

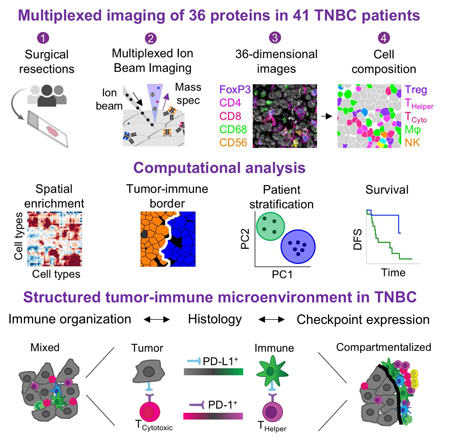
Abstract
The immune system is critical in modulating cancer progression, but knowledge of immune composition, phenotype, and interactions with tumor is limited. We used Multiplexed Ion Beam Imaging by Time-of-Flight (MIBI-TOF) to simultaneously quantify in-situ expression of 36 proteins covering identity, function and immune regulation at sub-cellular resolution in 41 triple-negative breast cancer patients. Multi-step processing, including deep-learning-based segmentation, revealed variability in the composition of tumor-immune populations across individuals, reconciled by overall immune infiltration and enriched co-occurrence of immune subpopulations and checkpoint expression. Spatial enrichment analysis showed immune mixed and compartmentalized tumors, coinciding with expression of PD1, PD-L1 and IDO in a cell-type- and location-specific manner. Ordered immune structures along the tumor-immune border were associated with compartmentalization and linked to survival. These data demonstrate organization in the tumor-immune microenvironment that is structured in cellular composition, spatial arrangement, and regulatory-protein expression and provide a framework to apply multiplexed imaging to immune oncology.
Introduction
Cancer progression is a complex process that depends on the interplay between individual cells in the tumor, the microenvironment, and the immune system, which can act both to promote and suppress tumor growth and invasion (Chen and Mellman, 2017). Immunotherapy, and in particular antibodies that target regulators of immune activation such as CTLA-4, PD1, and PD-L1, have significantly improved survival in renal cell carcinoma, lung adenocarcinoma, and melanoma (Ribas and Wolchok, 2018). Triple-negative breast cancer (TNBC) is an aggressive form of invasive breast cancer, negatively defined by lack of appreciable expression of the therapeutic targets estrogen receptor, progesterone receptor, and Her2 (Denkert et al., 2017). Consequently, radiation and chemotherapeutic neoadjuvant therapy are the current standard-of-care treatment. Several studies have shown increased tumor infiltrating lymphocytes (TILs) in TNBC compared to other breast cancers and correlations between specific immune subsets and prognosis (Andre et al., 2013; Denkert et al., 2010; Disis and Stanton, 2015; Teng et al., 2015), leading to clinical studies of several immunotherapeutic agents in TNBC (Kwa and Adams, 2018). Although preliminary, overall response rates typically range 15–30%, similar to observations in gastric and head and neck cancers (Muro et al., 2016; Seiwert et al., 2016). Many diagnostics have been suggested to delineate responders from non-responders, including expression of immuno-suppressive proteins, such as PD-L1 (Patel and Kurzrock, 2015), infiltration by CD8+ T cells (Tumeh et al., 2014), and expression of inflammatory genes, such as IFN-γ (Gao et al., 2016; Zaretsky et al., 2016). So far, however, no single biomarker has been sufficient for adequate patient stratification, presumably due to the complexity of the immune response to cancer (Maleki Vareki et al., 2017). To date, there is limited understanding of the tumor immune landscape: which immune cell types are found in the tumor, which cells express distinct immunoregulatory proteins, and how these vary between patients.
Tumors are spatially organized ecosystems that are comprised of distinct cell types, each of which can assume a variety of phenotypes defined by co-expression of multiple proteins. Thus, to interrogate solid tumor biology and response to treatment it is necessary to gauge the expression of a multitude of proteins with single-cell or even subcellular resolution while preserving spatial information. However, until recently, routine laboratory assays could only satisfy one of these two requirements – either measuring expression of one or two proteins in situ or many genes in cell suspensions from dissociated tissues (Chevrier et al., 2017; Matos et al., 2010). This disparity between our conceptual understanding of the tumor ecosystem and the experimental tools at our disposal has driven the recent developments of multiplexed imaging modalities (Chen et al., 2015; Coskun and Cai, 2016; Giesen et al., 2014; Huang et al., 2013; Lee et al., 2014).
We have previously reported Multiplexed Ion Beam Imaging (MIBI), a method that uses secondary ion mass spectrometry to image antibodies tagged with isotopically pure elemental metal reporters in intact tissue sections (Angelo et al., 2014). We have since constructed a purpose-built instrument that utilizes high brightness primary ion sources, novel ion extraction optics, and time-of-flight mass spectrometry (TOF) to increase channel multiplexing and decrease acquisition times by 50-fold (Methods). MIBI-TOF now enables fully automated, 40-plex imaging of large fields of view (up to 1mm2) at resolutions down to 260nm. It is compatible with a wide-range of sectioned tissues including formalin-fixed paraffin-embedded (FFPE) specimens, the primary preservation method of solid tissue biopsies in clinical anatomic pathology.
In this study, we leveraged MIBI-TOF to identify salient features demonstrating a structured tumor-immune microenvironment in a retrospective cohort of 41 TNBC patients. We imaged 36 proteins, including tumor and immune antigens and immunoregulatory proteins, and developed an extensible deep-learning-based pipeline for standardized processing, segmentation, and quantification of cell-types. We captured large differences between patients in both the composition and total number of immune cells. While the size of immune infiltrate spanned over two orders of magnitude, it correlated with tumor vascularity and immune composition. Co-occurrence of distinct immune populations across patients, in accordance with infiltrate size, suggest a lineage-specific interdependence where the presence of one cell type is frequently accompanied by the other. Simultaneously, we also evaluated the expression of four immunotherapy targets (PD-1, PD-L1, Lag3, and IDO) by distinct immune and tumor cell subsets and found differential enrichments across patients, highlighting the importance of multiplexed analysis. For example, PD-1 was primarily expressed on CD4+ T cells in some patients and on CD8+ T-cells in others. Expression of multiple immunoregulatory proteins by the same cell was statistically enriched, and patients that express one immunosuppressive pathway were more likely to express another, with potential implications for combination immunotherapies. In addition, we developed a data-driven approach to assess the spatial proximity of cell types in tissues. We found that tumors with similarly-sized immune infiltrates had different spatial organizations and divided the tumors into ‘Cold’ (no infiltrate), ‘Mixed’ (immune cells mixed with tumor cells) and ‘Compartmentalized’ (immune cells spatially separated from tumor cells). These subtypes were associated with distinct tumor and immune populations expressing immunoregulatory proteins. Highly ordered structures with PD-L1 and IDO along the tumor-immune border served as a hallmark of tumor compartmentalization and could be further linked to overall survival. Altogether, this work elucidates features of the tumor-immune microenvironment in TNBC and provides a starting framework for applying multiplexed imaging to uncover relational features of tumor, stromal, and immune cells in standard clinical specimens.
Results
Development of a multiplexed imaging assay for the tumor-immune microenvironment
The workflow for MIBI-TOF is comparable with that of conventional IHC (Fig. 1A). Clinical FFPE tissue specimens were placed on a slide and stained overnight using a single master mix of elementally-labeled primary antibodies (Bendall et al., 2011). All targets are stained together without the need for cyclical steps or enzymatic reactions. The slide is placed in the MIBI-TOF mass spectrometer (Fig. S1A), and the tissue is subjected to a nanometer-scale, rasterizing oxygen duoplasmatron primary ion beam. As this ion beam strikes the sample, elemental reporters conjugated to the antibodies are liberated as secondary ions, which are measured and quantified by a time-of-flight mass spectrometer. Thus, for each physical pixel in the tissue, a mass spectrum is recorded, representing the abundance of the antigens in that location (Methods).
Figure 1: Development of a multiplexed imaging assay for the tumor-immune microenvironment.
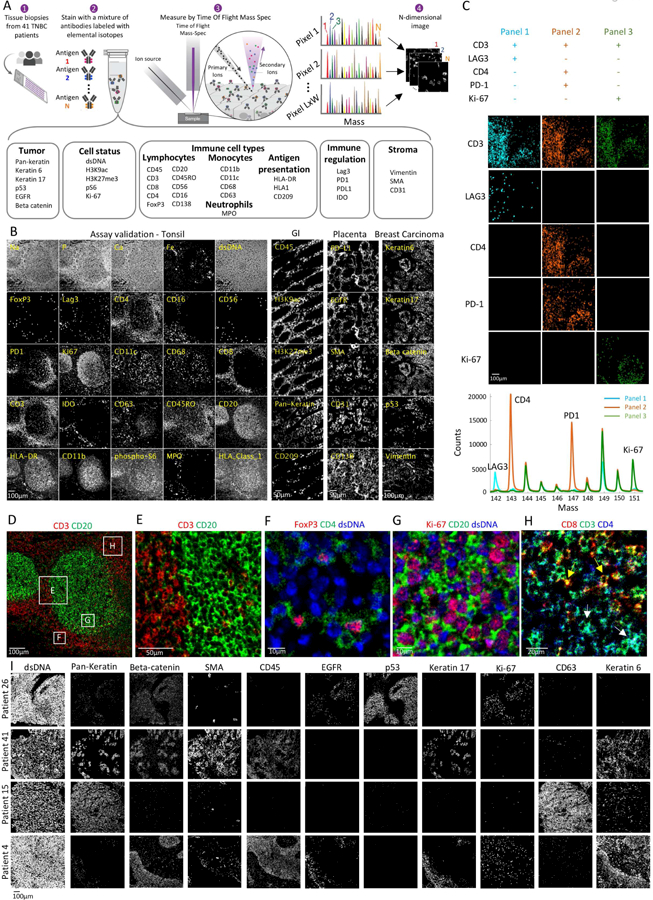
(A) Experimental workflow for MIBI-TOF. Clinical tissue specimens are stained using a mixture of antibodies labeled with elemental mass tags. The samples are rasterized by a primary ion beam that releases lanthanide adducts of the bound antibodies as secondary ions, which are recorded by a TOFMS. This results in an n-dimensional image depicting protein expression in the field. (B) MIBI-TOF images of samples stained with the panel in (A). (C) Serial sections of lymph node were stained with three sub-panels, each including 30 out of the 36 antibodies. Bottom: integrated counts across the image as a function of mass for the three panels. (D-H) Assay validation: Color overlays of tonsil show expected patterns of histologic architecture, subcellular staining (membrane/nucleus), and protein co-expression. (I) MIBI-TOF images of four TNBC patients.
We devised an antibody panel to interrogate 36 features of the tumor-immune microenvironment in TNBC (Fig. 1A, Table S1). The panel included tumor-related proteins (e.g. EGFR, p53), functional markers for proliferation and metabolic activity (e.g. Ki-67, pS6), immune-related proteins (e.g. CD4, FoxP3) and four immunoregulatory proteins (PD-1, PD-L1, LAG3 and IDO) currently targeted in the clinic or undergoing immunotherapy drug trials.
We performed several experiments to gauge assay sensitivity, specificity and repeatability. Antibody staining for each target was validated using six tissue types. Representative panel staining for tonsil, placenta, gastrointestinal tract and breast carcinoma are shown in Fig. 1B. Each antibody was tested across five or more titers with final titers selected to maximize the separation between signal and background (Methods). We first evaluated whether the staining of each antibody was specific to the known histologic distribution for each target and matched conventional imaging using immunohistochemistry (Fig. S1B). For example, bright epithelial staining for CK6 and CK17 was observed in morphologically distinct tumor cells in breast carcinoma, while very little was observed in the germinal or interfollicular regions in tonsil (negative staining not shown). CD20 primarily stained cells in the germinal center where B cells are the predominant population, while CD3 and CD4 primarily stained cells in the mantle zone, populated with T lymphocytes. (Fig. 1B,D–E). Second, we evaluated the subcellular localization of the different antigens. Expectedly, we find that transcription factors (e.g. Ki-67, FoxP3), are predominantly nuclear whereas membrane proteins (e.g. CD4, CD20) have low overlap with the nucleus (<10%, Fig. 1F,G). Third, we assessed the co-expression of proteins known to be expressed either on the same cell type or on different cell types. For example, consistent with T cell differentiation as either a helper (CD3+CD4+, Fig. 1H white arrows) or cytotoxic (CD3+CD8+, Fig. 1H yellow arrows) phenotype, we find that less than 3% of the CD3+CD4+ pixels are also CD8+ (Fig. 1H, S1C). Consistent with a T regulatory (Treg) phenotype, 99% of the FoxP3+ cells were positive for CD4 (Fig. 1F). In order to quantify cross-talk between markers we stained three serial sections from tonsil using four sub-panels, each one with six distinct empty channels. For each sub-panel we verified that the empty channels gave less than 0.1% of the original signal (Fig. 1C). Finally, we asserted the high reproducibility of our system by staining serial sections from human lymph node (R=0.9, P<10−20 Fig. S1D). Altogether, our assay allows to robustly image 36 antigens at sub-cellular resolution with a dynamic range sufficient for quantifying both high and low-abundance proteins in intact FFPE human tissues.
Automated image analysis pipeline delineates ordered immune composition in TNBC
We applied MIBI-TOF to profile a tissue microarray of 41 TNBC patients treated at Stanford hospital between 2002–2015 (Table S2). All samples were stained simultaneously using a single antibody master-mix, and scanned together to reduce technical variability. For each sample, we profiled a region of 8002μm2, 2048×2048 pixels, at 500nm resolution, obtaining information on thousands of cells (3,000–10,000 across patients). All the data, encompassing 1763 images, is available at https://mibi-share.ionpath.com, in a user-friendly interface, facilitating browsing. An initial overview identified large inter-patient heterogeneity in tumor protein expression, such as Keratins, EGFR and HLA-DR (Fig. 1I, S1E–J).
We developed a computational pipeline for data analysis (Methods). For each field of view (FOV), the counts for each reporter in each pixel were extracted from the mass spectra and converted to a multi-dimensional TIFF which was background subtracted using a blank channel. Noise was filtered using a k-nearest-neighbor approach and batch effects were removed using quantile normalization (Fig. S2A–F). To segment the cells from the images we leveraged the high-dimensionality of our data to adapt DeepCell (Van Valen et al., 2016), a convolutional neural network (CNN) for nuclear segmentation (Fig. 2A, Methods). To train the network, we manually segmented the nuclei of two patients (~8M pixels). The network achieved a per-pixel accuracy score of 74% on the training set and 73% on the validation (Fig. S2G). We applied the trained network to all patients in the cohort and output probability scores were post-processed to generate the final segmentation mask. The CNN approach yielded dramatically improved results compared with alternative methods tested (Fig. S2H). Following segmentation, we extracted the single-cell expression values for all the markers and clustered the cells from all patients (Van Gassen et al., 2015) (Fig. 2A, S2I).
Figure 2: Automated image analysis pipeline delineates ordered immune composition in TNBC.
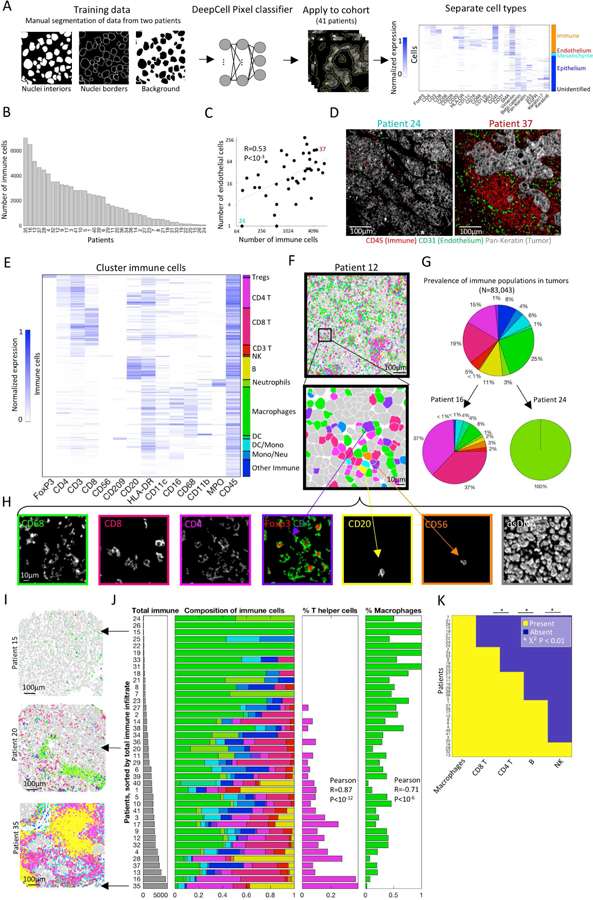
(A) Analytical pipeline for multiplexed imaging data. Cell segmentation is performed using a pixel-based convolutional neural network. Following segmentation, cellular features are extracted and cells are clustered into distinct populations. (B) The number of immune cells per patient. (C) The number of immune cells is correlated with the number of endothelial cells across patients. (D) Staining for tumor (Pan-Keratin), immune (CD45) and endothelium (CD31) in two patients. (E) Immune cells clustered by protein expression. Expression values for each protein are scaled from zero to one. (F) Pseudo-coloring of patient 12. Immune cells are color-coded as in E. Other cell types are colored gray. (G) Immune composition in all patients, and specific patients, color-coded as in E. (H) Expression of seven markers in the region shown in F. (I) Pseudo-coloring of three patients, color-coded as in E. (J) Left: Patients, sorted by number of immune cells (gray bars). Right: Immune composition normalized from zero to one (x-axis) in all patients (y-axis). Percentage of CD4+ T cells (magenta) increases whereas percentage of macrophages (green) decreases with total immune number. (K) For five immune populations shown is their presence or absence across patients.
To chart the immune landscape in TNBC we quantified the number of immune cells per area across patients and found large variability, spanning more than two orders of magnitude (Fig. 2B). This variability was confirmed by pathological TIL scoring on H&E staining of the entire cohort, as well as H&E staining of an additional core for patients 1–14, to establish low contribution of intra-patient variability in immune infiltration to these results (Fig. S3A–B, Table S2). To control for differences in absolute cell counts, we also quantified the fraction of immune cells in the field, which accordingly ranged from 1% to 91% (Fig. S4A). Total immune content was highly correlated with the amount of CD31+ vascular endothelium cells (Fig. 2C,D). However, there was no positive correlation between the number of immune cells or endothelial cells with the number of tumor cells, indicating no systematic differences between samples (Fig. S4B,C). These results highlight a dual role of angiogenesis in tumor biology. While vasculature is essential for tumor growth and survival by providing nutrients, and is accordingly a therapeutic target (Weis and Cheresh, 2011), it is also associated with increased immune infiltration into the tumor.
To identify the immune populations within tumors we clustered the immune cells by canonical markers (Fig. 2E, S4D, Methods). We found that our analytical pipeline was able to accurately classify immune cells, even when those with opposing identifiers were located in close proximity to each other (Fig. 2F,H). The frequency of immune subsets across all patients identified populations with a high (e.g. macrophages, 25%) and low prevalence (e.g. NK cells, <1%) where B-cells (11%), CD4+ (15%); CD8+ (19%), and regulatory (1%) T cells fell in between (Fig. 2G). However, across patients, we found large differences in both the variety and composition of the immune cells (Fig. 2G, Fig. S4G). Interestingly, the composition of immune populations across patients was highly correlated with the total number of immune cells. For example, patients that have more immune cells tended to have a larger fraction of CD4+ T cells (Pearson R=0.87, P<10−12) and a smaller fraction of macrophages (Pearson R=0.71, P<10−6) (Fig. 2I,J. Fig. S4E shows absolute numbers). Assuming that the level of immune infiltration could serve as a surrogate for time, these results may suggest a structured ordering of subsets entering the tumor.
To further gauge ordering in immune infiltration we analyzed the co-occurrence of immune populations across patients. For each patient, we classified the different immune populations as either present or absent in the sample, and evaluated their co-occurrence usinga chi-squared test. We found striking interdependence between the immune populations across patients (Fig. 2K). For example, all patients that had B cells also had CD4+ T cells and CD8+ T cells ( Χ2 P<0.005, P<0.0001 respectively). All patients that had NK cells, also had B cells (P<0.01). These results were robust to changing the thresholds for defining a population as present or absent (Fig. S4F). Similar correlations between one or two cell types have been observed previously (Yeong et al., 2017), but multiplexed imaging allowed us to evaluate the extent of this phenomenon across all major immune cell types simultaneously. Together, these results support a coordination in the immune response to tumors, whereby specific immune cells may be recruited to the tumor site in a context dependent manner.
Spatial analysis of TNBC reveals a hierarchy of organization of tumor and immune cells
To evaluate the spatial organization of the tumor-immune landscape in TNBC we developed a method for assessing spatial proximity enrichment for pairs of markers that accounts for differential tissue structure across varying cell numbers and composition (Fig. 3A). We quantified the number of positive cells for marker X that are located up to 100 pixels (39µm) from cells positive for marker Y. We repeatedly randomized the locations of Y+ cells, to generate a null-distribution of X-Y interactions and calculate a z-score representing the enrichment of X+ cells close or far from Y+ cells (Methods). Results were robust to ranging the distance and the number of randomizations (Fig. S5A). We applied this approach across the cohort and clustered the resulting pairwise-interactions. We found two types of prototypical spatial enrichment maps. In some patients, we observe a clear organization into two clusters – one encompassing tumor markers and the other encompassing immune markers. This corresponds to a physical separation of predominantly-immune regions and predominantly-tumor regions (Fig. 3B left). Alternatively, other patients exhibited less structure, with no predominant clustering of tumor and immune markers, indicating increased mixing of tumor and immune cells (Fig. 3B right).
Figure 3: Spatial analysis reveals a hierarchy of organization of tumor and immune cells.

(A) Assessment of enriched proximity/distance between two cell types, red and green. Location of red cells is randomized to generate a null distribution of red-green interactions. The actual number is compared to the distribution to calculate a z-score. (B) Top: Heat maps depicting spatial enrichment z-scores between pairs of proteins. Orange and blue boxes mark immune-and tumor-related proteins, respectively. Bottom: Pseudo coloring of immune and tumor cells. (C) Left: Patients, sorted by the number of immune cells and colored by their spatial architecture. Right: Pseudo coloring of immune and tumor cells in three patients. (D) Context-dependent spatial enrichment (CDSE) analysis controls for hierarchical tissue organization. The location of red cells is randomized only within the tumor compartment. (E) Heatmap of CDSE z-scores between pairs of proteins in patient 16. Squares are highlighted in F and G. (F) Pseudo-coloring of cell populations highlighted in E and G. (G) Protein staining in boxed regions in F. (H) Heatmap of CDSE z-scores between pairs of proteins in patient 6. Zoom-in on p53 shows its proximity to immune cells. (I) Pseudo-coloring of patient 6 showing increased proximity of p53+ tumor cells to immune cells. (J) CDSE z-scores, grouped for all patients. Patients are ordered as in K. (K) Zoom-in on a few interactions in J displaying variability (CD45xp53), depletion (Pan-KeratinxCD20) or enrichment (rest) across patients. Gray denotes patients with less than 10 positive cells for either maker.
To quantify the degree of mixing between tumor and immune cells we devised a mixing score, defined as the number of immune-tumor interactions divided by the number of immune interactions (Fig. S5B). We identified three archetypical subtypes of tumor-immune interactions: Cold– with low immune infiltrate, Mixed – with high mixing between tumor and immune cells, and Compartmentalized – in which there are regions comprised predominantly of either immune or tumor cells. Notably, tumors with similar numbers of immune cells can differ in their spatial organization and degree of mixing (Fig. 3C).
While the aforementioned analysis can identify global organizational patterns within the tissue, the same global patterns may hinder the analysis when probing expression of specific proteins. For example, if a protein is primarily expressed by endothelial cells, its z-score will be highly influenced by the spatial enrichments of endothelial cells. To control for such biases, we repeated our spatial enrichment analysis in a context-dependent manner. The analysis was as described above with the additional constraint that when performing randomizations, the total number of positive cells for each category (immune, epithelial, mesenchymal and endothelial) must be preserved. Thus, the expression of a protein that was expressed by endothelial cells was randomized only within endothelial cells, such that the final z-score reflected the spatial enrichment of that particular protein, and not of endothelial cells (Fig. 3D, Methods).
Applying this analysis to all patients revealed multiple degrees of spatial organization (Fig. 3E, S5C). First, we found enriched proximity of cells from the same lineage. For example, we identified tertiary lymphoid structures, comprised of clustered B cells (Fig. 3E,F,G1). Dendritic cells and neutrophils also tend to be spatially enriched next to cells of the same type (Fig. S5D). We also found that cells that were not necessarily from the same lineage, but shared similar expression features tended to be enriched in spatial proximity. For example, Ki-67+ tumor cells clustered together (Fig. 3E,F,G2) and IDO+ immune cells spatially clustered together, irrespective of their lineage (Fig. 3E,F,G3), which suggests that the functions associated with these proteins could largely be driven by the microenvironment. Finally, we identified enriched proximity of cells with distinct phenotypes. For example, patient 16 was enriched for neighboring cells expressing IDO and PD-1 (Fig. 3E,F,G4). Patient 6 had clusters of p53+ tumor cells, previously suggested to exert immunoregulatory activities (Di Minin et al., 2014), close to the tumor immune interface (Fig. 3H,I,S5E).
To identify spatial interactions that were either shared or unique between patients, we pooled the interactions from all patients together and performed hierarchical clustering (Fig. 3J, table S3). This showed that while some spatial features were represented with a subset of individuals (e.g. p53+ tumor cells near immune cells) others were more broadly conserved, suggesting design principles in tumor-immune organization (Fig. 3K). For example, we found conserved enrichment of neutrophils and depletion of B cells around tumor cells across patients. Immune cells consistently exhibited perivascular localization. Interestingly, while immunoregulatory proteins were not expressed in all patients, when present they were enriched to be located in close proximity. Altogether, we find that TNBC tumors and their immune microenvironment are spatially structured, with multiple layers of organization conserved across individuals; highlighting the importance of spatial information for providing context to single cell expression data.
Enriched co-expression of immunoregulatory molecules by specific cell types and patients
To chart the landscape of immune suppression in TNBC, we targeted four immunomodulatory proteins, PD-1, PD-L1, IDO and LAG3 - all approved or currently undergoing clinical trials as immunotherapy targets (Andrews et al., 2017; Kwa and Adams, 2018; Yu et al., 2017). We found that PD-1 and LAG3 were predominantly expressed by immune cells. In contrast, PD-L1 and IDO were expressed by both tumor and immune in similar proportions (Fig 4A). Examining the nature of immune cell types expressing each protein, we found that they can be expressed by a variety of cell types, consistent with some previous reports (Herbst et al., 2014) (Fig. 4B, S6A). For example, PD-L1 was expressed on CD4+ T cells, macrophages and dendritic cells. However, there was enrichment for specific cell types to express specific immunoregulatory proteins (Methods, Fig. 4C). PD-1 was enriched on both CD4+ and CD8+ T cells, whereas LAG3 was enriched on CD8+ T cells and Tregs, and PD-L1 and IDO were enriched on monocytes. The ability to easily distinguish differences in the expression of immunoregulatory proteins between cells types may provide a promising avenue for addressing inter-observer variability and discordance in PD-L1 scoring (Troncone and Gridelli, 2017).
Figure 4: Enriched co-expression of immunoregulatory proteins by cell types and patients.
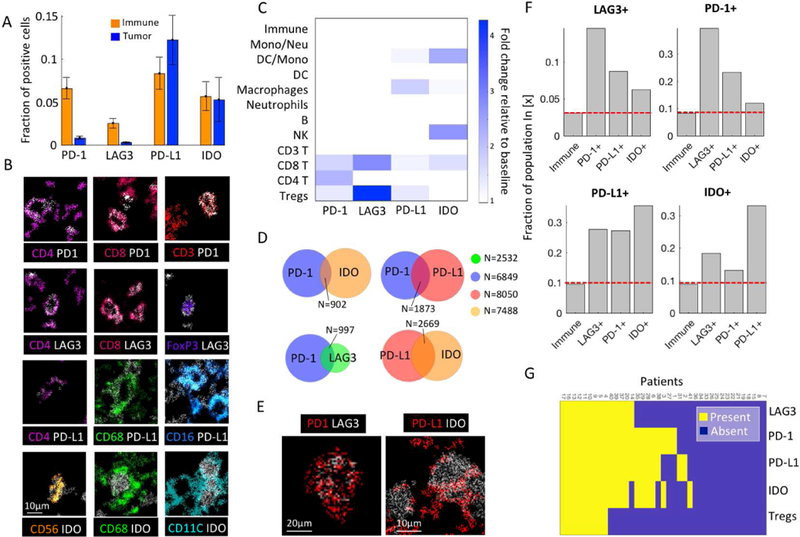
(A) Fraction of immune and tumor cells expressing immunoregulatory proteins (IRPs). Mean and SE across patients are shown. (B) Color overlays of immune lineage proteins (rainbow) and IRPs (white). (C) For each IRP, shown is the log2 fold change relative to the baseline in representation of the different immune populations. (D) Cellular co-expression of IRPs. (E) Co-expression of IRPs. (F) For each IRP, shown is the fraction of positive cells out of all immune cells, and out of immune cells positive for other IRPs. Left bar is consistently the lowest, indicating increased probability for IRP co-expression. (G) Presence or absence of IRPs and Tregs across patients.
Beyond individual marker patterns, our multiplexed analysis revealed cells that expressed combinations of immunoregulatory proteins (Fig. 4D,E, S6B). For example, out of 2,532 LAG3+ cells, 997 (39%) also expressed PD-1, in agreement with previous reports (Burugu et al., 2017; Woo et al., 2012). To assess the degree of co-expression, for each regulatory protein we compared the fraction of positive cells out of the general immune population, and the fraction of positive cells out of immune cells positive for each of the other regulatory molecules, and found consistently higher fractions for the latter (Fig. 4F). Specifically, we observed significant co-expression for IDO and PD-L1 (35%), and LAG3 and PD1 (39%). We confirmed the significance of these co-expressions even when controlling for enriched expression of different regulatory proteins by specific immune cell types (P<10−10, Methods, Fig. S6C). Interestingly, the relationship is not symmetrical (e.g. the proportion of LAG3+ cells out of PD-1+ cells is 0.15 and the proportion of PD-1+ cells out of LAG3+ cells is 0.39), which may suggest ordered or differentially-regulated protein expression. Certainly, these results indicate that expression of one immunoregulatory protein increased the probability that other regulatory proteins would be present. Recalling that immune cells positive for distinct immunoregulatory proteins were also enriched to reside in spatial proximity across patients (Fig. 3J–K) these combined observations reinforce that environmentally-derived signals may have pleiotropic effects in driving overall localization and coordination of immune inhibition.
In line with quantifying the coincidence of immune cell populations within an individual tumor (Fig. 2J–K) we also capture the co-expression of immunoregulatory proteins across patients (Fig. 4G). We classified each immunoregulatory protein as either present or absent in an individual, and clustered the resulting matrix (Fig. 4G). We found that patients that express one immunosuppressive pathway are more likely to express another. Most patients had either no expression of immunoregulatory proteins, or they expressed several, with only 5 patients expressing only a single regulatory protein. These results were robust to analysis of background probabilities, selection of thresholds, and to analysis of either absolute numbers or percentages (Fig. S6D–F). Expression of distinct immunoregulatory proteins was highly correlated; for example, all patients that had Tregs, also had expression of LAG3, PD-1, PD-L1 and IDO (Χ2 P<10−7, P=0.003, P=0.002, P<0.001 respectively). These observations may reflect the ability of IDO to drive the differentiation of naive CD4+ T cells toward an inducible Treg phenotype (Munn, 2011). Altogether, the landscape of immune regulation in this cohort suggest that combination immunotherapies would likely increase the total pool of targeted cells and could be synergistic in blockade of double positive cells in patients that respond to monotherapy, as demonstrated in both mice (Woo et al., 2012) and humans (Ott et al., 2017; Postow et al., 2015). However, due to the interdependence of immunoregulatory protein expression here, combination immunotherapies may provide only a modest increase to the pool of responsive patients, as observed in anti-PD-1 anti-CTLA-4 combination therapies in melanoma (Larkin et al., 2015).
Expression patterns of immunoregulatory proteins coincide with TNBC architecture
We assessed inter-patient heterogeneity in immunoregulatory expression profiles. While we generally observed similar enrichments across patients (e.g. enrichment of PD-1 expression on T cells), we identified several pronounced differences between patients in the cell types expressing different regulatory proteins (Fig. S6G,H). We found that some patients have predominantly PD1+ CD4+ T-cells whereas others have predominantly PD1+ CD8+ T-cells. For example, patient 35 has 368 PD1+ immune cells. Of these, 68% are CD4+ and 8% are CD8+. Similarly, patient 14 has 390 PD1+ cells. However, of these, 7% are CD4+ and 78% are CD8+ (Fig. 5A). Expression of PD-1 on either Cytotoxic (CD8+) or T-helper cells (CD4+) may be crucial for tumor-immune interactions and immunotherapy outcome and is difficult to assess by current, IHC-based scoring criteria (Kwa and Adams, 2018).
Figure 5: Expression patterns of immunoregulatory proteins coincide with TNBC architecture.
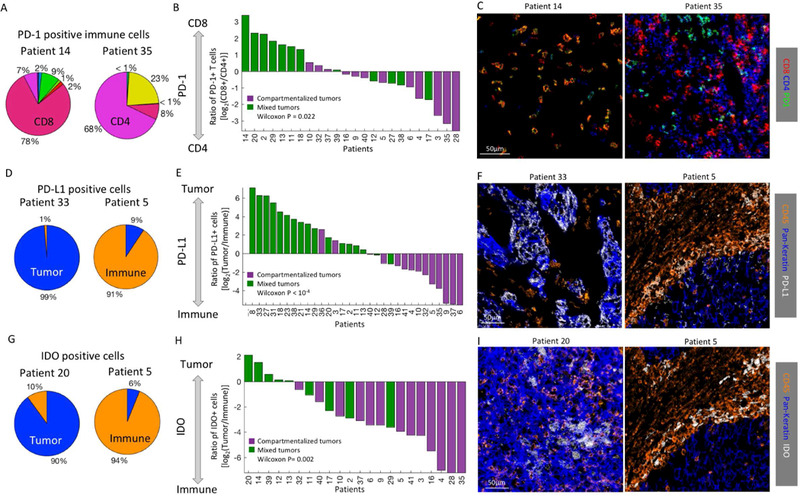
(A) PD-1+ immune cell composition in two patients. Immune populations are color-coded as in 2E. (B) For all patients with over 20 PD-1+ cells (x-axis) shown is the log ratio of PD-1+CD8+ and PD-1+CD4+ T-cells. Patients are colored by their spatial architectures. Mixed tumors tend to have more PD-1+CD8+ T cells than PD-1+CD4+ T cells, as determined by Wilcoxon rank-sum test. (C) Color overlay of CD8, CD4 and PD-1. (D) PD-L1+ cell composition in two patients. (E) Same as B, showing the log ratio of PD-L1+ tumor and immune cells. (F) Color overlay of CD45 (immune), Pan-keratin (tumor) and PD-L1. (G) IDO+ cell composition in two patients. (H) Same as B, showing the log ratio of IDO+ tumor and immune cells. (I) Color overlay of CD45 (immune), Pan- keratin (tumor) and IDO.
Interestingly, we found that expression of immunoregulatory proteins by distinct cellular subtypes correlates with the spatial architecture of the tissue as mixed and compartmentalized (Fig. 3B–C). Predominance of PD1 expression by either CD4+ or CD8+ T-cells correlates with compartmentalized or mixed TNBC, respectively (Wilcoxon Rank Sum test P=0.022, Fig. 5A–C, Methods). This relationship was not driven by the ratio of total CD8+ and CD4+ T cells in compartmentalized and mixed tumors (Fig. S6I,J), nor was it affected by ranging the threshold used to include patients in the analysis between 10 and 100 positive cells (Fig. S7A–C).
We observed similar architectural restrictions for PD-L1 and IDO. Here, individuals exhibited a wide range of enriched PD-L1 or IDO expression on tumor versus immune cells (Fig. 5D–F). At the same time, lineage enrichment of immunoregulatory proteins could be linked to the tumor architecture where mixed tumors had PD-L1 and IDO expression primarily on tumor cells and compartmentalized tumors had PD-L1 and IDO expression predominantly on immune cells (Wilcoxon Rank Sum test, IDO P=0.002 , PD-L1 P<10−4). These results relate molecular expression profiles in a cell-specific manner to histological attributes of tumors, highlighting the multiple layers of information that can be gleaned from multiplexed imaging.
The tumor border has complex multicellular structures of immune regulation
To further explore the relationship between molecular expression and tissue architecture, we focused on the tumor-immune boundary, which has been previously implicated to play a prognostic role in tumor progression (Fortis et al., 2017). We developed a computational approach to automatically identify the tumor-immune border from the clustering annotations of the cells, and applied it to the compartmentalized tumors (Methods). Using a threshold of 100 pixels (39μm), for each tumor we classified the tumor and immune cells as those that resided close or far from the border (Fig. 6A). To identify differential expression along the border, for each marker in every patient we extracted the expression distribution on cells close and far from the border and compared the distributions using Wilcoxon’s Rank Sum test. For example, in patients 4 and 9 we found a gradient of histone H3 methylation to acetylation levels on tumor cells perpendicular to the tumor-immune border (Fig. 6B (white arrows), C, Wilcoxon P=10−20). Since H3K9ac was shown to be correlated with open chromatin and H3K27me3 with closed chromatin (Kouzarides, 2007), these global changes in their ratios may indicate that cells on the tumor border are more transcriptionally active than those in the tumor center.
Figure 6: The tumor border has complex multicellular structures of immune regulation.
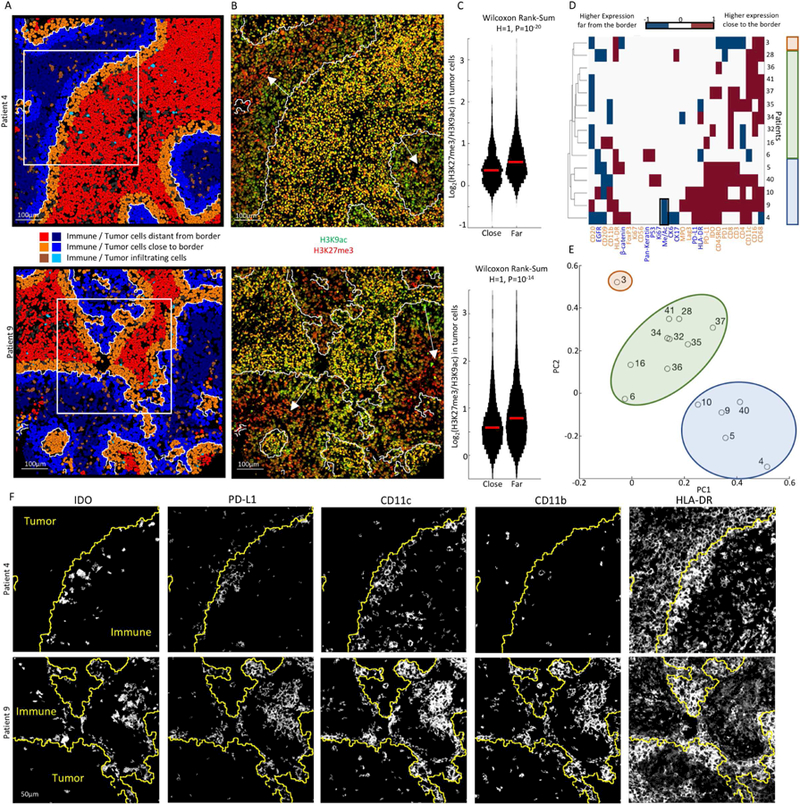
(A) Computationally-derived tumor-immune border (white line) in patients 4 and 9. Tumor and immune cells are pseudo-colored by their location relative to the border. Insets are shown in F. (B) Color overlays for H3K9ac and H3K27me3. The tumor-immune border is white. Arrows show a gradient of HH3 acetylation to methylation in tumor cells from the boundary inwards. (C) Distribution of the log-ratio of H3K27me3 to H3K9ac in tumor cells close or far from the tumor- immune border. Median is marked in red. (D) For each protein in either tumor (blue) or immune (orange) cells (x-axis), shown is the H-value from a Wilcoxon rank-sum test evaluating differential expression in cells close or far from the border for compartmentalized patients (y- axis). Patients and proteins are sorted by hierarchical clustering. Colored rectangles show groups identified by clustering and principle component analysis (PCA). Black box highlights results in C. (E) PCA of the data in D. Patients are plotted on the first and second PCs. (F) Staining for different proteins in the boxed regions in A. The tumor-immune boundary is in yellow. Immune cells near the border co-express IDO, PD-L1, CD11c and CD11b. Tumor cells express HLA-DR, with a gradient from the boundary inwards.
To more broadly assess recurrent spatial expression within compartmentalized TNBC patients, we combined significant results (Bonferroni corrected, P<10−5) across patients and markers and subjected the resulting data to hierarchical clustering (Fig. 6D) and principal component analysis (PCA, Fig. 6E). In accordance with our previous results, we found that B cells are consistently depleted along the tumor boundary across patients (Fig. 6D, 3J). In addition, we identify a subset of patients, which exhibit unique properties along the border (patients 4,5,9,10 and 40). This cluster of patients displayed increased expression of PD-L1, PD-1, and IDO by CD11c+ CD11b+ immune cells, a phenotype suggestive of myeloid derived suppressor cells (Gabrilovich and Nagaraj, 2009). These patients also tended to have HLA-DR positive tumors, with higher expression close to the boundary (Fig. 6F). This expression pattern may be suggestive of localized production of cytokines like IFN-γ (Carrel et al., 1985). These findings indicate that the tumor-immune border is a unique site of immune inhibition with altered expression profiles by both tumor and immune cells. The shared expression profiles for multiple proteins in distinct patients suggest a prototypical spatial expression signature, which may stratify patients with similar tumor-immune interactions (Fig. 7A).
Figure 7: Tumor-Immune mixing and immune-regulatory expression patterns relate to overall survival.
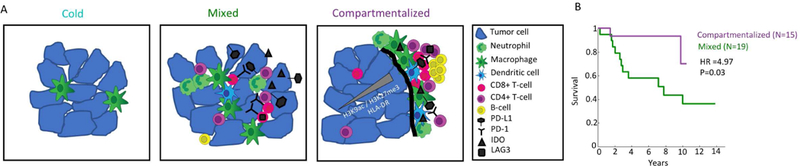
(A) Cartoon depicting three archetypes of tumor-immune composition and organization in TNBC. Cold tumors have few immune cells, mainly macrophages. Mixed tumors have tumor and immune cells mixed together. IDO and PD-L1, if expressed, are expressed primarily on tumor cells and PD-1 on CD8+ T cells. In compartmentalized tumors, the immune and tumor cells are spatially segregated. Neutrophils are enriched near the border, whereas B-cells form secondary lymphoid structures further away. IDO and PD-L1 are expressed primarily on immune cells and PD-1 on CD4+ T cells. A subset of tumors express HLA-DR, with a gradient of HLA-DR and H3K9ac/H3K27me3 from the border towards the tumor center. (B) Kaplan-Meier curves showing survival as a function of time for patients with compartmentalized or mixed tumor- immune organizations. Hazards ratio (HR) and p-value (P) were calculated using Cox regression analysis.
Finally, we examined whether the tumor-immune landscape is relevant to prognosis. We partitioned the patients into ‘Cold’, ‘Compartmentalized’ and ‘Mixed’ as described above. Since the ‘Cold’ group had only 5 patients, we discarded it from further analysis. Compartmentalized organization was associated with increased survival (Cox regression HR=4.97, P=0.03 Fig. 7B), regardless of the specific threshold used to separate compartmentalized and mixed phenotypes, and independently from pathological TIL scoring (Fig. S7D–H). This suggests that the clinical outcome of TNBC is influenced by the tumor immune microenvironment and emphasizes the clinical importance of the immune system even in non-immunotherapeutic settings.
Discussion
We developed MIBI-TOF, a multiplexed imaging platform, and applied it to interrogate the tumor-immune landscape by simultaneously imaging the expression of 36 proteins in archival tissues of 41 patients. This approach allowed us to describe how cell identity and phenotype (who) is related to tissue architechure (where) in TNBC. We found that the tumor-immune microenvironment can be categorized into prototypical archetypes with structured cellular composition, spatial arrangement, and expression of regulatory proteins that related to overall survival. Interrelated presence of distinct immune cell populations in the tumor region sheds light on how different immune cells come together and operate as a system to mount the immune response, and the co-dependence between different cell types. The connection observed between the presence of immune subsets and infiltrate size (Fig. 2) may suggest temporal ordering in immune infiltration into the tumors. Temporal ordering of the immune response has been shown in various processes, including viral infections (Dunn et al., 2009) and inflammation (Bagaitkar, 2014), and may play a role here. Further investigations, including longitudinal data, are necessary to further delineate ordering and immune cell co-dependence in cancer.
We also identified conserved design principles in the relative spatial organization of tumor and immune cells (Fig. 3). These included enriched proximity of cells from the same lineage, as well as cells from distinct lineages with similar phenotypes (e.g. proliferating cancer cells or checkpoint-positive immune cells) and specific distinct phenotypes (e.g. cells positive for different immunoregulatory proteins). These implicate a prominent role of the microenvironment in driving cell phenotype, cell recruitment, or both (Oelkrug and Ramage, 2014). In this sense, multiplexed imaging is uniquely positioned to decouple microenvironmental effects from cell-intrinsic properties.
Given the recent successes of checkpoint blockade in establishing durable remissions in multiple malignancies (Ribas and Wolchok, 2018), we evaluated the expression of immunoregulatory proteins across patients (Fig. 4). We found that expression of one protein increased the probability that other regulatory proteins would be present, both at the cellular level and at the patient level. In conjunction with the enriched spatial proximity of distinct regulatory proteins, these results support overlapping microenvironmental cues in inducing immune inhibition. These mechanisms could include shared signaling pathways and positive feedback between immunoregulatory proteins (Nirschl and Drake, 2013; Woo et al., 2012), and may be a valuable avenue for future mechanistic exploration.
Expression of immunoregulatory proteins differed across patients not only in quantity, but also with respect to the identity of the cell types expressing them. For example, PD-L1 was primarily expressed by tumor cells in some patients and by immune cells in others (Fig. 5). Importantly, these distinctions represent different tumor-immune states, which are currently not captured in patient stratification to clinical trials. Interestingly, these patterns of expression coincided with the tumor-immune architecture in patients as Cold, Mixed and Compartmentalized. Thus, the observations here allow us to relate single cell expression patterns to broader histological attributes of the tumors. This relationship was especially striking in a subset of compartmentalized patients, which exhibited ordered expression patterns by both immune and tumor cells at the tumor-immune border (Fig. 6). These included increased expression of PD-L1 and IDO by monocytes and increased expression of HLA-DR and H3K9ac by the tumor cells at the boundary. We note that the distinction between compartmentalized and mixed is not clear cut, as we observe a continuum of mixing scores between tumor and immune cells across patients. Since the compartmentalized histology correlated with improved overall survival, we propose that further interrogation into additional patients displaying similar expression properties will prove valuable in the context of standard-of-care chemotherapy and emerging immunotherapy regimens. Linking these micro-environmental immune features to existing molecular data on TNBC (Burstein et al., 2015; Lehmann et al., 2016) may provide future avenues for patient stratification and therapeutic design. Emerging cohorts, in which several regions are profiled for each patient, are needed to investigate the intra-patient heterogeneity in the composition and architecture of the tumor-immune microenvironment and its relationship to the molecular attributes of the tumor.
We developed a multi-step pipeline to analyze MIBI-TOF data, including low-level data processing, segmentation, extraction of counts, clustering, and spatial analysis. Each of these steps could be algorithmically improved, and we anticipate this to be an active field of research in the near future. We note that applying deep learning for nuclear segmentation outperformed all other methods in both segmentation results, scalability, and ease of use. Follow-up work which fully exploits the power of MIBI-TOF, such as extending this method to include additional cellular compartments as well as multi-cellular structures and 3-D data, will facilitate quantification of finer attributes of protein expression and regulation. Our spatial analysis revealed multiple layers of organization within tumors. This hierarchical spatial organization poses a confounding factor when evaluating proximity enrichment and may lead to misleading results if not properly controlled.
Taken together, our application of MIBI-TOF has revealed how tumor expression and immune composition are interrelated within a histological context that correlates with overall survival in TNBC. A better understanding of the spectrum and dynamics of immune states in cancer is critical for establishing faithful models and advancing therapeutic strategies that address the complexity of this disease. As clinical trials of immunotherapeutic agents and their various combinations progress, this approach should provide the insights needed to develop reliable guidelines for drug selection that will increase therapeutic response.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Angelo (mangelo0@stanford.edu).
Experimental model and subject details
Samples of TNBC patients of no special type (ER and PR positivity less than 1%, HER2 unamplified and Her2 not 3), as well as tonsil, lymph node, placenta, gastrointestinal tract and breast carcinoma controls were obtained from Stanford Pathology department. TNBC biopsies were reviewed by expert pathologists and representative 1mm cores were compiled into a tissue microarray (TMA). For one of the patients two cores were included in the dataset (#15 and #22). H&E staining was performed on all cores as previously described. For patients 1–14, an additional core was used for H&E staining, to assess the intra-patient variability in the amount of immune infiltration (Fig. S3). TIL scores were generated by a pathologist using H&E stained sections of the tissue microarray cores, according to recommended guidelines (Salgado et al., 2015). A four-tier scoring system was used such that TIL score 1, 2, 3, and 4 corresponded to TIL cellularity of <25%, 25–49%, 50–75%, and >75%, respectively. Data regarding age, tumor grade, TNM, clinical stage, TIL enumeration and survival for all patients are provided in table S2.
Method details
Gold Slide preparation
Slide preparation was performed at the Stanford Nano Shared Facility (SNSF). Superfrost plus glass slides (Electron Microscopy Sciences, Hatfield, PA) were soaked in water with dish detergent and rinsed extensively with distilled water and then acetone (Avantor-Macron Fine Chemicals, Center Valley, PA). Acetone was immediately evaporated under a stream of air to avoid trace of residues. Slides were coated with Tantalum (Ta, 30nm) and Gold (Au, 100nm). The slides were placed ten at a time in adjustable rails in an e-beam evaporation station and pumped down for about two hours prior to deposition. The base pressure before the deposition was ~4 × 10−7 Torr. The distance between the source and the substrate was 30cm.The Ta was evaporated at a rate of ~10 Angstroms per second. The Au was evaporated immediately after at a rate of 10–15 Angstroms per second. Gold-coated slides were silanized in acetone with 3-aminopropyltriethoxysilane for 30 min then washed with acetone and air dried (Vactabond, Burlingame, CA). Slide were baked at 70°C for 30 min and kept dry at room temperature until use.
Antibody conjugation
A summary of antibodies, reporter isotopes, and concentrations can be found in Table S1. Metal conjugated primary antibodies were prepared as described previously (Bendall et al., 2011). Following labeling, antibodies were diluted in Candor PBS Antibody Stabilization solution (Candor Bioscience GmbH, Wangen, Germany) to 0.2 mg mL−1 and stored long-term at 4 °C.
Staining
Tissue sections (4 μm thick) were cut from FFPE tissue blocks of the TNBC tissue microarray (TMA) or control tissues using a microtome, mounted on silanized-gold slides for MIBI-TOF analysis. Slide-tissue sections were baked at 70°C for 20 min. Tissue sections were deparaffinized with 3 washes of fresh-xylene. Tissue sections were then rehydrated with successive washes of ethanol 100% (2×), 95% (2×), 80% (1×), 70% (1×), and distilled water. Washes were performed using a Leica ST4020 Linear Stainer (Leica Biosystems, Wetzlar, Germany) programmed to 3 dips per wash for 30 sec each. The sections were then immersed in epitope retrieval buffer (Target Retrieval Solution, pH 9, DAKO Agilent, Santa Clara, CA) and incubated at 97°C for 40 min and cooled down to 65˚C using Lab vision PT module (Thermofisher Scientific, Waltham, MA). Slides were washed with a wash buffer made with PBS IHC Tween buffer (Cell Marque, Rocklin, CA) containing 0.1% (w/v) BSA (Thermofisher Scientific, Waltham, MA). Endogenous avidin, biotin binding proteins were blocked using Avidin/Biotin blocking systems (Biolegend, San Diego, CA). Sections were treated successively with avidin and biotin blocking solutions for 10 min and washed for 5 min in wash buffer. Sections were then blocked for 1h with 3% (v/v) donkey serum (Sigma-Aldrich, St Louis, MO) diluted in TBS IHC wash buffer (Cell Marque, Rocklin, CA). Metal-conjugated antibody mix was prepared in 3% (v/v) donkey serum TBS IHC wash buffer and filtered using centrifugal filter, 0.1 μm PVDF membrane (Ultrafree-MC, Merck Millipore, Tullagreen Carrigtowhill, Ireland). Two panels of antibody mix were prepared where the first panel contained the majority of the metal-conjugated antibodies and a biotinylated anti-PD- L1 antibody. This antibody mix was incubated overnight at 4°C in humid chamber. The second mix contained antibodies of structural genes with strong signal and an anti-biotin antibody. After overnight incubation, slides were washed on orbital shaker for 5 min in wash buffer (Table S1). The second, antibody mix was then applied and incubated for 1h at 4°C. Slides were then washed twice 5 min in wash buffer and fixed for 5 min in diluted glutaraldehyde solution 2% (Electron Microscopy Sciences, Hatfield, PA) in PBS-low barium. Slides were then rinsed briefly in PBS-low barium. Tissue sections were then dehydrated with successive washes of Tris 0.1 M (pH 8.5), (3×), distilled water (2×), and ethanol 70% (1×), 80%(1×), 95% (2×), 100% (2×). Slides were immediately dried in a vacuum chamber for at least 1 h prior to imaging.
IHC
The protocol for IHC closely followed the MIBI-TOF staining protocol, with a few changes. Before blocking, endogenous peroxidase activity was quenched by incubation in 3% H2O2 for 30 min and sections were washed with H2O on orbital shaker for 5 min. Sections were stained using the ImmPRESS universal (Anti-Mouse/Anti-Rabbit) kit (Vector labs) according to the manufacturer’s guidelines.
Tissue controls and titer selection
For each antibody, appropriate positive and negative tissue controls were selected. Tissues included tonsil, lymph node, placenta, gastrointestinal tract, skin, breast and breast carcinoma. The antibody panel was mixed according to the manufacturers’ suggested titers and then serially diluted 2-fold five times. For each antibody, the working titer was determined as the titer that yielded maximal separation between signal and noise across tissues (See section ‘Noise Removal’ for a description of how signal and noise were separated).
MIBI-TOF
Quantitative imaging was performed using a custom designed MIBI-TOF mass spectrometer equipped with a duoplasmatron ion source. O2+ primary ions are focused onto the sample at a working distance of 15 mm and 45° incident angle using a two lens, 30KV ion column operating in crossover mode. For each pixel in the image, corresponding positions on the tissue are sequentially sputtered with the primary ion beam using a fly back raster pattern. Secondary ions collected using a +68V sample bias and −125V extraction field subsequently pass through an electrostatic analyzer (ESA) with 5eV energy acceptance that has been tuned for preferential transmission of monoatomic ions relative to polyatomic organic species.The secondary ions then enter an orthogonal time of flight mass spectrometer with a mass range of 1–200 m/z+ and mass resolution of 1000 m/Δm operating at 100KHz repetition rate.Ion events are recorded using a time to digital converter (TDC) with 500ps time resolution.Lastly, multiple TOF spectra for each pixel are summed and saved to a data file.The experimental parameters used in acquiring all imaging data are as follows:
Pixel dwell time:6.4ms
Image size:800um2 at 2048 × 2048 pixels (for cohort data) or 400um2 at 1024×1024 pixels (for controls).
Probe size:500nm
Primary ion current:3.5 nA O2+
Ion dose per unit area:147 pC/μm2
Primary ion current was monitored continuously via an in stage current monitor, which demonstrated maximal variations of approximately +/− 1% during continuous operation over a 24 hour period.Using the acquisition settings aforementioned, this cohort of 41 TNBC patients was acquired across 13 days of continuous operation using autonomous software-based acquisition from pre-defined user FOVs.A total of ~180 million pixels were acquired in that time, generating a total of 1763 images.
Quantification and statistical analysis
Image analysis pipeline
Spectra calibration and file conversion.
For each field of view, Mass-spec pixel data was converted to a multi-dimensional TIFF. Counts for each mass were defined according to whether they fell between the ‘Start’ and ‘Stop’ values defined in table S1. For example, for FoxP3, conjugated to Pr 141, masses between 140.8 and 141.2 were considered. Time of Flight data was calibrated using the Sodium (Na, mass 22.99) and Gold (Au, mass 196.97) peaks (Fig. S2A).
Background subtraction.
Background across all channels was highly correlated to bare spots (with no tissue) on the slide. Background was filtered using a blank channel (mass 128–132). The background image was smoothed using a Gaussian kernel with R=3 pixels and then thresholded (T=0.07) to obtain a binary mask. For all other channels, positive pixels in the background channel were subtracted by two counts. Following the subtraction, negative values were converted to zeros. We found that this removal was sufficient to remove background-related noise, while preserving real signal. Channel 169 (CD45) exhibited additional background signal, highly correlated with gold, and so for all fields of view, this channel underwent similar background removal using the gold channel (T=0.3). Channel 144 (CD16) displayed non-specific nuclear signal, and underwent similar background removal using channel 89 (dsDNA) (T=0.2) (Fig. S2B).
Necrosis.
Necrotic regions were automatically identified using the dsDNA channel and verified using pathological inspection of H&E serial sections. The dsDNA image was smoothed using a Gaussian kernel with R=2, and subjected to morphological closing and opening (matlab imclose, imopen), and connected components of size < 10,000 pixels were removed. Necrotic regions were removed from the channels used for segmentation prior to segmentation (Fig. S2C).
Batch Normalization.
Quantile normalization was performed for Pan-keratin in patients 1–29 and 31–41, which were stained individually and exhibit systematic differences in intensity. Counts for each pixel in each batch were sorted and subsampled to create equally sized samples between the two batches. A transfer function mapped each value in batch 2 to the correspondingly ranked value in batch 1 (Fig. S2D).
Noise Removal.
MIBI-TOF data has two properties, which make it a unique challenge for denoising: (1) Low intensity values. Pixel intensity values vary between antigens and elements and can range from hundreds (e.g. for Au coming from the slide) to single counts for low abundant antigens. (2) For low abundant antigens, signal is sparse and pixelated. Therefore, there is more information about positive signal in the density of positive pixels, rather than their intensity. To filter noise by signal density a k-nearest-neighbor approach was used. Each count was given a density-determined confidence score, by calculating the average distance to the 25 nearest positive counts. Pixels with counts larger than one were treated as several counts with distance 0 from each other. Removal thresholds were determined as the crossing points in the bimodal distributions and low-confidence counts were removed. Removal thresholds for each channel are specified in table S1. Patient 30 had particularly noisy data and was therefore removed from all further analysis (Fig. S2E).
Aggregates removal.
In addition to the real staining, some channels exhibited dense, localized staining, which appeared like ‘antibody aggregates’. To remove aggregates, denoised images were smoothed using a Gaussian kernel with R=1 and the resulting image was binarized using Otsu’s method. Pixels of small connected components were set to zero. Size thresholds for each channel are specified in Table S1 (Fig. S2F).
Image segmentation.
Nuclear segmentation was performed by adapting DeepCell (Van Valen et al., 2016), a CNN-based approach for single-cell image segmentation to MIBI-TOF data. Generating training data. To generate training data, the channels of dsDNA, H3K27me3 and H3K9ac were summed, to generate a strong and robust nuclear image. Color overlays of the nuclear channel (blue), an immune membrane channel (CD45 and HLA-DR, green), and a cancer membrane/cytosol channel (Pan-Keratin and Beta-catenin, red) were also used to guide the manual segmentation in ambiguous cases. The nuclear images of patients 1 and 2 were manually segmented in ImageJ (Schneider et al., 2012) using a Wacom Intuos Draw graphics tablet as previously described (Van Valen et al., 2016). For each patient, three binary label images were generated, marking pixels classified as either nuclear interiors, nuclear borders or background. Edges were augmented by a radial dilation with R=1 to increase their prevalence. Training data was generated for the images of dsDNA, H3K27me3 and H3K9ac. Patches of 61×1×3 pixels around each annotated pixel were taken for 106 pixels, randomly sampled to maintain equal representation of the three labels. Training images were normalized by subtracting a 61×61 averaging filter and dividing by the std of the whole image. Training images were augmented by a random rotation of 0 to 180 degrees and reflection about the vertical and horizontal axes. Training the network. The architecture of the network was previously described (Van Valen et al., 2016). The data was split into training (90%) and test (10%). For training, the batch size was set to 256 and ran for 5 epochs. The learning rate was set to 0.01, the decay to 0.95 and the Nesterov momentum to 0.9. Segmenting new images. The trained network was run on the images of all patients in the cohort. The images were normalized in the same fashion as the training images. Post processing. The probability map for the ‘nuclear interior’ was thresholded using Otsu’s method to define nuclei borders. Regional maxima were identified (matlab imextendedmax, H=0.1) and adjacent nuclei were separated by watershedding (Meyer, 1994). Small nuclei (<40 pixels) were fused to adjacent nuclei. Cell Borders were defined as a 3-pixel radial expansion around the nuclei.
Expression assignment.
For each cell, for each maker, the total intensity per cell was computed. Values were divided by the cell size, Arcsinh transformed, and standardized across markers using std normalization.
Clustering.
Cells with low expression across all markers used for clustering (sum < 0.1) were removed prior to clustering. Cells from all patients were clustered in a hierarchical scheme. Initially, cells were clustered into ‘Immune’ and ‘Non-immune using 16 markers (CD45, FoxP3, CD4, CD8, CD3, CD20, CD16, CD68, MPO, HLA-DR, Pan-Keratin, Keratin17, Keratin6, p53, Beta catenin, EGFR). FlowSOM (Van Gassen et al., 2015) was used to cluster the data into 100 clusters, and then similar clusters (cosine distance < 0.05) were merged by hierarchical clustering. The same approach was used to cluster Non-immune cells into Epithelial, Mesenchymal, Endothelial and Unidentified using 8 channels (Vimentin, SMA, CD31, Beta-catenin, EGFR, Keratin 17, Keratin 6, Pan-keratin). Immune cells were clustered into 12 groups (Fig. 2E) using 13 channels (CD4, CD16, CD56, CD209, CD11c, CD68, CD8, CD3, CD20, HLA-DR, CD11b, MPO and FoxP3). The clustering scheme is illustrated in Fig. S2I.
Co-occurrence of immune populations.
Immune categories were truncated to those that were clearly defined (CD4 T, CD8 T, NK, B, Macrophages), and for each patient the number of immune cells in each category was extracted. A threshold of 30 cells was used to decide whether an immune population was present or absent in a patient. Changing the threshold up to 130 cells did not have a significant effect on the results (Fig. S4F). Significance of co-occurrence was assessed using a Χ2 test.
Spatial analysis.
Cells were considered positive for a protein if their scaled expression level was >0.5. For each cell, the physical distance to all other cells in the core was calculated and stored as an interaction matrix. For each pairwise combination of proteins (X and Y), the distance matrix was truncated to include only interactions between X-positive and Y-positive cells. Interactions with distance smaller than 100 pixels (39µm) were counted as close. To evaluate whether the number of close interactions is significant given the total number of cells in the tissue, the tissue architecture and the number of cells positive for X and Y, a bootstrap approach was used. The location of Y-positive cells was randomized, while keeping their overall number constant and the number of close interactions was quantified as described above. This process was repeated 500 times to generate a null distribution, and a z-score was calculated to assess the deviation of the actual number from the null distribution. Results were robust to ranging the distance between 100 and 500 pixels, and increasing the number of randomizations to 500 (Fig. S5A). Importantly, the approach yields different values when calculating the spatial enrichment of protein X close to protein Y, and when calculating the spatial enrichment of protein Y close to protein X. A context-dependent spatial enrichment analysis was used to control for the hierarchical organization of the tissue. The analysis was as described above with the additional constraint that when performing randomizations, the total number of Y-positive cells for each category (Immune, Epithelial, Mesenchymal and Endothelial) must be preserved. Pairwise interaction z-scores for all patients are provided in table S3.
Tumor-Immune mixing.
A mixing score was developed to quantify the degree of mixing between tumor and immune cells. The neighbors of each cell were defined as cells with direct contact with the cell of interest and were identified using Haralick’s gray-level co-occurrence matrix (Haralick et al., 1973). The mixing score for a patient was defined as the proportion of immune cells touching tumor cells and was formally calculated as the number of immune- tumor interactions divided by the number of immune-immune interactions in the neighbors’ matrix. Patients with less than 250 immune cells (N=6) were defined as ‘Cold’. Patients with a mixing score < 0.22 (N=15) were defined as compartmentalized and the rest of the patients (N=20) were defined as Mixed. The threshold was chosen to minimize the intra-group variability and maximize the inter-group variability. All results were robust to the specific selection of the threshold, as demonstrated in figure 6K for the survival analysis. The relationship between the mixing score and the number of immune cells in the sample as quantified both by MIBI-TOF and by pathological TIL analysis is shown in figure S7E–H.
Analysis of immunoregulatory protein expression.
To assess the significance of co-expression, for each pair of immunoregulatory proteins X and Y, the background probability for an immune cell to express protein X, P(X) was calculated and compared to P(X|Y), the conditional probability of expressing X given expression of Y. Significance of co-occurrence was assessed using a Χ2 test. A bootstrapping approach was used to further validate that pairs of immunoregulatory proteins were enriched for co-expression when controlling for enriched expression of different regulatory proteins by specific immune cell types. For each pair of proteins, X and Y, the number of double-positive cells was quantified. The identity of Y-positive cells was randomized while preserving the total number of positive cells within each immune category, and the number of X-Y-double-positive cells was quantified. Randomizations were repeated 500 times to generate a null distribution, and a z-score was calculated to assess the deviation of the actual number from the null distribution (Fig. S6C,D).
To examine co-occurrence of expression of immunoregulatory proteins across patients, for each patient the number of positive cells for each regulatory protein was extracted. A threshold of 30 positive cells was used to decide whether a protein was expressed or not in a patient. Ranging the threshold from 20 to 50 cells did not have a significant effect on the results (Fig. S6E). The analysis was repeated, thresholding according to the fraction of positive cells, rather than their absolute number. Ranging the threshold between 1%−4% did not have a significant effect on the results (Fig. S6F). Significance of co-occurrence was assessed using a Χ2 test.
Analysis of the tumor-immune border.
Analysis of the tumor-immune border was restricted to compartmentalized tumors (see above). Identification of the tumor-immune boundary. To identify the border, two binary images were generated, marking immune and tumor cells. Both images were dilated using a squared structuring element of size 10 pixels, to generate large connected components. Overlaps between tumor and immune signal resulting from the dilation were set to zero. Connected components of both immune and tumor signal were identified (Matlab bwconncomp) and connected components of size < 5000 were removed. Images were closed using a disc-shaped structuring element with a radius of 10 pixels to smoothen the edges. Holes were removed by inverting the images and removing connected component of size < 10000 pixels. Edges of immune and tumor areas were identified (Matlab edge) and the edges on the border of the image were removed. Edges identified by both tumor and immune images were classified as the tumor-immune border. For each cell, it’s distance to the boundary was defined as the shortest distance from the center of the cell to one of the boundary pixels (Matlab knnsearch). Cells were defined as either close (distance < 100 pixels), far (distance > 100 pixels) or Infiltrating (immune cells in tumor side of the boundary or tumor cells in immune side of the boundary). Analysis of protein expression close/far from border. This analysis was performed for every compartmentalized patient, for tumor and immune cells separately. For each protein, the expression distribution of cells close to the boundary was compared with the expression distribution of cells far from the boundary. Proteins for which there were less than 50 positive cells overall (expression < 0.5) were discarded from the analysis. The distributions were compared using Wilcoxon’s Rank-Sum test. A Bonferroni- corrected p-value of 0.05 was used to define significant changes. H-values were modified such that H=1 represents an interaction that is enriched close to the border and H=−1 represents an interaction that is enriched far from the border. H-values for tumor and immune cells were combined and subjected to hierarchical clustering and principle component analysis (PCA).
Survival Analysis.
De-identified data on tumor size, tumor grade, recurrence, date of diagnosis, and date of death was obtained from Stanford hospital. Association between tumor type and survival was evaluated using univariate Cox regression.
Supplementary Material
(A) Image of the MIBI-TOF machine. Tissue-mounted slides, stained with metal-conjugated antibodies, are inserted into the slide chamber. The sample is rasterized, pixel-by-pixel by a primary ion beam, releasing metal lanthanides that are measured by a time-of-flight mass spectrometer. (B) Comparison between MIBI-TOF (top) and IHC (bottom). For MIBI-TOF, FFPE human tonsil was stained using a panel of 36 antibodies and visualized using MIBI-TOF. Staining of CD20, CD3, CD4, PD-1 and Ki-67 is shown. (C) Left: Color overlay of CD8 (red), CD3 (green) and CD4 (blue) of the tonsil shown in figure 1C–H. Top right: Illustration depicting lineage-specific patterns of coexpression of CD3, CD4 and CD8 in T cells. Bottom right: Quantification of pixel color shows high coexpression for CD3 and CD4, and for CD3 and CD8, but not for CD4 and CD8. (D) Serial sections of FFPE human lymph node were stained using a panel of 36 antibodies and visualized using MIBI-TOF. Color overlay of CD3, CD209 and CD68 show high reproducibility (R=0.9, P<10-20) between sections. (E) Distributions of HLA-DR expression in tumor cells (y-axis) is plotted for all patients (1–41) and three normal breast controls (42–44) (x-axis). Patients and controls are sorted by their median HLA-DR expression in tumor cells. Gray bar indicates the 5th and 95th percentiles of the normal specimens pooled together. (F) Histograms of HLA-DR expression in tumor cells are plotted for three patients 2 (blue), 3 (red) and 9 (yellow). (G) Staining for tumor cells (Pan-keratin, purple) and HLA-DR (green). In patient 3 (left) the tumor cells are negative for HLA-DR, whereas in patient 9 (right) the tumor cells express HLA-DR. (H) Same as (E), for the log-ratio of H3K27me3 and H3K9ac. (I). Histograms of log2(H3K27me3/H3K9ac) in patient 10 tumor cells (blue), patient 10 immune cells (cyan), patient 32 tumor cells (red) and patient 32 immune cells (yellow). While the distributions for the immune cells in both patients are narrow and overlap, there is higher methylation in the tumor cells of patient 32 and higher acetylation in a large subset of the tumor cells of patient 10. (J) Staining for H3K27me3 (red) and H3K9ac (green). In patient 10 (left) tumor cells are green, whereas in patient 32 (right) tumor cells are red. Immune cells are yellow in both patients.
(A) Shown is the mass spectrum for masses 155–160 for an entire image. Dashed red lines indicate the mass range that will be considered as positive for each one of the channels. Values for all channels are specified in Table S1. For each pixel, mass spectra values are converted to counts for each one of the channels. (B) Left: Shown is an example of the signal on a background channel (mass window 128–132). Signal represents non-specific background and is highly correlated with regions of bare slide. Second from the left: Binary mask of the background channel, generated by convolving the image with a Gaussian kernel (R=3) and thresholding (t=0.07). Second from the right: Image of the CD45 channel before background subtraction. Arrow indicates the non-specific background signal. Right: Image of the CD45 channel following background subtraction. To subtract background, the value of each positive pixel in the background mask was subtracted by two counts. This method reduces background, while allowing to preserve real signal. (C) Left: Image of the dsDNA channel in patient 25. Arrow denotes necrotic region, conferred by H&E staining. Middle: Binary mask of the necrotic region, generated by morphological opening and closing (R=5) and removing small connected components (size < 10,000 pixels). Right: Image of dsDNA following necrosis subtraction. The value of each positive pixel in the necrosis mask was subtracted by ten counts. (D) Left: Images of the Pan-keratin channel in six patients, stained in either the first (top) or second (bottom) batch. Middle: Histogram of Pan-keratin-positive pixel counts in patients stained in the first (blue) or second (red) batch, confirming overall higher counts in the second batch. Right: Shown are the ranked counts per pixel in the first batch (x-axis) and second batch (y-axis). The resulting non-linear transformation was used to normalize Pan-keratin counts in batch 2 to batch 1. (E) Left: Image of the CD8 channel before noise removal. Second from the left: Illustration of noise removal method that is well suited for sparse, low-intensity data and makes use of both intensity and density information. For each positive pixel (red square), the distances to the 25 nearest neighbors are calculated and averaged. Pixels with values > 1 are counted multiple times, according to their value. Second from the right: Histogram of the average distance to the 25 nearest neighbors. Small distances represent counts with high density (signal) and large distances represent counts with low density (noise). Dashed red line indicates the threshold used for de-noising. Right: Image of CD8 after noise removal by thresholding and setting low- density counts to zero. (F) Left: Image of the p53 channel after de-noising. Arrow indicates ‘aggregates’. Middle: Binary mask of the p53 channel generated by thresholding using Otsu’s method. Right: Image of the p53 channel after aggregate removal, generated by removing connected components with size < 100 pixels. (G) Model error for DeepCell segmentation (y- axis) as a function of Epoch (x-axis) on training (blue) and test (orange) datasets. (H) Staining for dsDNA (grayscale) from patient 8 overlaid with segmentation obtained using Ilastik + post- processing (red) or DeepCell + post-processing (green). Post-processing was identical for both procedures (Methods). (I) Schematic of clustering process into distinct cell subtypes.
(A) Shown are H&E stains for the entire dataset. (B) For patients 1–14 an additional core was stained by H&E, in addition to the core used for MIBI-TOF analysis, to evaluate intra-patient heterogeneity in immune infiltration. Visual inspection of these duplicate cores suggests that for the most part, intra-patient heterogeneity in lymphocyte infiltration and mixing is lower than inter-patient heterogeneity.
(A) For all patients (x-axis), shown is the fraction of immune cells out of all the cells (y-axis). Values range from 0.01–0.91. (B) For all patients, shown is the number of tumor cells (x-axis) vs. the number of immune cells (y-axis). (C) For all patients, shown is the number of tumor cells (x- axis) vs. the number of endothelial cells (y-axis). (D) Left: Shown are expression values for all Immune cells (y-axis) clustered by their expression of thirteen proteins (x-axis). Expression values per protein are scaled from zero to one. Right: Immune cells (y-axis) are sorted as in left. Shown are their expression values for an additional 23 markers. Immune cells are negative for tumor-related proteins, such as keratins, EGFR and p53. (E) Immune composition (x-axis) is shown for all patients (y-axis). Patients are sorted by total number of immune cells. Immune populations are color coded as in G. (F) For five immune cells types (x-axis) shown is their presence (yellow) or absence (blue) in each patient (y-axis). Presence of immune population in a patient was defined as having at least 30 cells (top left), 70 cells (top right), 100 cells (bottom left) or 130 cells (bottom right). Co-existence of immune populations is robust to the threshold used. (G) fPseudo-coloring of immune populations in all patients. Patient 30 was excluded from the analysis due to high noise.
(A) Heatmap depicting spatial enrichment z-scores between pairs of proteins in patient 16. Shown are results for different run parameters. Z-score matrix was clustered by hierarchical clustering for the parameters used in the text (100 randomizations, distance of 100 pixels). Z- score matrices from all other runs are plotted in the same order. Ranging the distance threshold between 100–500 pixels and increasing the number of randomizations to 500 did not affect the results.(B) Mixing score between tumor and immune cells (y-axis) for all patients (x- axis). Red dashed line depicts partitioning between compartmentalized and mixed phenotypes. (C) Heatmaps depicting context-dependent spatial enrichment z-scores between pairs of proteins in all patients. (D) Pseudo-coloring of immune populations in patients 17, 20, 40 and 4 show close proximity between immune cells from the same lineage. (E) Left: Staining for immune cells (CD45, grayscale), tumor cells (beta-catenin, red) and p53 (green) in patient 6. Green arrows depict regions of p53-positive cells, in close proximity to immune cells. Red arrows depict regions of p53-negative tumor cells, distal from immune cells. Right: zoom-in on the marked inset.
(A) Immune composition normalized from zero to one (y-axis) for either all cells (left bar) or cells positive for distinct immunoregulatory proteins (x-axis). (B) For each immunoregulatory protein (x-axis), shown are positive cells (y-axis) from distinct immune subtypes. Parentheses describe the percentage of cells displayed from each immune subtype. Only cells positive for expression of at least one immunoregulatory protein are displayed. (C) A bootstrapping approach was used to validate that pairs of immunoregulatory proteins were enriched for co- expression when controlling for enriched expression of different immunoregulatory proteins by specific immune cell types. Shown are results for PD-1 and Lag3 (top), and for PD-L1 and IDO (bottom). For each pair of proteins, X and Y, the number of double-positive cells was quantified (red bar). The identity of Y-positive cells was randomized while preserving the total number of positive cells within each immune category, and the number of X-Y-double-positive cells was quantified. Randomizations were repeated 500 times to generate a null distribution (black). A z- score was calculated to assess the deviation of the actual number from the null distribution. (D) For each pair of immunoregulatory proteins X and Y, the fraction of patients expressing X out of the patients expressing Y is quantified. The diagonal displays the proportion of patients expressing X out of all patients. For each column, the diagonal has the lightest color, indicating that once a patient expresses one type of immunoregulatory protein, it has increased propensity to express another. (E-F) Presence (yellow) or absence (blue) of expression of four immunoregulatory proteins and Tregs (y-axis) in each patient (x-axis). In (E) expression of a protein in a patient was defined as having at least 20 (left), 30 (middle) or 50 (right) positive cells. In (F) expression of a protein in a patient was defined as having at least 1% (left), 2% (middle) or 4% (right) positive cells. Coexistence of immune populations is robust to using percentages or actual numbers and to the threshold used. (G) Immune composition normalized from zero to one (y-axis) across patients (x-axis) for cells positive for LAG3 (left), PD-1 (second to left), PD-L1 (second to right) and IDO (right). Patients are sorted by number of positive cells. (H) Immune composition (y-axis) across patients (x-axis) for cells positive for PD-1 (top left), Lag3 (top right), PD-L1 (bottom left) and IDO (bottom right). (I) Correlation between the log2- ratio of CD8+ to CD4+ T cells across patients (x-axis) and the log2-ratio of PD-1+CD8+ to PD- 1+CD4+ T cells across patients (y-axis). (J) For all patients (x-axis) shown is the log2 ratio of CD8+ to CD4+ T cells. Tumors with a compartmentalized spatial organization are colored purple and tumors with a mixed spatial organization are colored green. There are no significant differences between mixed and compartmentalized, as determined by Wilcoxon rank-sum test.
(A) For all patients with over 10 (left), 50 (middle) or 100 (right) PD-1+ cells (x-axis) shown is the log ratio of PD-1+CD8+ T-cells and PD-1+CD4+ T-cells. Tumors with a compartmentalized spatial organization are purple and tumors with a mixed spatial organization are green. Regardless of the threshold, mixed tumors tend to have more PD-1+CD8+ T cells than PD-1+CD4+ T cells, as determined by Wilcoxon rank-sum test. (B) Same as (A) for the log ratio of PD-L1+ tumor cells and immune cells. (C) Same as (A) for the log ratio of IDO+ tumor cells and immune cells. (D) Kaplan-Meier curves showing survival (y-axis) as a function of time (a-axis, years) for patients with compartmentalized (purple) or mixed (green) tumor-immune organizations, using different thresholds for partitioning the compartmentalized and mixed groups. Left: T=0.16. Middle: T=0.3. Right: The analysis was restricted to the twenty patients (ten from each group), with the most extreme mixing scores. Patients with intermediate mixing scores were removed from the analysis. Hazards ratio (HR) was calculated using Cox regression analysis. (E) High correlation between TIL score (x-axis) and total immune infiltrate as quantified from MIBI-TOF images (y-axis) for 25 TNBC patients in the cohort with available TIL data (black points). Robust least squares regression (black line) was performed by regressing all the data and removing points over 3 standard deviations away from the line (N=1). (F) Scatter of TIL score (x-axis) and tumor- immune mixing score (y-axis) for 20 non-cold TNBC patients in the cohort with available TIL data (black points). R2 denotes squared pearson correlation coefficient. Mixing score is generally anti-correlated to TIL score (R2=0.44), but captures independent information. This is evident by many cases in which patients with the same TIL score exhibit a wide range of mixing scores. (G) Kaplan-Meier curves showing survival (y-axis) as a function of time (a-axis, years) for patients with high (>2.5, dark blue, N=8) or low (<=2.5, light blue, N=12) TIL scores. Hazards ratio (HR) was calculated using Cox regression analysis. (H) Same patients as in G were partitioned according to their mixing scores into compartmentalized (<0.22, purple, N=10) and mixed (>0.22, green, N=10) phenotypes. Shown are survival (y-axis) as a function of time (a-axis, years) and HR for the two groups.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| A full list of antibodies is provided in Table S1 | ||
| Biological Samples | ||
| TNBC TMA was compiled by the Stanford Pathology Department |
||
| Chemicals, Peptides, and Recombinant Proteins | ||
| TBS IHC Wash Buffer with Tween 20 | Cell Marque | Cat#935B-09 |
| PBS IHC Wash Buffer with Tween 20 | Cell Marque | Cat#934B-09 |
| Target Retrieval Solution, pH 9, (3:1) | Agilent (Dako) | Cat#S2375 |
| Avidin/Biotin Blocking Kit | Biolegend | Cat#927301 |
| Gelatin (cold water fish skin) | Sigma-Aldrich | Cat#G7765–250 |
| Xylene Histological grade | Sigma-Aldrich | Cat#534056–500 |
| Glutaraldehyde 8% Aqueous Solution EM Grade | EMS | Cat#16020 |
| Normal Donkey serum | Sigma-Aldrich | Cat#D9663–10ML |
| Bovine Albumin (BSA) | Fisher | Cat#BP1600–100 |
| Centrifugal filters (0.1 μm) | Millipore | Cat#UFC30VV00 |
| Critical Commercial Assays | ||
| Maxpar X8 Antibody labeling kit | Fluidigm | Cat#2011XXX |
| MIBItag Conjugation Kit | IONpath | Cat#600XXX |
| ImmPRESS UNIVERSAL (Anti-Mouse/Anti-Rabbit) IgG KIT (HRP) |
Vector Laboratories | Cat#MP-7500–15 |
| ImmPACT DAB (For HRP Substrate) | Vector Laboratories | Cat#SK-4105 |
| Deposited Data | ||
| https://mibi-share.ionpath.com | All the data of the study |
|
| Software and Algorithms | ||
| Data analysis was done using Matlab 2016 | Mathworks | |
| https://github.com/lkeren/MIBIAnalysis | Analysis code | |
| https://hub.docker.com/r/vanvalen/deepcell-mibi | Segmentation code | |
Highlights.
Multiplexed Imaging of subcellular expression of 36 proteins in 41 TNBC patients
Enriched co-occurrence of immune populations define a structured immune environment
Mixing of tumor and immune cells varies from Compartmentalized to Mixed patients
Expression of PD-1 and PD-L1 by distinct cells relate to tumor histology and survival
Acknowledgements
The authors thank Tom Carver, Dana Pe’er, Tyler Risom, David Glass, Erin McCaffrey and Felix Hartmann for discussions and comments. L.K. is supported by the Damon Runyon Cancer Research Foundation (DRG-2292-17) and EMBO Long-Term fellowship (ALTF 1128-2016). D.V.V is supported by an NIH F32 Postdoctoral Fellowship, a Burroughs Wellcome PDEP award, The Allen Discovery Center at Stanford University, and a Crowdflower A.I. for everyone award. R.W is supported by NIH 5R01CA18390402. S.C.B. is supported by a gift from Christy and Bill Neidig, the Damon Runyon Cancer Research Foundation (DRG-2017-09), NIH 1DP2OD022550-01, 1-R00-GM104148-01, 5U19AI116484-02, and U19 AI104209. M.A is supported by 1-DP5- OD019822. S.C.B and M.A. are supported by NIH 1R01AG056287-01, 1R01AG057915-01, and 1U24CA224309-01, the Bill and Melinda Gates Foundation, and a Translational Research Award from the Stanford Cancer Institute. The Stanford Nano Shared Facility is supported by NSF ECCS-1542152.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
M.A. and S.C.B have patents relating to MIBI technology, and are board members, shareholders and consultants in Ionpath Inc.
Data and software availability
All the data described in this work, including channel images, segmentation masks and cell identities can be accessed through a web interface and downloaded at https://mibi-share.ionpath.com. The code for the analysis can be downloaded at https://github.com/lkeren/MibiAnalysis. All the information required for cell segmentation including the manual training data, trained neural networks, and code for training and running DeepCell are available at https://hub.docker.com/r/vanvalen/deepcell-mibi.
References
- Andre F, Dieci MV, Dubsky P, Sotiriou C, Curigliano G, Denkert C, and Loi S (2013). Molecular pathways: involvement of immune pathways in the therapeutic response and outcome in breast cancer. Clin. Cancer Res 19, 28–33. [DOI] [PubMed] [Google Scholar]
- Andrews LP, Marciscano AE, Drake CG, and Vignali DAA (2017). LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev 276, 80–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, Levenson RM, Lowe JB, Liu SD, Zhao S, et al. (2014). Multiplexed ion beam imaging of human breast tumors. Nat. Med 20, 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagaitkar J (2014). Cellular dynamics of resolving inflammation. Blood 124, 1701–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir ED, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. (2011). Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SAW, Savage MI, Osborne CK, Hilsenbeck SG, Chang JC, et al. (2015). Comprehensive Genomic Analysis Identifies Novel Subtypes and Targets of Triple-Negative Breast Cancer. Clin. Cancer Res 21, 1688–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burugu S, Gao D, Leung S, Chia SK, and Nielsen TO (2017). LAG-3+ tumor infiltrating lymphocytes in breast cancer: clinical correlates and association with PD-1/PD-L1+ tumors. Ann. Oncol [DOI] [PubMed] [Google Scholar]
- Carrel S, Schmidt-Kessen A, and Giuffrè L (1985). Recombinant interferon-γ can induce the expression of HLA-DR and -DC on DR-negative melanoma cells and enhance the expression of HLA-ABC and tumor-associated antigens. Eur. J. Immunol 15, 118–123. [DOI] [PubMed] [Google Scholar]
- Chen DS, and Mellman I (2017). Elements of cancer immunity and the cancer–immune set point. Nature 541, 321–330. [DOI] [PubMed] [Google Scholar]
- Chen KH, Boettiger AN, Moffitt JR, Wang S, and Zhuang X (2015). Spatially resolved, highly multiplexed RNA profiling in single cells. Science (80-. ) 348, aaa6090-aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, Ries CH, Ailles L, Jewett MAS, Moch H, et al. (2017). An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 169, 736–749.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun AF, and Cai L (2016). Dense transcript profiling in single cells by image correlation decoding. Nat. Methods 13, 657–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denkert C, Loibl S, Noske A, Roller M, Müller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R, Hanusch C, et al. (2010). Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol 28, 105–113. [DOI] [PubMed] [Google Scholar]
- Denkert C, Liedtke C, Tutt A, and von Minckwitz G (2017). Molecular alterations in triple- negative breast cancer—the road to new treatment strategies. Lancet 389, 2430–2442. [DOI] [PubMed] [Google Scholar]
- Di Minin G, Bellazzo A, Dal Ferro M, Chiaruttini G, Nuzzo S, Bicciato S, Piazza S, Rami D, Bulla R, Sommaggio R, et al. (2014). Mutant p53 Reprograms TNF Signaling in Cancer Cells through Interaction with the Tumor Suppressor DAB2IP. Mol. Cell 56, 617–629. [DOI] [PubMed] [Google Scholar]
- Disis ML, and Stanton SE (2015). Triple-Negative Breast Cancer: Immune Modulation as the New Treatment Paradigm. Am. Soc. Clin. Oncol. Educ. B 35, e25–e30. [DOI] [PubMed] [Google Scholar]
- Dunn C, Peppa D, Khanna P, Nebbia G, Jones M, Brendish N, Lascar RM, Brown D, Gilson RJ, Tedder RJ, et al. (2009). Temporal Analysis of Early Immune Responses in Patients With Acute Hepatitis B Virus Infection. Gastroenterology 137, 1289–1300. [DOI] [PubMed] [Google Scholar]
- Fortis SP, Sofopoulos M, Sotiriadou NN, Haritos C, Vaxevanis CK, Anastasopoulou EA, Janssen N, Arnogiannaki N, Ardavanis A, Pawelec G, et al. (2017). Differential intratumoral distributions of CD8 and CD163 immune cells as prognostic biomarkers in breast cancer. J. Immunother. Cancer 5, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, and Nagaraj S (2009). Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol 9, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 167, 397–404.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, and Saeys Y (2015). FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part A 87, 636–645. [DOI] [PubMed] [Google Scholar]
- Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schüffler PJ, Grolimund D, Buhmann JM, Brandt S, et al. (2014). Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422. [DOI] [PubMed] [Google Scholar]
- Haralick RM, Shanmugam K, and Dinstein I (1973). Textural Features for Image Classification. IEEE Trans. Syst. Man. Cybern SMC-3, 610–621. [Google Scholar]
- Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Hennrick K, and Drew S (2013). A colorful future of quantitative pathology: validation of Vectra technology using chromogenic multiplexed immunohistochemistry and prostate tissue microarrays. Hum. Pathol 44, 29–38. [DOI] [PubMed] [Google Scholar]
- Kouzarides T (2007). Chromatin Modifications and Their Function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Kwa MJ, and Adams S (2018). Checkpoint inhibitors in triple-negative breast cancer (TNBC): Where to go from here. Cancer 124, 2086–2103. [DOI] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. (2015). Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med 373, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, Terry R, Jeanty SSF, Li C, Amamoto R, et al. (2014). Highly Multiplexed Subcellular RNA Sequencing in Situ. Science (80-. ) 343, 1360–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Jovanović B, Chen X, Estrada MV, Johnson KN, Shyr Y, Moses HL, Sanders ME, and Pietenpol JA (2016). Refinement of Triple-Negative Breast Cancer [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS One 11, e0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki Vareki S, Garrigós C, and Duran I (2017). Biomarkers of response to PD-1/PD-L1 inhibition. Crit. Rev. Oncol. Hematol 116, 116–124. [DOI] [PubMed] [Google Scholar]
- Matos L.L. de, Trufelli DC, de Matos MGL, and da Silva Pinhal MA (2010). Immunohistochemistry as an important tool in biomarkers detection and clinical practice. Biomark. Insights 5, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer F (1994). Topographic distance and watershed lines. Signal Processing 38, 113–125. [Google Scholar]
- Munn DH (2011). Indoleamine 2,3-dioxygenase, Tregs and cancer. Curr. Med. Chem 18, 2240–2246. [DOI] [PubMed] [Google Scholar]
- Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S, Eder JP, Golan T, Le DT, Burtness B, et al. (2016). Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol 17, 717–726. [DOI] [PubMed] [Google Scholar]
- Nirschl CJ, and Drake CG (2013). Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin. Cancer Res 19, 4917–4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelkrug C, and Ramage JM (2014). Enhancement of T cell recruitment and infiltration into tumours. Clin. Exp. Immunol 178, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott PA, Hodi FS, Kaufman HL, Wigginton JM, and Wolchok JD (2017). Combination immunotherapy: a road map. J. Immunother. Cancer 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, and Kurzrock R (2015). PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther 14, 847–856. [DOI] [PubMed] [Google Scholar]
- Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. (2015). Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N. Engl. J. Med 372, 2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, Wienert S, Van den Eynden G, Baehner FL, Penault-Llorca F, et al. (2015). The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann. Oncol 26, 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, et al. (2016). Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol 17, 956–965. [DOI] [PubMed] [Google Scholar]
- Teng MWL, Ngiow SF, Ribas A, and Smyth MJ (2015). Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res 75, 2139–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troncone G, and Gridelli C (2017). The reproducibility of PD-L1 scoring in lung cancer: can the pathologists do better? Transl. Lung Cancer Res 6, S74–S77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Valen DA, Kudo T, Lane KM, Macklin DN, Quach NT, DeFelice MM, Maayan I, Tanouchi Y, Ashley EA, and Covert MW (2016). Deep Learning Automates the Quantitative Analysis of Individual Cells in Live-Cell Imaging Experiments. PLOS Comput. Biol 12, e1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis SM, and Cheresh DA (2011). Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med 17, 1359–1370. [DOI] [PubMed] [Google Scholar]
- Woo S-R, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, et al. (2012). Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res 72, 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeong J, Thike AA, Lim JCT, Lee B, Li H, Wong S-C, Hue SSS, Tan PH, and Iqbal J (2017). Higher densities of Foxp3+ regulatory T cells are associated with better prognosis in triple-negative breast cancer. Breast Cancer Res. Treat 163, 21–35. [DOI] [PubMed] [Google Scholar]
- Yu L-Y, Tang J, Zhang C-M, Zeng W-J, Yan H, Li M-P, and Chen X-P (2017). New Immunotherapy Strategies in Breast Cancer. Int. J. Environ. Res. Public Health 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Image of the MIBI-TOF machine. Tissue-mounted slides, stained with metal-conjugated antibodies, are inserted into the slide chamber. The sample is rasterized, pixel-by-pixel by a primary ion beam, releasing metal lanthanides that are measured by a time-of-flight mass spectrometer. (B) Comparison between MIBI-TOF (top) and IHC (bottom). For MIBI-TOF, FFPE human tonsil was stained using a panel of 36 antibodies and visualized using MIBI-TOF. Staining of CD20, CD3, CD4, PD-1 and Ki-67 is shown. (C) Left: Color overlay of CD8 (red), CD3 (green) and CD4 (blue) of the tonsil shown in figure 1C–H. Top right: Illustration depicting lineage-specific patterns of coexpression of CD3, CD4 and CD8 in T cells. Bottom right: Quantification of pixel color shows high coexpression for CD3 and CD4, and for CD3 and CD8, but not for CD4 and CD8. (D) Serial sections of FFPE human lymph node were stained using a panel of 36 antibodies and visualized using MIBI-TOF. Color overlay of CD3, CD209 and CD68 show high reproducibility (R=0.9, P<10-20) between sections. (E) Distributions of HLA-DR expression in tumor cells (y-axis) is plotted for all patients (1–41) and three normal breast controls (42–44) (x-axis). Patients and controls are sorted by their median HLA-DR expression in tumor cells. Gray bar indicates the 5th and 95th percentiles of the normal specimens pooled together. (F) Histograms of HLA-DR expression in tumor cells are plotted for three patients 2 (blue), 3 (red) and 9 (yellow). (G) Staining for tumor cells (Pan-keratin, purple) and HLA-DR (green). In patient 3 (left) the tumor cells are negative for HLA-DR, whereas in patient 9 (right) the tumor cells express HLA-DR. (H) Same as (E), for the log-ratio of H3K27me3 and H3K9ac. (I). Histograms of log2(H3K27me3/H3K9ac) in patient 10 tumor cells (blue), patient 10 immune cells (cyan), patient 32 tumor cells (red) and patient 32 immune cells (yellow). While the distributions for the immune cells in both patients are narrow and overlap, there is higher methylation in the tumor cells of patient 32 and higher acetylation in a large subset of the tumor cells of patient 10. (J) Staining for H3K27me3 (red) and H3K9ac (green). In patient 10 (left) tumor cells are green, whereas in patient 32 (right) tumor cells are red. Immune cells are yellow in both patients.
(A) Shown is the mass spectrum for masses 155–160 for an entire image. Dashed red lines indicate the mass range that will be considered as positive for each one of the channels. Values for all channels are specified in Table S1. For each pixel, mass spectra values are converted to counts for each one of the channels. (B) Left: Shown is an example of the signal on a background channel (mass window 128–132). Signal represents non-specific background and is highly correlated with regions of bare slide. Second from the left: Binary mask of the background channel, generated by convolving the image with a Gaussian kernel (R=3) and thresholding (t=0.07). Second from the right: Image of the CD45 channel before background subtraction. Arrow indicates the non-specific background signal. Right: Image of the CD45 channel following background subtraction. To subtract background, the value of each positive pixel in the background mask was subtracted by two counts. This method reduces background, while allowing to preserve real signal. (C) Left: Image of the dsDNA channel in patient 25. Arrow denotes necrotic region, conferred by H&E staining. Middle: Binary mask of the necrotic region, generated by morphological opening and closing (R=5) and removing small connected components (size < 10,000 pixels). Right: Image of dsDNA following necrosis subtraction. The value of each positive pixel in the necrosis mask was subtracted by ten counts. (D) Left: Images of the Pan-keratin channel in six patients, stained in either the first (top) or second (bottom) batch. Middle: Histogram of Pan-keratin-positive pixel counts in patients stained in the first (blue) or second (red) batch, confirming overall higher counts in the second batch. Right: Shown are the ranked counts per pixel in the first batch (x-axis) and second batch (y-axis). The resulting non-linear transformation was used to normalize Pan-keratin counts in batch 2 to batch 1. (E) Left: Image of the CD8 channel before noise removal. Second from the left: Illustration of noise removal method that is well suited for sparse, low-intensity data and makes use of both intensity and density information. For each positive pixel (red square), the distances to the 25 nearest neighbors are calculated and averaged. Pixels with values > 1 are counted multiple times, according to their value. Second from the right: Histogram of the average distance to the 25 nearest neighbors. Small distances represent counts with high density (signal) and large distances represent counts with low density (noise). Dashed red line indicates the threshold used for de-noising. Right: Image of CD8 after noise removal by thresholding and setting low- density counts to zero. (F) Left: Image of the p53 channel after de-noising. Arrow indicates ‘aggregates’. Middle: Binary mask of the p53 channel generated by thresholding using Otsu’s method. Right: Image of the p53 channel after aggregate removal, generated by removing connected components with size < 100 pixels. (G) Model error for DeepCell segmentation (y- axis) as a function of Epoch (x-axis) on training (blue) and test (orange) datasets. (H) Staining for dsDNA (grayscale) from patient 8 overlaid with segmentation obtained using Ilastik + post- processing (red) or DeepCell + post-processing (green). Post-processing was identical for both procedures (Methods). (I) Schematic of clustering process into distinct cell subtypes.
(A) Shown are H&E stains for the entire dataset. (B) For patients 1–14 an additional core was stained by H&E, in addition to the core used for MIBI-TOF analysis, to evaluate intra-patient heterogeneity in immune infiltration. Visual inspection of these duplicate cores suggests that for the most part, intra-patient heterogeneity in lymphocyte infiltration and mixing is lower than inter-patient heterogeneity.
(A) For all patients (x-axis), shown is the fraction of immune cells out of all the cells (y-axis). Values range from 0.01–0.91. (B) For all patients, shown is the number of tumor cells (x-axis) vs. the number of immune cells (y-axis). (C) For all patients, shown is the number of tumor cells (x- axis) vs. the number of endothelial cells (y-axis). (D) Left: Shown are expression values for all Immune cells (y-axis) clustered by their expression of thirteen proteins (x-axis). Expression values per protein are scaled from zero to one. Right: Immune cells (y-axis) are sorted as in left. Shown are their expression values for an additional 23 markers. Immune cells are negative for tumor-related proteins, such as keratins, EGFR and p53. (E) Immune composition (x-axis) is shown for all patients (y-axis). Patients are sorted by total number of immune cells. Immune populations are color coded as in G. (F) For five immune cells types (x-axis) shown is their presence (yellow) or absence (blue) in each patient (y-axis). Presence of immune population in a patient was defined as having at least 30 cells (top left), 70 cells (top right), 100 cells (bottom left) or 130 cells (bottom right). Co-existence of immune populations is robust to the threshold used. (G) fPseudo-coloring of immune populations in all patients. Patient 30 was excluded from the analysis due to high noise.
(A) Heatmap depicting spatial enrichment z-scores between pairs of proteins in patient 16. Shown are results for different run parameters. Z-score matrix was clustered by hierarchical clustering for the parameters used in the text (100 randomizations, distance of 100 pixels). Z- score matrices from all other runs are plotted in the same order. Ranging the distance threshold between 100–500 pixels and increasing the number of randomizations to 500 did not affect the results.(B) Mixing score between tumor and immune cells (y-axis) for all patients (x- axis). Red dashed line depicts partitioning between compartmentalized and mixed phenotypes. (C) Heatmaps depicting context-dependent spatial enrichment z-scores between pairs of proteins in all patients. (D) Pseudo-coloring of immune populations in patients 17, 20, 40 and 4 show close proximity between immune cells from the same lineage. (E) Left: Staining for immune cells (CD45, grayscale), tumor cells (beta-catenin, red) and p53 (green) in patient 6. Green arrows depict regions of p53-positive cells, in close proximity to immune cells. Red arrows depict regions of p53-negative tumor cells, distal from immune cells. Right: zoom-in on the marked inset.
(A) Immune composition normalized from zero to one (y-axis) for either all cells (left bar) or cells positive for distinct immunoregulatory proteins (x-axis). (B) For each immunoregulatory protein (x-axis), shown are positive cells (y-axis) from distinct immune subtypes. Parentheses describe the percentage of cells displayed from each immune subtype. Only cells positive for expression of at least one immunoregulatory protein are displayed. (C) A bootstrapping approach was used to validate that pairs of immunoregulatory proteins were enriched for co- expression when controlling for enriched expression of different immunoregulatory proteins by specific immune cell types. Shown are results for PD-1 and Lag3 (top), and for PD-L1 and IDO (bottom). For each pair of proteins, X and Y, the number of double-positive cells was quantified (red bar). The identity of Y-positive cells was randomized while preserving the total number of positive cells within each immune category, and the number of X-Y-double-positive cells was quantified. Randomizations were repeated 500 times to generate a null distribution (black). A z- score was calculated to assess the deviation of the actual number from the null distribution. (D) For each pair of immunoregulatory proteins X and Y, the fraction of patients expressing X out of the patients expressing Y is quantified. The diagonal displays the proportion of patients expressing X out of all patients. For each column, the diagonal has the lightest color, indicating that once a patient expresses one type of immunoregulatory protein, it has increased propensity to express another. (E-F) Presence (yellow) or absence (blue) of expression of four immunoregulatory proteins and Tregs (y-axis) in each patient (x-axis). In (E) expression of a protein in a patient was defined as having at least 20 (left), 30 (middle) or 50 (right) positive cells. In (F) expression of a protein in a patient was defined as having at least 1% (left), 2% (middle) or 4% (right) positive cells. Coexistence of immune populations is robust to using percentages or actual numbers and to the threshold used. (G) Immune composition normalized from zero to one (y-axis) across patients (x-axis) for cells positive for LAG3 (left), PD-1 (second to left), PD-L1 (second to right) and IDO (right). Patients are sorted by number of positive cells. (H) Immune composition (y-axis) across patients (x-axis) for cells positive for PD-1 (top left), Lag3 (top right), PD-L1 (bottom left) and IDO (bottom right). (I) Correlation between the log2- ratio of CD8+ to CD4+ T cells across patients (x-axis) and the log2-ratio of PD-1+CD8+ to PD- 1+CD4+ T cells across patients (y-axis). (J) For all patients (x-axis) shown is the log2 ratio of CD8+ to CD4+ T cells. Tumors with a compartmentalized spatial organization are colored purple and tumors with a mixed spatial organization are colored green. There are no significant differences between mixed and compartmentalized, as determined by Wilcoxon rank-sum test.
(A) For all patients with over 10 (left), 50 (middle) or 100 (right) PD-1+ cells (x-axis) shown is the log ratio of PD-1+CD8+ T-cells and PD-1+CD4+ T-cells. Tumors with a compartmentalized spatial organization are purple and tumors with a mixed spatial organization are green. Regardless of the threshold, mixed tumors tend to have more PD-1+CD8+ T cells than PD-1+CD4+ T cells, as determined by Wilcoxon rank-sum test. (B) Same as (A) for the log ratio of PD-L1+ tumor cells and immune cells. (C) Same as (A) for the log ratio of IDO+ tumor cells and immune cells. (D) Kaplan-Meier curves showing survival (y-axis) as a function of time (a-axis, years) for patients with compartmentalized (purple) or mixed (green) tumor-immune organizations, using different thresholds for partitioning the compartmentalized and mixed groups. Left: T=0.16. Middle: T=0.3. Right: The analysis was restricted to the twenty patients (ten from each group), with the most extreme mixing scores. Patients with intermediate mixing scores were removed from the analysis. Hazards ratio (HR) was calculated using Cox regression analysis. (E) High correlation between TIL score (x-axis) and total immune infiltrate as quantified from MIBI-TOF images (y-axis) for 25 TNBC patients in the cohort with available TIL data (black points). Robust least squares regression (black line) was performed by regressing all the data and removing points over 3 standard deviations away from the line (N=1). (F) Scatter of TIL score (x-axis) and tumor- immune mixing score (y-axis) for 20 non-cold TNBC patients in the cohort with available TIL data (black points). R2 denotes squared pearson correlation coefficient. Mixing score is generally anti-correlated to TIL score (R2=0.44), but captures independent information. This is evident by many cases in which patients with the same TIL score exhibit a wide range of mixing scores. (G) Kaplan-Meier curves showing survival (y-axis) as a function of time (a-axis, years) for patients with high (>2.5, dark blue, N=8) or low (<=2.5, light blue, N=12) TIL scores. Hazards ratio (HR) was calculated using Cox regression analysis. (H) Same patients as in G were partitioned according to their mixing scores into compartmentalized (<0.22, purple, N=10) and mixed (>0.22, green, N=10) phenotypes. Shown are survival (y-axis) as a function of time (a-axis, years) and HR for the two groups.