

Abstract
A three-component coupling of aryl bromides, arylboron reagents, and alkenylarenes is presented. The method tolerates a variety of substitution patterns on all of the components. In particular, 1,2-disubstituted alkenylarenes are suitable and undergo highly diastereoselective diarylation.
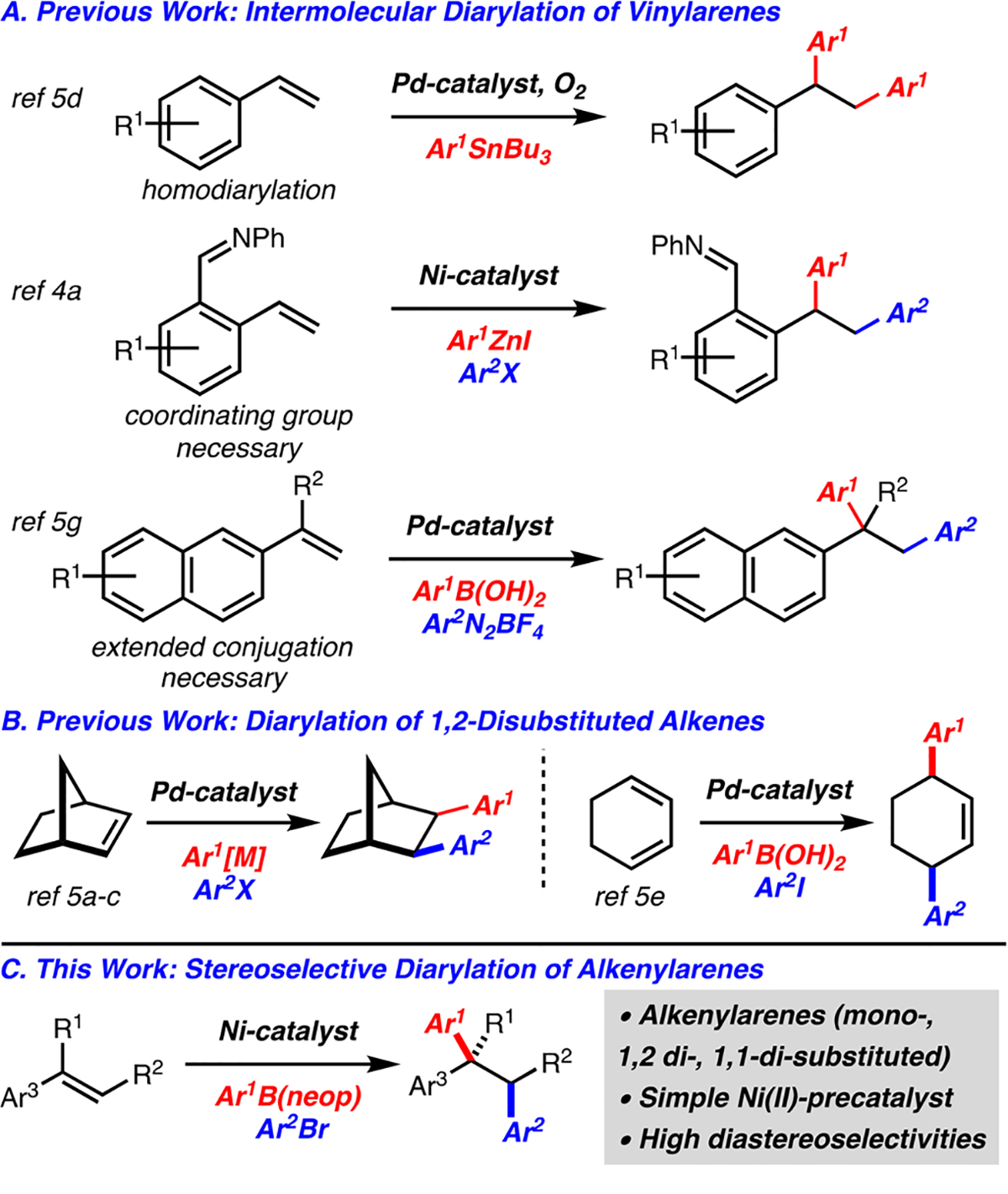
Alkenes represent an appealing class of molecules for chemical synthesis because of their wide availability and ease of preparation.1 While alkene difunctionalization is known, reactions that incorporate two distinct carbon-based groups are more rare.2 In particular, addition of two different aryl groups across an alkene represents an attractive goal for method development because of the prevalence of polyarylalkanes in natural products and pharmaceutical agents.
One strategy to achieve alkene diarylation is to interrupt a cross-coupling reaction with a migratory insertion event.2 However, challenges with this strategy lie in competing direct cross-coupling and β-hydride elimination of alkylmetal intermediates (Mizoroki−Heck reaction). To mitigate these concerns, methods involving intramolecular variants,2,3 sub strates with coordinating groups,4 and/or use of select classes of activated alkenes have been developed.5 Several examples of related intermolecular aryl/alkenylation of activated alkenes are known.6 In addition, intermolecular aryl/alkylation by metalloradical processes have been reported.7
As noted above, while several classes of activated alkenes are known to undergo diarylation, three examples of diarylation of vinylarenes have been reported. The first is a homodiarylation of vinylarenes with aryltin reagents.5d The other two examples involve heterodiarylation; while one requires the use of a coordinating group,4a the second necessitates substrates with extended conjugation (Scheme 1A), thus limiting the reaction scope.5g Furthermore, reactions of 1,2-alkenylarenes are not known yet particularly important, as the opportunity for stereocontrol becomes apparent. Only two examples of diarylation of 1,2-disubstituted alkenes are known and are limited to norbornene derivatives5a–c and 1,3-cyclohexadiene (Scheme 1B).5e It is evident that despite the significant advances outlined above, heterodiarylation of simple styrene derivatives and reactions of 1,2-disubstituted alkenylarenes are lacking. Herein we disclose a stereoselective Ni-catalyzed diarylation of mono-, 1,1-di-, and 1,2-disubstituted alkenylarenes with aryl bromides and arylboron reagents (Scheme 1C).8 The method is notable in that it is stereoselective, a range of substitution patterns are tolerated, and gram-scale synthesis can be achieved.
Scheme 1.
Intermolecular 1,2-Diarylation of Alkenes
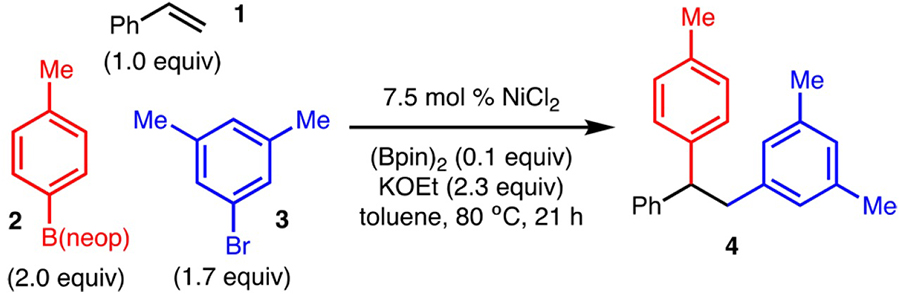
In the context of recent efforts from our lab on Ni-catalyzed carboboration of unactivated alkenes9 and Cu-catalyzed alkene diarylation reactions,10 we discovered a novel diarylation of styrene (1) with p-tolB(neop)11 (2) and 3,5-dimethylbromo-benzene (3). The optimized reaction conditions (Table 1, entry 1) utilize inexpensive NiCl2 at 80 °C in toluene. Under these conditions, triarylethane 4 was formed in 77% yield, accompanied by the direct cross-coupling product in ∼25% yield (<2% yield of products derived from β-hydride elimination was observed).12 Key to the development of this reaction was the identification of 0.1 equiv of (Bpin)2 in the presence of KOEt as a suitable reductant for NiCl2, presumably to generate a Ni(0) complex.13 Use of the common reductants Zn and Mn (Table 1, entry 4) was ineffective. In addition, use of Ni(COD)2 (with or without (Bpin)2) allowed for product formation, albeit in reduced yield, thus confirming that Ni(0) is a competent catalyst (Table 1, entries, 5 and 6). Phosphineand amine-based ligands suppressed the alkene functionalization and allowed only direct cross-coupling to occur (Table 1, entries 7 and 8). When lower amounts of ArBr or ArB(neop) were employed, slightly lower yields were observed (compare Table 1, entries 10 and 11 with entry 1). Finally, for reasons that are unclear at this time, the reaction is sensitive to the counterion of the base, as NaOEt was significantly less effective than KOEt (compare Table 1, entry 12 with entry 1).
Table 1.
Change from Standard Conditions a
| entry | change from standard conditions | Yield (%)b |
|---|---|---|
| 1 | no change | 77 |
| 2 | no (Bpin)2 | <2d |
| 3 | 0.1 equiv of (Bneop)2 instead of 0.1 equiv of (Bpin)2 | 70 |
| 4 | 1.0 equiv of Zn0 or Mn0 instead of 0.1 equiv of (Bpin)2 | <2c |
| 5 | Ni(COD)2 instead of NiCl2 and (Bpin)2 | 53 |
| 6 | Ni(COD)2 instead of NiCl2 | 54 |
| 7 | NiCl2(PCy3)2 instead of NiCl2 | <2d |
| 8 | NiCl2(bpy) instead of NiCl2 | <2d |
| 9 | NiCl2(DME) instead of NiCl2 | 73 |
| 10 | 1.2 equiv of ArBr instead of 1.7 equiv of ArBr | 68 |
| 11 | 1.8 equiv of ArB(neop) instead of 2.0 equiv of ArB(neop) | 72 |
| 12 | NaOEt instead of KOEt | 16d |
The reactions were run on a 0.2 mmol scale.
Yields were determined by GC analysis with a calibrated internal standard. The conversion of styrene was >98%, except where noted otherwise.
>80% of the styrene was recovered.
~50% of the styrene was recovered.
After the optimized conditions were established, the scope of the diarylation was evaluated. With respect to the alkene component, styrene derivatives bearing electron-donating (products 7 and 10) and electron-withdrawing substituents (products 8 and 9) worked well (Scheme 2). Sterically demanding 2-methylstyrene was tolerated; however, the yield was slightly diminished (product 11). Substrates that have extended conjugation also allowed for product formation (products 12 and 13).
Scheme 2.

Evaluation of Various Alkenesa
aThe reactions were run on a 0.5 mmol scale. “NMR yield” refers to yields determined by1H NMR analysis of the unpurified reaction mixtures with an internal standard; “yield” refers to yields of isolated products after silica gel column chromatography. Diastereomeric ratios were determined by 1H NMR analysis of the unpurified reaction mixtures. The conversion of styrene was >98%, except where noted.bAr1 = 4-MeC6H and Ar3 4 = 4-OMeC6H4.c27% of the alkene was recovered.
One of the most notable aspects of this method is that 1,1-and 1,2-disubstituted alkenylarenes were also tolerated (Scheme 2). For example, reaction of α-methylstyrene allowed for formation of 14, which bears a quaternary carbon (see also products 15 and 16). Reaction of 1,2-disubstituted alkenylarenes occurred with control of the stereoselectivity; in all cases, syn addition of the aryl group was observed as the major product regardless of the alkene geometry (compare products 21 and 22, from trans- and cis-stilbene, respectively). In the case of cis-stilbene, slightly lower levels of diastereoselectivity were observed relative to trans-stilbene. Substitution at the β-position could be varied to a silyl-protected hydroxyalkyl group (product 19), i-Pr (product 20) or Ph (products 21 and 22). With respect to known limitations, attempted diarylation of a trisubstituted alkene and an unactivated alkene did not lead to product formation.12
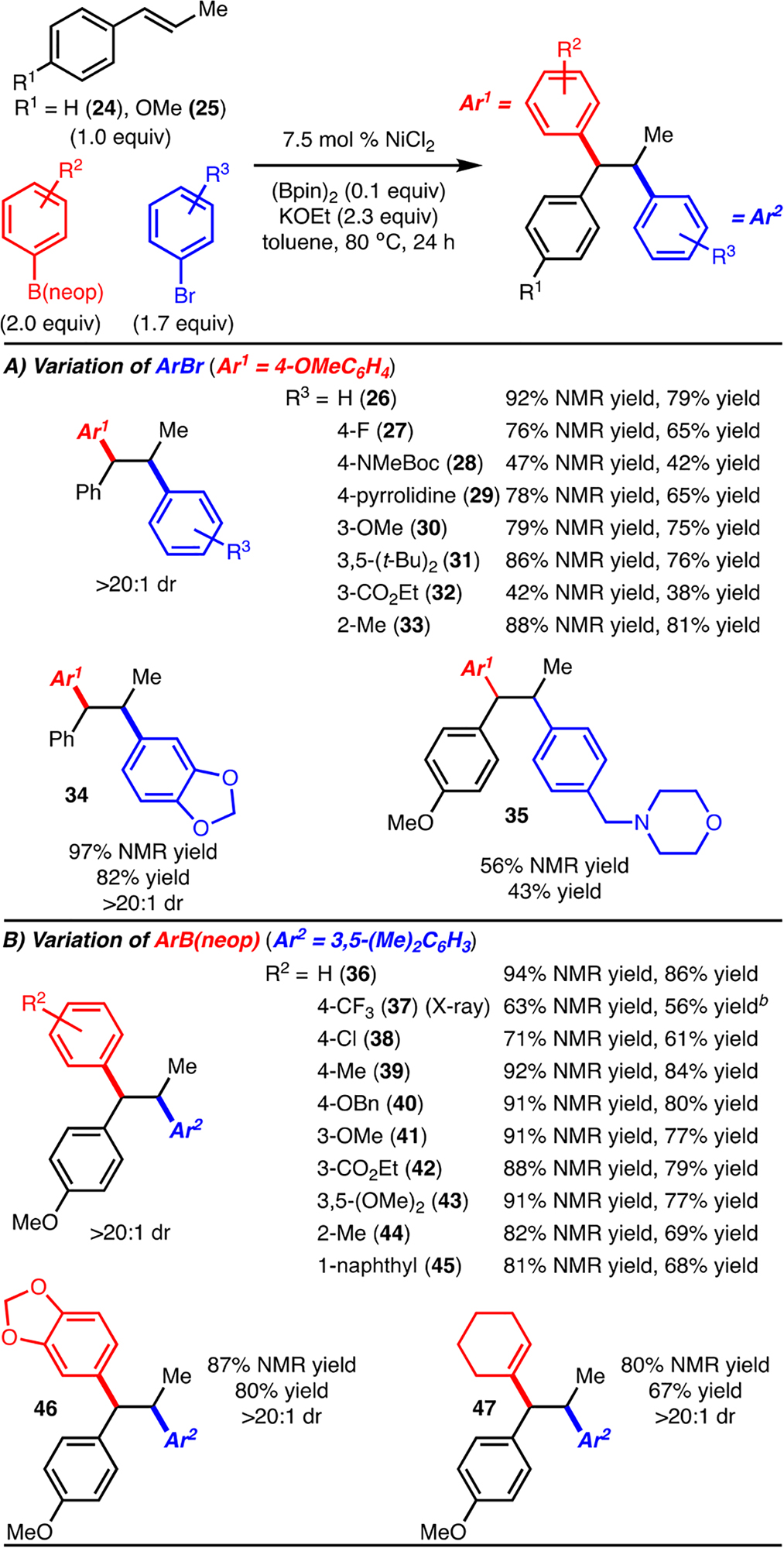
Reactions with a variety of aryl bromides were investigated and found to tolerate electron-donating (products 29, 30, and 34), electron-withdrawing (products 27 and 32), and sterically demanding (product 33) substituents (Scheme 3A). The functional group tolerance was also evaluated and included esters (product 32), amides (product 28), and tertiary amines (products 29 and 35). The moderate yield observed in the formation of 35 is likely due to the Lewis basic nitrogen, which partially inhibits catalysis. It should also be noted that heterocyclic aryl bromides, such as 3-bromopyridine, are not tolerated in the reaction, likely for similar reasons.12 A similar scope with respect to the arylboron reagent compared to the aryl bromide was observed, with electron-donating, electron-withdrawing, and sterically demanding substituents being tolerated (Scheme 3B). In addition, an alkenylB(neop) could be used to access 47, thus demonstrating an example of aryl/alkenylation. However, mono- or disubstituted alkenylboranes did not function well and resulted only in direct cross-coupling.12
Scheme 3.
Evaluation of ArBr and ArB(neop)a
aSee footnote a of Scheme 2. b24 was used instead of 25.
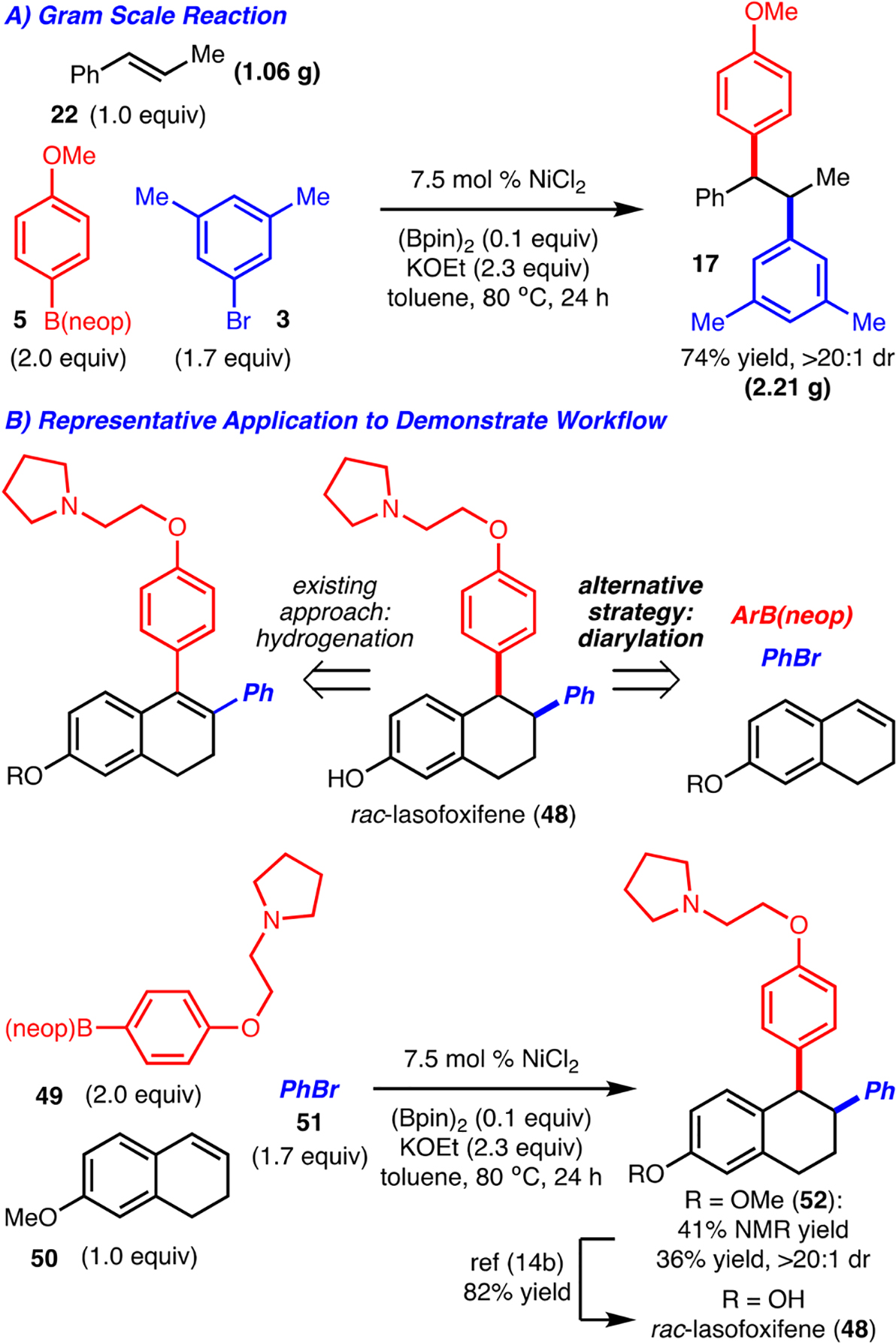
The reaction was performed on a gram scale with similar yield and selectivity as in the smaller-scale reactions (Scheme 4A). It should be emphasized that the reaction conditions employ inexpensive and readily available reagents, making for a practical synthesis of polyarylalkanes.
Scheme 4.
Utility of the Method
To compare this method with existing strategies for polyarylalkane synthesis, rac-lasofoxifene (48) was chosen as a representative target (Scheme 4B). The established approach toward rac-lasofoxifene involves hydrogenation of a tetrasubstituted double bond, wherein the exocyclic aryl groups are installed at an earlier stage in the synthesis.14,15 The strategy outlined herein is orthogonal but has the advantage that the exocyclic aryl groups are installed at a late stage by the convergent assembly of simple components. This is significant when modular approaches to molecules are desired, such as in early-stage drug discovery chemistry. In practice, diarylation of 6-methoxy-3,4-dihydronaphthalene (50) with PhBr and ArB-(neop) 49 allowed for the formation of a known precursor to rac-lasofoxifene with >20:1 dr in 36% yield. As noted previously with product 35, the moderate yield observed for the formation of 52 is likely due to the presence of the Lewis basic nitrogen.
A reasonable catalytic cycle for the diarylation reaction is illustrated in Scheme 5.4a It is proposed that NiCl2 is reduced to Ni(0) with (Bpin)2 and KOEt, perhaps via reductive elimination of EtOBpin from complex 54.13 Oxidative addition of Ni(0) and Ar2Br generates NiIIBrAr2 (56), which upon migratory insertion provides π-benzyl–Ni complex 57 (this is likely why alkenyl arenes, as opposed to unactivated alkenes, are required). Transmetalation with Ar1B(neop) assisted by KOEt generates 58, which undergoes reductive elimination to form the diarylation product. The process likely does not involve radical intermediates, as reaction of (E)- and (Z)-alkene isomers gave rise to different major diastereomers resulting from syn diarylation (Scheme 2, products 21 and 22). It should be noted that this process might involve a heterogeneous/surface reaction, as strongly donating ligands are absent.
Scheme 5.
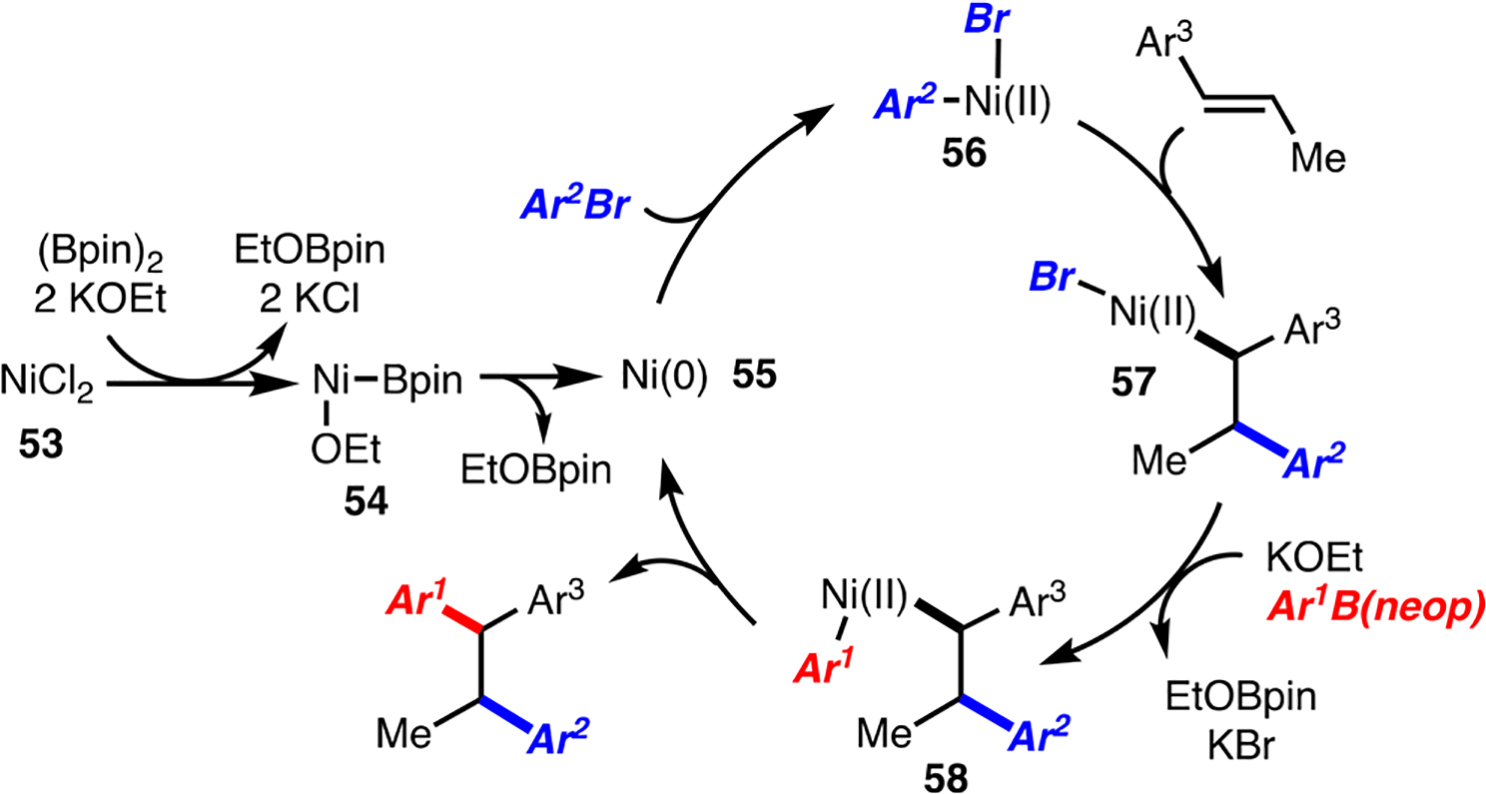
Proposed Catalytic Cycle
The conditions outlined herein allow for diarylation in preference to direct cross-coupling or Mizoroki–Heck reaction, likely for several reasons. The first is the absence of strongly donating ligands (e.g., phosphines and amines), which allows for alkene coordination and subsequent migratory insertion. In the presence of strongly donating ligands, alkene coordination is likely to be inhibited, and thus, direct cross-coupling dominates. The second is the use of arylboronic esters, which reduces the rate of cross-coupling because of slower transmetalation relative to other nucleophiles. For example, if an arylzinc reagent is used in place of ArB(neop), the cross-coupling product is the major product observed.12 In addition, the use of 1.7 equiv of ArBr and 2.0 equiv of ArB(neop) is required not because of competing direct cross-coupling, as the cross-coupling product is formed in <25% yield, but rather to ensure rapid capture of intermediates 55 and 57 prior to off-cycle reactions (e.g., Ni–C bond homolysis or Ni aggregation). Finally, use of Ni catalysts results in the formation of π-benzyl–Ni complexes, which are less prone to β-hydride elimination compared with π-benzyl–Pd complexes.16 In the latter case, β-hydride elimination is known to be rapid.5g
In summary, a Ni-catalyzed diarylation of alkenylarenes has been developed. The method represents a substantial departure from known methods for reaction of vinylarenes in that specialized substrates are not required and the process is uniquely effective for diarylation of 1,2-disubstituted alkenylarenes. Such advances allow for the efficient and modular synthesis of a wide variety of polyarylalkanes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Indiana University and the NIH (5R01GM114443) for financial support. P.G. thanks the China Scholarship Council and the National Natural Science Foundation of China (21602168) for generous financial support. This project was partially funded by the Vice Provost for Research through the Research Equipment Fund.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b05680.
Crystallographic data for 21 (CIF)
Crystallographic data for 22 (CIF)
Crystallographic data for 37 (CIF)
Experimental procedures and analytical data for all compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).For reviews, see: Saini V; Stokes BJ; Sigman MS Angew. Chem., Int. Ed 2013, 52 (43), 11206–11220.Coombs JR; Morken JP Angew. Chem., Int. Ed 2016, 55 (8), 2636–2649.
- (2).For reviews, see: Jensen KH; Sigman MS Org. Biomol. Chem 2008, 6 (22), 4083–4088.Giri R; KC SJ Org. Chem 2018, 83 (6), 3013–3022.Derosa J; Tran VT; van der Puyl VA; Engle KM Aldrichimica Acta 2018, 51, 21–32.
- (3).Zeni G; Larock RC Chem. Rev 2006, 106 (11), 4644–4680. [DOI] [PubMed] [Google Scholar]
- (4)(a).Shrestha B; Basnet P; Dhungana RK; KC S; Thapa S; Sears JM; Giri R J. Am. Chem. Soc 2017, 139 (31), 10653–10656. [DOI] [PubMed] [Google Scholar]; (b) Derosa J; Tran VT; Boulous MN; Chen JS; Engle KM J. Am. Chem. Soc 2017, 139 (31), 10657–10660. [DOI] [PubMed] [Google Scholar]; (c) Li W; Boon JK; Zhao Y Chem. Sci 2018, 9, 600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Thapa S; Dhungana RK; Magar RT; Shrestha B; KC S; Giri R Chem. Sci 2018, 9, 904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5)(a).Catellani M; Chiusoli GP; Concari S Tetrahedron 1989, 45 (16), 5263–5268. [Google Scholar]; (b) Kang S-K; Kim J-S; Choi S-C; Lim K-H Synthesis 1998, 1998 (09), 1249–1251. [Google Scholar]; (c) Shaulis KM; Hoskin BL; Townsend JR; Goodson FE; Incarvito CD; Rheingold AL J. Org. Chem 2002, 67 (16), 5860–5863. [DOI] [PubMed] [Google Scholar]; (d) Urkalan KB; Sigman MS Angew. Chem., Int. Ed 2009, 48 (17), 3146–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhang X; Larock RC Tetrahedron 2010, 66 (24), 4265–4277. [Google Scholar]; (f) Stokes BJ; Liao L; de Andrade AM; Wang Q; Sigman MS Org. Lett 2014, 16, 4666–4669. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kuang Z; Yang K; Song Q Org. Chem. Front 2017, 4 (7), 1224–1228. [Google Scholar]
- (6)(a).Liao L; Jana R; Urkalan KB; Sigman MS J. Am. Chem. Soc 2011, 133 (15), 5784–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McCammant MS; Liao L; Sigman MS J. Am. Chem. Soc 2013, 135 (11), 4167–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McCammant MS; Sigman MS Chem. Sci 2015, 6, 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kuang Z; Yang K; Song Q Org. Lett 2017, 19 (10), 2702–2705. [DOI] [PubMed] [Google Scholar]
- (7)(a).Gu J-W; Min Q-Q; Yu L-C; Zhang X Angew. Chem., Int. Ed 2016, 55 (40), 12270–12274. [DOI] [PubMed] [Google Scholar]; (b) García-Domínguez A; Li Z; Nevado CJ Am. Chem. Soc 2017, 139 (20), 6835–6838. [DOI] [PubMed] [Google Scholar]
- (8).For a review regarding Ni catalysis, see: Tasker SZ; Standley EA; Jamison TF Nature 2014, 509 (7500), 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Logan KM; Sardini SR; White SD; Brown MK J. Am. Chem. Soc 2018, 140 (1), 159–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10)(a).You W; Brown MK J. Am. Chem. Soc 2014, 136 (42), 14730–14733. [DOI] [PubMed] [Google Scholar]; (b) You W; Brown MK J. Am. Chem. Soc 2015, 137 (46), 14578–14581. [DOI] [PubMed] [Google Scholar]
- (11).neop = neopentylglycolate.
- (12).See the Supporting Information for details.
- (13)(a).Fujita T; Arita T; Ichitsuka T; Ichikawa J Dalton Trans 2015, 44 (45), 19460–19463. [DOI] [PubMed] [Google Scholar]; (b) For a potential reductive elimination of MeOBpin from a Ni(III) complex, see: Xu H; Zhao C; Qian Q; Deng W; Gong H Chem. Sci 2013, 4 (10), 4022–4029. [Google Scholar]
- (14)(a).Umareddy P; Arava VR Synth. Commun 2016, 46 (4), 309–313. [Google Scholar]; (b) Khan MA; Trivedi NR; Kandasamy NN; Patil DG; Pandey VK; Sinha S Process for the Preparation of Lasofoxifene Tartrate. U.S. Patent US8937188B2, 2015. [Google Scholar]
- (15) (–).-Lasofoxifene is obtained from the racemate by fractional crystallization with D-tartaric acid (see ref 14b). However, a recent report details an enantioselective hydrogenation approach. See: Zanotti-Gerosa A; Gazic Smilovic I;Časar Z Org. Chem. Front 2017, 4 (12), 2311–2322. [Google Scholar]
- (16).Lin; Liu L; Fu Y; Luo S-W; Chen Q; Guo Q-X Organometallics 2004, 23 (9), 2114–2123. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.