viability of the envisioned system was validated with coumaric acid derivatives 3a–j bearing analogous substituents to vinylphenols 4a–j (except for analogues of poorly accepted 4d and 4h) (Scheme 3, Table 2). To estimate the rate of racemization observed in the previous time study in the presence of different substituents, the reaction progress was evaluated after 3.5 and 24 h (Table 2, Scheme 3). All products were initially formed with a moderate to very good ee, which however decreases upon continued incubation for 24 h and a correlation between conversion, and rate of ee loss can be observed. Electron-withdrawing halogen substituents in position 2 (5b, 5f, 5g) seem to accelerate both the water addition and decline of ee in the order of Br > Cl > F due to deactivating polarization, whereas with an electron-donating group (5c, 5e) or no substitution in this position (5a) this effect is less pronounced. Halogens in position 3 support the formation of the quinone-methide form by inductive and mesomeric effects and hydration, and selectivity decline is more pronounced with a fluorine (5i) than a chlorine substituent (5j). Although stereoselectivity slightly suffered in the two-step setup, conversions comparable to the direct hydration of 4 were observed, highlighting the viability of this system.
Scheme 3. Two-Step Decarboxylation/Hydration Cascade.
Table 2. Results of the Substrate Screening for the Decarboxylation/Hydration Cascadea.
| substrate | product | R | 3.5 h conv (ee) [%]b | 24 h conv (ee) [%]b |
|---|---|---|---|---|
| 3a | 5a | H | 30 (91) | 93 (84) |
| 3b | 5b | 2-Cl | 91 (95) | 96 (66) |
| 3c | 5c | 2-OMe | 6 (54) | 93 (42) |
| 3e | 5e | 2-Me | 58 (91) | 94 (78) |
| 3f | 5f | 2-F | 61 (85) | 96 (63) |
| 3g | 5g | 2-Br | 94 (93) | 98 (64) |
| 3i | 5i | 3-F | 51 (93) | 94 (75) |
| 3j | 5j | 3-Cl | 14 (96) | 86 (87) |
Reaction conditions: purified FDC_Es wt (10 μM), purified FDC_Es V46E variant (100 μM), substrates 3 (10 mM, supplied as 100 mM stock in iPrOH, 10% v/v) in potassium phosphate buffer (50 mM, pH 6.0), incubation for 3.5 and 24 h at 25 °C, and 700 rpm shaking (orbital shaker).
The (S)-enantiomer was formed throughout.
The most straightforward way to prepare the benzylic alcohol products would be the direct hydroxyethyl functionalization of simple substituted phenols. In the present setup we envision telescoping a recently developed vinylation cascade19 by a fourth biocatalytic hydration step to gain direct access to chiral benzylic alcohols starting from phenols (Scheme 1B). An engineered PLP-dependent tyrosine phenol lyase (TPL) from Citrobacter freundii catalyzes the coupling of phenol with pyruvate and ammonia in the first step of the cascade to give the corresponding tyrosine derivative.28 The α-amino group of the tyrosine is eliminated (and thus formally recycled) by the action of a tyrosine ammonia lyase (TAL, from Rhodococcus sphaeroides)29 in the second step of the cascade, arriving at the substituted coumaric acid as the intermediate. The final decarboxylation drives the cascade toward complete conversion into hydroxystyrene derivatives, which in turn serve as substrates for enzymatic hydration. The redox-neutral net reaction of this system therefore represents a formal hydroxyethylation of phenols with pyruvate as cosubstrate and CO2 as sole side product, which does not require costly redox cofactors and associated recycling systems (Scheme 4).
Scheme 4. Four-Step Vinylation/Hydration (Redox-Neutral Hydroxyethylation) Cascade (Net Equation).
A reaction pH of 8.0 was shown to be crucial for the vinylation cascade to work efficiently,19 but the optimal pH of the hydration process was found at pH 6.0, above which the selectivity of the hydratase decreases.13 To address this compatibility issue, 2-chlorophenol (1b) was used as a model substrate in a time study of the overall vinylation/hydration cascade at pH 8.0 (Figure 1b).
A steady conversion of phenol 1b to tyrosine 2b and further conversion of the latter to the corresponding coumaric acid 3b were observed. Due to its rapid decarboxylation, the amount of 3b was ≤1% over time (not shown in Figure 1b), and only formation of the hydroxystyrene 4b could be detected after 1 h, reaching a steady state at around 15% after 5 h. The benzylic alcohol 5b accumulated over time, reaching 75% conversion after 24 h. As in the two-step decarboxylation/hydration cascade, the ee of the chiral (S)-alcohol product drops linearly over time (87% after 24 h), albeit at a lower rate (Figure 1b). Even though the one-pot cascade was operated at pH 8.0 to optimize the performance of the vinylation cascade, a good stereoselectivity for the styrene hydration could be maintained, and the decline of ee was less pronounced. Attempts to improve the performance of the cascade by changing to a sequential mode (providing an option for pH adjustment) did not lead to significantly better results [see Table S3, Supporting Information (SI)].
In order to evaluate the biocatalytic scope of the multienzyme system, a representative set of substituted phenols (1) were applied as substrates (Table 3). All phenols were successfully converted into the corresponding benzylic alcohols (5) via the four-enzyme cascade, except for o-cresol (1e), whose conversion mainly stalled at the stage of the corresponding amino acid (2e). Slightly lower conversions to 5 compared to the two-step decarboxylation/hydration cascade (Table 2) can be explained by the presence of additives required for the preceding vinylation step (NH4Cl, pyruvate, and PLP) and the nonoptimal reaction pH. However, stereoselectivity was generally improved with this system.
Table 3. Results of the Phenol Hydroxyethylation Cascadea.
| substrate | product | R | convb (ee) [%] | yieldd (ee) [%] |
|---|---|---|---|---|
| 1a | 5a | H | 73 (92) | – |
| 1b | 5b | 2-Cl | 85 (83) | – |
| 1e | 5e | 2-Me | 12c (95) | – |
| 1f | 5f | 2-F | 91 (81) | 84 (78) |
| 1g | 5g | 2-Br | 84 (87) | 58 (88) |
| 1i | 5i | 3-F | 80 (87) | 72 (85) |
| 1j | 5j | 3-Cl | 36 (95) | 29 (92) |
Reaction conditions analytical scale: Lyophilized E. coli whole cells containing the heterologously expressed TPL_Cf M379 V (10 mg mL–1, 448 mU), TAL_Rs (40 mg mL–1, 35 mU), FDC_Es wt (2 mg mL–1, 3.2 U), and purified FDC_Es V46E variant (100 μM, 1 mol %) in reaction buffer [potassium phosphate buffer (50 mM, pH 8.0), sodium pyruvate (92 mM), NH4Cl (180 mM), and PLP (80 μM), pH adjusted to 8.0] with substrate phenol (10 mM, 50 μL of a 200 mM stock in iPrOH, 5% v/v). Incubation at 30 °C and 850 rpm for 24 h.
HPLC conversion: the corresponding vinylphenol 4 was detected as the major remaining intermediate.
Phenol 1e and tyrosine 2e were detected as major constituents.
Isolated yield after column chromatography.
Finally, the one-pot cascade was performed on a 30–40 mg scale (0.2 mmol) with substrates 1f, 1g, 1i, and 1j (20 mM), followed by isolation and characterization of the products (for yields, see Table 3). Comparable results to the analytical scale reactions without significant erosion of the ee were achieved (see Table S4, SI).
In conclusion, we evaluated the substrate scope of two vinylphenol hydratases that were rationally designed from a ferulic acid decarboxylase and found them to perform the (S)-selective addition of water with high conversion and stereoselectivities although restricted to vinylphenols bearing various substituents on the aromatic core. These hydratases lack the dependency on bicarbonate required as the hydration cofactor by the wild-type decarboxylase, thus facilitating the implementation of this biotransformation into multienzyme cascades. Two redox-neutral systems aiming at circumventing the use of delicate vinylphenol substrates were investigated for the synthesis of (S)-benzylic alcohols starting from either substituted coumaric acids or simple phenols as substrates.
Acknowledgments
We thank Philipp Neu and Klaus Zangger (Institute of Chemistry, University of Graz) for the measurement of high-resolution mass and NMR spectra, respectively. Eduardo Busto (Department of Organic Chemistry, Complutense University of Madrid) is cordially thanked for sharing his know-how on the vinylation cascade. Funding by the Austrian Science Fund (FWF project P26863-N19) and the Austrian BMWFW, BMVIT, SFG, Standortagentur Tirol, Government of Lower Austria, and ZIT through the Austrian FFG-COMET-Funding Program is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.8b02058.
Experimental details and supplementary results, preparation of substrates and reference material, and compound characterization (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sheldon R. A.; Woodley J. M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. 10.1021/acs.chemrev.7b00203. [DOI] [PubMed] [Google Scholar]
- Schrittwieser J. H.; Velikogne S.; Hall M.; Kroutil W. Artificial Biocatalytic Linear Cascades for Preparation of Organic Molecules. Chem. Rev. 2018, 118, 270–348. 10.1021/acs.chemrev.7b00033. [DOI] [PubMed] [Google Scholar]
- Sperl J. M.; Sieber V. Multienzyme Cascade Reactions—Status and Recent Advances. ACS Catal. 2018, 8, 2385–2396. 10.1021/acscatal.7b03440. [DOI] [Google Scholar]
- Wu S.; Li Z. Whole-Cell Cascade Biotransformations for One-Pot Multistep Organic Synthesis. ChemCatChem 2017, 10, 2164–2178. 10.1002/cctc.201701669. [DOI] [Google Scholar]
- Nasário F. D.; Cazetta T.; Moran P. J. S.; Rodrigues J. A. R. Deracemization of 1-Phenylethanol via Tandem Biocatalytic Oxidation and Reduction. Tetrahedron: Asymmetry 2016, 27, 404–409. 10.1016/j.tetasy.2016.03.008. [DOI] [Google Scholar]
- Neupert A.; Ress T.; Wittmann J.; Hummel W.; Gröger H. Enantioselective Biocatalytic Reduction of Non-protected Hydroxyacetophenones. Z. Naturforsch., B: J. Chem. Sci. 2010, 65, 337–340. 10.1515/znb-2010-0317. [DOI] [Google Scholar]
- Stampfer W.; Kosjek B.; Faber K.; Kroutil W. Biocatalytic Asymmetric Hydrogen Transfer Employing Rhodococcus ruber DSM 44541. J. Org. Chem. 2003, 68, 402–406. 10.1021/jo026216w. [DOI] [PubMed] [Google Scholar]
- Ewing T. A.; Kühn J.; Segarra S.; Tortajada M.; Zuhse R.; van Berkel W. J. H. Multigram Scale Enzymatic Synthesis of (R)-1-(4′-Hydroxyphenyl)ethanol Using Vanillyl Alcohol Oxidase. Adv. Synth. Catal. 2018, 360, 2370–2376. 10.1002/adsc.201800197. [DOI] [Google Scholar]
- Schnapperelle I.; Hummel W.; Gröger H. Formal Asymmetric Hydration of Non-activated Alkenes in Aqueous Medium Through a ″Chemoenzymatic Catalytic System″. Chem. - Eur. J. 2012, 18, 1073–1076. 10.1002/chem.201102579. [DOI] [PubMed] [Google Scholar]
- Wuensch C.; Gross J.; Glueck S. M.; Faber K.. Asymmetric Hydration of 4-Hydroxystyrene Derivatives Employing Decarboxylases. Int. Patent WO 2013/186358 A1, 2013, CAN160:99774.
- Wuensch C.; Gross J.; Steinkellner G.; Gruber K.; Glueck S. M.; Faber K. Asymmetric Enzymatic Hydration of Hydroxystyrene Derivatives. Angew. Chem., Int. Ed. 2013, 52, 2293–2297. 10.1002/anie.201207916. [DOI] [PubMed] [Google Scholar]
- Sheng X.; Himo F. Theoretical Study of Enzyme Promiscuity: Mechanisms of Hydration and Carboxylation Activities of Phenolic Acid Decarboxylase. ACS Catal. 2017, 7, 1733–1741. 10.1021/acscatal.6b03249. [DOI] [Google Scholar]
- Payer S. E.; Pollak H.; Glueck S. M.; Faber K. A Rational Active-Site Redesign Converts a Decarboxylase into a C = C Hydratase: ″Tethered Acetate″ Supports Enantioselective Hydration of 4-Hydroxystyrenes. ACS Catal. 2018, 8, 2438–2442. 10.1021/acscatal.7b04293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W.; Li X.; Huang J.; Duan Y.; Meng Z.; Zhang K.-Q.; Yang J. Cloning, Sequencing, and Overexpression in Escherichia coli of the Enterobacter sp. Px6–4 Gene for Ferulic Acid Decarboxylase. Appl. Microbiol. Biotechnol. 2011, 89, 1797–1805. 10.1007/s00253-010-2978-4. [DOI] [PubMed] [Google Scholar]
- Tang P.-L.; Hassan O.; Maskat M. Y.; Badri K. Production of Monomeric Aromatic Compounds from Oil Palm Empty Fruit Bunch Fiber Lignin by Chemical and Enzymatic Methods. BioMed Res. Int. 2015, 2015, 891539. 10.1155/2015/891539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulkarnain A.; Bahrin E. K.; Ramli N.; Phang L. Y.; Abd-Aziz S. Alkaline Hydrolysate of Oil Palm Empty Fruit Bunch as Potential Substrate for Biovanillin Production via Two-Step Bioconversion. Waste Biomass Valorization 2018, 9, 13–23. 10.1007/s12649-016-9745-4. [DOI] [Google Scholar]
- Sun Z.; Fridrich B.; de Santi A.; Elangovan S.; Barta K. Bright Side of Lignin Depolymerization: Toward New Platform Chemicals. Chem. Rev. 2018, 118, 614–678. 10.1021/acs.chemrev.7b00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinikul R.; Chenprakhon P.; Maenpuen S.; Chaiyen P. Biotransformation of Plant-Derived Phenolic Acids. Biotechnol. J. 2018, 13, 1700632. 10.1002/biot.201700632. [DOI] [PubMed] [Google Scholar]
- Busto E.; Simon R. C.; Kroutil W. Vinylation of Unprotected Phenols Using a Biocatalytic System. Angew. Chem., Int. Ed. 2015, 54, 10899–10902. 10.1002/anie.201505696. [DOI] [PubMed] [Google Scholar]
- Demidoff F. C.; de Souza F. P.; Netto C. D. Synthesis of Stilbene-Quinone Hybrids through Heck Reactions in PEG-400. Synthesis 2017, 49, 5217–5223. 10.1055/s-0036-1589095. [DOI] [Google Scholar]
- Falk A.; Cavalieri A.; Nichol G. S.; Vogt D.; Schmalz H.-G. Enantioselective Nickel-Catalyzed Hydrocyanation using Chiral Phosphine-Phosphite Ligands: Recent Improvements and Insights. Adv. Synth. Catal. 2015, 357, 3317–3320. 10.1002/adsc.201500644. [DOI] [Google Scholar]
- Movahhed S.; Westphal J.; Dindaroğlu M.; Falk A.; Schmalz H. G. Low-Pressure Cobalt-Catalyzed Enantioselective Hydrovinylation of Vinylarenes. Chem. - Eur. J. 2016, 22, 7381–7384. 10.1002/chem.201601283. [DOI] [PubMed] [Google Scholar]
- Sovish R. Notes: Preparation and Polymerization of p-Vinylphenol. J. Org. Chem. 1959, 24, 1345–1347. 10.1021/jo01091a606. [DOI] [Google Scholar]
- Kodaira K.; Onishi Y.; Ito K. An Oligomerization of 2-Methoxy-4-vinylphenol. Makromol. Chem., Rapid Commun. 1980, 1, 427–431. 10.1002/marc.1980.030010704. [DOI] [Google Scholar]
- Kaneko M.; Noguchi T.; Oka N.. Process for preparing vinyl phenol polymers and stabilized compositions of vinyl phenol-containing polymerization raw material. European patent EP 0864590 A2, 2000, CAN129:231167.
- Frank A.; Eborall W.; Hyde R.; Hart S.; Turkenburg J. P.; Grogan G. Mutational Analysis of Phenolic Acid Decarboxylase from Bacillus subtilis (BsPAD), Which Converts Bio-derived Phenolic Acids to Styrene Derivatives. Catal. Sci. Technol. 2012, 2, 1568–1574. 10.1039/c2cy20015e. [DOI] [Google Scholar]
- Pesci L.; Baydar M.; Glueck S.; Faber K.; Liese A.; Kara S. Development and Scaling-Up of the Fragrance Compound 4-Ethylguaiacol Synthesis via a Two-Step Chemo-Enzymatic Reaction Sequence. Org. Process Res. Dev. 2017, 21, 85–93. 10.1021/acs.oprd.6b00362. [DOI] [Google Scholar]
- Seisser B.; Zinkl R.; Gruber K.; Kaufmann F.; Hafner A.; Kroutil W. Cutting Long Syntheses Short: Access to Non-Natural Tyrosine Derivatives Employing an Engineered Tyrosine Phenol Lyase. Adv. Synth. Catal. 2010, 352, 731–736. 10.1002/adsc.200900826. [DOI] [Google Scholar]
- Gloge A.; Zoń J.; Kövári Á.; Poppe L.; Rétey J. Phenylalanine Ammonia-Lyase: The Use of Its Broad Substrate Specificity for Mechanistic Investigations and Biocatalysis—Synthesis of L-Arylalanines. Chem.–Eur. J. 2000, 6, 3386–3390. 10.1002/1521-3765(20000915)6:18<3386::AID-CHEM3386>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.