

Abstract
Inflammatory bowel disease (IBD) is a group of chronic diseases of the digestive tract affecting millions of people worldwide. Genetic, environmental and microbial factors have been implicated in onset and exacerbation of IBD. However, the mechanisms associating gut microbial dysbioses and aberrant immune responses remain largely unknown. The integrative Human Microbiome Project (iHMP) seeks to close these gaps by examining the dynamics of microbiome functionality in disease by profiling the gut microbiomes of more than 100 individuals sampled over a one year period. Here, we present the first results based on 78 paired fecal metagenomes/metatranscriptomes and 222 additional metagenomes from 59 Crohn’s disease (CD), 34 ulcerative colitis (UC), and 24 non-IBD control patients. We demonstrate several cases in which measures of microbial gene expression in the inflamed gut can be informative relative to metagenomic profiles of functional potential. First, while many microbial organisms exhibited concordant DNA and RNA abundances, we also detected species-specific biases in transcriptional activity, revealing predominant transcription of pathways by individual microbes per host (e.g. by Faecalibacterium prausnitzii). Therefore, a loss of these organisms in disease may have more far-reaching consequences than suggested by their genomic abundances. Further, we identified organisms that were metagenomically abundant but inactive or dormant in the gut with little or no expression (e.g. Dialister invisus). Lastly, certain disease-specific microbial characteristics were more pronounced or only detectable at the transcript level, such as pathways predominantly expressed by different organisms in IBD patients (e.g. Bacteroides vulgatus and Alistipes putredinis). This provides potential insights into gut microbial pathway transcription that can vary over time, inducing phenotypic changes complementary to those linked to metagenomic abundances. The study’s results highlight the strength of analyzing both the activity and presence of gut microbes to provide insight into the role of the microbiome in IBD.
Inflammatory bowel disease (IBD) is a group of chronic inflammatory disorders that affects all or part of the digestive tract and incidence rates are increasing worldwide1. IBD is a lifelong disease with no effective long-term treatment options, and an estimated 25-30% of all patients present with symptoms before the age of 202,3. Several human genetic mutations are implicated in an increased susceptibility to IBD; however, not everyone who carries these mutations develops IBD, indicating that additional exposures are also involved. Recognizing that the gut microbiome is one such factor altered in Crohn’s disease (CD) and ulcerative colitis (UC) patients, the two main forms of IBD, is one of the most significant developments in the field of IBD in the last decade and provides a wealth of opportunities for the discovery of diagnostic and therapeutic approaches4,5.
A gut microbial dysbiosis exists at the community-level in patients with IBD, combining a general decrease in alpha diversity with clade-specific enrichments and depletions6,7. However, microbial taxonomic profiles can be highly divergent among patients, making it difficult to implicate specific microbial species or strains in disease onset and progression. Some broad patterns do apply: taxa from the Enterobacteriaceae family are generally increased, whereas members of the Firmicutes phylum are decreased8–10. Further, several bacterial taxa have been suggested to have protective effects in IBD, such as Lactobacillus and Faecalibacterium11–13.
Most such findings are based on samples from cross-sectional cohorts, emphasizing the need for longitudinal studies to explore changes in the gut microbiome within individual patients. Indeed, gut microbiome composition is known to vary over time within individuals, and such variations may be more pronounced within IBD patients14,15. The links between metagenomic functional potential and realized functional activity (gene expression, or other molecular products such as proteins or metabolites) remain almost completely unexplored in IBD. While the overall functional potential of the gut microbiome tends to be stable relative to taxonomic composition16, it can vary over time and across phenotypes. For example, short chain fatty acid (SCFA) producing bacteria are depleted in IBD; SFCAs are metabolites that are broadly anti-inflammatory in a typical gut17,18. Furthermore, an increase in oxidative stress pathways and a decrease in carbohydrate metabolism and amino acid biosynthesis have also been consistently reported6, each affecting multiple underlying taxa in different individuals. Notably, the functional potential of an organism (i.e. the genes and pathways encoded in its genome) provides only indirect information about the level or extent to which these functions are active. Such measures of functional activity are critical for understanding the mechanisms associating gut microbial dysbioses and aberrant immune responses, which to date remain largely unknown. Alterations in transcriptional activity in IBD have been established based on rRNA expression and indicate that some bacterial populations are active in IBD patients while other groups are inactive or dormant in disease19. However, the specific bacterial species and metabolic pathways remain to be elucidated.
In order to close these gaps, we compared the functional potential of gut microbial communities (from shotgun metagenomics) to direct measures of functional activity (from metatranscriptomics) in a longitudinal cohort. Fecal samples were collected biweekly from approximately 100 patients over the course of one year and subjected to shotgun metagenomic (DNA) and metatranscriptomic (RNA) sequencing. Here, we present the results based on 78 paired metagenomes and metatranscriptomes and an additional 222 metagenomes. For many species, functional potential was well-correlated with functional activity. However, we were able to identify species-specific shifts in transcript levels indicating that some organisms (e.g. Faecalibacterium prausnitzii) and pathways may play a more central role in maintaining gut health than their genomic abundances indicate. We also detected organisms that were metagenomically present but with low or non-existent gene expression (e.g. Dialister invisus), suggesting that the organism is either dead or inactive and hence of questionable importance in the gut community. In addition, we uncovered disease-specific changes in microbial gene expression that were either more pronounced or only detectable on the RNA level (e.g. metabolic pathways contributed by Bacteroides vulgatus and Alistipes putredinis). Together, our findings highlight that crucial insight into microbial community dynamics can be gained through integrated analysis of metatranscriptomic and metagenomic profiles of microbial community structure and function. This approach will lead to a better understanding of the underlying mechanisms of gut microbial dysbioses and their role in IBD.
Results
A longitudinal IBD cohort profiled using metagenomic and metatranscriptomic sequencing
As part of the Integrative Human Microbiome Project (iHMP or HMP2), the goal of the IBD Multi’omics Database (IBDMDB, http://ibdmdb.org) is to assemble longitudinal multi’omic profiles of IBD patients to gain insight into the mechanisms of microbial dysbioses and their effects on disease onset and progression (Fig. 1). Participants provided biweekly stool samples over the course of one year from which we generated shotgun metagenomic and metatranscriptomic sequencing data. In this one of several resulting datasets and studies, we analyzed 78 paired metagenomes/metatranscriptomes and an additional 222 metagenomes from 117 individuals: 59 CD patients, 34 with UC, and 24 non-IBD controls (Fig. 1a). These datasets yielded high-resolution profiles of gut microbial community composition (taxonomy), functional potential, and functional activity.
Figure 1: Longitudinal metagenomes and metatranscriptomes in inflammatory bowel disease.
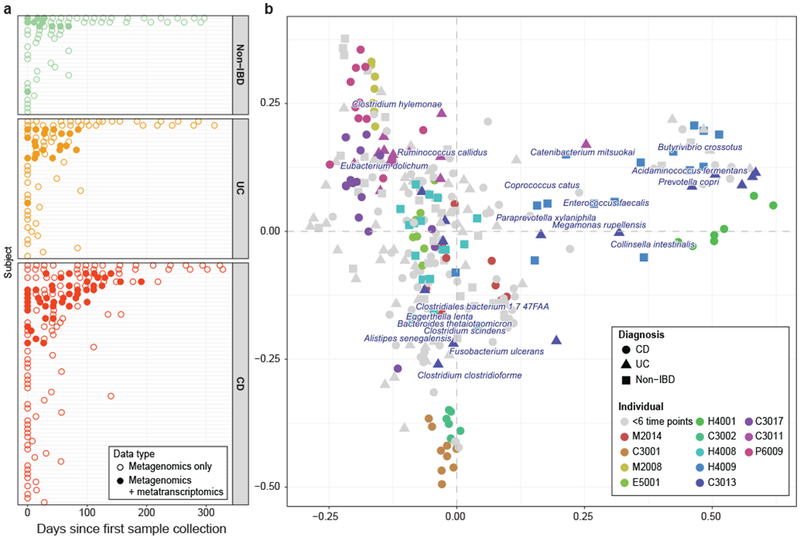
(a) Bi-weekly stool samples were collected from 117 individuals (59 Crohn’s disease (CD), 34 ulcerative colitis (UC), and 24 non-IBD controls) over the course of one year each. The resulting data comprise 78 paired stool metagenomes/metatranscriptomes and 222 additional metagenomes. While most samples are part of short time courses, two long time courses per disease phenotype (with up to 17 time points) were also included. (b) Principal Coordinate Analysis (PCoA) on Bray-Curtis distance at the species level for all 300 metagenomic taxonomic profiles from 117 patients. In addition, 19 species are overlaid where their position represents the species’ weighted average score and indicates that samples in close proximity are likely to contain higher abundances of the respective organism. While the time courses highlight that inter-individual effects dominate the variation in taxonomic composition, longitudinal shifts were also observed, motivating the investigation of inter- and intra-personal dynamic patterns of microbial species.
Consistent with previous studies, taxonomic shifts in microbial composition inferred from the metagenomic data accounted for significant, but modest effect size, separation among the three phenotypes (Fig. 1b). Longitudinal profiling further emphasized that variation in microbial community composition is dominated by inter-individual effects, as samples from the same subject tended to cluster tightly. However, we also observed taxonomic shifts in community composition over time that coincided with changes in disease severity [i.e. as measured by the Harvey-Bradshaw Index (HBI)] and antibiotic treatment (Fig. S1). This highlights the importance of examining longitudinal profiles in order to establish a better understanding of species dynamics within and across patients.
Inter- and intra-personal dynamic patterns of microbial species
In order to better understand the variability in microbial species composition within and across individuals, we first examined the taxonomic profiles from six long time courses (i.e. 2 CD, 2 UC and 2 non-IBD patients with at least 12 samples each over the one-year sampling period; Fig. S2a). Three general patterns were observed: 1) intra-personal stability, 2) global stability, and 3) inter- and intra-personal variability. Intra-personal stability refers to species that were only encountered in individual patients or a subset of the patients and represented permanent members of their gut microbial community (Fig. S2b). The relative abundances of these species often remained fairly stable over the course of the year. Among these patient-specific microbial organisms were several Bacteroides species, suggesting that these closely related organisms can contribute similar functions in different patients.
In contrast, we also encountered many examples of microbial organisms that were universally present in all patients at high abundance (1-10%), including Faecalibacterium prausnitzii and Bacteroides vulgatus, two species that are implicated in gut inflammation and IBD specifically (Fig. S2c)13,17,20,21. Their tendency to be present in all patients irrespective of disease phenotype suggests that their abundance or transcriptional activity rather than presence/absence plays a role in gut inflammation. Finally, some species displayed inter- and intra-personal variability patterns, intermittently disappearing and reappearing in all six patients over time (Fig. S2d). Among these were prominent IBD-associated organisms such as Ruminococcus gnavus and Roseburia intestinalis, highlighting that taxonomic variability is not only observed between patients but also within a patient over time22,23. Furthermore, spikes of R. gnavus were observed in some IBD patients in the overall cohort, with relative abundances of up to 19%.
Functional potential is often, but not always, proportional to metatranscriptomic expression in the gut microbiome
In order to compare and contrast community functional potential and functional activity, we profiled the 78 paired metagenomes and metatranscriptomes with HUMAnN224, which outputs per-sample pathway abundance, stratified according to individual species’ contributions (Methods). Averaging first within and then across patients, we found that species contributing more pathway copies to the total pool of microbial genomic DNA (i.e. more metagenomically abundant species) also tended to contribute more pathway transcripts (Fig. 2a).
Figure 2: Metatranscriptomic activities assigned to specific microbial species and disease phenotypes.
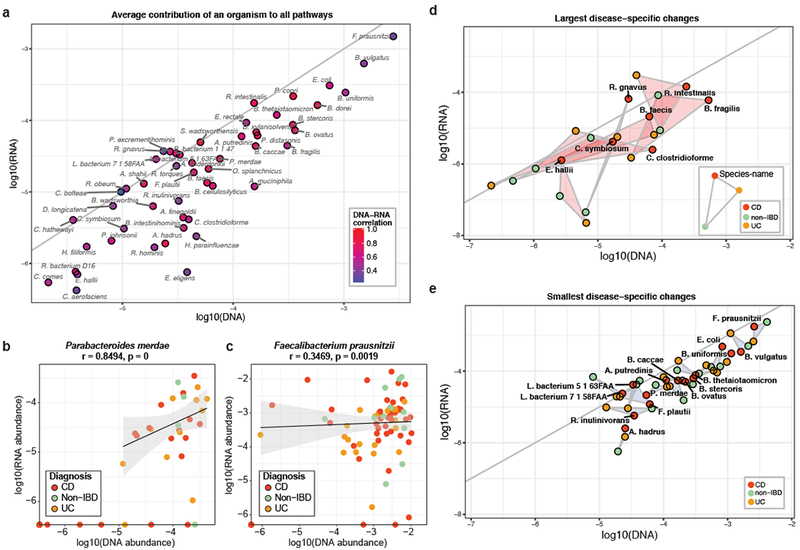
(a) Summary of metagenomic and metatranscriptomic activities across all analyzed pathways as assigned to individual species (n=78). The activity of each species is first averaged within and subsequently across participants for DNA and RNA samples, respectively; only species that were detected in at least 10% of the samples are shown. Comparisons of DNA and RNA pathway levels per species are summarized for each point, with examples expanded in (b) and (c): Parabacteroides merdae (high DNA-RNA correlation, Spearman r=0.85) and Faecalibacterium prausnitzii (low correlation, Spearman r=0.35). Here, each point represents one sample (n=78)., indicating the species’ overall contribution to all pathways in its metagenome and metatranscriptome, including a linear fit with a 95% confidence interval. (d) and (e): Species that exhibited the largest and smallest disease-specific changes, respectively, after repeating the analysis described in (a) for each disease group separately. Results are summarized as triangles for each species connecting vertices representing the values for each disease group (CD n=46, UC n=21, non-IBD n=11). Overall, while functional potential is indicative of functional activity for many microbial organisms, others exhibited low DNA-RNA-correlation or disease-specific differences in functional activity.
Moreover, among metagenomically abundant species (which are less sensitive to undersampling), mean pathway abundance at the DNA and RNA levels tended to correlate well across samples. This correlation was particularly strong for Parabacteroides merdae, a mucin-degrading, short-chain fatty acid (SCFA) producer (Spearman r=0.85, Fig. 2b)25,26. Such a strong correlation suggests that the total transcript output of P. merdae is relatively constant across samples, and hence samples with a larger P. merdae cell fraction (i.e. metagenomic relative abundance) coincide with an increase in contributions from P. merdae in the total transcript pool. This behavior is in contrast with that of Dialister invisus: a bacterium predominantly associated with the human oral cavity, which has also been detected in the gut and been implicated in diseases such as IBD and type-1 diabetes22,27,28. While the average DNA abundance of D. invisus was comparable to that of P. merdae, and varied across samples, D. invisus was largely absent from our metatranscriptomic data (Fig. S3a). This suggests that D. invisus is not actively transcribing in the gastrointestinal tract, consistent with a dead or non-growing population. While we infer that organisms such as D. invisus are not transcriptionally active when we do not observe any transcripts from those organisms, it is technically possible that different microbial species might have different RNA stability resulting in faster degradation of RNA from one organism relative to another. Nevertheless, such dramatic differences in transcriptional behavior between metagenomically similar species underscore the importance of measuring functional potential and activity in tandem.
Faecalibacterium prausnitzii exhibited a third distinct pattern of behavior: poor correlation in total DNA and RNA abundances across samples despite being abundant on average across both data types (Fig. 2c). F. prausnitzii is a known producer of butyrate, a SCFA that plays a role in maintaining gut health and barrier function, and previous studies have found the species to be depleted in IBD patients13,17,20. While F. prausnitzii remained fairly stably abundant in IBD patients with long time courses (Fig. S2c), the metatranscriptomic data suggests that the metagenomic abundance of this species is not predictive of its relative transcriptional activity (Fig. 2c).
Disease-specific differences in functional activity of microbial organisms
The patterns of species-specific microbial transcription introduced above can be further stratified to identify interactions with IBD phenotype (Fig. S4). More specifically, if we define a dysbiosis as a shift in a species’ mean pathway contributions between IBD phenotypes (UC or CD) and non-IBD controls, a species can be dysbiotic at the DNA level, the RNA level, or both, and in any combination of directions. The seven species that exhibited the largest such dysbioses are summarized in Fig. 2d (for a complete list see Table S1). Of these, two exhibited a more pronounced dysbiosis in their functional activity compared to their functional potential. Ruminococcus gnavus exhibited the largest amplification of disease-specific dysbiosis on the RNA level, with greatly increased RNA abundance in both CD and UC patients compared to non-IBD controls (~3 orders of magnitude) in a background of a smaller increase in DNA abundance (~1 order of magnitude). Hence, small changes in the abundance of R. gnavus may be more consequential than previously assumed.
The two species that exhibited the largest difference in community DNA compared to RNA were Clostridium symbiosum and Bacteroides faecis. One possible implication may be that the impact of these species on disease progression is less critical than suggested by the metagenomic data alone. While the preceding examples focused on dysbioses that were consistent across the two IBD subtypes, this was not always the case. For example, Bacteroides fragilis was less abundant in DNA in UC patients compared to non-IBD controls, while it was more abundant in CD patients. On the other hand, the species’ mean RNA abundance was similar between CD and non-IBD patients but markedly lower in UC. Furthermore, we observed many examples of species whose overall expression was comparable across disease phenotypes with similar DNA and RNA pathway abundances (Fig. 2e). These organisms are thus unlikely to play a role in disease onset and progression.
Contrasting metabolic functional potential and functional activity
To compare the functional potential and activity of the entire microbial community, we next investigated overall metabolic pathway abundances in both data types (Fig. 3). We used contributional alpha diversity as a measurement to compare the diversity of organisms contributing metagenomically and metatranscriptomically to each pathway (Methods). This allowed us to distinguish pathways contributed by a single or few microbial organisms, representing specialized metabolic processes, from pathways that are contributed by a multitude of organisms, representing more essential metabolic processes (Fig. 3a).
Figure 3: Comparing species-specific metagenomic functional potential with metatranscriptomic functional activity.
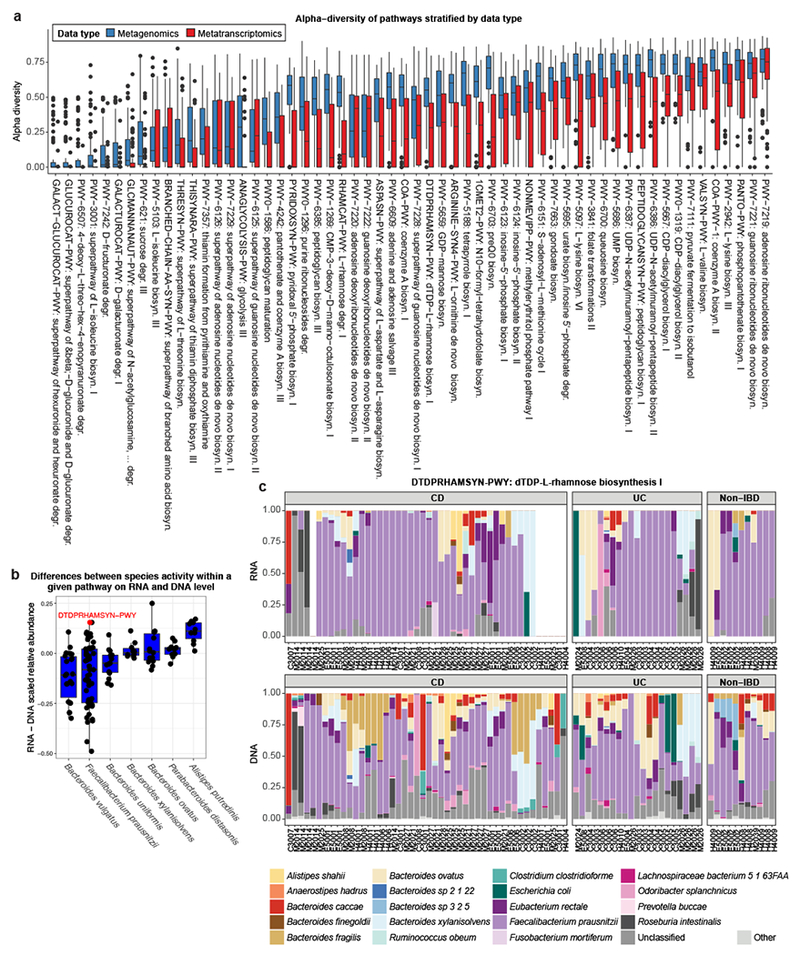
(a) Contributional alpha diversity of species represented in DNA (metagenomic) and RNA (metatranscriptomic) pathway profiles, calculated across all 78 paired samples. The mean, first and third quartiles are displayed. Pathways are ordered by the sum of their median DNA- and RNA-level diversity measures. While diversity was generally lower on the transcriptional level, as expected, we also observed that many specialized pathways were contributed by only one or few microbial species. (b) Differences in per-pathway DNA versus RNA contributions of microbial species, each point representing one pathway averaged first within samples from the same patient and subsequently across patients. The mean, first and third quartiles are displayed. Only pathways detected in at least 20% of metagenomic and metatranscriptomic samples (n=78) were included for a particular species, and only species contributing to at least 8 pathways across samples are displayed. Some microbial organisms exhibited a general tendency for over- or under-transcription, while others displayed pathway-specific activity patterns. (c) Example where pathway transcription is dominated by a single species. Relative contributions of the top 20 species in metagenomes (DNA) versus metatranscriptomes (RNA) for the dTDP-L-rhamnose biosynthesis I pathway (n=78). This pathway was one of the most over-transcribed pathways of F. prausnitzii, the species with the largest range in pathway-specific over- and undertranscription.
The two pathways with the lowest contributional diversity on the DNA and RNA levels were almost entirely from F. prausnitzii: 1) GALACT−GLUCUROCAT−PWY, superpathway of galacturonate and glucuronate degradation [synonym: superpathway of hexuronide and hexuronate degradation (Fig. S5a)] and 2) GLUCUROCAT−PWY, superpathway of beta-D-glucuronide and D-glucuronate degradation. A subset of samples broke this trend, with Escherichia coli appearing as the dominant transcriber of these pathways. E. coli has been previously shown to be able to use beta-D-glucuronides and the hexuronates D-glucuronate and D-fructuronate as the sole carbon source for growth. Our data suggest that F. prausnitzii is the main utilizer of these sugars, even in samples where E. coli is present (Fig. S5a).
In contrast, the most ubiquitous pathways with the greatest contributional alpha diversities were two biosynthesis pathways for the ribonucleotides adenosine and guanosine (PWY−7219 and PWY−7221), which are involved in numerous basic biochemical processes (including functioning as extracellular signaling molecules). These essential functions were contributed by a multitude of organisms and generally all organisms that encoded the pathway were also expressing it (Fig. S5b + S6a). Adenosine is an important modulator of inflammation with anti-inflammatory effects and therefore a potential therapeutic target in IBD29,30. Further, guanosine can inhibit LPS-induced pro-inflammatory responses in the context of neuroinflammatory-related diseases31.
Many pathways exhibited similar alpha diversity patterns in DNA and RNA, as illustrated by the examples above. However, for a subset of pathways, a lower diversity of contributing species was detected on the RNA level, with transcription often dominated by a single species. The species with the greatest discrepancies in DNA and RNA transcriptional profiles included four Bacteroides species (B. vulgatus, B. uniformis, B. ovatus, B. xylanisolvens), Faecalibacterium prausnitzii, Parabacteroides distasonis and Alistipes putredinis (Fig. 3b). As an example, F. prausnitzii showed the highest degree of variation in DNA-RNA differences and also contributed to the largest number of pathways.
One of the pathways where transcription was dominated by F. prausnitzii was dTDP-L-rhamnose biosynthesis I (Fig. 3c). The resulting deoxysugar β-L-rhamnopyranose functions as a building block of the glycan component of the O-antigens, which is a major target for the immune systems and the target of many vaccine development studies32–35. Regulation of this biosynthetic pathway has been previously studied in the context of Pseudomonas aeruginosa, in which it is transcriptionally regulated specifically by quorum sensing systems36. Quorum sensing is a mechanism by which regulation (within or among species) can achieve multi-stability, i.e. activate or deactivate expression only after a certain concentration of signaling molecule is achieved. As this type of multi-stability is exhibited by many pathways in the metatranscriptome - that is, only one of several possible organisms upregulated, and others downregulated - we hypothesize that these, and specifically the expression of F. prausnitzii for this pathway, may be regulated through quorum-sensing-like mechanisms, constituting a potential example of interspecies interactions in the gut.
Disease-specific transcriptional microbial signatures
Differences between pathway encoding versus transcription were particularly evident in the pathway contributions of Alistipes putredinis and Bacteroides vulgatus and these transcriptional effects were also disease-associated (Fig. 4). A. putredinis exhibited the highest discrepancy between functional potential and functional activity across all of its pathways (Fig. 3b). One example is the methylerythritol phosphate (MEP) pathway, which was consistently overtranscribed by A. putredinis (Fig. 4a). The product of this pathway, isopentenyl diphosphate (IPP), is used by organisms in the biosynthesis of terpenes and terpenoids: a group of natural products that have been increasingly mined for drug discovery, in particular for cancer. In E. coli, the MEP pathway is also involved in the production of phosphate-containing antigens recognized by human gamma delta T lymphocytes, which are suggested to play an important role in the immune response to microbial organisms37. Furthermore, IPP can be converted to the more-reactive electrophile dimethylallyl pyrophosphate (DMAPP), which has been shown to induce acute inflammation38.
Figure 4: Dynamic changes in IBD-specific metatranscription over time.
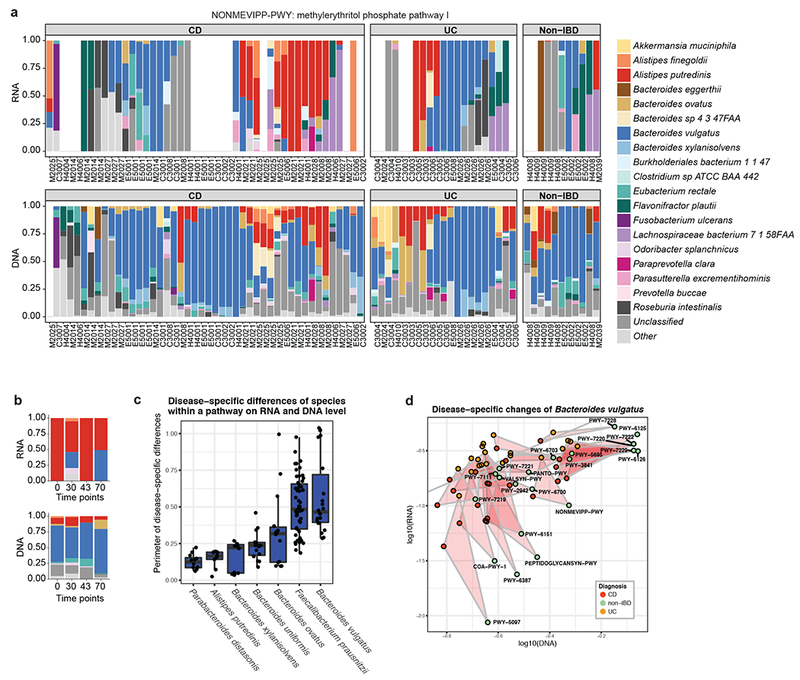
(a) Relative contribution of organisms to the methylerythritol phosphate pathway I (NONMEVIPP-PWY), including all time points and individuals (n=78).. Overtranscription is visible for A. putredinis in the absence of metagenomic disruption. (b) Relative contribution of organisms to the same pathway, restricted to the samples from one CD patient (M2021) over time. A. putredinis consistently over-transcribed this pathway within this individual. Increases in the HBI of this patient were observed at time points 2 and 4, uniquely where pathway transcription was partially contributed by Bacteroides vulgatus. (c) Differences in per-pathway DNA versus RNA contributions of microbial species across disease phenotypes, each point representing the perimeter of the disease-specific pathway triangles described in Fig. 2d+e. The mean, first and third quartiles are displayed. Only pathways detected in at least 20% of metagenomic and metatranscriptomic samples (n=78) were included for a particular species, and only species contributing to at least 8 pathways across samples are displayed. On average, Bacteroides vulgatus exhibited the largest disease-specific difference. (d) Changes in relative contribution of Bacteroides vulgatus to its transcribed pathways stratified by IBD phenotype. For this organism, IBD-specific dysbioses were particularly pronounced on the transcriptional level, with many pathways upregulated both in UC and in CD.
Finally, disease-associated transcriptional effects became evident when examining patient time courses. For one CD patient, for example (Fig. 4b), we observed fairly constant proportions of all microbial species contributing the MEP pathway at the DNA level. However, among RNA data, A. putredinis monopolized MEP pathway transcription, and it was strikingly the sole contributor at time points 1 and 3. At time points 2 and 4, B. vulgatus contributed transcriptional activity for the MEP pathway. Interestingly, this coincided with changes in disease severity for this patient, with HBI scores increased at both time points where B. vulgatus was a pathway contributor (Fig. S3b). Both species exhibited an overall correlation with disease severity, with A. putredinis negatively correlated with disease severity and B. vulgatus positively correlated (Fig. S6b+c). We hypothesize that this is likely a sufficient but not necessary covariation with disease. Indeed, this correlation was not induced on a population-level. This example highlights the importance of examining changes in metatranscription over time within individual patients: such changes may correlate variation in disease severity, thus suggesting a mechanistic relationship that would be masked in DNA data alone.
We further examined disease-associated transcriptional changes that generalized across IBD patients. The most pronounced IBD-specific transcriptional changes were observed for Bacteroides vulgatus (Fig. 4c). More specifically, most B. vulgatus pathways were more DNA-abundant in non-IBD patients, but many of these pathways were considerably more RNA-abundant among UC and CD patients (Fig. 4d). This suggests that B. vulgatus follows a different transcriptional program in IBD patients, possibly triggered by disease-specific environmental changes in the gut (e.g. inflammation or increased levels of oxygen).
Discussion
Our findings highlight that directly measuring functional activity of the gut microbiome through metatranscriptomics reveals important insights that are only partially observable in metagenomic functional potential, including disease-linked observations. For some pathways, a dominant transcribing organism was identified in a background of mixed metagenomic contributions. Several striking examples of this phenomenon involved, for example, Faecalibacterium prausnitzii, Bacteroides vulgatus or Alistipes putredinis, which often dominated pathway transcription in IBD even when not the most abundant organism in a sample. Furthermore, several species displayed altered expression profiles in IBD.
Importantly, many IBD-specific signals were either more pronounced or only detectable on the RNA level, such as pathways that were substantially upregulated in disease and species that displayed altered expression profiles in IBD patients. These altered expression profiles are potentially the result of changes in the gut environment in IBD patients, which include increased levels of inflammation (resulting in an aggravated immune response), higher concentrations of oxygen (which may be toxic to obligate anaerobes), and a diminished mucus layer39. Metatranscriptomics circumvents the challenges of assaying diverse biochemical products dynamically in situ (e.g. mucus40,41 or oxygen6,42) and enables us to study the effects of environmental changes on microbial expression patterns in vivo for large human populations.
In addition to the direct benefits of measuring community functional activity, coupling such measurements with longitudinal sampling enables association of modulated activity with disease progression. This longitudinal design resulted in most samples corresponding to a minority of patients, thus not appropriate for most cross-sectional analyses. For example, our data highlight cases where microbial genomic contributions to a particular pathway remained stable over time, while the corresponding expression patterns varied with disease severity. Therefore, microbial dysbioses impacting disease progression and severity may be mechanistically related to changes in the transcriptional programs of an otherwise stable community, thus making metatranscriptomic profiling an important tool for understanding such mechanisms. While both RNA and DNA abundances can change in microbial communities, they of course do so at very different time scales (minutes or less, versus hours or more). This underlying biological difference represents another way in which the measurement types may capture complementary microbial processes as they relate to host phenotypes, such as disease flares or changes in inflammation.
We hypothesize that behavior such as that of F. prausnitzii involves multi-species bistability (or more accurately multi-stability), in which inter-microbial interactions converge on a single dominant transcriber for some functions that can differ between individuals. This type of behavior in microbial communities is best known from quorum sensing, which itself has been mostly studied in the context of biofilm formation and pathogenic bacteria. For example, the quorum sensor gene lasR of Pseudomonas aeruginosa (an opportunistic pathogen) produces the molecule N-3-oxo-dodecanoyl-l-homoserine lactone (C12), which allows a microbial subpopulation to bistably activate (or deactivate) regulation after reaching a critical threshold43,44. Other examples include Staphylococcus epidermis, a bacterium that uses quorum sensing to evade human innate immune defense mechanisms45. Further, quorum sensing molecules have also been shown to affect gut microbial community composition in mice, where increased levels of the quorum-sensing signal autoinducer-2 (AI-2) favored an expansion of Firmicutes following antibiotic treatment46. Since many transcriptional systems in the human gut appear to be regulated in a manner that is multi-stable among microbes and individuals, it remains to be determined whether formal quorum sensing molecules or other regulatory mechanisms are responsible, particularly in the context of IBD.
Some technical limitations apply to RNA-based measurements in stool. Fecal metatranscriptomics captures RNA that is extractable, not degraded during the extraction procedure or in the cells beforehand and restricted to the organisms that are present in stool samples. While this is a subset of total biological regulatory activity, the same kind of caveats and technical limitations apply to any kind of RNA-based measurements of transcriptional regulation in other systems. Some of these technical limitations also apply to fecal metagenomics. While biopsies may be more representative of microbial abundance and expression at the colonic mucosa, frequent longitudinal sampling is implausible due to the invasive nature of this procedure, and extracting sufficient amounts of bacterial nucleotides for metagenomics or metatranscriptomics is challenging due to the predominance of host tissue. Differences may also arise due to variation in transit time among subjects. Furthermore, in this and most studies, samples were processed uniformly, ensuring that the same technical limitations apply to all phenotypes and that disease-specific differential expression is likely to reflect underlying biological differences.
In conclusion, metagenomics and metatranscriptomics can provide complementary insights into community interactions and disease-specific alterations in population-scale human microbiomes, here demonstrated in the IBD gut microbial community. In particular, disease-related changes in the gut environment may specifically affect microbial expression patterns, in different organisms and pathways among individuals, and in some cases without altering metagenomically-measured functional potential. In order to understand the underlying mechanisms associating microbial dysbiosis with aberrant immune responses, we need to understand how the behavior of individual organisms, as well as the gut community as a whole, changes in disease. Furthermore, disease-specific changes may be patient-specific and the specific microbial organisms in a patient’s gut may react differently to environmental changes, resulting in different short-term expression dynamics. Longitudinal, multi’omic, patient-focused studies will thus provide an important step towards understanding microbiome-related diseases and their roles in personalized medicine.
Methods
Experimental Model and Subject Details
Human Cohort
Patients at Massachusetts General Hospital [as part of the Prospective Registry in IBD Study (PRISM)], Emory University, and Cincinnati Children’s Hospital Medical Center, with a suspected diagnosis of IBD were approached for participation in the new-onset and pediatric portion of the study. Patients were consented prior to a screening colonoscopy, which separated them into confirmed IBD patients and non-IBD controls. Sampling and data gathering began at a later “baseline” visit no more than 6 months after their diagnosis was confirmed. New-onset patients were excluded if they were on an anti-TNF inhibitor. Established disease patients were recruited from the MLI cohort at the Cedars-Sinai IBD Center, and were required to have had a diagnosis of IBD for over 5 years. Participants in all groups were excluded if they were pregnant, had a known bleeding disorder, had taken antibiotics within the month preceding the screening visit, were actively being treated for a malignancy with chemotherapy, had an acute gastrointestinal infection, were diagnosed with an indeterminate colitis, or had had bowel/intestinal surgery other than an appendectomy or cholecystectomy. Non-IBD controls were further required to have no known immune-mediated disease (rheumatoid arthritis, lupus, or type 1 Diabetes mellitus).
In total, 117 patients participated in the study, with 59 CD patients, 34 UC patients and 24 non-IBD controls. This includes 55 pediatric patients (age ≤ 17 years; 13 non-IBD patients) with new-onset disease (13 UC, 29 CD) and 62 adults (age ≥ 18 years, 11 non-IBD patients), which were divided into new-onset (9 UC, 13 CD) and those with established disease (12 UC, 17 CD). Gender was balanced across all cohorts, with 57 male and 60 female patients overall, and no more than a difference of 2 patients between genders for any disease type. Stool samples were self-collected biweekly for one year from each patient according to the protocol established in47, starting from the baseline visit. Disease severity was monitored using the Harvey-Bradshaw Index48 (HBI) for CD patients and the Simple Clinical Colitis Activity Index49 (SCCAI) for UC patients.
Ethics statement
Subject recruitment and study procedures were approved by and carried out in accordance with the Research Ethics Boards of Massachusetts General Hospital (IRB for adult cohort: 2013P002215, IRB for pediatric cohort: 2014P001115), Cincinnati Children’s Hospital Medical Center (IRB: 2013-7586), Emory University (IRB: IRB00071468), and Cedars-Sinai Medical Center (IRB: 3358). In compliance with the Research Ethics Board study approval, informed consent was obtained from all study participants immediately prior to the initial sample collection. Further, all experimental methods are compliant with the Helsinki Declaration.
Method Details
Shotgun Sequencing
For metagenomic sequencing, the total genomic DNA content of the sample was sequenced, allowing us to infer functional potential of the community and taxonomic composition at species-level. For metatranscriptomics, messenger RNA (mRNA) was extracted, reverse transcribed into complementary DNA (cDNA), and subsequently sequenced. DNA was extracted from 300 samples spanning all 117 participants, and RNA from a subset of 78 samples spanning 28 participants. Illumina HiSeq sequencing yielded a total of 4.59 Gnt and 1.06 Gnt of paired-end reads (2×100 nt) of metagenomic and metatranscriptomic sequencing, respectively. Metagenomes averaged 30,581,993 reads ± 12,567,915 reads (mean ± s.d.) per sample before quality filtering (see below) and 28,242,423 reads ± 12,437,200 reads afterward. Meanwhile, metatranscriptomes averaged 27,211,997 reads ± 21,831,783 reads and 20,050,758 reads ± 16,301,242 reads before and after quality control, respectively.
Quantification and Statistical Analysis
Preprocessing and Quality Control
Sequence reads were processed with the KneadData v0.5.1 quality control (QC) pipeline (http://huttenhower.sph.harvard.edu/kneaddata), which uses the Trimmomatic50 and BMTagger51 filtering and decontamination algorithms to remove low-quality read bases and host (human) reads, respectively. Trimmomatic was run with parameters MAXINFO:80:0.5, and Phred quality scores were thresholded at <20. Trimmed non-human reads shorter than 50 nt were discarded. Potential human contamination was filtered by removing reads that aligned to the human genome (reference genome hg19). Additionally, metatranscriptomic reads were filtered against the human transcriptome and the SILVA database52. After QC, samples averaged 28 million and 20 million reads in MGX and MTX respectively (minimum 2 million).
Taxonomic and Functional Profiling
Taxonomic profiling was performed using the MetaPhlAn2 classifier53, which unambiguously classifies metagenomic reads to taxonomies based on a database of clade-specific marker genes derived from 17,000 microbial genomes (corresponding to >7,500 bacterial, viral, archaeal, and eukaryotic species). Functional profiling of metagenomes and metatranscriptomes was performed using HUMAnN224 version 0.9.6 (http://huttenhower.sph.harvard.edu/humann2). Briefly, the MetaPhlAn2 taxonomic profile generated from a metagenome is used to identify the set of organisms present in a sample. Metagenomic and metatranscriptomic reads are then mapped using Bowtie254 to sample-specific pangenomes including all gene families in any microbe present. A translated search using DIAMOND55 then maps unmapped reads against UniRef9056. Hits are counted per gene family and normalized for length and alignment quality. Gene family abundances from both the nucleotide and translated searches are then combined into structured pathways from MetaCyc57 and sum-normalized to relative abundances. We ran HUMAnN2 with the MinPath58 and gap filling options. As a result, 385 pathways had non-zero abundance in at least one metagenome, and 331 pathways had non-zero abundance in at least one metatranscriptome. The nucleotide search identified 182 species contributing these pathways in metagenomes, and 134 species in metatranscriptomes (a subset of the MGX species).
All of our datasets consisted of at least 2 million reads (corresponding to at least 20 observed reads per species) and the majority of them were in excess of 10 million reads (corresponding to at least 100 observed reads per species), ensuring that species calls were well supported (Fig. S2E).
Measuring Activity of Microbial Species
(Fig. 2A+D+E, 4E, S3C): The total contribution of each species to the functional profile was computed by summing their contributions over all pathways. Only the 51 species, which contributed at least one pathway in DNA and RNA level in >10% of samples (8 samples) were considered. Species contributions were first averaged over all samples where the species was detected within each patient and then across patients. Fig. 2B+C: For all species we computed Spearman correlation coefficients between their total pathway contributions to the metagenomes and metatranscriptomes across all samples.
Sample Order in Stacked Bar Plots
Stacked bar plots presented in Figs. 3, 4, S3, and S4 were ordered to maximize the similarity of species contributions to the pathway’s abundance between adjacent samples. For this, we calculated Bray-Curtis dissimilarities between the sum-normalized species contributions to the pathway in a given sample for both the metatranscriptomic and metagenomic data. The two dissimilarity matrices were combined by a weighted mixture, with metagenomic dissimilarities weighted at 1/100th of metatranscriptomic dissimilarities. The final sample order was determined by running solve_TSP from the R package TSP on the mixture dissimilarity matrix. To determine which sample to place first, we included a “fence” sample with zero dissimilarity to all other samples in the above procedure. The fence sample was then placed in the first position by rotating the final sample order appropriately, before finally removing it.
Contributional Alpha Diversity of Pathways
We quantified the contributional alpha diversity of species contributing to the abundance of a MetaCyc pathway in DNA or RNA by the Gini-Simpson index of alpha diversity. Pathways were first excluded if they had non-zero abundance in DNA in less than 95% of the samples, or if more than 25% of the pathway was attributed to unclassified organisms (from HUMAnN2’s translated search) in more than 25% of the samples. For each of the 58 remaining pathways, we then computed the Gini-Simpson index from the relative contribution of each species to the pathway for each sample (excluding unclassified organisms). The pathway’s alpha diversity was then defined as the mean alpha diversity of samples with non-zero abundance.
Data and Software Availability
Data Resources
All sequencing data and metadata is available on www.ibdmdb.org. The metagenomic and metatranscriptomic sequencing data is also available on SRA (BioProject: PRJNA389280).
Supplementary Material
Acknowledgements
We thank the participants from Massachusetts General Hospital, Emory University, Cedars-Sinai IBD Center, and Cincinnati Children’s Hospital Medical Center, who made this study possible. Further, we acknowledge Bahar Sayoldin for making the data available through the Short Read Archive (SRA) and our collaborators throughout the Integrative Human Microbiome Consortium. This work was supported by National Institutes of Health (NIH) grants U54DK102557 (CH, RJX), STARR Cancer Consortium (CH), CCFA 20144126 (RX) and R01DK92405 (RX), U01DK062413 (DPBM), P01DK046763 (DPBM), UL1TR001881 (JB), and The Leona M. and Harry B. Helmsley Charitable Trust (DPBM).
Footnotes
Competing interests
DPBM is consulting for Cidara. The authors declare no other competing financial interests.
References
- 1.Burisch J, Jess T, Martinato M & Lakatos PL The burden of inflammatory bowel disease in Europe. J Crohns Colitis 7, 322–37 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Inflammatory bowel disease in children and adolescents: recommendations for diagnosis--the Porto criteria. J Pediatr Gastroenterol Nutr 41, 1–7 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Kaplan GG The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol 12, 720–7 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Fava F & Danese S Intestinal microbiota in inflammatory bowel disease: friend of foe? World J Gastroenterol 17, 557–66 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hold GL et al. Role of the gut microbiota in inflammatory bowel disease pathogenesis: what have we learnt in the past 10 years? World J Gastroenterol 20, 1192–210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan XC et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13, R79 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gevers D et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–92 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lupp C et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2, 119–29 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Frank DN et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis 17, 179–84 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kostic AD, Xavier RJ & Gevers D The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146, 1489–99 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Llopis M et al. Lactobacillus casei downregulates commensals’ inflammatory signals in Crohn’s disease mucosa. Inflamm Bowel Dis 15, 275–83 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Sokol H et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis 15, 1183–9 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Sokol H et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 105, 16731–6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halfvarson J et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol 2, 17004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis JD et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 18, 489–500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhernakova A et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Machiels K et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63, 1275–83 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Nagao-Kitamoto H & Kamada N Host-microbial Cross-talk in Inflammatory Bowel Disease. Immune Netw 17, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rehman A et al. Transcriptional activity of the dominant gut mucosal microbiota in chronic inflammatory bowel disease patients. J Med Microbiol 59, 1114–22 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Dorffel Y, Swidsinski A, Loening-Baucke V, Wiedenmann B & Pavel M Common biostructure of the colonic microbiota in neuroendocrine tumors and Crohn’s disease and the effect of therapy. Inflamm Bowel Dis 18, 1663–71 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Bloom SM et al. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe 9, 390–403 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joossens M et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 60, 631–7 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Hoffmann TW et al. Microorganisms linked to inflammatory bowel disease-associated dysbiosis differentially impact host physiology in gnotobiotic mice. Isme j 10, 460–77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abubucker S et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 8, e1002358 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng Q et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun 6, 6528 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Zitomersky NL et al. Characterization of adherent bacteroidales from intestinal biopsies of children and young adults with inflammatory bowel disease. PLoS One 8, e63686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Downes J, Munson M & Wade WG Dialister invisus sp. nov., isolated from the human oral cavity. Int J Syst Evol Microbiol 53, 1937–40 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Maffeis C et al. Association between intestinal permeability and faecal microbiota composition in Italian children with beta cell autoimmunity at risk for type 1 diabetes. Diabetes Metab Res Rev 32, 700–709 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Ye JH & Rajendran VM Adenosine: an immune modulator of inflammatory bowel diseases. World J Gastroenterol 15, 4491–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antonioli L, Blandizzi C, Pacher P & Hasko G Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer 13, 842–57 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Bellaver B et al. Guanosine inhibits LPS-induced pro-inflammatory response and oxidative stress in hippocampal astrocytes through the heme oxygenase-1 pathway. Purinergic Signal 11, 571–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pier GB Pseudomonas aeruginosa lipopolysaccharide: a major virulence factor, initiator of inflammation and target for effective immunity. Int J Med Microbiol 297, 277–95 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santos MF et al. Lipopolysaccharide as an antigen target for the formulation of a universal vaccine against Escherichia coli O111 strains. Clin Vaccine Immunol 17, 1772–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Wang Q & Reeves PR The variation of O antigens in gram-negative bacteria. Subcell Biochem 53, 123–52 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Kintz E et al. Salmonella enterica Serovar Typhi Lipopolysaccharide O-Antigen Modification Impact on Serum Resistance and Antibody Recognition. Infect Immun 85(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aguirre-Ramirez M, Medina G, Gonzalez-Valdez A, Grosso-Becerra V & Soberon-Chavez G The Pseudomonas aeruginosa rmlBDAC operon, encoding dTDP-L-rhamnose biosynthetic enzymes, is regulated by the quorum-sensing transcriptional regulator RhlR and the alternative sigma factor sigmaS. Microbiology 158, 908–16 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Feurle J et al. Escherichia coli produces phosphoantigens activating human gamma delta T cells. J Biol Chem 277, 148–54 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Bang S, Yoo S, Yang TJ, Cho H & Hwang SW Nociceptive and pro-inflammatory effects of dimethylallyl pyrophosphate via TRPV4 activation. Br J Pharmacol 166, 1433–43 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naughton J, Duggan G, Bourke B & Clyne M Interaction of microbes with mucus and mucins: recent developments. Gut Microbes 5, 48–52 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skoog EC et al. Human gastric mucins differently regulate Helicobacter pylori proliferation, gene expression and interactions with host cells. PLoS One 7, e36378 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tu QV, McGuckin MA & Mendz GL Campylobacter jejuni response to human mucin MUC2: modulation of colonization and pathogenicity determinants. J Med Microbiol 57, 795–802 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Albenberg L et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 147, 1055–63.e8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LaFayette SL et al. Cystic fibrosis-adapted Pseudomonas aeruginosa quorum sensing lasR mutants cause hyperinflammatory responses. Sci Adv 1(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glucksam-Galnoy Y et al. The bacterial quorum-sensing signal molecule N-3-oxo-dodecanoyl-L-homoserine lactone reciprocally modulates pro- and anti-inflammatory cytokines in activated macrophages. J Immunol 191, 337–44 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao Y et al. Characterization of the Staphylococcus epidermidis accessory-gene regulator response: quorum-sensing regulation of resistance to human innate host defense. J Infect Dis 193, 841–8 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Thompson JA, Oliveira RA, Djukovic A, Ubeda C & Xavier KB Manipulation of the quorum sensing signal AI-2 affects the antibiotic-treated gut microbiota. Cell Rep 10, 1861–71 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Franzosa EA et al. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci U S A 111, E2329–38 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harvey RF & Bradshaw JM A simple index of Crohn’s-disease activity. Lancet 1, 514 (1980). [DOI] [PubMed] [Google Scholar]
- 49.Walmsley RS, Ayres RC, Pounder RE & Allan RN A simple clinical colitis activity index. Gut 43, 29–32 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.A framework for human microbiome research. Nature 486, 215–21 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quast C et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Truong DT et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods 12, 902–3 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buchfink B, Xie C & Huson DH Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Suzek BE, Huang H, McGarvey P, Mazumder R & Wu CH UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23, 1282–8 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Caspi R et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 40, D742–53 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ye Y & Doak TG A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput Biol 5, e1000465 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.