

Key Clinical Message
We present a girl born with a frontal bossing, short neck, bell‐shaped thorax, short limbs with prominent joints, and a tail‐like coccygeal appendage. Genetic screening of TRPV4 identified a novel de novo heterozygous missense variant. We believe the variant causes the severe form of metatropic dysplasia in this patient.
Keywords: genetic diseases, metatropic dysplasia, rare diseases, TRPV4
1. INTRODUCTION
Metatropic dysplasia is a skeletal dysplasia named after the striking reversal of body composition between birth and childhood from the Greek word metatropos. At birth, the limbs are disproportionately short due to metaphyseal abnormalities. In childhood, scoliosis and/or kyphosis makes the trunk relatively shorter than limbs.1
Affected individuals present with narrow thorax, prominent joints and occasionally tail‐like coccygeal appendage.2 Radiological hallmarks are narrow thorax with short ribs, severe platyspondyly, flared ilia, and metaphyseal enlargement of the long bones.2 Other skeletal findings include poor joint range of motion, contractures, and torticollis 1 and occasionally sensorineural hearing loss.3 There is a very broad range of clinical severity among patients.4
Metatropic dysplasia is yet seen only with TRPV4 variants demonstrating locus homogeneity for this disease. The TRPV4‐gene is located on chromosome 12q24.11 comprising 15 exons and codes for a transient receptor potential cation channel, subfamily V, member 4 (OMIM # 605427), which mediates calcium influx in response to physical, chemical, and hormonal stimuli.5
TRPV4 variants are implicated in autosomal dominant diseases in two systems—a range of skeletal dysplasias as well as a range of peripheral neuropathies.6
Both skeletal dysplasias and peripheral neuropathies are mainly caused by heterozygous missense gain‐of‐function variants.1, 7, 8 Some individuals have a mixed phenotype with both skeletal dysplasia and peripheral neuropathy.9 There seem to be no fundamental differences in the position or pattern of amino acid changes in TRPV4 between variants causing the two disease spectrums.9
2. CLINICAL REPORT
A now 3‐year‐old girl is the second child of Caucasian unrelated healthy parents. Ultrasound measurements at 20 weeks of gestation of the tubular bones were below the mean but within normal range. Breech presentation was detected in late pregnancy, and an ultrasound sonography was detected short limbs (tubular bones minus 6‐8 standard deviations [SD]) with a suspicion of fractures, a small bell‐shaped thorax with possible lung hypoplasia and an estimated fetal weight of minus 32%. Cesarean section was performed at gestational age 38 + 3 due to breech presentation, risk of pulmonary in compensation, and the possibility of a nonviable fetus.
A live‐born girl with birthweight 3250 g (−1.5 SD), length 48 cm (−1.4 SD), and head circumference 34.6 cm (+1 SD) was born.
Postnatal examination confirmed ultrasound findings. The patient was found to have a frontal bossing, short limbs, prominent joints, long hands and feet, a very short neck, prominent proximal sternal bone, and a tail‐like coccygeal appendage (Figure 1). Pictures were submitted to the Skeletal Dysplasia Network, and this gave a tentative diagnosis of metatropic dysplasia within 12 hours. A genetic analysis was thus initiated the day after birth, and the diagnosis was genetically confirmed by Centogene, Germany.
Figure 1.
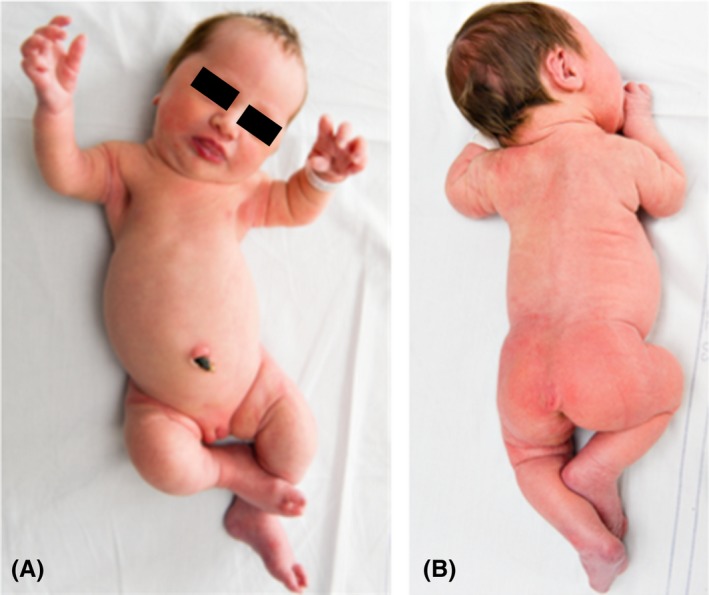
Two pictures of our patient newborn depicting a prominent forehead, short limbs, prominent joints, a very short neck, and a tail‐like coccygeal appendage
Early in life positioning in cervical opisthotonus was required, due to the prominent sternum and the short neck, to avoid discomfort. At the age of 4 months, she presented with short afebrile tonic‐clonic seizures initiated during sleep. An MR scanning of cerebrum and the total vertebral column revealed normal cerebrum and foramen magnum, but relative spinal stenosis at the level of C2/C3 and distally in the thorax as well.
Physical examination revealed reduced muscle power, tone, and mobility of joints, but normal deep tendon reflexes in all 4 extremities. The lower extremities were more severely affected, and only few spontaneous movements were seen. In the upper extremities, a side difference was seen with more spontaneous movements of the right arm.
Due to the neurological and diagnostic imaging findings, surgery was performed with decompression of arcus posterior of C1 and removal of the coccygeal appendage at the age of 10 months. Improvement of especially the left arm was seen after surgery.
At the age of 18 months, a thoracolumbar scoliosis was significant and an MR scanning revealed recurrence of cervical spinal stenosis and increasing cervical kyphosis (Figure 2). The cervical kyphosis further reduced the length and mobility of the neck and impaired breathing. Surgery was considered, but the Skeletal Dysplasia Network did not recommend this. At this age, weight was 7.2 kg (>−5 SD), height was 62.5 cm (>−8 SD), and head circumference was 46.5 cm (median).
Figure 2.
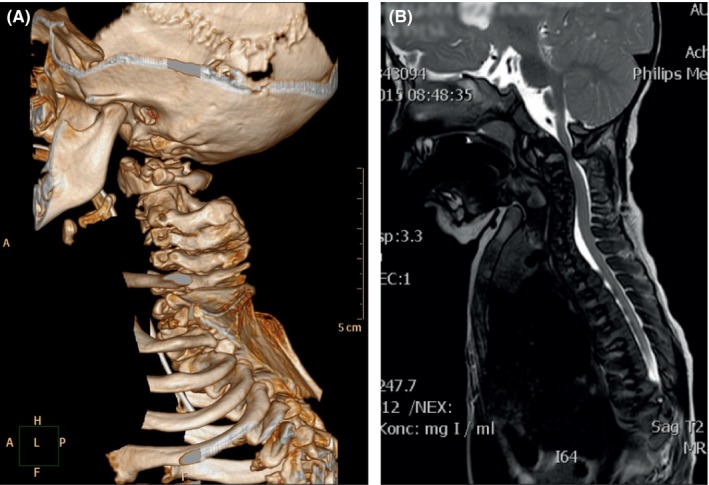
A, 3D CT of cervical column at age 1y 10 m depicting dysplastic vertebrae, spinal stenosis C1/C2, and malformation and conjoining vertebrae C2 and C3. B, T2 MRI of thoracic column at age 1y 2 m depicting high cervical and low thoracic spinal stenosis
Pneumonia was diagnosed at the age of 21 months causing severe respiratory distress and resulted in cardiac arrest due to hypoxia. Owing to rapid treatment, the patient was evaluated to be without sequelae after the cardiac arrest. At the age of 22 months, decompression surgery of C1 and Th10 was performed due to increasing medullopathy and increasing hypotonia, hyperreflexia, and clonus in the lower extremities.
After surgery, a neurogenic bladder and increasing obstipation were noticed and a CT scanning revealed narrow passage in the pelvic region. Tracheostomy was considered but deselected due to the anatomically narrow conditions.
At present (Figure 3), the patient has a need for continuous positive airway pressure. Voluntary movements are sparse in the upper limbs and almost absent in the lower limbs. Electromyography was never performed because the patient could not cooperate, and anesthesia was considered too risky. At present, weight is 8.47 kg (>−5 SD), height is 65 cm (>−8 SD), and head circumference is 49 cm (median). Cognitive function is thought to be within the normal range, and her verbal skills are improving, even though not at the same level as her peers.
Figure 3.
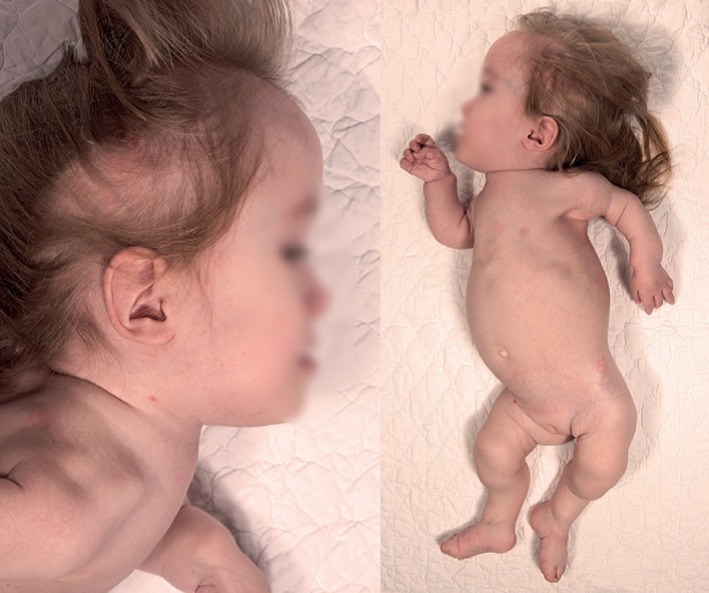
Two pictures of our patient at age 3 years depicting mild prognathia, midfacial hypoplasia, very superiorly located sternum, small thorax, and short limbs
3. METHODS
The phenotype raised suspicion of a TRPV4 variant. DNA from a blood sample was analyzed. In addition to quantitative PCR assay (qPCR), the entire coding region of TRPV4 and flanking intronic regions were sequenced at Centogene AG, Germany.
4. RESULTS
A novel heterozygous missense variant in exon 5 of TRPV4 (NM_021625.4 c.838G>A, p.Gly280Ser) was detected (Figure 4). The identified variant has been reported in dbSNP rsID (rs763354006). The variant is located in a highly conserved nucleotide and amino acid position. There are small physiochemical differences between the amino acids glycine and serine. Software prediction (PolyPhen‐2, SIFT, Mutation Tester and Align‐GVGD) predicts the variant to be probably damaging.10 Combined Annotation Dependent Depletion (CADD) score was 25. The parents do not carry the variant in their blood.
Figure 4.
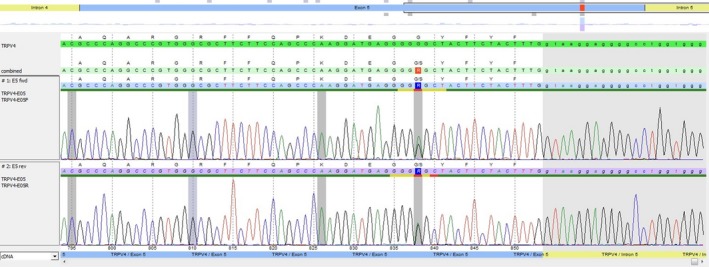
Electropherogram of the detected TRPV4 variant
5. DISCUSSION
We present a severe case of metatropic dysplasia caused by a novel missense variant in TRPV4. The clinical diagnosis was suspected with the help of the Skeletal Dysplasia Network.
TRPV4 variants are implicated in a range of skeletal dysplasias as well as a range of peripheral neuropathies with a considerable overlap between the two phenotypes. The phenotypic spectrum in patients with both skeletal and neurological affection is wide. Despite the presence of unequivocal radiographic changes, patients with both skeletal and peripheral nervous system affection have typically been reported to be relatively mildly affected in their skeleton with heights within the normal range.1 However, fetal akinesia as the presenting feature of severe metatropic dysplasia has been reported, suggesting that certain TRPV4 variants can cause both a severe skeletal and a severe neuropathic phenotype.11
Severe skeletal manifestations are likely to cause neurological symptoms due to compression of nerves and to discern neurological symptoms caused by a TRPV4 variant in patients with a severe skeletal dysplasia is difficult. In this patient, we have not found any clear indications of TRPV4‐related peripheral nervous system affection independently of the skeletal manifestations. Medullopathy and consequently peripheral neurological sequelae (partial tetraplegic) are believed to be a consequence of medullary compression. However, electromyography was not possible to perform.
The vast majority of the known TRPV4 pathogenic variants are missense variants fitting a gain‐of‐function mechanism.1 The theory that at least the TRPV4 pathogenic variants associated with skeletal dysplasias may cause a gain of channel function is supported by studies reporting that whole‐gene or contiguous‐gene deletion of TRPV4 causing any of the recognized phenotypes has not been described.1 Moreover, studies on mice have shown that TRPV4 knockout mice lack a comparable phenotype,12 an increased amount of wild‐type protein can be tolerated, but an activating variant is required to produce a skeletal dysplasia phenotype.13 This supports the pathogenic potential of a missense variant as found in this case. However, exceptions have been reported, as at least one frameshift variant has been reported to cause skeletal dysplasia.14 Hence, at the current state of knowledge, it appears that the phenotypic diversity of TRPV4 variants cannot be explained exhaustively by a simple gain‐of‐function mechanism.6
For the most part, the TRPV4 variants reported in the neurological phenotype patients are different than those reported in the patients with skeletal disorders. There is no clear explanation as to why the variants should have such distinct effects.6 Different variants leading to different abnormal transcripts with divergent functions is one possible explanation of the wide phenotypic spectrum seen with TRPV4 variants. This theory is reminiscent of the situation with the FLNA‐gene. Variants in FLNA can produce variable phenotypes including either a skeletal phenotype or a neurological one, but one patient has been described with both the neurological and skeletal phenotypes caused by a particular FLNA variant, and it has been proposed that the variant led to two different abnormal transcripts with divergent functions.11
6. CONCLUSION
We believe the c.838G>A is a new variant in TRPV4 causing the severe form of metatropic dysplasia in this patient. Early diagnosis is of major importance in the management of complex patients with rare diseases. The Skeletal Dysplasia Network was crucial in this diagnostic process.
CONFLICT OF INTEREST
The authors have no conflict of interest to disclose.
AUTHOR CONTRIBUTION
LG: involved in developing the idea, drafting the article, critical revision of the article, and final approval of the version to be published. AH, BNH, KKP, VG, and GG: involved in the clinical examinations and developing the idea, critical revision of the article, and final approval of the version to be published. IV: involved in developing the idea and the data analysis, critical revision of the article, and final approval of the version to be published. PAG: involved in the clinical examinations, developing the idea and the data analysis, critical revision of the article, and final approval of the version to be published.
ACKNOWLEDGMENTS
We would like to thank the family for participation. We also send our gratitude to the Skeletal Dysplasia Network for valuable input in diagnosis and treatment in this patient.
Graversen L, Haagerup A, Andersen BN, et al. Novel TRPV4 variant causes a severe form of metatropic dysplasia. Clin Case Rep. 2018;6:1774–1778. 10.1002/ccr3.1598
REFERENCES
- 1. Schindler A, Sumner C, Hoover‐Fong JE. TRPV4‐Associated Disorders In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews(R). Seattle (WA): University of Washington, 1993:1993‐2018. [Google Scholar]
- 2. Dai J, Kim OH, Cho TJ, et al. Novel and recurrent TRPV4 mutations and their association with distinct phenotypes within the TRPV4 dysplasia family. J Med Genet. 2010;47:704‐709. [DOI] [PubMed] [Google Scholar]
- 3. Camacho N, Krakow D, Johnykutty S, et al. Dominant TRPV4 mutations in nonlethal and lethal metatropic dysplasia. Am J Med Genet A. 2010;152A:1169‐1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andreucci E, Aftimos S, Alcausin M, et al. TRPV4 related skeletal dysplasias: a phenotypic spectrum highlighted byclinical, radiographic, and molecular studies in 21 new families. Orphanet J Rare Dis 2011;6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Everaerts W, Nilius B, Owsianik G. The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog Biophys Mol Biol. 2010;103:2‐17. [DOI] [PubMed] [Google Scholar]
- 6. Dai J, Cho TJ, Unger S, et al. TRPV4‐pathy, a novel channelopathy affecting diverse systems. J Hum Genet. 2010;55:400‐402. [DOI] [PubMed] [Google Scholar]
- 7. Kang SS, Shin SH, Auh CK, Chun J. Human skeletal dysplasia caused by a constitutive activated transient receptor potential vanilloid 4 (TRPV4) cation channel mutation. Exp Mol Med. 2012;44:707‐722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deng HX, Klein CJ, Yan J, et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat Genet. 2010;42:165‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cho TJ, Matsumoto K, Fano V, et al. TRPV4‐pathy manifesting both skeletal dysplasia and peripheral neuropathy: a report of three patients. Am J Med Genet A. 2012;158A:795‐802. [DOI] [PubMed] [Google Scholar]
- 10. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Unger S, Lausch E, Stanzial F, et al. Fetal akinesia in metatropic dysplasia: the combined phenotype of chondrodysplasia and neuropathy? Am J Med Genet A. 2011;155A:2860‐2864. [DOI] [PubMed] [Google Scholar]
- 12. Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4‐/‐ mice. Proc Natl Acad Sci U S A. 2003;100:13698‐13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weinstein MM, Tompson SW, Chen Y, Lee B, Cohn DH. Mice expressing mutant Trpv4 recapitulate the human TRPV4 disorders. J Bone Miner Res. 2014;29:1815‐1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nishimura G, Dai J, Lausch E, et al. Spondylo‐epiphyseal dysplasia, Maroteaux type (pseudo‐Morquio syndrome type 2), and parastremmatic dysplasia are caused by TRPV4 mutations. Am J Med Genet A. 2010;152A:1443‐1449. [DOI] [PubMed] [Google Scholar]