

Abstract
Duchenne muscular dystrophy is a recessive X‐linked disease characterized by progressive muscle wasting; cardiac or respiratory failure causes death in most patients by the third decade. The disease is caused by mutations in the dystrophin gene that lead to a loss of functional dystrophin protein. Although there are currently few treatments for Duchenne muscular dystrophy, previous reports have shown that upregulating the dystrophin paralog utrophin in Duchenne muscular dystrophy mouse models is a promising therapeutic strategy. We conducted in silico mining of the Connectivity Map database for utrophin‐inducing agents, identifying the p38‐activating antibiotic anisomycin. Treatments of C2C12, undifferentiated murine myoblasts, and mdx primary myoblasts with anisomycin conferred increases in utrophin protein levels through p38 pathway activation. Anisomycin also induced utrophin protein levels in the diaphragm of mdx mice. Our study shows that repositioning small molecules such as anisomycin may prove to have Duchenne muscular dystrophy clinical utility.
STUDY HIGHLIGHTS
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ Duchenne muscular dystrophy is caused by mutations in the Duchenne muscular dystrophy gene that lead to a loss of functional dystrophin protein in skeletal and cardiac muscle. This results in progressive muscle wasting and loss of ambulation. Currently, there are no effective treatments for Duchenne muscular dystrophy.
WHAT QUESTION DID THIS STUDY ADDRESS?
✓ As previous reports have shown that upregulating the dystrophin paralog utrophin in DMD mouse models may serve as a therapeutic strategy, we conducted in silico mining of the Connectivity Map database searching for utrophin‐inducing agents, verifying one of our hits.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
✓ We identified the p38‐activating antibiotic anisomycin as a candidate compound. Anisomycin treatments of C2C12, undifferentiated murine myoblasts, and mdx primary myoblasts conferred significant increases in utrophin protein levels through p38 pathway activation. We also observed that anisomycin induced utrophin protein levels in mdx mice in the diaphragm.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
✓ Our study shows that repositioning small molecules such as anisomycin may prove to have Duchenne muscular dystrophy clinical utility.
Duchenne muscular dystrophy (DMD) is an X‐linked recessive disorder with a prevalence of 1 in 5,000 to 1 in 6,500 boys in the United States.1 DMD is caused by mutations in the dystrophin gene (DMD) and the resulting reduction of dystrophin protein.2, 3 Dystrophin is a 427 kDa, multidomain protein primarily found in skeletal and cardiac muscle. In muscle, dystrophin is localized to the sarcolemma and forms an essential part of the dystrophin‐associated glycoprotein complex (DGC).4 The function of this protein complex is to connect the extracellular matrix to the intracellular actin cytoskeleton, thereby maintaining sarcolemma stability.5 Thus, loss of dystrophin in skeletal muscle results in instability of the sarcolemma. Membrane fragility causes the loss of calcium maintenance across the membrane and increased intracellular calcium. This initiates an array of events that lead to contraction‐mediated injury, widespread muscle necrosis, and degeneration.6
DMD is characterized by progressive muscle wasting, proximal muscle weakness, and significantly elevated serum creatine kinase levels.7 Increasing lower limb muscle weakness leads to the patient needing a wheelchair for mobility, on average by age 12.8 The leading cause of DMD mortality is progressive respiratory failure, which usually develops in the second decade.9 The respiratory muscle most severely affected in DMD is the diaphragm. This may devolve from the body's dependency on the diaphragm to drive ventilation while at rest.10 Animal models of DMD, such as the mdx mouse, show more severe damage in the diaphragm compared with other skeletal muscles. Although the mouse phenotype is milder than human DMD, there is a reduction in lifespan, thought to be due to respiratory or cardiac failure.11 In fact, the diaphragm is abnormal in both structure and function, making it a natural tissue to assess when testing DMD therapies.12, 13
Despite our growing understanding of the genetic and molecular pathogenesis of the condition, no effective treatment exists for DMD. Pharmacologic upregulation of the structural and functional dystrophin paralog, utrophin, represents one potential therapeutic avenue. Utrophin is a ubiquitously expressed 400 kDa protein that primarily localizes to the neuromuscular junction in normal skeletal muscle. Utrophin upregulation is a promising DMD treatment strategy because, in addition to its functional similarity to dystrophin, when overexpressed a widespread “dystrophin‐like” sarcolemmal distribution in muscle is observed in DMD.14, 15, 16 Several studies have established utrophin as a developmental precursor to dystrophin and have shown that increased utrophin partly compensates for low dystrophin at the sarcolemma in DMD.17, 18
The identification of agents that induce utrophin would clearly be of potential therapeutic value. The advent of massively parallel transcriptional profiling has enabled the creation of system‐wide databases of human gene expression changes elicited by compounds including US Food and Drug Administration (FDA)‐approved drugs allowing in silico drug screens. One such database is the Broad Institute's Connectivity Map (Cmap; https://www.broadinstitute.org/cmap/) which conducted Affymetrix array‐based analysis on different cell lines employing 1,600 compounds and is currently the most comprehensive database of clinic‐tested drug‐induced transcriptional responses in live cells.19 Using Cmap, we identified a list of small molecule compounds that increased utrophin mRNA. Anisomycin, a p38‐activating antibiotic, was among the most promising utrophin mRNA upregulating agents identified.20 Here we show that anisomycin induces utrophin protein levels both in vitro and in vivo. We also confirm that p38 MAPK activity is essential to the utrophin‐inducing effect of anisomycin.
METHODS
Reagents
Anisomycin and the p38 inhibitor SB239063 (Sigma, St. Louis, MO), utrophin antibody (N‐terminus; clone DRP3/20C5) (Vector Labs, Burlingame, CA), tubulin monoclonal antibody (Abcam, Cambridge, MA), phospho‐p38 mitogen‐activated protein kinase (MAPK; Thr180/Tyr182) rabbit monoclonal antibody, and total‐p38 antibody (Cell Signaling Technology, Beverly, MA) were used.
Connectivity map analysis
Cmap is a database of drug–gene interactions employing the AffymetrixGeneChip U133‐A microarray (Santa Clara, CA). Three cell lines (HeLa, PC3, and MCF7) were individually treated with ∼1,300 drugs for 6 hours at a concentration of 10 μM and RNA was isolated, reverse‐transcribed, and used to probe the microarray. Two cDNA probes for utrophin found on the U133‐A GeneChip (213022_s_at and 213023_s_at) resulted in two utrophin gene expression lists. We used these two lists, which ranked the 1,300 drugs in terms of utrophin induction potency, to identify the strongest suitable for further validation. In order to reduce the number of false positives, only drugs that were tested a minimum of four times for each probe set were considered as potential utrophin‐inducing compounds. Drugs were ranked based on an average of their effect on the utrophin gene between both probe sets.
Tissue culture and drug treatment conditions
C2C12 culture
C2C12 mouse myoblast cells were grown at 37°C with 5% CO2 in Dulbecco's modified eagle medium (DMEM) with freshly added penicillin, streptomycin, and 10% fetal calf serum. For the utrophin upregulation and p38 inhibition assays, C2C12 cells were plated at a density of 100,000 cells per well and cultured overnight. Cells were treated with anisomycin at 0.1, 1.0, and 10 nM. For the p38 inhibition assay, cells were treated with SB239063 (3 μM) for 2.5 hours and then treated with anisomycin (1 nM) for 24 hours. Following incubation, cells were washed with 1x phosphate‐buffered saline (PBS) two times and then lysed using radioimmunoprecipitation assay (RIPA) buffer with phenylmethanesulfonyl fluoride (PMSF), aprotinin, and leupeptin. Antiphosphatase molecules β‐glycerol phosphate (5 mM), sodium fluoride (50 mM), sodium orthovanadate (0.2 mM) were added to the RIPA lysis buffer for the p38 phosphorylation assay. Cell lysates were centrifuged at 13,000 rpm for 15 minutes and protein supernatant collected and quantified using Bradford reagent.
Mdx myoblast culture
Murine primary myoblasts (mdx cells) were isolated from gastrocnemius and quadriceps muscles of adult female mdx mice. Cells were cultured in DMEM (ATCC, Rockville, MD) with freshly added penicillin, streptomycin, 20% fetal calf serum, 10% equine serum, 10 ng/ml basic fibroblast growth factor (bFGF; Cell Signaling Technology), and 2 ng/ml hepatocyte growth factor (HGF; PreproTech, Rocky Hill, NJ). Fresh growth factors were added daily to the growth media. Cells were monitored closely for indications of differentiation. Mdx cells were plated at a density of 2 × 105 cell per well in 6‐well plates. Cells were then treated with 1 nM anisomycin for 24 hours and subsequently processed as outlined for C2C12 culture.
Western blot analysis
Protein samples from cells and murine tissue were run on a biphasic gel system composed of a lower 15% acrylamide and an upper 6% acrylamide gel. Utrophin DRP2 monoclonal antibody (Vector Labs), tubulin, phospho‐p38, and total‐p38 monoclonal antibodies were used to immunoblot the membranes. Antimouse horseradish peroxidase (HRP)‐linked secondary antibody was used for utrophin, tubulin, and total‐p38, whereas antirabbit HRP‐linked secondary antibody was used for phoshpo‐p38. Membranes were exposed using enhanced chemiluminescence‐prime (ECL‐prime) or ECL (tubulin) and X‐ray films. All results were quantified using ImageJ (NIH, Bethesda, MD).
Animals
The Animal Care and Veterinary Services and Ethics Committee of the University of Ottawa approved all protocols and all experiments were performed in accordance with the Canadian Institute of Health Research. Adult male and female C57/BL10ScSn Dmdmdx (mdx) mice from Jackson Laboratories (Bar Harbor, ME) were used to start a colony yielding 10‐day‐old mice for the 30‐day anisomycin treatment experiments.
Anisomycin administration
For the 30‐day trial, 12–14 postnatal day 10 (P10) male mdx mice were treated with 2, 20, and 200 μg/kg anisomycin or vehicle (0.2% dimethyl sulfoxide (DMSO) in 1x PBS). These doses were chosen based on pharmacokinetic data of anisomycin serum concentration in mice to approximate the concentration of drug used for in vitro studies and are in line with previously published studies on efficacy and tolerability of anisomycin in vivo. 37 Mice were dosed daily by intraperitoneal (i.p.) injection using 30½ gauge needles for 30 days until P40. All treatments were diluted so that 2 μl/g was the constant volume administered. At the end of each experiment, mice were anesthetized with isoflurane and sacrificed by cervical dislocation. Tissue samples from diaphragm were flash‐frozen in liquid nitrogen for immunoblotting. All tissues were kept at –80°C for long‐term storage. Tissue samples were pulverized with a Bessman Tissue Pulverizer and then lysed in RIPA lysis buffer. Protein lysates were extracted and quantified and Western blotting performed as previously described.
RESULTS
Connectivity map analysis; Overrepresentation of p38 pathway modulators
Analysis of the two Cmap utrophin expression builds identified the top 10 utrophin‐inducing molecules that were tested a minimum of four times (Table 1; ranked by impact on utrophin transcript level). The greatest utrophin‐inducing compound was anisomycin, conferring a 1.5‐fold upregulation. Interestingly, three of the top 10 utrophin‐inducing molecules were p38 activators (anisomycin, ementine, cycloheximide) while two of the top utrophin‐inducing molecules were repressors of the p38 pathway (tacrine, alexidine).
Table 1.
Identification of in vitro utrophin mRNA inducers from Connectivity Map
| No. | Drug | UTRN fold‐expression (Cmap data) | p38 MAPK effect | References for p38 effect |
|---|---|---|---|---|
| 1 | Anisomycin | 1.50 | + | Liu et al. (2014); Farooq et al. (2009); Xiong et al. (2006) |
| 2 | Emetine | 1.47 | + | Kim et al. (2015); Islam et al. (2006) |
| 3 | Cycloheximide | 1.44 | + | Oksvold et al. (2012); Itani et al. (2003) |
| 4 | Chlorzoxazone | 1.40 | ? | |
| 5 | Chelidonine | 1.38 | ? | |
| 6 | Loxapine | 1.38 | ? | |
| 7 | Piperacetazine | 1.38 | ? | |
| 8 | Tacrine | 1.36 | – | Li et al. (2005) |
| 9 | Alexidine | 1.35 | – | Zhu et al. (2015) |
| 10 | Lobeline | 1.33 | ? |
In vitro validation: Anisomycin‐induced utrophin upregulation
The utrophin upregulating effect of the top‐ranking compound from the Cmap screen, anisomycin, was further investigated in C2C12 mouse myoblasts. The Cmap used standardized 10 μM dosing for their drug treatments, however, given the therapeutic serum level of 100 nM for anisomycin, we initially performed a dose curve in these cells to study the effects of anisomycin at low (0.1 and 1 nM) and therapeutic ranges (10, 25 and 75 nM) and the Cmap dose (10 μM). The C2C12 cells appear to be more sensitive to anisomycin at higher doses, as cellular toxicity was observed with the 75 nM dose after 24‐hour treatment (data not shown), confirming previous reports in the literature regarding the dose response and toxicity of anisomycin.34, 36 However, a dose–response curve on cells treated with 0.1, 1, and 10 nM anisomycin for 24 hours showed a significant utrophin upregulation with 0.1 and 1 nM anisomycin ( Figure S1 a). Additional biological replicates of C2C12 cells treated with 1 nM anisomycin for 24 hours confirmed an approximate 2.5‐fold increase in utrophin protein (Figure 1 a). The effect of low‐dose anisomycin treatment on utrophin protein expression was further studied in mdx mouse primary myoblasts. A more modest, 1.4‐fold, induction was observed in utrophin protein with 1 nM anisomycin treatment (Figure 1 b).
Figure 1.
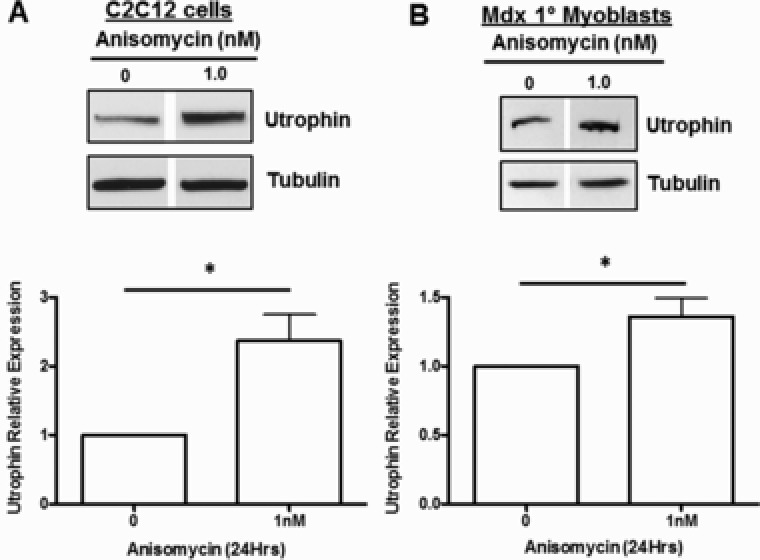
Anisomycin upregulates utrophin protein in vitro. (a) C2C12 myoblasts treated with 1 nM anisomycin or vehicle (DMSO) and harvested at 24 hours (n = 9). (b) Mdx primary myoblasts treated with 1 nM anisomycin or vehicle (DMSO) and harvested at 24 hours (n = 4). Immunoblotting for utrophin shows dramatic upregulation with respect to the housekeeper protein tubulin. Error bars show SEM of average (*P < 0.01, two‐tailed unpaired Student's t‐test).
p38‐MAPK is required for anisomycin‐induced utrophin upregulation
Anisomycin is known to activate the p38 MAPK pathway, which has been implicated in the regulation of the utrophin gene.19, 21 Two of the strongest utrophin mRNA‐inducing agents identified in the Cmap data, emetine and anisomycin, were p38 MAPK pathway activators (Table 1). To explore the potential role of the p38 pathway in the anisomycin‐mediated upregulation of utrophin, analysis of the phosphorylation of p38 and a p38 MAPK inhibition assay were performed. C2C12 cells were treated with 1 nM anisomycin and harvested at 0, 2, 4, and 6 hours for Western blot analysis. There was a significant increase in phosphorylation of p38 following 6 hours of anisomycin treatment (Figure 2 a). The experiments were repeated in the presence of the p38 inhibitor SB239063 to assess the involvement of this pathway in anisomycin‐induced utrophin upregulation. SB239063 moderately reversed the anisomycin‐induced utrophin induction (Figure 2 b). Interestingly, the inhibitor alone increased utrophin protein expression as much as did anisomycin treatment, possibly consistent with the presence of p38 inhibitors, tacrine and alexidine, in the Cmap list of utrophin inducers.
Figure 2.
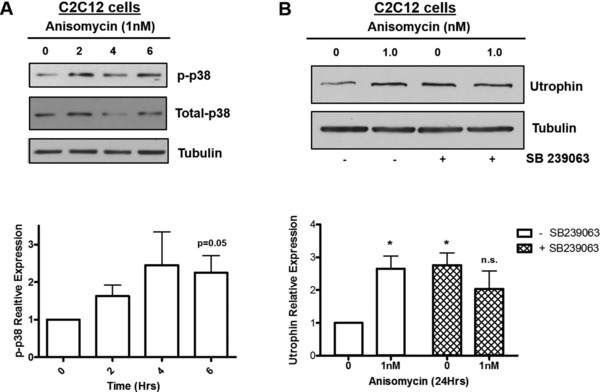
Activation and inhibition of p38‐MAPK induces utrophin expression. (a) C2C12 myoblasts were treated with 1 nM anisomycin for 0, 2, 4, and 6 hours. Immunoblotting for phospho‐p38 and total‐p38 indicates upregulation of p38 phosphorylation in comparison to total‐p38 (n = 3). (b) C2C12 myoblasts were treated with 3μM SB239063 (p38 inhibitor) for 2.5 hours. Cells were then treated for 24 hours with 1 nM anisomycin. (n = 3) Error bars show SEM of average. The untreated control cells were used as the reference for all treatment groups (*P < 0.05, two‐tailed unpaired Student's t‐test).
In vivo small molecule therapy
The mdx mouse is a well‐described model of DMD used for in vivo testing of potential DMD treatment strategies.22 For our analysis, we focused on the diaphragm, as it is the most involved muscle in the mdx mouse and therefore a good proxy for human DMD.12 Male mdx mice were treated daily with 2, 20, and 200 μg/kg i.p. anisomycin starting at postnatal day 10. After 30 days, mice were sacrificed and diaphragms were collected. Western blot of whole protein lysate extracted from diaphragm showed an ∼2‐fold upregulation of utrophin protein in mice treated with 20 μg/kg anisomycin ( Figure S1 b). With the addition of nine biological replicates (thus 12 in all), ∼1.5‐fold utrophin upregulation was observed in mdx diaphragm with 20 μg/kg anisomycin treatment for 30 days (Figure 3 a).
Figure 3.
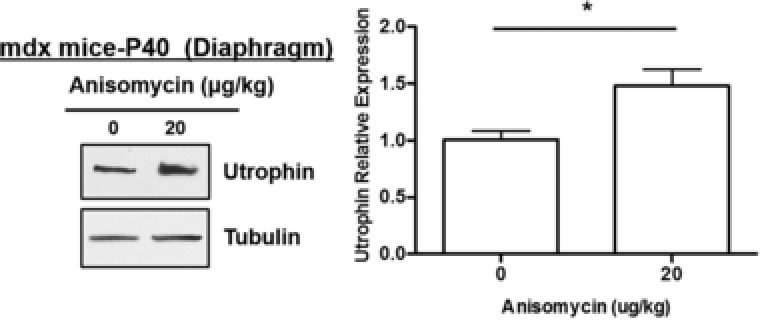
Anisomycin upregulates utrophin protein levels in mdx mouse diaphragm. Male mdx mice received daily i.p. injections of vehicle (0.2% DMSO in PBS) or anisomcyin (20 μg/kg) from P10 for 30 days. Mice were sacrificed at P40. Representative blots are shown for diaphragm (n = 12–14) (*P < 0.01, two‐tailed unpaired Student's t‐test).
DISCUSSION
DMD is a life‐limiting neuromuscular disorder for which there is currently no effective treatment. Respiratory failure is typically the cause of mortality in patients with DMD, due to degeneration of, among other muscles, the diaphragm.9 Therefore, the restoration or preservation of diaphragmatic function is an important aspect of any effective DMD therapy.
One potential therapeutic approach for DMD is to increase levels of the dystrophin paralog utrophin either through transcriptional induction and/or transcript stabilization. Increased levels of utrophin have been shown to compensate, in part, for the loss of dystrophin protein and thus reduce mdx disease severity.17, 23 Previous in vivo studies 15 have shown that very modest inductions of utrophin transcript and protein levels can lead to impressive functional rescue.
In this article we report drug hits mined from the Cmap database; however, given the large variation in transcriptional expression between different tissue culture systems and the rate of false‐positives reported by Mears et al.,24 the list of drug hits for utrophin would likely be different if sourced from another database. However, we believe the identification of p38‐activating compounds as utrophin‐inducing in other databases is still likely given our demonstration here of this relationship as well as previous reports documenting this.
Anisomycin was shown to be the top utrophin‐inducing compound identified by in silico screening of the Cmap. In this study we documented a 2.5‐fold upregulation of utrophin in vitro in C2C12 cells and a 1.4‐fold upregulation in mdx myoblasts with 1 nM anisomycin treatment. A dose of 20 μg/kg in mice resulted in an ∼50% increase in utrophin protein levels in the diaphragm. This in vivo anisomycin dosing falls well below that used in human trials for infections with amoebas (amoebiasis).25 Our results are similar to those found in other studies that have been used in preclinical and clinical trials. For example, the nonsteroidal antiinflammatory drug nabumetone upregulates utrophin protein expression 1.2‐fold in C2C12 cells.26 SMTC1100, a drug tested in phase I clinical trials, upregulated utrophin twofold in human DMD cells and showed an ∼75% increase in expression in the diaphragm of mdx mice.27
Previous work has shown that the 3’UTR of utrophin A transcript contains a number of AU‐rich elements (AREs) that are crucial to transcript stability.28, 29 These cis‐acting elements are well‐recognized destabilizing sections of mRNA.30 The interaction of trans‐acting ARE‐binding proteins at these sites can either increase mRNA stability or enhance its degradation.31 Amirouche et al. 21 showed that p38‐mediated phosphorylation of the KH‐type splicing regulatory protein leads to increased stabilization of utrophin A mRNA that in turn caused an increase in utrophin protein (C2C12 cells). Our lab and others have established anisomycin as a potent p38 activator even at low doses.32, 33
Interestingly, when we employed the p38 inhibiting molecule SB239063 to establish the pathway of ansomycin‐mediated utrophin induction, we found that the inhibitor alone caused significant upregulation of utrophin. This is the first time that inhibition of the p38 pathway has been linked to utrophin upregulation. The Cmap results provide corroborating evidence for a potential link between p38 downregulation and utrophin upregulation, as two of the top 10 utrophin‐inducing molecules (tacrine, alexidine) have been linked to p38 inhibition.34, 35 These data indicate that the role of the p38 pathway in utrophin expression may be more complex than previous reports suggest. Further work would be required to elucidate the mechanism that causes utrophin upregulation when the p38 pathway is inhibited.
Several anisomycin doses were used in C2C12 cells to determine the concentration for optimal utrophin induction in cultured myoblasts. The most consistent utrophin protein induction was seen with 1 nM anisomycin in both C2C12 cell and primary mdx myoblasts. This concentration is four orders of magnitude lower than the concentration used in the Cmap. Anisomycin doses even two orders of magnitude lower than the dose initially used in the Cmap (10 μM) were highly toxic to C2C12 cells. Other reports have shown that in many cell types anisomycin at micromolar levels completely inhibit protein translation, blocks cell proliferation, and induces apoptosis.36, 37 The apparent bimodal utrophin‐inducing nature of anisomycin could be due to a bimodal agonistic effect on the p38 MAPK pathway; at translation‐inhibiting levels it acts via the ribotoxic stress response pathway, but may act in a more direct p38‐activating fashion at lower than inhibitory concentrations.38, 39, 40, 41 The upregulation of utrophin mRNA observed with 10 μM anisomycin by the Broad Institute was probably caused by ribotoxic stress‐mediated p38 induction, whereas the utrophin protein upregulation reported in this work may result from direct p38 activation by low‐dose anisomycin, which we have also observed in our previous work.32
The use of utrophin as a disease‐rescuing dystrophin paralog has been studied in the mdx mouse, in which transgene experiments revealed that a twofold increase in sarcolemmal utrophin completely rescues dystrophin‐deficient muscle mechanical function and effectively normalizes classical markers of DMD‐related muscle damage.15, 42, 43 Several reports have also shown that pharmacological induction of utrophin mRNA and protein in mdx muscle reduces systemic and muscle‐specific hallmarks of DMD.27, 44 In this report we show that daily treatment of mdx mice with low‐dose anisomycin leads to a significant increase in utrophin protein in the diaphragm. The diaphragm pathology in mdx mice most closely resembles the skeletal muscle pathology seen in human DMD patients, and is the best target muscle to test therapeutic agents. Rescuing the diaphragm muscle alone in mdx mice can restore cardiac function through increasing both utrophin and dystrophin expression. This finding potentially increases the relevance of our anisomycin findings to treatment of humans, as it may prove to reduce the respiratory and cardiac morbidities seen in DMD patients.6
Although our results show a utrophin protein upregulating effect of anisomycin in vitro and in vivo, it remains to be seen whether this upregulation would lead to physiologically significant results. Behavioral testing to assess grip strength of mice treated with anisomycin and exercise tolerance could be studied as well as the systemic effects of anisomycin by serum creatine kinase detection, which is a marker of muscle damage in DMD patients.
In conclusion, low‐dose anisomycin treatment is shown here to be effective for upregulating utrophin protein in vitro and at a preclinical level in DMD model mice. Further work will be necessary to determine the efficacy of anisomycin for treating DMD in humans.
Conflict of Interest
The authors declare no competing interests for this work.
Supporting information
Figure S1. Anisomycin in vitro and in vivo dose curves. (A) Utrophin protein levels in C2C12 myoblasts treated with DMSO, 0.1, 1, 10 nM Anisomycin (n = 4). (B) Utrophin protein levels in the diaphragms of male mdx mice that received daily intraperitoneal (i.p.) injections of vehicle (DMSO) or anisomcyin (2, 20, 200 μg/kg) from P10 for 30 days (n = 3). Odyssey CLX Imager was used to identify the protein of interest (western blot). The figure depicts preliminary results of utrophin induction by anisomycin at a range of doses. (*P < 0.05, two‐tailed unpaired Student's t‐test).
Acknowledgments
This work was performed under the Care4Rare Canada Consortium funded by Genome Canada, the Canadian Institutes of Health Research, the Ontario Genomics Institute (OGI‐049), Ontario Research Fund, Genome Quebec, Genome British Columbia, and CHEO Foundation.
Funding
This work was funded by Genome Canada, the Canadian Institutes of Health Research, the Ontario Genomics Institute (OGI‐049), Ontario Research Fund, Genome Quebec, Genome British Columbia, and the CHEO Foundation.
Author Contributions
J.H., S.S., and A.M. wrote the article; F.F. and A.M. designed the research; J.H., L.W., and K.M. performed the research; J.H., L.W., and K.M. analyzed the data.
References
- 1. Romitti, P.A. et al Prevalence of Duchenne and Becker Muscular Dystrophies in the United States. Pediatrics 135, 513–521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koenig, M. et al Complete cloning of the duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50, 509–517 (1987). [DOI] [PubMed] [Google Scholar]
- 3. Monaco, A.P. et al Localization and cloning of Xp21 deletion breakpoints involved in muscular dystrophy. Hum. Genet. 75, 221–227 (1987). [DOI] [PubMed] [Google Scholar]
- 4. Pilgram, G.S. et al The roles of the dystrophin‐associated glycoprotein complex at the synapse. Mol. Neurobiol. 41, 1–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lapidos, K.A., Kakkar, R. & McNally, E.M. The dystrophin glycoprotein complex: Signaling strength and integrity for the sarcolemma. Circ. Res. 94, 1023–1031 (2004). [DOI] [PubMed] [Google Scholar]
- 6. Blake, D.J. et al, Function and genetics of dystrophin and dystrophin‐related proteins in muscle. Physiol Rev. 82, 291–329 (2002). [DOI] [PubMed] [Google Scholar]
- 7. Jennekens, F.G. et al Diagnostic criteria for duchenne and becker muscular dystrophy and myotonic dystrophy. Neuromuscul. Disord. 1, 389–391 (1991). [DOI] [PubMed] [Google Scholar]
- 8. McMillan, H.J., Campbell, C. & Mah, J.K. Canadian Paediatric Neuromuscular Group. Duchenne muscular dystrophy: Canadian paediatric neuromuscular physicians survey. Can. J. Neurol. Sci. 37, 195–205 (2010). [DOI] [PubMed] [Google Scholar]
- 9. Gozal, D. Pulmonary manifestations of neuromuscular disease with special reference to Duchenne muscular dystrophy and spinal muscular atrophy. Pediatr. Pulmonol. 29, 141–150 (2000). [DOI] [PubMed] [Google Scholar]
- 10. Mead, A.F. et al Diaphragm remodeling and compensatory respiratory mechanics in a canine model of Duchenne muscular dystrophy. J. Appl. Physiol. 116, 807–815 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chamberlain, J.S. et al Dystrophin‐deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 21, 2195–2204 (2007). [DOI] [PubMed] [Google Scholar]
- 12. Stedman, H.H. et al The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352, 536–539 (1991). [DOI] [PubMed] [Google Scholar]
- 13. Ishizaki, M. et al Mdx respiratory impairment following fibrosis of the diaphragm. Neuromuscul. Disord. 18, 342–348 (2008). [DOI] [PubMed] [Google Scholar]
- 14. Blake, D.J. , Tinsley, J.M. & Davies, K.E. Utrophin: A structural and functional comparison to dystrophin. Brain Pathol. 6, 37–47 (1996). [DOI] [PubMed] [Google Scholar]
- 15. Guiraud, S. et al Second‐generation compound for the modulation of utrophin in the therapy of DMD. Hum. Mol. Genet. 24, 4212–4224 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moorwood, C. et al A cell‐based high‐throughput screening assay for posttranscriptional utrophin upregulation, J. Biomol. Screen. 18, 400–406 (2013). [DOI] [PubMed] [Google Scholar]
- 17. Janghra, N. et al Correlation of utrophin levels with the dystrophin protein complex and muscle fibre regeneration in Duchenne and Becker muscular dystrophy muscle biopsies. PLoS One 11, e015818 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ljubicic, V. & Jasmin, B.J. Metformin increases peroxisome proliferator–activated receptor γ Co‐activator‐1α and utrophin‐a expression in dystrophic skeletal muscle. Muscle Nerve 52, 139–142 (2015). [DOI] [PubMed] [Google Scholar]
- 19. Lamb, J. et al The connectivity map: Using gene‐expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006). [DOI] [PubMed] [Google Scholar]
- 20. Vergarajauregui, S., San Miguel, A. & Puertollano, R. Activation of p38 mitogen‐activated protein kinase promotes epidural growth factor receptor internalization. Traffic 7, 686–698 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amirouche, A. et al Activation of p38 signalling increases utrophin A expression in skeletal muscle via the RNA‐binding protein KSRP and inhibition of AU‐rich element‐mediated mRNA decay: Implications for novel DMD therapeutics. Hum. Mol. Genet. 22, 3093–3111 (2013). [DOI] [PubMed] [Google Scholar]
- 22. Manning, J. & O'Malley, D. What has the mdx mouse model of duchenne muscular dystrophy contributed to our understanding of this disease? J. Musc. Res. Cell Motil. 36, 155–167 (2015). [DOI] [PubMed] [Google Scholar]
- 23. Miura, P. & Jasmin, B.J. Utrophin upregulation for treating Duchenne or Becker muscular dystrophy: How close are we? Trends Mol. Med. 12, 122–129 (2006). [DOI] [PubMed] [Google Scholar]
- 24. Mears, A. et al Mining the transcriptome for rare disease therapies: A comparison of the efficiencies of two data mining approaches and a targeted cell‐based drug screen. Genomic Med. 2, 14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gonzalez Constandse, R. Anisomycin in intestinal amebiasis; study of 30 clinical cases. Prensa. Med. Mex. 21, 114–115 (1956). [PubMed] [Google Scholar]
- 26. Moorwood, C. et al Drug discovery for Duchenne muscular dystrophy via utrophin promoter activation screening. PLoS One 6, e26169 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tinsley, J.M. et al Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One 6, e19189 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gramolini, A.G., Belanger, O. & Jasmin, B.J. Distinct regions in the 3' untranslated region are responsible for targeting and stabilizing utrophin transcripts in skeletal muscle cells. J. Cell. Biol. 154, 1173–1183 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chakkalakal, J.V. , Miura, P. , Belanger, G. , Michel, R.N. & Jasmin, B.J. Modulation of utrophin A mRNA stability in fast versus slow muscles via an AU‐rich element and calcineurin signaling. Nucleic Acids Res. 36, 826–838 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khabar, K.S. The AU‐rich transcriptome: More than interferons and cytokines, and its role in disease. J. Interferon Cytokine Res. 25, 1–10 (2005). [DOI] [PubMed] [Google Scholar]
- 31. Eberhardt, W. et al Modulation of mRNA stability as a novel therapeutic approach. Pharmacol. Ther. 114, 56–73 (2007). [DOI] [PubMed] [Google Scholar]
- 32. Farooq, F. et al p38 mitogen‐activated protein kinase stabilizes SMN mRNA through RNA binding protein HuR. Hum. Mol. Genet. 18, 4035–4045 (2009). [DOI] [PubMed] [Google Scholar]
- 33. Islam, Z. , Gray, J.S. & Pestka, J.J. p38 mitogen‐activated protein kinase mediates IL‐8 induction by the ribotoxin deoxynivalenol in human monocytes. Toxicol. Appl. Pharmacol. 213, 235–244 (2006). [DOI] [PubMed] [Google Scholar]
- 34. Li, W. et al Novel dimeric acetylcholinesterase inhibitor bis7‐tacrine, but not donepezil, prevents glutamate‐induced neuronal apoptosis by blocking N‐methyl‐D‐aspartate receptors. J. Biol. Chem. 280, 18179–18188 (2005). [DOI] [PubMed] [Google Scholar]
- 35. Zhu, X. et al Alexidine dihydrochloride attenuates osteoclast formation and bone resorption and protects against LPS‐induced osteolysis. J. Bone Miner. Res. 31, 560–372 (2016). [DOI] [PubMed] [Google Scholar]
- 36. Tang, Z. et al In vivo toxicological evaluation of anisomycin. Toxicol. Lett. 208, 1–11 (2012). [DOI] [PubMed] [Google Scholar]
- 37. Croons, V. et al The protein synthesis inhibitor anisomycin induces macrophage apoptosis in rabbit atherosclerotic plaques through p38 mitogen‐activated protein kinase. J. Pharmacol. Exp. Ther. 329, 856–864 (2009). [DOI] [PubMed] [Google Scholar]
- 38. Grollman, A.P . Inhibitors of protein biosynthesis. II. mode of action of anisomycin. J. Biol. Chem. 242, 3226–3233 (1967). [PubMed] [Google Scholar]
- 39. Bunyard, P. et al Ribotoxic stress activates p38 and JNK kinases and modulates the antigen‐presenting activity of dendritic cells. Mol. Immunol. 39, 815–827 (2003). [DOI] [PubMed] [Google Scholar]
- 40. Monaghan, D. et al Inhibition of protein synthesis and JNK activation are not required for cell death induced by anisomycin and anisomycin analogues. Biochem. Biophys. Res. Commun. 443, 761–767 (2014). [DOI] [PubMed] [Google Scholar]
- 41. Torocsik, B. & Szeberenyi, J. Anisomycin uses multiple mechanisms to stimulate mitogen‐activated protein kinases and gene expression and to inhibit neuronal differentiation in PC12 phaeochromocytoma cells. Eur. J. Neurosci. 12, 527–532 (2000). [DOI] [PubMed] [Google Scholar]
- 42. Gilbert, R. et al Adenovirus‐mediated utrophin gene transfer mitigates the dystrophic phenotype of mdx mouse muscles. Hum. Gene Ther. 10, 1299–1310 (1999). [DOI] [PubMed] [Google Scholar]
- 43. Squire, S. et al Prevention of pathology in mdx mice by expression of utrophin: Analysis using an inducible transgenic expression system. Hum. Mol. Genet. 11, 3333–3344 (2002). [DOI] [PubMed] [Google Scholar]
- 44. Krag, T.O. et al Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc. Natl. Acad. Sci. U. S. A. 101, 13856–13860 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu, Y. et al Low‐dose anisomycin sensitizes glucocorticoid‐resistant T‐acute lymphoblastic leukemia CEM‐C1 cells to dexamethasone‐induced apoptosis through activation of glucocorticoid receptor and p38‐MAPK/JNK. Leuk. Lymphoma 55, 2179–2188 (2014). [DOI] [PubMed] [Google Scholar]
- 46. Xiong, W. et al Anisomycin activates p38 MAP kinase to induce LTD in mouse primary visual cortex. Brain Res. 1805, 68–76 (2006). [DOI] [PubMed] [Google Scholar]
- 47. Kim, J.H. et al Emetine inhibits migration and invasion of human non‐small‐cell lung cancer cells via regulation of ERK and p38 signaling pathways. Chem. Biol. Interact. 242, 25–33 (2015). [DOI] [PubMed] [Google Scholar]
- 48. Oksvold, M.P. et al Effect of cycloheximide on epidermal growth factor receptor trafficking and signaling. FEBS Lett. 586, 3575–3581 (2012). [DOI] [PubMed] [Google Scholar]
- 49. Itani, O.A. et al Cycloheximide increases glucocorticoid‐stimulated alpha ‐ENaC mRNA in collecting duct cells by p38 MAPK‐dependent pathway. Am. J. Physiol. Renal Physiol. 284, F778–787 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Anisomycin in vitro and in vivo dose curves. (A) Utrophin protein levels in C2C12 myoblasts treated with DMSO, 0.1, 1, 10 nM Anisomycin (n = 4). (B) Utrophin protein levels in the diaphragms of male mdx mice that received daily intraperitoneal (i.p.) injections of vehicle (DMSO) or anisomcyin (2, 20, 200 μg/kg) from P10 for 30 days (n = 3). Odyssey CLX Imager was used to identify the protein of interest (western blot). The figure depicts preliminary results of utrophin induction by anisomycin at a range of doses. (*P < 0.05, two‐tailed unpaired Student's t‐test).