

Abstract
Cisplatin-based chemotherapy is commonly used in the treatment of solid tumors; however, this agent is limited by its adverse effects on normal tissues, including the kidneys, ears and peripheral nerves. Mechanisms of cisplatin nephrotoxicity are proposed to involve oxidative stress, inflammation, cellular apoptosis and cell cycle regulation. Sirtuin 3 (Sirt3) is a member of the sirtuin family of NAD+-dependent enzymes with homology to Saccharomyces cerevisiae gene silent information regulator 2. Sirt3 is located in mitochondria and is involved in mitochondrial energy metabolism and function; however, the role of Sirt3 in cisplatin nephrotoxicity remains unclear. In the present study, whether Sirt3 has anti-inflammatory and anti-apoptotic effects on cisplatin-induced nephrotoxicity was investigated in mice. Sirt3 knockout mice (Sirt3(−/−)) and corresponding wild type mice were employed in the present study. Cisplatin nephrotoxicity was induced by intraperitoneal injection of cisplatin (20 mg/kg). After 3 days following cisplatin treatment, blood and kidney tissues were harvested. Renal function and histology were evaluated. Tubular apoptosis, cell adhesion molecule expression, and inflammatory cells were evaluated by immunohistochemistry and western blot analysis. Following the induction of cisplatin nephrotoxicity, renal function was significantly aggravated in Sirt3 knockout (KO) mice. Tubular injury and inflammatory cell infiltration were significantly increased in Sirt3KO mice compared with wild type mice. Terminal deoxynucleotidyl transferase-mediated dUTP nick-end label-positive tubular cells and renal monocyte chemoattractant protein-1 expression levels were increased in Sirt3KO mice compared with in wild type mice. In summary, the absence of Sirt3 aggravated in renal injury by increasing renal inflammation and tubular apoptosis. The results of the present study suggested that Sirt3 may have an important role in cisplatin-induced nephrotoxicity.
Keywords: cisplatin, acute kidney injury, sirtuin 3, inflammation, apoptosis
Introduction
Cisplatin is a highly effective chemotherapeutic agent that has been approved for the treatment of testicular and ovarian cancer, non-small cell lung cancer, and squamous cell carcinoma of the head and neck (1); however, clinical limitations of this treatment occur due to its nephrotoxicity, ototoxicity and neurotoxicity (2). Renal toxicity is enhanced by underlying host risk factors, including older age, sex, congestive heart failure, cancer-induced nephrotic syndrome, hepatic dysfunction, obstructive jaundice and acute or chronic kidney disease (3). Currently, recommendations for the prevention of cisplatin nephrotoxicity include the maintenance of euvolemia by adequate hydration with normal saline during cisplatin chemotherapy (4). Preventive efforts have been proposed; however, oncologists commonly encounter acute cisplatin-associated kidney damage. Therefore, novel therapeutic or preventive approaches for cisplatin nephrotoxicity are required.
The sirtuin family comprises evolutionarily conserved proteins with NAD+-dependent protein deacetylase and ADP-ribosyltransferase activities (5,6). There are seven homologous mammalian sirtuins (Sirts), Sirt1-7, which function act as non-histone deacetylases (7). Sirt3 is highly expressed in mitochondrion-rich tissues, including the brain, heart, liver, brown adipose and the kidney. Sirt3 regulates global mitochondrial lysine acetylation (8,9). The known roles of Sirt3 include the regulation of mitochondrial biogenesis (10), redox status (11), fatty acid metabolism (12) and aging (13). In addition, impaired Sirt3 activity may have an important role in myocardial hypertrophy and dysfunction (14). In an obesity and diabetic cardiomyopathy animal model, downregulation of Sirt3 in the heart was reported to be associated with metabolic stress, and contribute to myocardial mitochondrial dysfunction and oxidative stress (15,16). However, controversial results in regulation of Sirt3 on cellular apoptosis have been reported (17,18). Sirt3 has been demonstrated to serve a pro-apoptotic role in B-cell lymphoma-2 (Bcl-2) or c-Jun N-terminal kinase (JNK)-induced apoptosis within tumorigenic and normal cells under non-stress conditions (17). In renal proximal tubular cells, high glucose levels decrease Sirt3 mRNA and protein expression, but increase cellular apoptosis (18). Overexpression of Sirt3 in renal tubular cells reduces high glucose-induced apoptosis by regulating oxidative stress and the protein kinase B/forkhead box protein O signaling pathway (18). Koyama et al (19) reported that overexpression of Sirt3 decreased palmitate-induced lipotoxicity and reactive oxygen species (ROS)-associated inflammation in renal tubular cells; however, further investigation into the role of Sirt3 in cisplatin nephrotoxicity via regulation of cellular apoptosis and inflammation is required.
It has been suggested that cisplatin induces renal tubular apoptosis and inflammation by activating the p53 tumor suppressor protein, the nuclear factor-κB (NF-κB) signaling pathway, and by inducing the production of ROS (20–22). Our previous studies suggested that Sirt1 has a protective role in cisplatin nephrotoxicity via deacetylation of p53 and NF-κB p65 (23,24). Morigi et al (25) suggested that Sirt3 exhibits protective effects in acute kidney injury by modulating mitochondrial dynamics. In the present study, whether Sirt3 exhibits anti-inflammatory and anti-apoptotic effects on cisplatin nephrotoxicity were investigated in mice.
Materials and methods
Animal experiments
The animal experimental protocol was reviewed and approved by the Institutional Animal Care and Use Committee of Chonbuk National University (Jeonju, Korea; CBU 2014–0018). Sirt3 knockout mice (129-Sirt3tm1.1Fwa/J; n=30, 8–10 weeks-old) and wild type control mice (129S1/SvlmJ; n=30, approximately 20–25 g) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and housed under controlled conditions: Temperature (23±1°C), humidity (50±10%) under a 12 h light/dark cycle with free access to water and standard chow. The mice were divided into four groups: Vehicle-treated wild type (WT) mice (n=15), vehicle-treated Sirt3 knockout (Sirt3KO) mice (n=15), cisplatin-treated WT mice (n=15), and cisplatin-treated Sirt3KO mice (n=15). The dose of cisplatin and duration of treatment were selected based on our previous study (21). PBS was used as the vehicle treatment and 200 µl of PBS was injected intraperitoneally. Maximal renal injury was observed at 72 h after a single intraperitoneal injection of cisplatin (20 mg/kg; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) by functional and histologic assessments as described below. On the final experimental day, mice were anesthetized with a mixture of ketamine (100 mg/kg; Huons Co., Ltd., Seoul, Korea) and xylazine (10 mg/kg; Bayer Korea Ltd., Seoul, Korea) via an intraperitoneal injection. A total of 800 µl blood was collected by intracardiac puncture and the kidneys were harvested to evaluate changes in renal morphology and degree of tubular apoptosis at 72 h after treatment with vehicle or cisplatin. Following collection of the blood and kidney samples, mice were sacrificed by CO2 inhalation. For functional analysis, blood urea nitrogen (BUN) and creatinine levels were measured by an enzymatic assays using an automatic analyzer (Hitachi 7180; Hitachi, Ltd., Tokyo, Japan).
Renal histologic examination
Kidneys were fixed in 4% paraformaldehyde for 24 h at 4°C and embedded in paraffin. Blocks were cut into 5-µm sections and stained with Periodic acid-Schiff (PAS) by using 0.5% Periodic acid solution for 5 min and Schiff reagent for 15 min at room temperature. Immunohistochemical staining was performed as described previously (26). Tissue sections were deparaffinized with xylene, rehydrated via washes with graded ethanol in water, and rinsed in pure water. Following the previously described heat-induced antigen retrieval process (26) and treatment with blocking buffer (Protein Block Serum-Free Ready-to-use; cat. no. X0909; Dako; Agilent Technologies, Inc., Santa Clara CA, USA) for 10 min at room temperature (26), slides were incubated overnight at 4°C with rabbit anti-mouse monocyte chemoattractant protein-1 (MCP-1; 1:100; cat. no. 70R50662; Fitzgerald Industries International, Acton, MA, USA) and rat anti-mouse lymphocyte antigen 6 complex, locus G (Gr-1; 1:50; cat. no. 560453; BD Pharmingen; BD Biosciences, San Jose, CA, USA) antibodies. Subsequently, polyclonal goat anti-rabbit immunoglobulins/Biotinylated (1:500; cat. no E0432; Dako; Agilent Technologies, Inc.) for MCP-1 and polyclonal goat anti-rat immunoglobulins/Biotinylated (1:500; cat. no. E0468; Dako; Agilent Technologies, Inc.) for Gr-1 and incubated for 1 h at room temperature. The kidney sections were treated with chromogen (Dako AEC + High Sensitivity Substrate Chromogen Ready-to-Use; cat. no. K3469; Dako; Agilent Technologies, Inc.) to visualize immunocomplexes for 10 min at room temperature and then counterstained with 0.1% hematoxylin (Sigma-Aldrich; Merck KGaA) for 1 min at room temperature.
For immunofluorescence staining, freshly frozen renal tissues were fixed with 4% paraformaldehyde at 4°C for 24 h, and embedded in optimum cutting temperature (OCT) compound Tissue-Tek (Sakura Finetek, Tokyo, Japan) and cut into 10-µm thickness on Superfrost (Thermo Fisher Scientific, Inc., Waltham, MA, USA) slide. The slides were permeabilized in 1% Triton X-100 for 10 min at room temperature and then incubated with a blocking buffer (Protein Block Serum-Free Ready-to-use; cat. no. X0909; Dako; Agilent Technologies, Inc.) for 10 min at room temperature. The tissue samples were incubated with rat anti-mouse adhesion G protein-coupled receptor E4 (F4/80; 1:200; cat. no. 14-4801-82; eBioscience; Thermo Fisher Scientific, Inc.). Slides were exposed to a goat anti-rat Cy3-labeled secondary antibody (1:1,000; cat. no. A10522; Thermo Fisher Scientific, Inc.) for 1 h at room temperature. Nuclear staining was performed using DAPI (1:1,000, Molecular Probes; Thermo Fisher Scientific, Inc.) for 1 min at room temperature.
Analysis was conducted by two researchers in a blinded manner to evaluate the morphology of all slides using a Zeiss Z1 microscope for light and fluorescence microscopy (Zeiss AG, Oberkochen, Germany). For immunofluorescence, 543 nm was applied for the detection of Cy3-labled F4/80 (+) macrophages. Renal tubular injury and positive areas of immunohistochemical staining was assessed as previously described (21,26). Tubular injury was classified by a six-level scoring system based on the magnitude of tubular epithelial cell loss, necrosis, intratubular debris, and tubular cast formation in 10 randomly selected, non-overlapping fields (magnification, ×200) under light microscopy: 0, none; 0.5, <10%; 1, 10–25%; 2, 25–50%; 3, 50–75%; and 4, >75% (21,26). The MCP-1-positive area was measured in 10 randomly chosen, non-overlapping fields at a magnification of ×200 using ImageJ 1.47v software (National Institutes of Health, Bethesda, MD, USA). The number of F4/80-positive macrophages and Gr-1-positive neutrophils were counted in 10 randomly chosen, non-overlapping fields (magnification, ×400).
Detection of apoptosis
Apoptosis was assessed by a terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay, and the number of apoptotic cells, as defined by chromatin condensation or nuclear fragmentation (apoptotic bodies), was counted as previously described (21). Apoptosis in the specimens was detected using the ApopTag Plus Peroxidase In Situ Apoptosis Detection kit (EMD Millipore, Billerica, MA, USA) according to the manufacturer's protocols. Samples were fixed and embedded as aforementioned. The number of apoptotic cells in each section (n=10 sections per kidney) was calculated by counting the number of TUNEL-positive apoptotic cells in 10 random selected, nonoverlapping fields per slide (magnification, ×400).
Western blot analysis
Western blot analysis was performed as described previously (21). Kidney tissues were homogenized in radioimmunoprecipitation assay lysis and extraction buffer (Thermo Fisher Scientific, Inc.) with a protease inhibitor cocktail (EMD Millipore), and the protein concentration was quantified by a Bradford protein assay. Samples (20–30 µg protein per lane) were mixed with sample buffer, boiled for 7 min, separated by (8–15%) SDS-PAGE and electroblotted onto a nitrocellulose membrane (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The membrane was blocked with 5% non-fat dry milk in Tris-buffered saline [25 mmol/l Tris (pH 7.5), 150 mmol/l NaCl] with 0.1% Tween-20 buffer for 1 h at room temperature and then incubated overnight at 4°C with rabbit anti-mouse Sirt3 (1:1,000; cat. no. 5490, Cell Signaling Technology, Inc., Danvers, MA, USA) and rabbit anti-mouse caspase-3 (1:1,000; cat. no. 9662; Cell Signaling Technology, Inc.). Membranes were washed with PBS and incubated with horseradish peroxidase-conjugated anti-rabbit IgG antibodies (1:5,000; cat. no. 65-6120; Thermo Fisher Scientific, Inc.) for 1 h at room temperature. Bands were visualized with a chemiluminescent detection kit according to the manufacturer's protocols (Amersham ECL Prime Western Blotting Detection Reagent, GE Healthcare, Chicago, IL, USA). Membranes were then probed with an anti-β-actin antibody (1:10,000; cat. no. A5316; Sigma-Aldrich; Merck KGaA) to verify equal loadings of protein in each lane. All signals were analyzed by densitometric scanner (LAS-3000 pro software; FUJIFILM Corporation, Tokyo, Japan).
Statistical analysis
Data are expressed as the mean ± standard deviation. Graphs and statistical analyses were produced and conducted, respectively, using the SigmaPlot program (version 11; Systat Software, Inc., San Jose, CA, USA). Analysis was conducted in triplicate. Multiple comparisons were performed to investigate significant differences using one-way analysis of variance, followed by an individual comparison with a Tukey post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of Sirt3 on cisplatin-induced renal tubular damage
To address the role of Sirt3 in cisplatin-associated nephrotoxicity, the present study investigated Sirt3 protein expression following the administration of cisplatin. At 72 h following intraperitoneal injection of cisplatin (20 mg/kg), renal expression of Sirt3 was significantly decreased in cisplatin-treated WT mice compared within the vehicle group (Fig. 1A). Subsequently, Sirt3KO mice were employed to evaluate the effects of Sirt3 on renal function following treatment with cisplatin. Compared with in WT mice, Sirt3KO mice exhibited significantly increased BUN and serum creatinine levels at 72 h after the administration of cisplatin (Fig. 1B). Consistent with the aforementioned blood chemistry findings, renal histologic analysis revealed that Sirt3KO mice accumulated more intratubular debris, PAS-positive materials and cast formation in the tubular lumen; loss of the brush border and tubular epithelial cells, tubular dilatation, and inflammatory cell infiltration were also observed at 72 h following cisplatin treatment compared with in cisplatin treated WT mice (Fig. 1C). The tubular injury score was determined at 72 h after treatment with cisplatin in WT and Sirt3KO mice. The Sirt3KO mice that were injected with cisplatin exhibited significantly higher tubular injury scores than control WT and Sirt3KO mice (Fig. 1D). These data suggested that the absence of Sirt3 may be associated with aggravation of cisplatin-induced renal injury.
Figure 1.

Effects of Sirt3 on cisplatin-induced renal tubular damage. (A) Sirt3 expression in kidney tissue from WT mice injected with Veh or cisplatin (20 mg/kg body weight) was evaluated by western blotting. Data obtained from densitometric analysis are presented as the relative ratio of each protein to β-actin. The relative ratio of protein measured in kidneys from vehicle-injected mice is arbitrarily presented as 1. **P<0.01 vs. Veh. (B) Kidneys in Sirt3KO and WT mice were harvested 3 days after treatment with cisplatin (20 mg/kg body weight). Blood samples were collected 72 h following cisplatin treatment, and BUN and serum creatinine levels were measured (n=10 in each group). **P<0.01 vs. Veh; #P<0.05 vs. WT Cisplatin. (C) Representative PAS-stained sections of kidneys from WT and Sirt3KO mice treated with vehicle or cisplatin. Arrows indicate inflammatory cells; arrowheads indicate tubular epithelial cell loss and necrosis, and c represents intratubular debris and tubular cast formation. Scale bar, 100 µm. (D) Histological damage in PAS-stained kidney sections was scored by measuring the magnitude of tubular epithelial cell loss, necrosis, intratubular debris and tubular cast formation. ***P<0.001 vs. Veh, ##P<0.01 vs. WT Cisplatin. Data are expressed as the mean ± standard deviation. PAS, periodic acid-Schiff; Veh, vehicle; Sirt3, Sirtuin 3; BUN, blood urea nitrogen; WT, wild type; KO, knockout.
Effects of Sirt3 on cisplatin-induced renal inflammation
The infiltration of inflammatory cells, including neutrophils and macrophages, and MCP-1 expression were evaluated by immunofluorescence and immunohistochemical analysis, respectively, to investigate the effects of Sirt3 on cisplatin-induced renal inflammation. F4/80-positive macrophages were observed in the tubulointerstitial area at 72 h following cisplatin treatment in the WT and Sirt3KO mice; the number of F4/80-positive macrophages was significantly higher in Sirt3KO mice compared with WT mice (Fig. 2A and B). Additionally, neutrophil infiltration following cisplatin-induced renal injury was investigated in the present study. Gr-1-positive neutrophils notably infiltrated the tubulointerstitial area at 72 h following cisplatin treatment in WT and Sirt3KO mice compared with the vehicle treated groups (Fig. 3A). Cisplatin treated Sirt3KO mice exhibited significantly increased neutrophil infiltration compared with WT mice with cisplatin treatment (Fig. 3B). IgG control probed slides were not stained in both WT and Sirt3KO mice samples.
Figure 2.
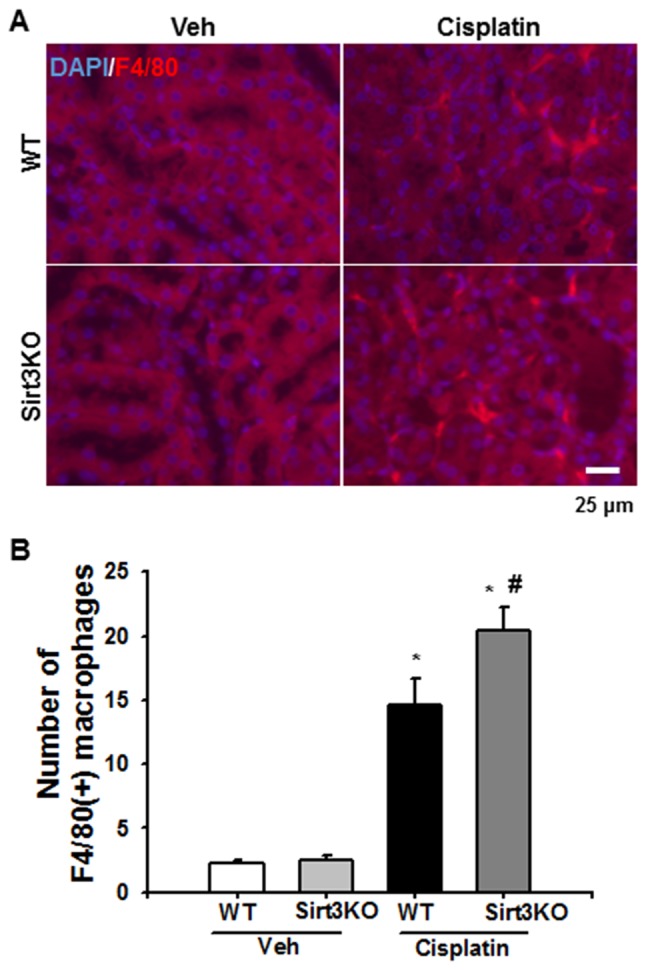
Effects of Sirt3 on cisplatin-induced macrophage infiltration. (A) Representative immunofluorescence staining for F4/80 in kidneys from WT and Sirt3KO mice treated with vehicle or cisplatin. Nuclear staining was performed using DAPI. Scale bar, 25 µm. (B) Bar graph of the number of F4/80-positive cells in the kidneys of WT and Sirt3KO mice. A total of 10 randomly selected, nonoverlapping fields were quantified (n=10 in each group; magnification, ×400). *P<0.05 vs. Veh; #P<0.05 vs. WT Cisplatin. Data are expressed as the mean ± standard deviation. Sirt3KO, NAD+-dependent protein deacetylase 3 knockout mice. Veh, vehicle; WT, wild type; Sirt3, Sirtuin 3; KO, knockout; F4/80, adhesion G protein-coupled receptor E4.
Figure 3.
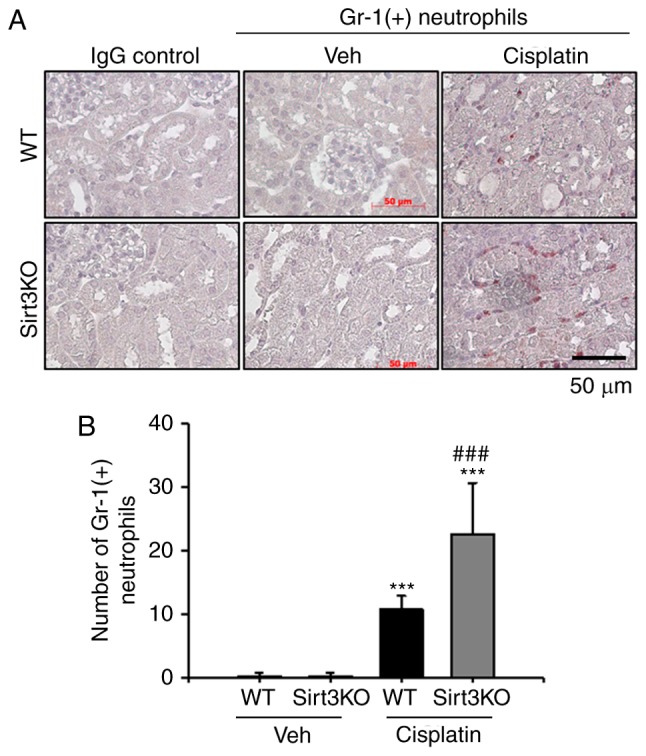
Effects of Sirt3 on cisplatin-induced neutrophil infiltration. (A) Representative Immunohistochemistry staining for Gr-1-positive neutrophils in the kidneys of WT and Sirt3KO mice treated with vehicle or cisplatin. Scale bar, 50 µm. (B) Bar graph of the number of Gr-1-positive cells in WT and Sirt3KO mice kidneys following treatment with vehicle or cisplatin. A total of 10 randomly selected, nonoverlapping fields were quantified (n=10 in each group; magnification, ×400). Data were expressed as the mean ± standard deviation. ***P<0.001 vs. Veh; ###P<0.001 vs. WT cisplatin. Gr-1, lymphocyte antigen 6 complex, locus G; Veh, vehicle; WT, wild type; Sirt3, Sirtuin 3; KO, knockout.
MCP-1 expression was significantly increased in the renal tubulointerstitial area at 72 h after the administration of cisplatin. The area of staining MCP-1 was significantly higher in Sirt3KO mice than WT mice following treatment with cisplatin, as well as that in the vehicle groups (Fig. 4A and B). IgG control probed slides were not stained in both WT and Sirt3 KO mice samples. These data suggested that the absence of Sirt3 may aggravate cisplatin-induced renal inflammation by inducing an increase in the infiltration of inflammatory cells.
Figure 4.
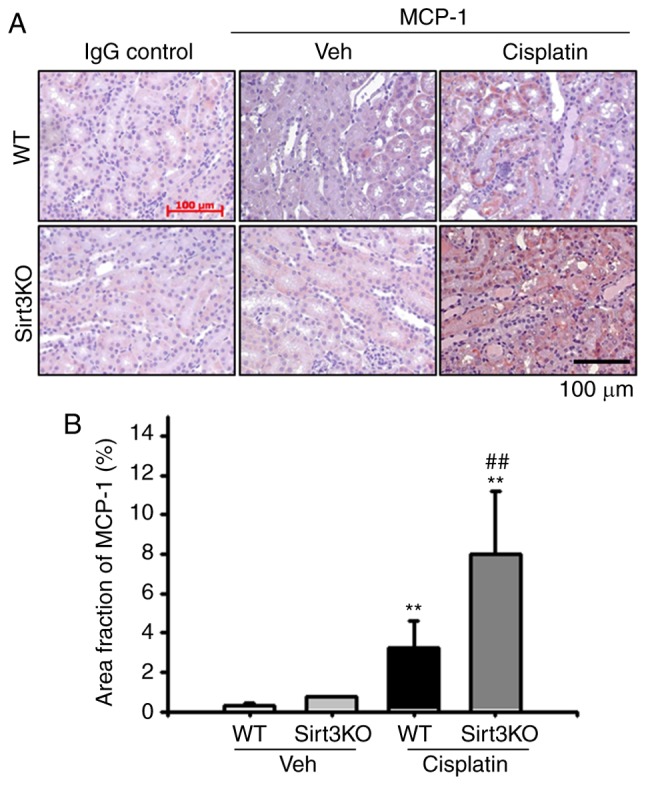
Effects of Sirt3 on cisplatin-induced tubular MCP-1 expression. (A) Representative immunohistochemistry staining for MCP-1 in the kidneys of WT and Sirt3KO mice treated with vehicle or cisplatin. Scale bar, 100 µm. (B) Bar graph demonstrated area fraction of MCP-1 in WT and Sirt3KO mice treated with vehicle or cisplatin. A total of 10 randomly selected, nonoverlapping fields at a magnification of ×200 were quantified (n=10 in each group). IgG control antibody was used for negative control for staining. **P<0.01 vs. Veh; ##P<0.01 vs. WT cisplatin. Data are expressed as the mean ± standard deviation. MCP-1, monocyte chemoattractant protein-1; Veh, vehicle; WT, wild type; Sirt3, Sirtuin 3; KO, knockout.
Effects of Sirt3 on cisplatin-induced renal tubular apoptosis
Renal tubular apoptosis is a suggested mechanism of cisplatin-induced nephrotoxicity (27). Cisplatin inhibits mitochondrial oxidative phosphorylation and membrane potentials, which results in apoptotic cell death (28). Therefore, cisplatin-induced renal tubular apoptosis was evaluated by a TUNEL assay in the present study. As presented in Fig. 5A, the number of TUNEL-positive cells significantly increased in Sirt3KO mice compared with in WT mice at 72 h after cisplatin treatment. Additionally, the number TUNEL-positive cells in both cisplatin-treated groups compared with the respective vehicle controls. The expression level of cleaved caspase-3 was significantly increased at 72 h after cisplatin treatment in Sirt3KO mice compared with in WT mice; a significant difference was observed between both the cisplatin treatment groups and the respective vehicle controls (Fig. 5B). These findings indicated that Sirt3 may serve a critical role in cisplatin-induced tubular apoptosis.
Figure 5.

Effects of Sirt3 on cisplatin-induced tubular apoptosis. (A) Kidneys from WT and Sirt3KO mice treated with vehicle or cisplatin were evaluated for tubular apoptosis by a TUNEL assay. The bar graph shows the number of TUNEL-positive cells in WT and Sirt3KO mice treated with vehicle or cisplatin. A total of 10 randomly selected, nonoverlapping fields were quantified (n=10 in each group; magnification, ×400). Data are expressed as the mean ± standard deviation. Scale bar, 50 µm. (B) Representative western blotting for caspase-3 and cleaved caspase-3 from the kidneys of WT and Sirt3KO mice treated with vehicle or cisplatin. Data from densitometric analysis were presented as the relative ratio of each protein to β-actin. The relative ratio measured in kidneys from WT mice treated with vehicle is arbitrarily presented as 1. Data are expressed as the4 mean ± standard deviation. **P<0.01 vs. Veh; ##P<0.01 vs. WT cisplatin. Veh, vehicle; WT, wild type; Sirt3, Sirtuin 3; KO, knockout; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
Discussion
Renal dysfunction is a common complication of cancer; renal dysfunction may be associated with the cancer itself, induced by cancer-associated treatment or due to secondary complications, including sepsis, nephrotoxic drugs or radiocontrast agent administration (29). Acute kidney injury following chemotherapy has been associated with increased morbidity and mortality in patients with cancer (3). As there are limited therapeutic options for treating cisplatin nephrotoxicity, the underlying mechanisms of toxicity require further investigation to identify novel therapeutic agents or targets. In the present study, it was demonstrated that Sirt3 may serve an essential role in protecting the kidney from cisplatin-induced renal inflammation and tubular apoptosis. In the current study, the absence of Sirt3 was been associated with increased renal macrophage infiltration and tubular MCP-1 expression in cisplatin-induced nephrotoxicity. In addition, Sirt3KO mice exhibited increased cisplatin-induced tubular apoptosis compared with WT mice in the present study.
Renal inflammation is an important mechanism of cisplatin nephrotoxicity (22,27). Several inflammatory mediators are involved in cisplatin nephrotoxicity. We previously reported that, following cisplatin injection, macrophages infiltrate the tubulointerstitial areas (22). In addition to inflammatory cell infiltration, cell adhesion molecules, including intercellular adhesion molecule-1 and MCP-1 were expressed in peritubular capillaries and injured tubules (21,22,30). Therefore, regulation of the cisplatin-induced inflammatory response may be important for the treatment and prevention of cisplatin nephrotoxicity. The present study revealed that the absence of Sirt3 in cisplatin-treated mice was associated with increased induction of renal inflammatory responses compared with in WT mice. These data support reports of the protective role of Sirt3 in cisplatin nephrotoxicity by regulating inflammation.
The renal toxicity of cisplatin may be associated with formation of DNA adducts, which can inhibit DNA replication and transcription, resulting in altered protein synthesis, mitochondrial injury, DNA damage and programmed cell death (31). We previously reported that cisplatin-induced activation of the p53 tumor suppression gene is one mechanism of cisplatin-induced renal tubular apoptosis, which was attenuated by treatment with luteolin (21). In the present study, it was demonstrated that the absence of Sirt3 in cisplatin-treated mice was associated with increased tubular apoptosis and cleaved caspase 3 expression levels compared with in cisplatin-treated WT mice. These data indicated that Sirt3 may serve an important role in cisplatin-induced renal tubular apoptosis.
Sirt3 is an NAD+-dependent deacetylase of the sirtuin family of proteins and is localized to the mitochondrial matrix (32). Sirt3 has a role in controlling respiratory chain activity, the tricarboxylic acid cycle, fatty acid β-oxidation and the antioxidant pathway (32). Sirt3 can regulate mitochondrial dynamics in cisplatin-induced proximal tubular injury and alter the NF-κB dependent inflammatory pathway and oxidative stress in proteinuric kidney disease (19,25). The findings of the present study revealed that cisplatin treatment was associated with downregulated Sirt3 expression in WT mice; however, Sirt3KO mice exhibited aggravated cisplatin-induced renal injury, potentially by increasing the induction of the inflammatory response and tubular apoptosis.
The role of Sirt3 on cellular apoptosis remains controversial. Allison and Milner (17) reported that Sirt3 is an essential pro-apoptotic mediator for the Bcl-2/p53- and JNK2/JNK1-regulated apoptosis signaling pathways in a colorectal carcinoma cell line. In leukemic cell lines, kaempferol was reported to increase auto-oxidation and generate ROS to induce cellular apoptosis by increasing Bcl-2-associated-X (Bax) and Sirt3 expression levels, and activating caspase-3 cascades (33). However, the results of the present study suggested that the absence of Sirt3 increased cisplatin-induced renal tubular apoptosis via increasing the expression levels of active caspase-3. Sundaresan et al (34) reported that Sirt3 augments Ku70-Bax interactions to prevent Bax translocation to the mitochondrial membrane, which protects cells from stress-induced apoptosis. This discrepancy may be due to variations in cell types, including tumorigenic and normal cells. Therefore, further investigation is required to identify the specific role of Sirt3 in cisplatin-associated nephrotoxicity.
Different pharmacological approaches have suggested that potential Sirt3 activators may increase Sirt3 expression or Sirt3 activity (14). Honokiol and resveratrol are natural products that increase Sirt3 expression and modulate Sirt3 activity, and have high bioavailability (14,35). These agents possess anti-inflammatory, anti-apoptotic and anti-oxidant properties, as well as AMP activated protein kinase activation and induction of autophagy abilities, which exert protective effects against cardiac hypertrophy, heart failure and myocardial ischemia-reperfusion injury (36–39). Therefore, a pharmacologic agent to activate Sirt3 may have beneficial effects on cisplatin-induced renal injury. Alhazzazi et al (40) reported that a novel Sirt3 inhibitor, LC-0296, exhibited anticancer effects in head and neck cancer via the regulation of cell proliferation and apoptosis. This agent may also have synergistic effects on cancer cells to increase sensitivity to radiation and cisplatin treatment. Therefore, modulation of Sirt3 may benefit patients with cancer as an anticancer strategy and preserve renal function.
A limitation of the present study was that a novel Sirt3 activator or inhibitor for the prevention of cisplatin-induced renal injury was not used. In. addition, the present study did not establish a tumor model to evaluate the role of a Sirt3 activator or inhibitor in chemotherapy-associated toxicity in the kidney and other tissues. Therefore, further investigation of novel Sirt3 activators and inhibitors for the treatment of chemotherapy-associated complications are required.
In summary, the present study reported that the cisplatin nephrotoxicity induced by loss of Sirt3 may occur via increased renal inflammation and tubular apoptosis. Therefore, Sirt3 may serve an important role protecting against cisplatin-induced nephrotoxicity; however, further investigation is required to understand the molecular mechanisms underlying this nephrotoxicity.
Acknowledgements
We thank Ms. Kieu Thi Thu Trang for the excellent technical assistance.
Funding
The present study was supported by the National Research Foundation of Korea funded by the Korean government (grant nos. NRF-2015R1D1A3A03015653 to KPK and NRF-2017R1D1A3B03035494, to SKP) and the research funds of Chonbuk National University Hospital (grant no. CUH2013-0047, to KPK).
Availability of data and materials
Not applicable.
Authors' contributions
DK, WP and KPK designed and performed the experiment of the present study. SL and WK contributed to the design of the present study, the analysis of the results and the final version of the manuscript. SKP and KPK made substantial contributions to the conception and design of the present study, conducted data analysis and writing of the manuscript.
Ethics approval and consent to participate
The present study was approved by the Institutional Animal Care and Use Committee of Chonbuk National University (Jeonju, Korea; CBU 2014-0018).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.dos Santos NA, Carvalho Rodrigues MA, Martins NM, dos Santos AC. Cisplatin-induced nephrotoxicity and targets of nephroprotection: An update. Arch Toxicol. 2012;86:1233–1250. doi: 10.1007/s00204-012-0821-7. [DOI] [PubMed] [Google Scholar]
- 2.Arany I, Safirstein RL. Cisplatin nephrotoxicity. Semin Nephrol. 2003;23:460–464. doi: 10.1016/S0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- 3.Perazella MA, Moeckel GW. Nephrotoxicity from chemotherapeutic agents: Clinical manifestations, pathobiology, and prevention/therapy. Semin Nephrol. 2010;30:570–581. doi: 10.1016/j.semnephrol.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Launay-Vacher V, Rey JB, Isnard-Bagnis C, Deray G, Daouphars M. European Society of Clinical Pharmacy Special Interest Group on Cancer Care: Prevention of cisplatin nephrotoxicity: State of the art and recommendations from the European society of clinical pharmacy special interest group on cancer care. Cancer Chemother Pharmacol. 2008;61:903–909. doi: 10.1007/s00280-008-0711-0. [DOI] [PubMed] [Google Scholar]
- 5.Bause AS, Haigis MC. SIRT3 regulation of mitochondrial oxidative stress. Exp Gerontol. 2013;48:634–639. doi: 10.1016/j.exger.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Haigis MC, Guarente LP. Mammalian sirtuins-emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 7.Sack MN. The role of SIRT3 in mitochondrial homeostasis and cardiac adaptation to hypertrophy and aging. J Mol Cell Cardiol. 2012;52:520–525. doi: 10.1016/j.yjmcc.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dang W. The controversial world of sirtuins. Drug Discov Today Technol. 2014;12:e9–e17. doi: 10.1016/j.ddtec.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis; Proc Natl Acad Sci USA; 2008; pp. 14447–14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 12.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, De Rango F, Greco V, Maggiolini M, Feraco E, Mari V, Franceschi C, et al. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics. 2005;85:258–263. doi: 10.1016/j.ygeno.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Koentges C, Bode C, Bugger H. SIRT3 in cardiac physiology and Disease. Front Cardiovasc Med. 2016;3:38. doi: 10.3389/fcvm.2016.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeng H, Vaka VR, He X, Booz GW, Chen JX. High-fat diet induces cardiac remodelling and dysfunction: Assessment of the role played by SIRT3 loss. J Cell Mol Med. 2015;19:1847–1856. doi: 10.1111/jcmm.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeng H, He X, Hou X, Li L, Chen JX. Apelin gene therapy increases myocardial vascular density and ameliorates diabetic cardiomyopathy via upregulation of sirtuin 3. American journal of physiology. Am J Physiol Heart Circ Physiol. 2014;306:H585–H597. doi: 10.1152/ajpheart.00821.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allison SJ, Milner J. SIRT3 is pro-apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle. 2007;6:2669–2677. doi: 10.4161/cc.6.21.4866. [DOI] [PubMed] [Google Scholar]
- 18.Jiao X, Li Y, Zhang T, Liu M, Chi Y. Role of Sirtuin3 in high glucose-induced apoptosis in renal tubular epithelial cells. Biochem Biophys Res Commun. 2016;480:387–393. doi: 10.1016/j.bbrc.2016.10.060. [DOI] [PubMed] [Google Scholar]
- 19.Koyama T, Kume S, Koya D, Araki S, Isshiki K, Chin-Kanasaki M, Sugimoto T, Haneda M, Sugaya T, Kashiwagi A, et al. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic Biol Med. 2011;51:1258–1267. doi: 10.1016/j.freeradbiomed.2011.05.028. [DOI] [PubMed] [Google Scholar]
- 20.Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110:835–842. doi: 10.1172/JCI200215606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang KP, Park SK, Kim DH, Sung MJ, Jung YJ, Lee AS, Lee JE, Ramkumar KM, Lee S, Park MH, et al. Luteolin ameliorates cisplatin-induced acute kidney injury in mice by regulation of p53-dependent renal tubular apoptosis. Nephrol Dial Transplant. 2011;26:814–822. doi: 10.1093/ndt/gfq528. [DOI] [PubMed] [Google Scholar]
- 22.Kang KP, Kim DH, Jung YJ, Lee AS, Lee S, Lee SY, Jang KY, Sung MJ, Park SK, Kim W. Alpha-lipoic acid attenuates cisplatin-induced acute kidney injury in mice by suppressing renal inflammation. Nephrol Dial Transplant. 2009;24:3012–3020. doi: 10.1093/ndt/gfp242. [DOI] [PubMed] [Google Scholar]
- 23.Jung YJ, Lee JE, Lee AS, Kang KP, Lee S, Park SK, Lee SY, Han MK, Kim DH, Kim W. SIRT1 overexpression decreases cisplatin-induced acetylation of NF-κB p65 subunit and cytotoxicity in renal proximal tubule cells. Biochem Biophys Res Commun. 2012;419:206–210. doi: 10.1016/j.bbrc.2012.01.148. [DOI] [PubMed] [Google Scholar]
- 24.Kim DH, Jung YJ, Lee JE, Lee AS, Kang KP, Lee S, Park SK, Han MK, Lee SY, Ramkumar KM, et al. SIRT1 activation by resveratrol ameliorates cisplatin-induced renal injury through deacetylation of p53. Am J Physiol Renal Physiol. 2011;301:F427–F435. doi: 10.1152/ajprenal.00258.2010. [DOI] [PubMed] [Google Scholar]
- 25.Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, Novelli R, Remuzzi G, Benigni A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest. 2015;125:715–726. doi: 10.1172/JCI77632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim D, Lee AS, Jung YJ, Yang KH, Lee S, Park SK, Kim W, Kang KP. Tamoxifen ameliorates renal tubulointerstitial fibrosis by modulation of estrogen receptor alpha-mediated transforming growth factor-β1/Smad signaling pathway. Nephrol Dial Transplant. 2014;29:2043–2053. doi: 10.1093/ndt/gfu240. [DOI] [PubMed] [Google Scholar]
- 27.Pabla N, Dong Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 28.Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int. 2011;80:29–40. doi: 10.1038/ki.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fischler R, Meert AP, Sculier JP, Berghmans T. Continuous renal replacement therapy for acute renal failure in patients with cancer: A well-tolerated adjunct treatment. Front Med (Lausanne) 2016;3:33. doi: 10.3389/fmed.2016.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sung MJ, Kim DH, Jung YJ, Kang KP, Lee AS, Lee S, Kim W, Davaatseren M, Hwang JT, Kim HJ, et al. Genistein protects the kidney from cisplatin-induced injury. Kidney Int. 2008;74:1538–1547. doi: 10.1038/ki.2008.409. [DOI] [PubMed] [Google Scholar]
- 31.Park MS, De Leon M, Devarajan P. Cisplatin induces apoptosis in LLC-PK1 cells via activation of mitochondrial pathways. J Am Soc Nephrol. 2002;13:858–865. doi: 10.1681/ASN.V134858. [DOI] [PubMed] [Google Scholar]
- 32.Perico L, Morigi M, Benigni A. Mitochondrial Sirtuin 3 and Renal Diseases. Nephron. 2016;134:14–19. doi: 10.1159/000444370. [DOI] [PubMed] [Google Scholar]
- 33.Marfe G, Tafani M, Indelicato M, Sinibaldi-Salimei P, Reali V, Pucci B, Fini M, Russo MA. Kaempferol induces apoptosis in two different cell lines via Akt inactivation, Bax and SIRT3 activation, and mitochondrial dysfunction. J Cell Biochem. 2009;106:643–650. doi: 10.1002/jcb.22044. [DOI] [PubMed] [Google Scholar]
- 34.Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. 2008;28:6384–6401. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pillai VB, Samant S, Sundaresan NR, Raghuraman H, Kim G, Bonner MY, Arbiser JL, Walker DI, Jones DP, Gius D, Gupta MP. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun. 2015;6:6656. doi: 10.1038/ncomms7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Averett C, Arora S, Zubair H, Singh S, Bhardwaj A, Singh AP. Molecular targets of honokiol: A promising phytochemical for effective cancer management. Enzymes. 2014;36:175–193. doi: 10.1016/B978-0-12-802215-3.00009-4. [DOI] [PubMed] [Google Scholar]
- 37.Kumar A, Kumar Singh U, Chaudhary A. Honokiol analogs: A novel class of anticancer agents targeting cell signaling pathways and other bioactivities. Future Med Chem. 2013;5:809–829. doi: 10.4155/fmc.13.32. [DOI] [PubMed] [Google Scholar]
- 38.Zordoky BN, Robertson IM, Dyck JR. Preclinical and clinical evidence for the role of resveratrol in the treatment of cardiovascular diseases. Biochim Biophys Acta. 2015;1852:1155–1177. doi: 10.1016/j.bbadis.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 39.Chen T, Li J, Liu J, Wang S, Liu H, Zeng M, Zhang Y, Bu P. Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF-β/Smad3 pathway. Am J Physiol Heart Circ Physiol. 2015;308:H424–H434. doi: 10.1152/ajpheart.00454.2014. [DOI] [PubMed] [Google Scholar]
- 40.Alhazzazi TY, Kamarajan P, Xu Y, Ai T, Chen L, Verdin E, Kapila YL. A Novel Sirtuin-3 Inhibitor, LC-0296, inhibits cell survival and proliferation, and promotes apoptosis of head and neck cancer cells. Anticancer Res. 2016;36:49–60. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.