

Abstract
Astrocytes are the most abundant cell type in the central nervous system (CNS). Once considered to be of fairly homogeneous phenotype throughout the brain and spinal cord, they are now understood to be heterogeneous in both structure and function. They are important in brain functions as diverse as ion and fluid balance in the interstitial space, contributing to integrity of the neurovascular unit (blood-brain barrier), neurotransmitter regulation, metabolism of energy substrates and possibly even axonal regeneration. After ischemic or hemorrhagic brain/spinal cord injury, formation of an astrocytic scar adjacent to the ‘lesion’ is a characteristic histopathologic feature, and this astrogliosis can be demonstrated by immunohistochemistry, usually using primary antibodies to glial fibrillary acidic protein (GFAP). Astrocytes interact with microglia and oligodendroglia in novel ways that will be discussed in this review.
This article is part of the Special Issue entitled ‘Cerebral Ischemia’.
Keywords: Astrocytes, Microglia, Oligodendroglia, Ischemia, Intracerebral hemorrhage, Brain injury - responses
1. Introduction
Neuroglia or glia are considered the ‘supporting’ cells of the central nervous system (CNS), including the brain and spinal cord—the ‘glue that holds the neurons together’, to be overly simplistic. Rudolf Virchow, widely credited with discovering neurogliadwhich he conceptualized as a “neuroglia connective tissue” or ‘Nervenkitt’, conceived of them as functioning to maintain nerve cell structure (Liddelow and Barres, 2017). They are of four primary phenotypes: astrocytes, oligodendroglia, ependymal cells and microglia (Ellison et al., 2013; Vinters and Kleinschmidt-DeMasters, 2015). Oligodendroglia are the myelin-forming cells of the CNS and will be considered briefly further in this review. Ependymal cells form the lining of the ventricular system (including aqueduct of Sylvius) and have a fairly limited repertoire of responses to injury: when there is an (unspecified) injury to the ependyma (e.g. secondary to a viral infection in the cerebrospinal fluid), there may be proliferation of underlying astrocytes to produce an ‘ependymal granulation’ or granular ependymitis, a lesion of unknown significance that is frequently encountered in autopsy brain sections sampled from the periventricular region. They will not be considered in detail. Microglia and astrocytes are the main cell types that respond to various types of brain injury, but their functions are increasingly recognized as being complex, frequently interactive (sometimes even synergistic), and often even beneficial (rather than deleterious) to optimal CNS functions (Wirenfeldt et al., 2011; Liddelow and Barres, 2017).
The origins of microglia and astrocytes, and the relationship between microglia and brain macrophages remain subjects of debate. Astrocytes, which constitute almost a third of cells in the CNS and vastly outnumber nerve cell bodies, are considered to be of neuroectodermal origin, whereas microglia are thought to originate from mesodermal precursors (Wirenfeldt et al., 2011). Circulating monocytes (bone marrow-derived) may enter the brain and therein behave as microglia or macrophages, especially in response to ischemia (see below).
Microglia and astrocytes are recognized in tissue sections by their morphology, but more commonly using special cytochemical techniques or immunohistochemistry. Microglia and astrocytes in their resting state may be inconspicuous in a tissue section unless it has been labelled with an immunohistochemical method. Activated microglia are ameboid or elongated cells with ramified processes; they are effectively immunolabelled with primary antibodies to various epitopesdthe one most commonly used in surgical specimens is CD68, which does not distinguish microglia from macrophages (see also below) (Wirenfeldt et al., 2011). In research settings, Iba1+ is a highly reproducible immunomarker for activated microglia, and the immunoreactive signal can be easily quantified using image analysis software (Wirenfeldt et al., 2009) [Fig. 1]. The intensity of CD45 immunoreactivity of cells within brain may reflect their origin from the circulation (blood-derived macrophages, high CD45) or within the brain (low CD45 expression). Reactive astrocytes are recognized, when activated, on routine tissue sections by characteristic coarse nuclear chromatin, prominent glassy eosinophilic cytoplasm (gemistocytic differentiation) and processes extending from the cytoplasm, though immunohistochemistry is usually employed to confirm cell phenotype. Antibodies to glial fibrillary acidic protein (GFAP) (one of the first brain-enriched proteins discovered in the early 1970s, though it is found in other loci throughout the body) remain a reliable way to immunolabel most astrocytes. Using this technique, labelled astrocytes often show a star-like appearance in tissue sections, with prominently labelled and non-overlapping processes radiating from the cell body [Fig. 1]. Astrocytic proliferation resulting in gliosis was, decades ago, highlighted by special stains that had an affinity for this cell type—these included the Holzer and phosphotungstic acid hematoxylin (PTAH) stains. In the modern era, gliosis is almost always demonstrated using GFAP immunohisto-chemistry; an important caveat, however, is that not all astrocytes stain with anti-GFAP, so that the severity of astrogliosis may be underestimated using this immunohistochemical approach. Conversely, it may be overestimated because of the tendency of injured (as by ischemia) CNS tissue to collapse, leading to an apparent increase in astrocytic densitydastrocytes being more resistant to ischemia than neurons. GFAP is an intermediate filament protein (others in this family include nestin and vimentin, which are also upregulated in reactive astrocytes) that is essential for the process of astrogliosis and scar formation but, of interest, is not essential for normal astrocytic function. There are isoforms of GFAP (alpha, beta, gamma, delta, and kappa). Recently it has been suggested that Sox9 is a reliable nuclear marker of astrocytes in ‘non-neurogenic’ brain regions (Sun et al., 2017). To quote from a review on astrocyte biology and pathologic reactions (Sofroniew and Vinters, 2010) “[they] tile the entire CNS in a contiguous and essentially non-overlapping manner that is orderly and well organized”.
Fig. 1.
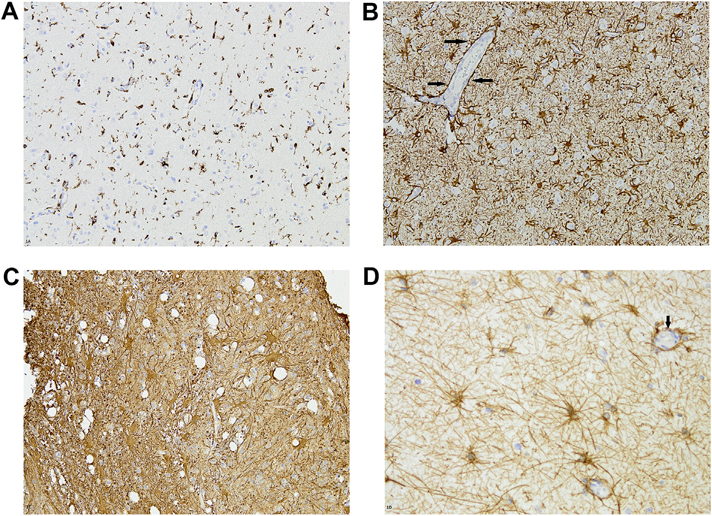
A. Activated microglia adjacent to a region of ischemia (surgical specimen), highlighted by immunohistochemistry using anti-CD68. Note variable morphologies, including rounded, triangular and ‘spindled’ cells. B. Astrocytes prominently labelled with anti-GFAP. Cells have variable amounts of cytoplasm and prominent processes. Arrows indicate a blood vessel (which itself is GFAP negative) surrounded by GFAP-immunoreactive astrocytic processes. (Panels A and B are micrographs from the same surgical specimen, photographed at identical magnifications) C. Prominently GFAP-immunoreactive cells with abundant cytoplasm (gemistocytes). D. More star-shaped astrocytes (GFAP immunostain) with relatively little cytoplasm. Arrow indicates a small vessel encircled by astrocytic processes.
Astrocytes are subclassified further by their location within the brain, and morphology. Protoplasmic astrocytes are found mainly within grey matter and show a morphology characterized by stem branches that give rise to many finely branching processes in a uniform globoid distribution. Their processes envelop synapses. Fibrous astrocytes populate the subcortical white matter and show many long fiber-like processes, which contact nodes of Ranvier. The intimate relationship between astrocytes and blood vessels (of many sizes, but especially capillaries) is notable on GFAP immuno-stained sections, which almost invariably show GFAP processes in close apposition to the abluminal aspects of blood vessels, sometimes entirely surrounding and ensheathing the vessel [Fig. 1]. Since the 1980s astrocytic influences on the cerebral microvasculature have been studied using various approaches (including tissue culture models) (Beck et al., 1984), so that the astrocyte is now recognized as being an important element of the ‘neurovascular unit or niche’—or the ‘extended blood-brain barrier’, i.e. cerebral capillary endothelium, pericytes (in the case of capillaries), smooth muscle cells (arterioles) and perivascular astrocytes (Carmichael, 2016; Sofroniew and Vinters, 2010). Astrocyte-like cells are seen outside the cerebral hemispheres and spinal cord, including Muller glia in the retina, Bergmann glia in the cerebellum, pituicytes in the neurohypophysis, and tanycytes at the base of the third ventricle. Bergmann glia react to ischemic cerebellar lesions, but usually not by developing a ‘gemistocytic’ phenotype, as in the cerebral hemispheres and spinal cord (see below).
Astrocytes do not (unlike neurons) propagate an action potential, but undergo changes in intracellular calcium concentration; these ‘regulated’ changes in calcium concentration may be important in astrocyte-neuron and astrocyte-astrocyte communication. Astrocyte functions in the ‘healthy’ CNS include uptake of potassium, water (astrocytes contain abundant aquaporin-4, a water channel; Moftakhar et al., 2010; Shi et al., 2017), and neurotransmitters (glutamate, GABA, glycine), and release of energy substrates (e.g. lactate), neurotransmitter precursors (e.g. glutamine), purines (ATP, adenosine), and growth factors (e.g. BDNF, TNFalpha). Aqua-porin 4-mediated glutamate-induced astrocytic swelling may be partially mediated through the activation of metabotropic gluta-mate receptor 5 (Shi et al., 2017). The tight apposition of astrocytes to cerebral microvessels, neuronal perikarya, axons (in particular nodes of Ranvier) and synapses make them a crucial player in CNS function, metabolism and dysfunction. Astrocytes contain abundant glycogen and such glycogen stores are prominent in areas of high synaptic density. Astrocytic glycogen utilization can sustain neuronal activity during hypoglycemia and periods of high neuronal activity. Astrocytic glycogen content can be modulated by neurotransmitters such as glutamate; glucose metabolites can be passed across gap junctions in a process that is regulated by neuronal activity and glutamate. There are numerous triggers of astrocytic ‘reactivity’, including interleukins, growth factors and cytokines—which in turn may originate in microglia, leukocytes, or astrocytes themselves. Cell injury or death (including that secondary to ischemia) are potent stimulators of astrocytic hypertrophy and proliferation (the two processes often go together), through various mechanisms that may involve reactive oxygen species (ROS) and nitric oxide (NO) (Sofroniew and Vinters, 2010). Recent data suggests that reactive astrocytes may even function as phagocytes after ischemia (Morizawa et al., 2017).
As indicated above, astrocytes and their processes make frequent and intimate contacts with blood vessel walls; interactions between them are bidirectional. Astrocytes produce and release molecular mediators of CNS blood flow and vascular diameter; these include prostaglandins, nitric oxide, and arachidonic acid. Because astrocytes (especially within the cortex) are also in close contact with synapses, they may titrate blood flow in relation to levels of neural and synaptic activity.
1.1. Observations in human tissues
The events surrounding the occurrence of an ischemic infarct (and the brain’s ‘attempt’ to recover from this insult or limit its severity) are extremely complex (for a recent mechanistic review, see Carmichael, 2016). Astrocytic proliferation occurs adjacent to both ischemic and hemorrhagic brain lesions. Ischemic infarcts and hemorrhages may be observed in biopsies (e.g. an edematous infarct that mimics a neoplasm on neuroimaging studies, a hemorrhagic lesion of unclear origin that is biopsied or resected to establish its etiology) but are more commonly observed in autopsy specimens (Vinters et al., 1998; Ellison et al., 2013; Vinters, 2001). Infarcts can be classified by size: microinfarcts are ones that are not seen on neuroimaging studies or gross inspection of a necropsy brain slice, but are identified on microscopy—often highlighted with the aid of CD68 or GFAP immunohistochemistry (Wang et al., 2012). Lacunar infarcts are seen grossly but are smaller than 1.0 cm in greatest dimension, and are usually visible on neuroimaging studies, most commonly in the deep central grey matter or pons. Cystic infarcts are larger than 1.0 cm and usually in the territory of a major artery or one of its branches. The sequence of cellular events occurring after irreversible brain ischemia is usually diapedesis of polymorphonuclear leukocytes (PMNs) (hours to 1–2 days after irreversible ischemia), followed by microglial activation and infiltration of blood-borne macrophage precursors (monocytes) (2–5 days after ischemia), then followed by astrocytic proliferation and hypertrophy (3–10 days post-ischemia). Reactive astrocytes with abundant cytoplasm may be referred to as gemistocytes. Hyper-trophy may be a more significant factor than astrocyte proliferation in astrocytosis, because of pronounced upregulation of GFAP production (after ischemic injury) and collapse of the tissue, leading to an apparent rather than a real increase in astrocyte density or numbers (Liddelow and Barres, 2017). Astrocytes can be especially abundant in the preserved neocortical layer I (molecular layer, often spared even in a large cortical infarct), but are easily identified at the interface between an infarct and surrounding brain, where they have historically been considered a barrier to axon regeneration [Fig. 2].
Fig. 2.
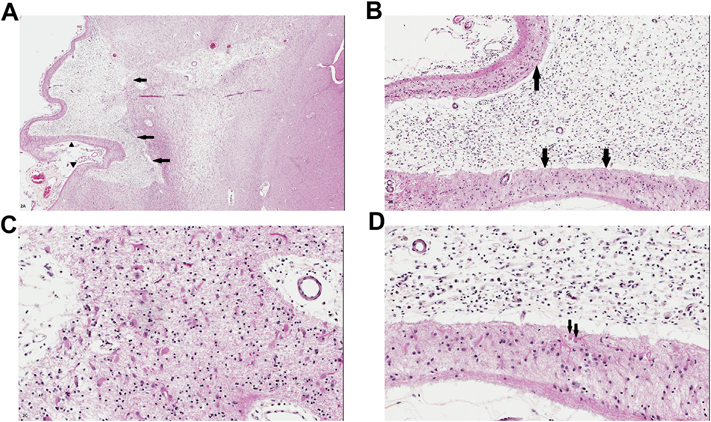
A. Remote or ‘old’ cystic infarct in cortex lateral to the basal ganglia (visible at right of the micrograph; all images are different views and magnifications of the same infarct). Arrows indicate a cystic cavity representing the infarct. Arrowheads indicate preserved layer I of the cortex, a characteristic finding in old ‘strokes’. B. Magnified view of the infarct shows the cystic cavity replete with lipid-laden macrophages; arrows indicate the preserved and clearly outlined layer I of the cortex, immediately underlying leptomeninges. C. Deep ‘margin’ of the cystic infarct shows abundant astrocytes, including gemistocytes, with prominent eosinophilic cytoplasm. D. Magnified view of the interface between preserved cortical layer I (bottom of the image) and lipid laden macrophages (at top of the image). Subarachnoid space is at the bottom edge of the micrograph. Arrows indicate two gemistocytes with prominent cell processes. [All images are from sections stained with hematoxylin and eosin/H&E].
Macrophages can persist in an infarct even after it has undergone cavitation, and may be found many years after necrosis has occurred, as is the residual astroglial rim or margin [Fig. 2]. Large or small brain hemorrhages (including microbleeds or BMBs) contain, by definition, abundant altered blood pigment, usually hemosiderin filling the cytoplasm of macrophages [Figs. 3 and 4]. The “dating” or “timing” of an infarct or brain hemorrhage (large or small) based upon the above histopathologic changes is not, however, a ‘precise science’. In most instances, the neuropathologist provides an opinion upon whether the appearance of an ischemic or hemorrhagic lesion is ‘consistent with’ its having occurred in a specific clinical time frame. This becomes highly problematic with microinfarcts or BMBs, since a given lesion by itself may not result in a definable neurologic deficit; rather, neurologic impairment is the result of progressive accumulation of microscopic lesions over months or years (microinfarcts and BMBs are now regarded as important substrates of ischemic-vascular dementia) (Ellison et al., 2013). The pathogenesis of microinfarcts is probably multifactorial (microemboli, atheroemboli, systemic factors such as hypotension, microvascular disease including amyloid angiopathy (Soontornniyomkij et al., 2010). A mouse model of multiple cerebral microinfarcts induced by internal carotid injection of 40–70 μm cholesterol particles/crystals has been developed, and the cellular dynamics associated with the microfoci of necrosis have been studied (Wang et al., 2012). The investigators found that the microinfarcts had a core of macrophages, surrounded by large regions of GFAP-immunoreactive astrocytes—i.e. similar to what is seen in human autopsy material [Fig. 2]. Astrocytes appeared to show ‘mislocalization’ of the water channel aquaporin 4, persisting for a significant time after the injury. Surrounding brain showed delayed neuronal loss as long as 28 days after the induced ‘strokes’.
Fig. 3.
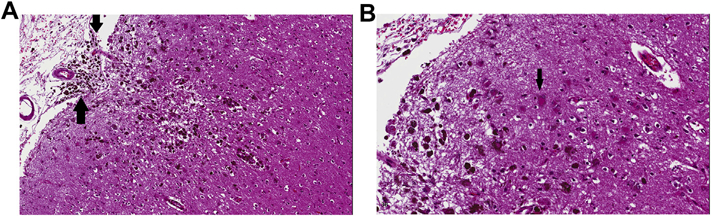
A. Old cortical microhemorrhage, probably related to amyloid angiopathy, in an elderly patient. The neuropil appears cystic, there is slight indentation of the pial surface (secondary to underlying tissue loss) and hemosiderin-laden macrophages are seen in abundance. Arrows indicate extension of the microhemorrhage into the subarachnoid space. B. Magnified view from edge of the brain microbleed (BMB). Arrow indicates a gemistocyte that has either phagocytosed altered blood pigment, or shows it encrusted on the cell membrane. (Images are from H&E-stained sections).
Fig. 4.
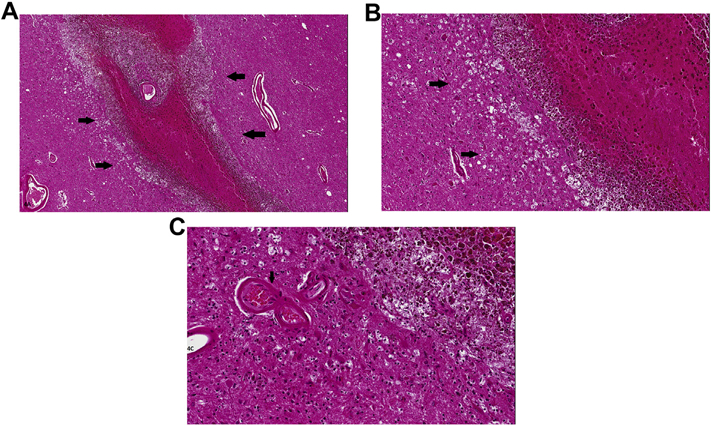
A. Linear thalamic hemorrhage, probably of hypertensive origin (indicated by arrows), in an elderly female subject. By history, the ‘stroke’ had occurred several weeks/months prior to autopsy. Large vessel at the center of the hematoma shows significant arteriosclerotic change and may have been the ‘vessel of origin’ of the bleed. Hematoma itself shows preserved erythrocytes at center of the hematoma, but significant hemosiderin-laden macrophages at its margins. B. Magnified view of edge of the hematoma highlighting hemosiderin-laden macrophages and numerous astrocytes. The ‘vacuolated’ cells indicated by arrows probably represent Wallerian degeneration in fiber tracts injured by the hematoma. C. Arrow indicates an arteriosclerotic vessel in close proximity to the hematoma. Note numerous reactive astrocytes; upper right of the micrograph shows macrophages containing altered blood pigment. (Images are from H&E-stained sections).
Oligodendrocytes, the myelin forming cells in the CNS, are especially susceptible to ischemia, particularly in the developing brain, and injury leads to demyelination, dysmyelination and disrupted axonal function (Dewar et al., 2003; Mifsud et al., 2014). In adult-onset strokes, oligodendrocyte damage manifests histologically as loss of myelin staining in the white matter, and injured oligodendrocytes have been shown to immunohistochemically express tau in response to head injury or ischemia (Dewar et al., 2003; Irving et al., 1996; Uchihara et al., 2000). In the immature brain, injury to subcortical fiber tracts and oligodendrocyte progenitors leads to periventricular leukomalacia and chronic demyelination (Dewar et al., 2003). Periventricular leukomalacia is the most common form of brain injury in preterm infants due to the immaturity of the vasculature, low cerebral blood flow in the white matter, and impaired cerebrovascular autoregulation in premature infants as well as the predominance of late oligodendrocyte progenitors vulnerable to ischemia during the high-risk period of approximately 23–32 postconceptional weeks (Back, 2017; Mifsud et al., 2014; Volpe, 2001). It is characterized neuropathologically by focal necrosis of the deep periventricular white matter, which may evolve into cysts, and diffuse reactive gliosis in the surrounding white matter (Back, 2017; Folkerth, 2006). The necrotic lesions appear to correlate with the spastic diplegia of cerebral palsy while the diffuse component may contribute to the cognitive and behavioral abnormalities seen in survivors of prematurity (Folkerth, 2006; Volpe, 2001).
2. Experimental approaches
Innovative approaches to studies of astrocytic biology have evolved since the 1980s. Experimental approaches have evolved from use of various injuries (cortical freeze injury, vascular occlusion) to induce glial responses, to transgenic animal approaches and sophisticated tissue culture methods, to modern molecular techniques such as gene expression profiling (transcriptomics; see below). Until recent decades astrocytes were thought to function as glial support cells, simply providing metabolic, trophic, and structural support to nerve cells (Becerra-Calixto and Cardona-Gomez, 2017). Initial experiments were predicated on the hypothesis that destructive lesions within the brain would induce factors that stimulated astrocytic cell division and DNA synthesis (Nieto-Sampedro et al., 1985). Observations in experimental animals showed a 3- to 10-fold increase in the activity of factors capable of stimulating astrocyte DNA synthesis and cell division in vitro, and maximum mitogenic activity 10–15 days after lesion induction. A major innovation in studying astrocytic physiology was achieved with the ability to culture glial cells (astrocytes, oligodendroglia)—by Jean De Vellis and his colleagues, and other laboratories, in the 1980s (Bologa et al., 1988). Astrocytic dysfunction in an ischemic lesion may result from abnormalities in ion buffering, uptake and synthesis of neurotransmitters, water transport, influences on cerebral blood flow, release of antioxidants, and even immunomodulation (Becerra-Calixto and Cardona-Gomez, 2017). Astrocytes are considered as supporting the energy needs of neurons, and there is evidence of ‘bi-directional’ communication between neurons and astrocytes under some ischemic conditions (Wu et al., 2017). Astrocytes show a high glycolytic rate and show high expression of relevant enzymes (e.g. 6-phosphofructose-2-kinase, fructose-2,6-biphosphatase-3). They are also vital for the extra-cellular regulation of glutamate, which can be highly toxic to neurons—through the action of receptors such as glutamate transporter 1 (GLT-1) and glutamate aspartate transporter (GLAST). Astrocytes are capable of synthesizing glutamate from glucose. In conditions of brain hypoxia-ischemia, GLAST and GLT-1 are downregulated, suggesting a mechanism that facilitates glutamate-related excitotoxicity, with obvious implications for possible therapeutic avenues aimed at astrocytes.
Astrocytes may also be involved in protection against oxidative stress—through mechanisms that involve glutathione, the antioxidant ascorbic acid, and the redox-sensitive transcription factor Nrf2. The Nrf2 pathway is activated in both tissue culture and in vivo models of ischemia. Astrocytes may have significantly greater ‘metabolic plasticity’ than neurons; e.g., they respond to NO with an increase in glucose metabolism through the glycolytic pathway, limiting the decline in ATP, a response that does not seem to be present in neurons.
3. Molecular mechanisms
The molecular pathways underlying the crucial functions of glial cells, astrocytes, microglia, and oligodendroglial lineage cells, which were in the past thought to have simply a supportive or “glue” - like function, in both the healthy and diseased central nervous system (CNS), are increasingly being elucidated (Jäkel and Dimou, 2017). Astrocytes have a multitude of essential functions in the CNS (see also above), providing neurotrophic support, promoting formation and maintenance of synaptic activity and transmission, regulating blood flow and determining some functions and properties of the blood-brain barrier (BBB) or neurovascular unit (NVU) (Sofroniew, 2009; Sofroniew and Vinters, 2010). They play a central role in the glymphatic system, the recently discovered CNS waste clearance system (Verkhratsky and Nedergaard, 2016).
Astrocytes are crucial in the formation, maintenance and elimination of synapses through the secretion of synaptogenic molecules such as brain-derived neurotrophic factor (BDNF), cholesterol, glypicans, thrombospondins, hevin (SPARCL1), and SPARC as well as via contact mediated signaling through the activation of neuronal integrin receptors and the protein kinase C signaling pathway (Clarke and Barres, 2013). In development, astrocytes induce the production of C1q in neuronal synapses; this tags them for removal via the classical complement pathway, whereby they are phagocytosed by microglia expressing high levels of C1q and the complement receptors CR3–this in turn mediates phagocytosis of complement-coated particles, and CR5 (Stephan et al., 2012). Microglia are the resident inflammatory cells of the CNS (Block et al., 2007). They survey the extracellular microenvironment in the healthy brain, and are not only central in the initial response to injury, clearance of toxic debris, and tissue repair but are also important in synaptic remodeling and programmed neuronal elimination as well as neuronal survival (Block et al., 2007; Nimmerjahn et al., 2005; Schafer et al., 2012).
Astrocytic processes are associated with pre- and postsynaptic terminals as part of the tripartite synapse and are essential in the clearance of K+ and glutamate after neuronal activity (Halassa et al., 2007; Perea et al., 2009). It has been estimated that one astrocyte ‘oversees’ over 100,000 synapses (Bushong et al., 2002; Halassa et al., 2007). Astrocytes express high densities of transporters for neurotransmitters including glutamate, GABA, and glycine (Sofroniew and Vinters, 2010). They are the primary regulators of extracellular glutamate concentration, being responsible for the majority of glutamate uptake in synaptic and non-synaptic areas (Matute et al., 2006; Rossi et al., 2007). Moreover, neurotransmitter release induces changes in astrocytic intracellular calcium concentrations which in turn stimulates the release of gliotransmitters such as glutamate, ATP and D-serine (Halassa et al., 2007). Astrocytic processes are also an integral part of the neurovascular unit (see above) (Abbott et al., 2006; Sofroniew and Vinters, 2010). Furthermore, astrocytes are extensively coupled via gap junctions, composed of connexin (CX) proteins predominantly of the CX-43 and CX-30 subtypes, which allow for intercellular movement of metabolites and ions over long distances (Charveriat et al., 2017; Pekny and Nilsson, 2005). The high density of aquaporin 4 (AQP4) water channels and ion transporters on astrocytic foot processes are important in fluid homeostasis as well as vasogenic and cytotoxic edema after ischemia, trauma, and inflammation (Zador et al., 2009).
Oligodendroglial lineage cells include oligodendrocytes and the oligodendrocyte precursors, NG2-glia (Jäkel and Dimou, 2017). Although the functions of these NG2 proteoglycan protein expressing glia are still unclear, they generate mature myelinating oligodendrocytes and are able to form functional synapses with neurons (Jäkel and Dimou, 2017; Nave, 2010). Oligodendrocytes are abundant in the grey and white matter of the brain and spinal cord and produce myelin to insulate CNS axons for rapid saltatory conduction and provide neurotrophic support (Dewar et al., 2003; Jäkel and Dimou, 2017). They contain abundant microtubules for transporting myelin proteins to their processes, and the microtubule associated protein tau.
Oligodendrocytes are more vulnerable to ischemia compared to other glial cells and, in certain regions of the brain and stages of development, more susceptible even than neurons (Petito and Olarte, 1998). Their increased vulnerability to oxidative stress during ischemia from anaerobic metabolism and lactic acidosis is due to their high rate of oxidative metabolism, high lipid and iron content, elevated permeability of glutamate receptors, and low levels of the antioxidant glutathione (Dewar et al., 2003; Mifsud et al., 2014). As early as 30 min after arterial occlusion, oligodendrocytes demonstrate cell swelling and alterations in the Golgi apparatus and endoplasmic reticulum, and increased tau within 40 min of the ischemic insult (Dewar et al., 2003). Neurotransmitter receptors for ATP and glutamate on oligodendrocytes that mediate signaling from axons to stimulate differentiation and myelination in normal conditions are overstimulated in ischemia, leading to Ca2+ overload and mitochondrial dysfunction (Mifsud et al., 2014). Glutamate excitotoxicity, as noted above, is one of the major causes of ischemic injury, and glutamate transporters on oligodendrocytes, astrocytes, and microglia that under normal conditions mediate uptake of extracellular glutamate to maintain low levels in the extracellular space, in ischemia release glutamate into the extracellular space (Mifsud et al., 2014). Furthermore, brain injury-induced astrocyte swelling is likely mediated by glutamate transporters and metabotropic glutamate receptors (Shi et al., 2017), and it is well known that oligodendrocytes are susceptible to excitotoxicity mediated by AMPA/kainate ionotropic glutamate receptors (Mifsud et al., 2014). There is significant developmental upregulation of these non-NMDA receptors, and it has recently been shown that selective upregulation and downregulation of specific subunits of the NMDA receptors in subjects with periventricular leukomalacia may underlie NMDA-mediated vulnerability in the developing brain (Jantzie et al., 2015; Mifsud et al., 2014).
In general, astrocytes are more resistant to ischemia and other stressors compared to neurons, although several groups have found subtypes of astrocytes that appear to be more susceptible, especially to acidosis (Chen and Swanson, 2003). Within a few minutes of ischemia or other injury to the CNS, neurons and glial cells in the lesion release cytokines such as transforming growth factor (TGF)-α, ciliary neurotrophic factor (CNTF), interleukin (IL)-1, and kallikrein-related peptidase 6 (KLK6) to activate astrocytes (Liu and Chopp, 2016). Early in the response there is increased uptake of K+, glutamate and lactate by astrocytes which can result in swelling and cytotoxic edema resulting from excess water uptake through AQP4 channels, whereas failure of AQP4 or astrocyte loss can lead to vasogenic edema (Liu and Chopp, 2016; Sofroniew and Vinters, 2010). Gap junction connections are decreased but remain open in ischemia and allow for distribution of potentially harmful substances such as apoptotic signals over long distances and expansion of lesion size (Liu and Chopp, 2016; Pekny and Nilsson, 2005). Within a few hours of injury, astrocytes respond by undergoing reactive astrogliosis, a process in which they undergo a spectrum of molecular, functional and morphologic changes (Chen and Swanson, 2003). Changes range from mild hypertrophy of the cell body and processes without cellular proliferation to the severe end of the spectrum with cellular proliferation, extensive overlapping of astrocytic processes, and glial scar formation derived essentially from newly proliferated astrocytes (Sofroniew, 2014). Mild morphologic and functional changes can be reversible while severe scar formation is long-lasting (Sofroniew, 2009).
In correlation with changes in morphology, reactive astrocytes increase expression of the structural proteins GFAP and vimentin, and re-expression of nestin (Lin et al., 1995; Pekny and Nilsson, 2005). Increased GFAP expression and cellular hypertrophy are relatively proportional to the degree of reactivity (Sofroniew, 2014). GFAP and vimentin are intermediate filament proteins of which there are over 60 different forms identified in humans; along with actin filaments and microtubules, they are major constituents of the cytoskeleton (Li et al., 2008; Pekny and Nilsson, 2005). Vimentin and nestin are abundantly expressed in immature astrocytes during neural development and normally downregulated in the mature CNS (Liu and Chopp, 2016; Pekny and Pekna, 2004). These structural proteins are induced by signaling pathways involving cAMP, STAT3, and NF-κB, while astrocytic proliferation is regulated by signals including epidermal growth factor (EGF), fibroblast growth factor (FGF), endothelin 1, sonic hedgehog, thrombin, and albumin (Sofroniew, 2014). Reactive astrocytes also produce matrix metal-loproteinases and inhibitors to remodel the glial scar and secrete extracellular matrix components for BBB repair (Chen and Swanson, 2003). Glial scars are thought to form a barrier function to contain released toxic substances and inflammatory cells but at the same time impair axon regeneration through the expression of inhibitors of axonal regeneration such as chondroitin sulfate proteoglycans (Liu and Chopp, 2016; Sofroniew and Vinters, 2010) and ephrin-5A, a membrane bound astrocyte growth inhibitor (Overman et al., 2012). However, recent studies provide some evidence that glial scars may facilitate rather than prevent regeneration of axons in the CNS (Anderson et al., 2016).
Reactive astrocytes also increase the expression of numerous other proteins involved in transcription and energy metabolism, signaling molecules, and antioxidant proteins such as copper-zinc superoxide dismutase, glutathione peroxidase, and metal-lothionein (Chen and Swanson, 2003; Liu and Chopp, 2016). Indeed, astrocytes contain the highest concentrations of antioxidants in the brain (Pekny and Nilsson, 2005). Recently, genomic profiling has shown that reactive astrocytes upregulate over 1000 genes, with those of the extracellular matrix, cytoskeleton, cytokines and genes induced in response to injury and wound healing in peripheral tissues, well represented, as are transporters, especially those involved in metal ion homeostasis, and signaling receptors (Zamanian et al., 2012). Although GFAP expression is maintained in glial scars for a prolonged period after injury (Sofroniew, 2009), most expression changes are transient. There may be an acute increase in expression after exposure to the stressor, and subsequent moderation, with neurotrophic cytokines and growth factors rapidly downregulated (Zamanian et al., 2012). However, proinflammatory cytokines remain persistently elevated and expression of STAT3, which plays a major role in reactive gliosis and scar formation, has been shown to increase two-fold and maintain 50% elevation over one week after both ischemic and inflammatory insults (Burda and Sofroniew, 2014; Zamanian et al., 2012).
Most recently, reactive astrocytes have been be divided into two groups, A1 and A2, based on the type of injury, inflammation or ischemia, respectively (Liddelow et al., 2017; Zamanian et al., 2012). Ischemia induces A2 reactive astrocytes which express elevated levels of neurotrophic factors and cytokines such as CLCF1, LIF, IL-6, and thrombospondins suggesting that A2 reactive astrocytes are protective, promoting neuronal survival and repair. On the other hand, neuroinflammation induces A1 reactive astrocytes which upregulate many genes of the classic complement cascade such as C1r, C1s, C3, and C4, which are involved in synaptic pruning and lead to loss of synapses, neurons and mature oligodendrocytes (Liddelow et al., 2017; Zamanian et al., 2012).
Triggers of reactive astrogliosis and astrocytic proliferation include a variety of molecular signals released upon any form of CNS injury or disease. These include substances released from dead and dying cells, inflammatory and glial cells, and molecules entering through a disrupted BBB; they include cytokines, ATP, endothelin, FGF2, thrombin, bone morphogenic proteins, and sonic hedgehog (Burda and Sofroniew, 2014; Sofroniew, 2009). Early triggers consist of pro-inflammatory cytokines and purines/pyrimidines such as ATP and nucleotides released from damaged cells and increased excitotoxic transmission, as ATP is co-released with neurotransmitters (Buffo et al., 2010). Later, growth factors and endothelin sustain the astrogliosis while inhibitory factors such as interferon β, IL-10 and erythropoietin modulate the response. Astrocytes upregulate a variety of trophic factors such as BDNF, nerve growth factor (NGF), ciliary neurotrophic factor (CNTF) a member of the IL-6 family of cytokines with neurotrophic and differentiating effects, FGF, vascular endothelial growth factor (VEGF), and platelet derived growth factor (PDGF), which support neurons and promote angiogenesis (Chen and Swanson, 2003; Seidel et al., 2015; Sofroniew, 2014). Recent studies have also implicated microRNAs (miRs) and miR regulatory enzymes such as Dicer in the modulation of astrogliosis (Sofroniew, 2014). ATP and analogues have been shown to promote astrocyte proliferation and growth of long and branched processes through G protein-coupled P2Y receptors (Buffo et al., 2010).
ATP also activates microglia which migrate and send processes to the injured site (Chen and Swanson, 2003; Davalos et al., 2005). Activated microglia transform from their resting state characterized by ramified morphology into an amoeboid shape and upregulate surface molecules including CD14, major histocompatibility complex (MHC) proteins, and chemokine receptors (Block et al., 2007). Although astrocytes release certain pro-inflammatory cytokines, microglia are the main source of cytokines in injured brain (Buffo et al., 2010). Primary mediators include tumor necrosis factor (TNF), IL-1β, and interferon γ, which lead to the production of secondary mediators such as arachidonic acid, NO and matrix metalloproteases. Liddelow et al. have demonstrated that activated microglia secrete IL-1α, TNF and C1q to induce A1 reactive astrocytes to release a yet uncharacterized neurotoxin that promotes death of subtypes of CNS neurons and mature oligodendrocytes (Liddelow et al., 2017). Furthermore, A1 reactive astrocytes are abundant in neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, and in acute active demyelinating plaques of multiple sclerosis (Liddelow et al., 2017).
The protective role of reactive astrogliosis in ischemia has been demonstrated by increased inflammation and tissue damage resulting from experimental blockade of astrogliosis in various types of CNS injury including ischemia, trauma, infection, autoimmune inflammation and neurodegeneration (Sofroniew, 2014). In GFAP and vimentin knockout mice in which there is absence of intermediate filaments in reactive astrocytes, induced ischemia results in larger infarct volumes without an increase in the number of astrocytes in the affected region (Li et al., 2008). This was associated with reduced inhibition of astrocyte gap-junctional communication, which is thought to promote propagation of injury, decreased glutamate uptake, and decreased levels of plasminogen activator inhibitor-1, which could enhance the neurotoxicity of tissue plasminogen activator, in the knockout mice (Li et al., 2008).
References
- Abbott NJ, Rönnbäck L, Hansson E, 2006. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci 7, 41–53. [DOI] [PubMed] [Google Scholar]
- Anderson MA, Burda JE, Ren Y, Ao Y, Coppola G, Khakh BS, Deming TJ, Michael V, Angeles L, 2016. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, 2017. White matter injury in the preterm infant: pathology and mechanisms. Acta Neuropathol [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra-Calixto A, Cardona-Gomez GP, 2017. The role of astrocytes in neuro-protection after brain stroke: potential in cell therapy. Front. Mol. Neurosci 10, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck DW, Vinters HV, Hart MN, Cancilla PA, 1984. Glial cells influence polarity of the blood-brain barrier. J. Neuropathol. Exp. Neurol 43, 219–224. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong J-S, 2007. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci 8, 57–69. [DOI] [PubMed] [Google Scholar]
- Bologa L, Cole R, Chiapelli F, Saneto RP, De Vellis J, 1988. Expression of glial fibrillary acidic protein by differentiated astrocytes is regulated by serum antagonistic factors. Brain Res 457, 295–302. [DOI] [PubMed] [Google Scholar]
- Buffo A, Rolando C, Ceruti S, 2010. Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol 79, 77–89. [DOI] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV, 2014. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH, 2002. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci 22, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael ST, 2016. Emergent properties of neural repair: elemental biology to therapeutic concepts. Ann. Neurol 79, 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charveriat M, Naus CC, Leybaert L, Sáez JC, Giaume C, 2017. Connexin-dependent neuroglial networking as a new therapeutic target. Front. Cell. Neurosci 11, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Swanson RA, 2003. Astrocytes and brain injury. J. Cereb. Blood Flow. Metab 23, 137–149. [DOI] [PubMed] [Google Scholar]
- Clarke LE, Barres BA, 2013. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci 14, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan W, 2005. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci 8, 752–758. [DOI] [PubMed] [Google Scholar]
- Dewar D, Underhill SM, Goldberg MP, 2003. Oligodendrocytes and ischemic brain injury. J. Cereb. Blood Flow. Metab 23, 263–274. [DOI] [PubMed] [Google Scholar]
- Ellison D, Love S, Chimelli L, Harding BN, Lowe JS, Vinters HV, Brandner S, Yong WY, 2013. Neuropathology A erence Text of CNS Pathology, third ed. Elsevier Mosby, Edinburgh. [Google Scholar]
- Folkerth RD, 2006. Periventricular leukomalacia: overview and recent findings. Pediatr. Dev. Pathol 9, 3–13. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG, 2007. The tripartite synapse: roles for glio-transmission in health and disease. Trends Mol. Med 13, 54–63. [DOI] [PubMed] [Google Scholar]
- Irving EA, Nicoll J, Graham DI, Dewar D, 1996. Increased tau immunoreactivity in oligodendrocytes following human stroke and head injury. Neurosci. Lett 213, 189–192. [DOI] [PubMed] [Google Scholar]
- Jäkel S, Dimou L, 2017. Glial cells and their function in the adult brain: a journey through the history of their ablation. Front. Cell. Neurosci 11, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantzie LL, Talos DM, Jackson MC, Park HK, Graham DA, Lechpammer M, Folkerth RD, Volpe JJ, Jensen FE, 2015. Developmental expression of N-methyl-D-aspartate (NMDA) receptor subunits in human white and gray matter: potential mechanism of increased vulnerability in the immature brain. Cereb. Cortex 25, 482–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Ståhlberg A, Aprico K, Larsson K, Yabe T, Moons L, Fotheringham A, Davies I, Carmeliet P, Schwartz JP, Pekna M, Kubista M, Blomstrand F, Maragakis N, Nilsson M, Pekny M, 2008. Protective role of reactive astrocytes in brain ischemia. J. Cereb. Blood Flow. Metab 28, 468–481. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA, 2017. Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung W-S, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA, 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RCS, Matesic DF, Marvin M, McKay RDG, Brüstle O, 1995. Re-expression of the intermediate filament nestin in reactive astrocytes. Neurobiol. Dis 2, 79–85. [DOI] [PubMed] [Google Scholar]
- Liu Z, Chopp M, 2016. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog. Neurobiol 144, 103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C, Domercq M, Sánchez-Gómez M-V, 2006. Glutamate-mediated glial injury: mechanisms and clinical importance. Glia 53, 212–224. [DOI] [PubMed] [Google Scholar]
- Mifsud G, Zammit C, Muscat R, Di Giovanni G, Valentino M, 2014. Oligodendrocyte pathophysiology and treatment strategies in cerebral ischemia. CNS Neurosci. Ther 20, 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moftakhar P, Lynch MD, Pomakian JL, Vinters HV, 2010. Aquaporin expression in the brains of patients with or without cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol 69, 1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizawa YM, Hirayama Y, Ohno N, Shibata S, Shigetomi E, Sui Y, Nabekura J, Sato K, Okajima F, Takebayashi H, Okano H, Koizumi S, 2017. Reactive astrocytes function as phagocytes after brain ischemia via ABCA-1 mediated pathway. Nat. Commun 8, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave K-A, 2010. Myelination and support of axonal integrity by glia. Nature 468, 244–252. [DOI] [PubMed] [Google Scholar]
- Nieto-Sampedro M, Saneto RP, de Vellis J, Cotman CW, 1985. The control of glial populations in brain: changes in astrocyte mitogenic and morphogenic factors in response to injury. Brain Res 343, 320–328. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F, 2005. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1319. [DOI] [PubMed] [Google Scholar]
- Overman JJ, Clarkson AN, Wanner IB, Overman WT, Eckstein I, Maguire JL, Dinov ID, Toga AW, Carmichael ST, 2012. A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc. Natl. Acad. Sci. U. S. A 109, E2230–E2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Nilsson M, 2005. Astrocyte activation and reactive gliosis. Glia 50, 427–434. [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M, 2004. Astrocyte intermediate filaments in CNS pathologies and regeneration. J. Pathol 204, 428–437. [DOI] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A, 2009. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32, 421–431. [DOI] [PubMed] [Google Scholar]
- Petito C, Olarte J, 1998. Selective glial vulnerability following transient global ischemia in rat brain. J. Neuropathol. Exp. Neurol 57, 231–238. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Brady JD, Mohr C, 2007. Astrocyte metabolism and signaling during brain ischemia. Nat. Neurosci 10, 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B, 2012. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel JL, Faideau M, Aiba I, Pannasch U, Escartin C, Rouach N, Bonvento G, Shuttleworth CW, 2015. Ciliary neurotrophic factor (CNTF) activation of astrocytes decreases spreading depolarization susceptibility and increases potassium clearance. Glia 63, 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Zhang W, Lu Y, Lu Y, Xu L, Fang Q, Wu M, Jia M, Wang Y, Dong L, Yan X, Yang S, Yuan F, 2017. Aquaporin 4-mediated glutamate-induced astrocyte swelling is partially mediated through metabotropic glutamate receptor 5 activation. Front. Cell. Neurosci 11, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, 2009. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32, 638–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, 2014. Astrogliosis. Cold spring harb. Perspect. Biol 7, a020420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV, 2010. Astrocytes: biology and pathology. Acta Neuropathol 119, 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soontornniyomkij V, Lynch MD, Mermash S, Pomakian J, Badkoobehi H, Clare R, Vinters HV, 2010. Cerebral microinfarcts associated with severe cerebral beta-amyloid angiopathy. Brain Pathol 20, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Barres BA, Stevens B, 2012. The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci 35, 369–389. [DOI] [PubMed] [Google Scholar]
- Sun W, Cornwell A, Li J, Peng S, Osorio MJ, Aalling N, Wang S, Benraiss A, Lou N, Goldman SA, Nedergaard M, 2017. SOX9 is an astrocyte-specific nuclear marker in the adult brain outside the neurogenic regions. J. Neurosci 37, 4493–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchihara T, Tsuchiya K, Nakamura A, Ikeda K, 2000. Appearance of tau-2 immunoreactivity in glial cells in human brain with cerebral infarction. Neurosci. Lett 286, 99–102. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Nedergaard M, 2016. The homeostatic astroglia emerges from evolutionary specialization of neural cells. Philos. Trans. R. Soc. B Biol. Sci 371, 20150428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinters HV, 2001. Cerebrovascular diseasedpractical issues in surgical and autopsy pathology In: Love S (Ed.), Neuropathology. A Guide for Practising Pathologists Springer-Verlag, Berlin, pp. 51–99. [Google Scholar]
- Vinters HV, Farrell MA, Mischel PS, Anders KH, 1998. Diagnostic Neuropathology Marcel Dekker, New York. [Google Scholar]
- Vinters HV, Kleinschmidt-DeMasters BK, 2015. General pathology of the central nervous system In: Love S, Budka H, Ironside JW, Perry A (Eds.), Green-field’s Neuropathology, ninth ed. CRC Press, Boca Raton, pp. 1–58. [Google Scholar]
- Volpe JJ, 2001. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr. Res 50, 553–562. [DOI] [PubMed] [Google Scholar]
- Wang MH, Iliff JJ, Liao YH, Chen MJ, Shinseki MS, Venkataraman A, Cheung J, Wang W, Nedergaard M, 2012. Cognitive deficits and delayed neuronal loss in a mouse model of multiple microinfarcts. J. Neurosci 32, 17948–17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirenfeldt M, Babcock AA, Vinters HV, 2011. Microgliadinsights into immune system structure, function, and reactivity in the central nervous system. Histol. Histopathol 26, 519–530. [DOI] [PubMed] [Google Scholar]
- Wirenfeldt M, Clare R, Tung S, Bottini A, Mathern GW, Vinters HV, 2009. Increased activations of Iba1(þ) microglia in pediatric epilepsy patients with Rasmussen’s encephalitis compared with cortical dysplasia and tuberous sclerosis complex. Neurobiol. Dis 34, 432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XM, Qian C, Zhou YF, Yan YC, Luo QQ, Yung WH, Zhang FL, Jiang LR, Qian ZM, Ke Y, 2017. Bi-directionally protective communication between neurons and astroctyes under ischemia. Redox Biol 13, 20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zador Z, Stiver S, Wang V, Manley GT, 2009. Role of aquaporin-4 in cerebral edema and stroke In: Handbook of Experimental Pharmacology. Aquaporins Springer-Verlag, Berlin, pp. 95–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA, 2012. Genomic analysis of reactive astrogliosis. J. Neurosci 32, 63915–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]