

Abstract
Development of an oral testosterone therapy has proven extremely challenging because of extensive and variable first‐pass metabolism. We investigated the in vivo metabolism of testosterone with increasing oral doses of testosterone, both alone and with the co‐administration of dutasteride (5α‐reductase inhibitor) by liquid‐chromatography tandem mass spectrometry (LC‐MS/MS). In eugonadal men prior to dosing, the circulating concentration of testosterone, androstenedione, etiocholanolone‐glucuronide, and androsterone‐glucuronide was 8.6, 20.9, 9.1, and 55.3%, respectively, of the total testosterone‐related species, whereas testosterone‐glucuronide was ∼1%. When testosterone was dosed orally to men with experimental hypogonadism, a proportion of testosterone‐glucuronide increased to 13%. Dutasteride treatment significantly decreased levels of androsterone and its metabolites. This work reveals extensive metabolism of orally dosed testosterone to androsterone glucuronide via androstenedione, with testosterone‐glucuronide appearing to be the second most important metabolite. This information is of importance in the development of an effective oral testosterone therapy and may have implications for testosterone doping research.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ Oral administration of testosterone has a very low bioavailability as it undergoes extensive and highly variable first‐pass metabolism. The development of an oral testosterone is an unmet need.
WHAT QUESTION DID THIS STUDY ADDRESS?
✓ This study investigated testosterone metabolite profiling after oral dosing with and without dutasteride by measuring circulating levels of testosterone and its metabolites by LC‐MS/MS.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
✓ The majority of oral testosterone is metabolized to androstenedione and subsequent metabolites, androsterone glucuronide. The testosterone glucuronide formation is the second most important pathway of testosterone metabolism. Orally administered testosterone exhibited dose nonlinearity, and dutasteride co‐administration increased circulating testosterone but significantly inhibited testosterone metabolism to dihydrotestosterone, androsterone, and androsterone glucuronide.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
✓ The findings of this study are important in developing oral testosterone therapy and may have implications in testosterone doping research.
Testosterone is the key male sex hormone. Testosterone plays an indispensable role in the development of the male reproductive system during fetal life, triggers the development of secondary sexual characteristics at puberty, and maintains sexual function and bone and muscle health during adulthood.1 Low concentrations of testosterone can occur for a variety of reasons. Men with low testosterone experience fatigue, depression, diminished libido, loss of muscle strength,2 osteoporosis,3 visceral obesity, and insulin resistance.4 Testosterone replacement therapy (TRT) is the first‐line treatment for testosterone deficiency and has been shown to improve bone density, alleviate depression, and maintain sexual function.5 Currently available options for TRT in the United States include intramuscular injections, transdermal patches, and gels.6, 7 The development of an oral TRT has been extremely challenging because of extensive first pass metabolism. As a result, the 2–4 million hypogonadal men in the United States requiring TRT mainly rely on intramuscular or topical testosterone dosage forms,6, 7 both of which are challenging in terms of compliance, and have suboptimal pharmacokinetics (PK) or variable absorption.8 Clearly, the development of a safe and effective formulation of oral testosterone is a major unmet need for these men.
Oral testosterone is readily absorbed from the small intestines by diffusion in humans,9 but the absorbed testosterone dose is metabolized in both the intestines10 and the liver11 before reaching the circulation system, resulting in a very poor bioavailability of 2–8%.12, 13, 14 Exogenously administered testosterone has previously been reported to be metabolized to multiple unconjugated and conjugated metabolites (Figure 1)15 where the major urinary metabolites are glucuronide conjugates, which are formed in the liver and intestines by various human UDP‐glucuronosyl transferases (UGTs).16 In particular, UGT2B17 is the major testosterone‐glucuronidation enzyme. UGT2B17 is of interest as it is highly polymorphic and copy number variations in its genes are associated with multiple potentially testosterone related pathologies (e.g., obesity,17 prostate cancer,18 osteoporosis,19 ankylosing spondylitis,20 and endometrial cancer).21 UGT2B17 is highly expressed in the intestines and the liver.22, 23 Because of this and its known role in testosterone metabolism, we hypothesized that the first‐pass metabolism of testosterone by UGT2B17 is the main reason for its poor or variable bioavailability. Therefore, to investigate the quantitative contribution of glucuronidation in the in vivo metabolism of testosterone, we quantified the serum levels of testosterone and its downstream conjugated and unconjugated metabolites in subjects with acyline‐induced experimental hypogonadism,24 who were administered oral testosterone alone or with the 5α‐reductase (5‐AR) inhibitor, dutasteride.25 We also compared the circulating metabolite profile after testosterone treatment with the physiological serum profile of healthy individuals. UGT2B17 polymorphisms also pose a significant challenge in detection of testosterone doping by athletes, where metabolite ratios are used to create individual biological passports.26, 27 Therefore, to test if the testosterone metabolite ratios are dose‐independent, we quantified complete testosterone metabolite panel after treating with two oral doses (200 and 800 mg).
Figure 1.
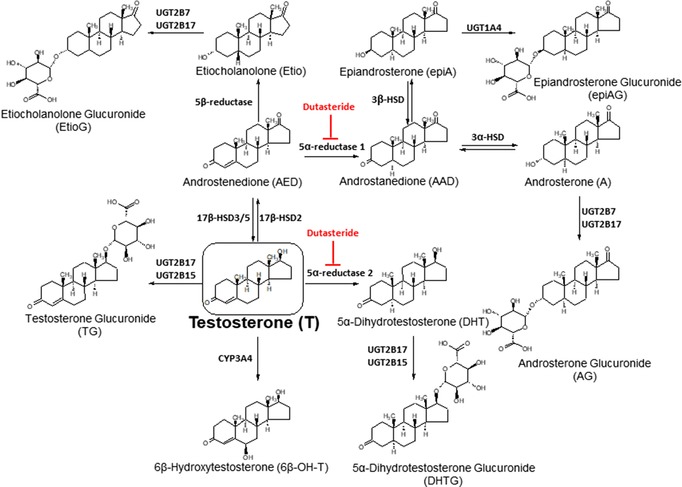
Major metabolite pathways of testosterone (T) disposition in vivo. All these metabolites were quantified by targeted metabolomics analysis. Testosterone is conjugated by UGT2B17 and UGT2B15 to testosterone glucuronide (TG). Testosterone is also converted to dihydrotestosterone (DHT), AED, and 6β‐hydroxytestosterone (6β‐OH‐T) by 5‐AR2, 17β‐HSD2, and CYP3A4, respectively. AED is then metabolized to the number of sequential metabolites, i.e., etiocholanolone (Etio), androstanedione, androsterone (A) and epiandrosterone (epiA). Both A and Etio are conjugated by UGT2B7 and UGT2B17 to A glucuronide (AG) and Etio glucuronide (EtioG), whereas epiA is glucuronidated by UGT1A4 to epiA glucuronide. All these metabolites (except epiA, epiAG, and 6β‐OH‐T) were quantifiable in both 200 and 800 mg dose in the following relative order: AG>TG>EtioG>AED>A>T>DHT>Etio. Dutasteride is a dual inhibitor of 5‐AR1 and 5‐AR2. Sulfate conjugates were not analyzed in this study.
Other than UGTs, 17β‐HSD2, 5‐AR, CYP3A4, and sulfotransferases can also metabolize testosterone, however, the quantitative contribution of these enzymes in testosterone first‐pass metabolism is unknown.28, 29, 30 Dutasteride is a selective inhibitor of the type 1 and type 2 isoforms of the 5‐AR enzyme (Figure 1). The effect of dutasteride on testosterone and dihydrotestosterone (DHT) serum level have been previously reported,31, 32 but its impact on the circulating testosterone metabolite profile has not been characterized. To fill this knowledge gap, we quantified the effect of dutasteride on testosterone metabolite profile at 200 and 800 mg testosterone oral doses.
METHODS
Materials
Liquid chromatography tandem mass spectrometry (LC‐MS/MS) grade acetonitrile, methanol, chloroform, and formic acid were purchased from Fisher Scientific (Fair Lawn, NJ). Unconjugated and conjugated steroid standards were purchased from Cerilliant Corporation (Round Rock, TX) and Steraloid (Newport, RI; Supplementary Table S1). Recombinant human UGT2B7, UGT2B15, and UGT2B17 superosomes were purchased from Corning Life Science (Corning, NY), and dutasteride for the in vitro experiments was from Sigma Aldrich (St Louis, MO). Iodoacetamide, dithiothreitol, and pierce trypsin protease (MS grade) were purchased from Thermo Fisher Scientific (Rockford, IL). Ammonium bicarbonate buffer (98% purity) was purchased from Acros Organics (Geel, Belgium). Human serum albumin and bovine serum albumin (BSA) were obtained from Calbiochem (Billerica, MA) and Thermo Fisher Scientific, respectively. Surrogate peptides were produced by Thermo Fisher Scientific. UDPGA and MgCl2 were purchased from Sigma‐Aldrich. Hepatocytes were provided gratis by Lonza (Walkersville, MD), hepatocyte thawing media (UCRM), hepatocyte plating media (UPCM), hepatocyte induction media (HIM) and hepatocyte quantification media (HQM) were procured from IVAL (Columbia, MD).
Clinical study
A targeted testosterone metabolomics study, which were previously collected and stored samples from seven subjects who received oral testosterone, were used.25 The brief clinical study design is discussed below. The study protocol was approved by the Institutional Review Board of the University of Washington (UW), and subjects gave written informed consent before screening. Healthy, normal male volunteers between 18 and 45 years of age were enrolled in the study. The inclusion criteria were no prior medical illnesses, normal physical examination, and routine hematology, blood chemistry, and liver function. Exclusion criteria included regular use of any medication; abnormal serum testosterone, DHT, or estradiol (E2); or previous or current ethanol, and illicit drug or anabolic steroid abuse. Baseline characteristic of study subjects are reported in Supplementary Table S2. The oral testosterone in sesame oil was manufactured by the compounding pharmacy at the UW, micronized testosterone (U.S.P. grade, Spectrum Quality Projects, Gardena, CA) was suspended at 100 mg/mL in sesame oil (N.F. grade; Spectrum Quality Projects) and mixed thoroughly to create a homogenous testosterone plus sesame oil emulsion. The emulsion was vigorously mixed by shaking with milk and administered to the subject. The drug exposure period lasted 11 days (Supplementary Figure S1). On day 0, subjects received a single injection of the gonadotropin‐releasing hormone antagonist acyline (300 μg/kg, subcutaneous), which has been shown to suppress testosterone production in normal men for a minimum of 15 days24 and confirmed in this study (Supplementary Table S3 and Supplementary Figure S2). After 1, 2, and 3 days of acyline administration, subjects were given 200, 400, or 800 mg testosterone orally. Subjects self‐administered dutasteride purchased by the UW investigational pharmacy (0.5 mg, orally, once daily) on days 5–10 after acyline injection, and doses of testosterone were repeated on days 8, 9, and 10, respectively. After each dose of testosterone, subjects had blood drawn via a heparin‐locked intravenous line at 0.5, 1, 2, 4, 6, 8, 10, 12, and 24 hours for measurement of serum testosterone and its unconjugated and conjugated metabolites. Serum was obtained from the blood by centrifugation and stored in a ‐80°C freezer until analysis by LC‐MS/MS.
Serum sample preparation for LC‐MS/MS metabolomics analysis
Serum was pooled from control subjects (before acyline dose, baseline) and from all time points after a single oral dose of testosterone at 200 mg (T200) and 800 mg (T800). Although the study was conducted with 200, 400, and 800 mg testosterone dose, only T200 and T800 were quantified and presented in this article. Testosterone and its metabolites were extracted from the human serum sample using a protein precipitation with ice cold methanol (containing internal standard mix; Supplementary Table S4) followed by solid‐phase extraction (SPE) in C18 HLB cartridges (Waters). Calibration curve, quality control, or test samples (500 μL) were transferred to 5 mL microcentrifuge tubes. Protein precipitation was carried out using a 1:4 by volume methanol (2 mL) containing internal standard mix. Samples were mixed for 1 minute by vortex mixing and then centrifuged for 10 minutes at 3,500 × g at 4°C. The supernatant was transferred to glass tubes and dried under nitrogen evaporator. The resulting dried residue was redissolved in 2 mL of 5% methanol containing 0.2% formic acid and was used for SPE. The SPE cartridges were mounted on the positive pressure manifold and activated with 2 mL methanol and conditioned with 2 mL 0.2% formic acid. Samples (2 mL) were loaded onto the cartridges and the flow‐through was discarded. Samples were washed with 1 mL of 5% methanol containing 0.2% formic acid to get rid of hydrophilic impurities. Testosterone metabolites were then eluted with 2 mL of methanol and collected in glass tubes. The eluate was then dried under nitrogen evaporator and reconstituted with 0.1 mL methanol: water (1:1, v/v) for LC‐MS/MS analysis.
Targeted LC‐MS/MS analyses of testosterone and its metabolites
LC‐MS/MS analyses were carried out on SCIEX 6500 triple quadrupole mass spectrometer coupled to a Waters Acquity UPLC system. Chromatographic separation of steroid metabolites was achieved using a reversed phase HSS T3 C18 column (2.1 × 100 mm, 1.8 μ particle size; Supplementary Figure S3). Mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) was used. The gradient was as follows: 0.0–1.0 minute (15% B), 1.0–2.5 minutes (15–40% B), 2.5–8.5 minutes (40–50% B), 8.5–12.0 minutes (50–98% B), and 12.0–12.8 minutes (98% B), followed by 0.2 minute gradient 15% B. The column was reconditioned at 15% B for 2.0 minutes. The mass spectrometer was operated in the positive ESI mode using the multiple reaction monitoring approach. Ion spray voltage was set at 4,500 V. The source temperature was optimized to 400°C. Curtain gas and collision gas pressures were 20 and 9 psi, respectively. Ion source gas1 and gas2 pressures were 45 and 35 psi, respectively. The DP and CE for each metabolite were optimized. For each metabolite, 2–3 fragments were selected and one is assigned as the quantifier based on the better response and other fragments were assigned as the qualifier fragments. All the multiple reaction monitoring transitions are reported in Supplementary Table S5. Data were acquired by Analyst 1.6 software. The LC‐MS/MS method was validated for linearity, precision, accuracy, extraction efficiency, and autosampler stability (Supplementary Tables S6–S9).
Effect of dutasteride on in vitro testosterone metabolism in human hepatocytes
Cryopreserved human hepatocytes were thawed and plated, as per the vendor's protocols, and is described in the Supplementary information. Because oral testosterone is completely absorbed but undergoes extensive first‐pass metabolism, only a minor fraction of the total dose is bioavailable and the plasma concentration of testosterone presumably does not reflect the concentration in the enterocytes and hepatocytes during the first‐pass. Therefore, to mimic high testosterone concentration exposed to drug metabolizing enzymes (DMEs) during the first‐pass, the plated human hepatocyte was co‐incubated with high testosterone concentration (50 μM) with 200 nM of dutasteride for 60 minutes at 37°C, 5% CO2. The reaction was quenched by isolating the quantification media (HQM) in microcentrifuge tubes and extracting the metabolites with 1 mL ice‐cold acetonitrile containing an internal standard cocktail. Samples were analyzed by LC‐MS/MS.
Inhibition assay in human liver microsome and recombinant human UGTs
Dutasteride was also screened for its ability to inhibit UGT2B17. For concentration of half inhibition determination, human liver microsomes (HLMs) or rUGT2B17 were incubated with increasing dutasteride concentrations (final concentrations, 0.02, 0.05, 0.1, 0.5, 1.0, 5.0, 10.0, and 20.0 μM). The incubation buffer consists of 5 mM MgCl2‐100 mM phosphate buffer at pH 7.4, alamethicin (0.1 mg/ml), BSA (0.1%) and substrate, testosterone (10 μM). The reaction mixture was incubated for 15 minutes on ice to allow alamethicin pore formation. Reaction was initiated by adding UDPGA (2.5 mM) and incubating for 30 minutes at 37°C. The reaction was quenched by acetonitrile containing isotope labelled internal standards (Supplementary Table S4) and the sample were centrifuged at 2,000 × g, 4°C for 5 minutes. The supernatant was analyzed by LC‐MS/MS to quantify substrate depletion and glucuronide formation. To evaluate the UGT2B17 inhibition, activities at different concentrations were compared with solvent control.
In vitro androgen UGT2B activity in HLM and recombinant systems (Supersomes) and prediction of fractional contribution of each UGT2Bs in androgen metabolism in intestines and liver
HLM samples were isolated from nine individual liver tissues from the UW, School of Pharmacy liver bank and quantified for UGT2B7, UGT2B15, and UGT2B17 protein abundance using a validated trypsin digestion protocol (Supplementary information, 33). Glucuronidation activity assay was performed in HLM (0.1 mg/mL total protein) or recombinant human UGTs (10 μg/ml total protein) containing 5 mM MgCl2‐100 mM phosphate buffer at pH 7.4, alamethicin (0.1 mg/mL), BSA (0.02%), and substrate, testosterone/androsterone (A)/etiocholanolone (Etio) (1 μM to 10 μM). The reaction mixture was incubated for 15 minutes on ice to allow alamethicin pore formation. Reaction was initiated by adding UDPGA (2.5 mM) and incubating for 30 minutes at 37°C. The reaction was quenched by acetonitrile containing internal standard and the samples were centrifuged at 2000 × g, 4°C for 5 minutes. The supernatant was analyzed by LC‐MS/MS to quantify substrate depletion and glucuronide formation.
We also extrapolated fractional contribution of each UGT in the glucuronidation of androgens in the intestinal and liver microsomes using the protein abundance data34 and the in vitro in vivo extrapolation approach are discussed elsewhere.35 Briefly, the intestinal and liver abundance of UGT2B7, UGT2B15, and UGT2B17 of 2.83, 0.0, and 8.87 vs. 36.6, 21.2, and 0.92 pmol/mg microsomal proteins, respectively, were considered for in vitro in vivo extrapolation of recombinant data to liver and intestinal microsomes.
RESULTS
Quantitative profiles of circulating testosterone and its metabolites in pooled serum samples at baseline and after testosterone oral doses
The relative concentrations of circulating testosterone metabolites in eugonadal men (i.e., baseline levels prior to the induction of hypogonadism) is depicted in (Figure 2 a, upper panel). As expected, glucuronides were observed as the major circulating testosterone metabolites; however, the relative predominance of androsterone glucuronide (AG) over TG was surprising. When absolute levels of individual metabolites were compared at baseline and after dosing with 200 or 800 mg of oral testosterone (T200 and T800) in the hypogonadal state (Figure 2 b,c), TG was observed to be significantly higher as compared with baseline by 78‐fold and 277‐fold at T200 and T800, respectively. When compared with baseline, absolute levels of etiocholanolone glucuronide (EtioG), AG, and DHT‐glucuronide were 13‐fold, 9.5‐fold, and 7.5‐fold at T200 vs. 39.5, 29.7, and 40.6‐fold higher at T800 doses, respectively. Similarly, the absolute levels of unconjugated metabolites (androstenedione (AED), A, Etio, and DHT) were 2.3‐fold, 1.2‐fold, 0.8‐fold, and 0.6‐fold (T200) and 14.9‐fold, 5.5‐fold, 3.5‐fold, and 2.2‐fold (T800) higher than that of baseline eugonadal state.
Figure 2.
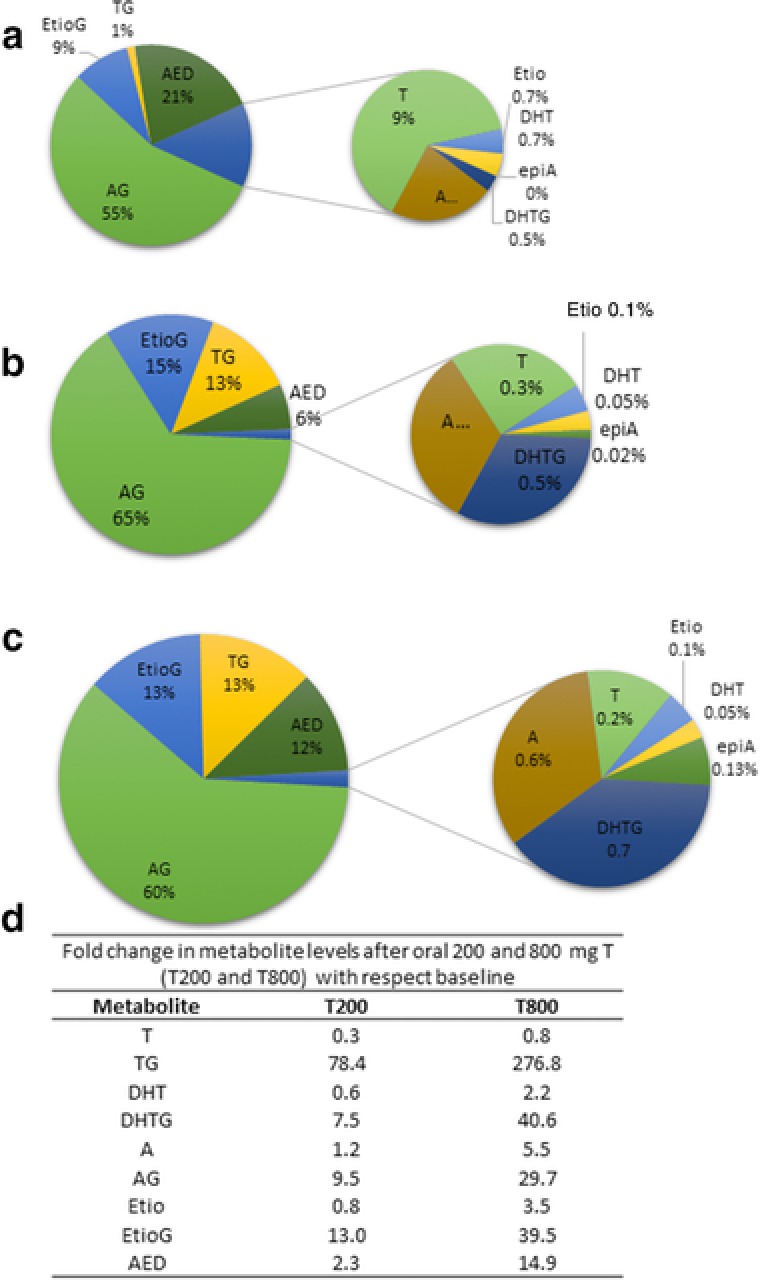
Quantitative profile of circulating testosterone (T) and its primary and secondary metabolites in pooled serum from control eugonadal subjects (n = 7) (before acyline dose, baseline) (a) and pools of all time‐points after single oral dose of testosterone at 200 mg (n = 7) (T200) (b) and 800 mg (n = 7) (T800) (c). Although the study was conducted with 200, 400, and 800 mg testosterone dose, only T200 and T800 were quantified. The table (d) represents fold change in the respective metabolites at T200 and T800 in experimental hypogonadism subjects as compared with the eugonadal subjects. Glucuronides (TG, AG, and EtioG) are the major circulating metabolites. AED, androstenedione; AG, androsterone glucuronide; DHT, dihydrotestosterone; DHTG, dihydrotestosterone glucuronide; EtioG, etiocholanolone glucuronide; T, testosterone; TG, testosterone glucuronide.
Circulating concentration‐time profiles of testosterone and its metabolites
The average serum testosterone area under the curve (AUC)0‐24h was 81.6 nmol/L*h at T200 (Figure 3 and Table 1). However, the major fraction of the oral testosterone dose (T200) was present as its glucuronide metabolites in the serum with average AUC0‐24h of 12,111 (AG), 2,160 (EtioG) and 857.2 (TG) nmol/L*h (Figures 3 and 4, 15). The average serum AUC0‐24h of the unconjugated metabolites were 26.5 (DHT), 373.7 (AED), 96.8 (A), and 11.2 (Etio) nmol/L‐h. At T800, the AUC0‐24h for testosterone, TG, DHT, AED, A, AG, Etio, and EtioG were 2.1‐fold, 16.5‐fold, 3.3‐fold, 4.2‐fold, 12.6‐fold, 5.4‐fold, 8.2‐fold, and 6.9‐fold, respectively, higher than that observed with T200, respectively (Figure 4 and Supplementary Figure S4). The 6β‐OH‐T was only detectable in a few samples, and hence not quantified in this study. Interestingly, multiple peaking phenomenon or humps in the serum concentration‐time profile were observed in the individual subject data (Supplementary Figure S5). The time of maximum plasma concentration (Tmax), peak plasma concentration (Cmax), and terminal half‐life values derived from serum concentration‐time profiles of testosterone and all detected metabolites are presented in Table 1. Interestingly, the dose‐normalized AUC0‐24h data (Supplementary Table S10) indicate that changing the dose from 200 to 800 mg results in substantial increase in TG/testosterone ratio by 9.3‐fold. In contrast, AUC0‐24h ratios (T200 vs. T800) were below 1 (AG/A and EtioG/A) or moderately increased (twofold to threefold; DHT/testosterone, AED/testosterone, A/AED, and Etio/AED).
Figure 3.
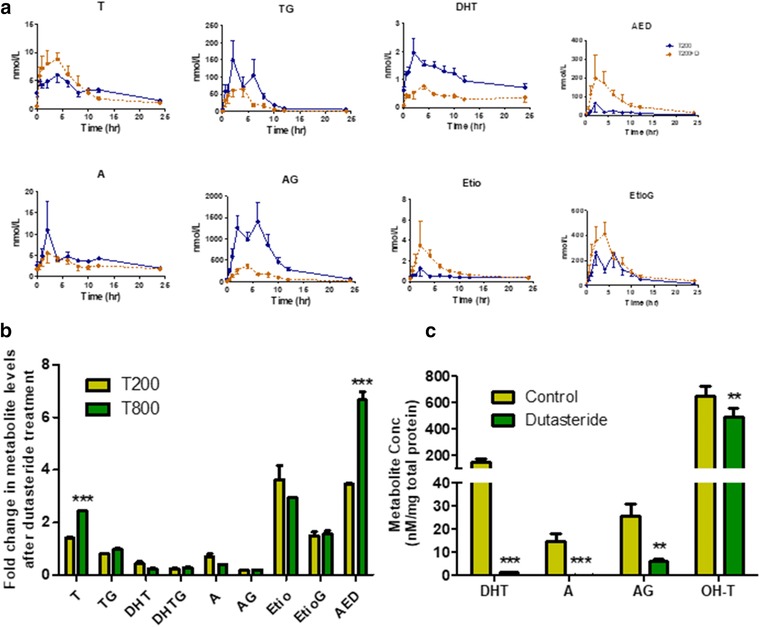
Effect of oral dutasteride on oral testosterone (T) metabolism in vivo and in vitro. (a) Pharmacokinetic profile of testosterone and its primary (testosterone glucuronide (TG), dihydrotestosterone (DHT), and androstenedione (AED)) and secondary (androsterone (A), androsterone glucuronide (AG), etiocholanolone (Etio), and etiocholanolone glucuronide (EtioG)) metabolites after single oral dose (200 mg) of testosterone with (dotted line) and without (solid line) dutasteride co‐administration (n = 7). Dots represent mean plasma concentration at individual sampling time points and bars represent SE. (b) Effect of co‐administered dutasteride on the levels of testosterone metabolites in the pooled serum (n = 7) indicating fold change at T200 and T800. (c) Effect of dutasteride cotreatment on the levels of DHT, A, and AG in human hepatocyte culture. Data for panels b and c were analyzed using Paired t test, **P value < 0.01, ***P value < 0.001.
Table 1.
Pharmacokinetic parameters of testosterone and its metabolites after oral dose of testosterone at 200 and 800 mg with and without dutasteride
| PK Parameter | Dose group | Testosterone | TG | DHT | AED | A | AG | Etio | EtioG |
|---|---|---|---|---|---|---|---|---|---|
| t1/2 (h) | T200 | 12.0 | 4.4 | 17.6 | 12.7 | 15.6 | 4.9 | 19.0 | 5.2 |
| T200+D | 7.0 | 4.3 | 24.0 | 8.3 | 15.2 | 4.5 | 11.3 | 10.5 | |
| T800 | 5.3 | 3.6 | 6.9 | 6.8 | 8.2 | 4.5 | 10.7 | 7.8 | |
| T800+D | 5.7 | 2.7 | 17.9 | 5.2 | 16.1 | 6.3 | 6.4 | 9.9 | |
| Tmax (h) | T200 | 4 | 2 | 2 | 2 | 2 | 6 | 2 | 2 |
| T200+D | 4 | 4 | 4 | 2 | 2 | 4 | 2 | 4 | |
| T800 | 4 | 1 | 2 | 1 | 2 | 4 | 1 | 1 | |
| T800+D | 2 | 2 | 4 | 2 | 2 | 4 | 2 | 2 | |
| Cmax (nmol/L) | T200 | 6.0 | 149.3 | 2.0 | 66.3 | 11.0 | 1404.4 | 1.2 | 266.9 |
| T200+D | 8.8 | 64.5 | 0.8 | 199.7 | 5.6 | 372.2 | 3.5 | 414.3 | |
| T800 | 16.8 | 1937.3 | 8.4 | 280.5 | 168.5 | 5649.1 | 12.4 | 1129.4 | |
| T800+D | 33.0 | 1174.1 | 1.8 | 2301.9 | 13.0 | 1346.5 | 33.9 | 2096.4 | |
| AUC0–24h (nmol/L*h) | T200 | 81.6 | 857.2 | 26.5 | 373.7 | 96.8 | 12110.8 | 11.2 | 2160.4 |
| T200+D | 84.7 | 373.4 | 9.6 | 1634.9 | 68.5 | 2582.7 | 24.4 | 3277.3 | |
| T800 | 168.9 | 14163.0 | 87.4 | 1551.8 | 1218.9 | 65427.8 | 91.7 | 14981.5 | |
| T800+D | 293.8 | 8589.7 | 26.6 | 10844.4 | 161.5 | 14867.7 | 220.2 | 27091.7 | |
| V/F_obs (L) | T200 | 110034 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| T200+D | 72890 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| T800 | 118392 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| T800+D | 72597 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Cl/F_obs (L/h) | T200 | 6351 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| T200+D | 7202 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| T800 | 15528 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| T800+D | 8831 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
A, androsterone; AED, androstenedione; AG, androsterone glucuronide; AUC, area under the curve; Cmax, peak plasma concentration; DHT, 5α‐dihydrotestosterone; Etio, etiocholanolone; EtioG, etiocholanolone glucuronide, t1/2, terminal half‐life; TG, testosterone glucuronide, Tmax, time of maximum plasma concentration.
Figure 4.
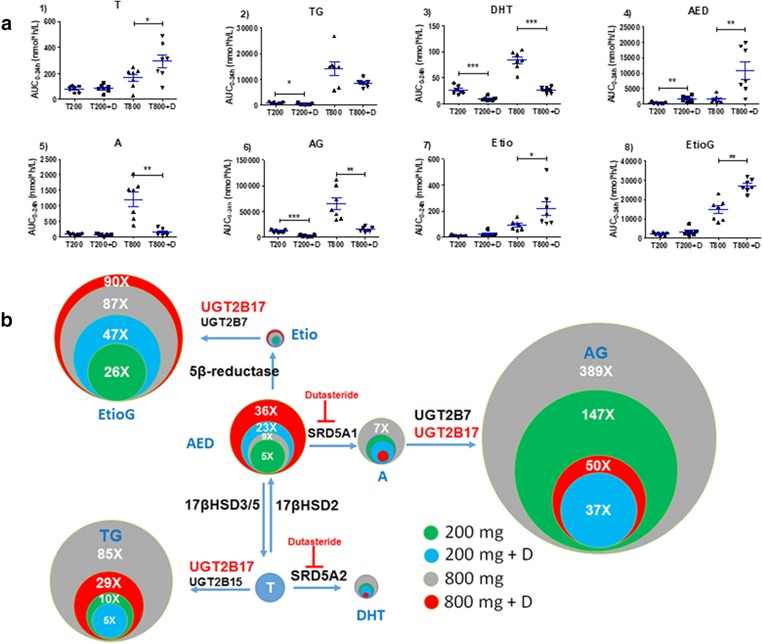
(a) Area under the curve (AUC)0–24h of testosterone and its primary and secondary metabolites in individual subjects after single oral dose of testosterone at T200 and T800 with and without dutasteride co‐administration (n = 7). Dots indicate individual data, horizontal line represents mean and error bars indicate SE. (b) Relative quantitative representation of mean AUC0–24h of testosterone, and its primary and secondary metabolites after single oral dose at T200 and T800 with and without dutasteride co‐administration (n = 7). Metabolic scheme and enzymes involved in testosterone metabolism are shown as reported in ref. 15. Size of the circle and number (nx) indicate relative mean AUC0–24h of individual metabolites as compared with testosterone in respective studies. Glucuronides are the major circulating metabolites of testosterone. Data were analyzed using Paired t test, *P value < 0.05, **P value < 0.01, ***P value < 0.001. AED, androstenedione; AG, androsterone glucuronide; DHT, dihydrotestosterone; Etio, etiocholanolone; EtioG, etiocholanolone glucuronide; T, testosterone; TG, testosterone glucuronide.
Effect of dutasteride co‐administration on circulating concentration‐time profiles of testosterone and its metabolites
The pooled serum testosterone levels were 1.4‐fold and 2.4‐fold higher after dutasteride co‐administration at T200 and T800, respectively, as compared with testosterone alone. The average AUC0‐24h was 3277, 2583, and 373.4 nmol/L*h for EtioG, AG, and TG, respectively, after 200 mg testosterone with dutasteride (T200+D), which were 151%, 21%, and 43% with respect to testosterone alone. The average AUC0‐24h at 800 mg testosterone with dutasteride (T800+D) were 180%, 22%, and 60% as compared with testosterone alone (Figure 3 and Table 1). The average AUC0‐24h for DHT with dutasteride was 64% and 70% lower in testosterone‐treated samples (T200 and T800, respectively) as compared with testosterone alone. The circulating A level was 30% (T200+D) and 87% (T800+D) lower in dutasteride‐treated samples as compared with testosterone alone. On the other hand, circulating levels for AED and Etio were 5.7‐fold and 2.3‐fold higher in T200+D and T800+D as compared with testosterone alone.
Effect of dutasteride on testosterone‐metabolism in human hepatocytes
Consistent with the in vivo data, co‐treatment of dutasteride with testosterone resulted in complete inhibition of 5‐AR types 1 and 2 mediated metabolism of testosterone in human hepatocytes. This resulted in significant decreases in DHT, A, AG, and 6β‐OH‐T formation (Figure 3 C; P < 0.01). No significant changes were observed in the levels of TG and AED (data not shown).
Fractional contribution of UGT2B7, UGT2B15, and UGT2B17 in testosterone, A, and Etio glucuronidation
In HLMs, testosterone is primarily glucuronidated by UGT2B17 with minor contribution from UGT2B15 (Figure 5 34, 36). For example, there was a good correlation between TG formation and UGT2B17 protein abundance in HLMs (r2 = 0.87; Figure 5, 34, 36). These results were consistent with the recombinant UGT data (Figure 5, 34, 36). AG formation is primarily mediated by UGT2B7 as confirmed by recombinant UGT and HLM data (Figure 5, 34, 36). Etio is metabolized by both UGT2B17 (r2 = 0.7; Figure 5 c) and UGT2B7 (r2 = 0.44; Figure 5). Scaling these data by using absolute UGT2B abundance in the intestine34 and the liver36 (Supplementary Table S11) predicts that UGT2B17 is the major contributor for the intestinal glucuronidation of testosterone (100%), Etio (90%), and A (70%), whereas UGT2B7 is the major contributor for hepatic glucuronidation of A (100%) and Etio (85%).
Figure 5.
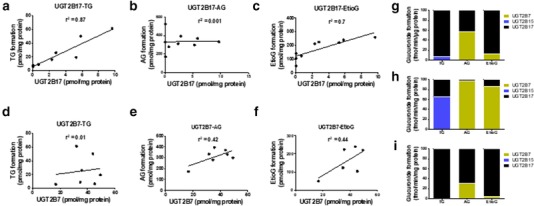
In vitro glucuronidation of testosterone, A and Etio using human liver microsomes (HLMs) and recombinant human UGT2B enzymes. (a‐c) These panels represent the correlation between UGT2B17 protein expression in HLM and glucuronidation activity toward testosterone, A and Etio, respectively. (d‐f) These panels represent the correlation between UGT2B7 protein expression in HLM and glucuronidation activity toward testosterone, A and Etio, respectively. (g) This panel represents glucuronidation activity of recombinant human UGT2B enzymes toward testosterone, A and Etio at 1 μM. (h and i) These panels show the estimated glucuronidation activity of UGT2Bs toward testosterone, A, and Etio in the human liver and the intestinal microsomes. The scaling was based on UGT2B protein abundance in the liver (36.6, 21.2, and 0.92 pmol/mg36 and the intestine (2.83, 0.0, and 8.87 pmol/mg) for UGT2B7, UGT2B15, and UGT2B17, respectively.34 AG, androsterone glucuronide; EtioG, etiocholanolone glucuronide; TG, testosterone glucuronide
DISCUSSION
Approximately 2–5% men are affected by hypogonadism, which requires TRT.37, 38 Because of high first‐pass metabolism, only nonoral testosterone formulations are available in the United States, and these formulations are frequently associated with poor patient compliance and variable response. In order to develop strategies to ensure optimum testosterone bioavailability, it is important to characterize mechanisms that affect first‐pass testosterone metabolism. In this study, first, we observed that the metabolism of testosterone differs between eugonadal men and those same men treated with oral testosterone after induction of hypogonadism. Particularly, distinctly high AG, TG, and EtioG concentrations in testosterone‐treated vs. untreated samples indicate that glucuronidation is the major mechanism of testosterone first‐pass elimination. Of particular note, we observed that, contrary to the prevailing thinking, glucuronidation of oral testosterone to AG seemed to be more prevalent than formation of TG. Although the clearance of TG and AG may differ (depending on differential renal clearance) and, therefore, individual contribution of each pathway (fm) cannot be calculated from the circulating metabolite concentration data, this finding suggests that “back‐formation” of AED via 17 β‐HSD2 is the primary route of metabolism of oral testosterone. Interestingly, relative proportions of AG and EtioG to all metabolites (Figure 2) were marginally different from their proportion at baseline; however the proportion of TG (baseline vs. treated) to all metabolites differs significantly. Although genotype data were not available to us in this study, high interindividual variability (Supplementary Tables S12 and S13) in TG PK profiles was consistent with polymorphic nature of UGT2B17.17
Although AED can also be biosynthesized from dehydroepiandrosterone (DHEA) by 3α‐HSD,39 testosterone to AED via 17β‐HSD2 was observed as the major testosterone metabolic pathway, which subsequently metabolized to the major metabolites, AG and EtioG. These data are corroborated by Bhasin et al.40 study, which showed the association of aldo‐keto reductase 1C3 (AKR1C3/17β‐HSD5) polymorphism (rs12529) with variable testosterone circulating levels in men. High levels of AED and its metabolites with oral testosterone is presumably due to interaction of testosterone with 17β‐HSDs and AED with 5α/β‐reductase (5‐AR/5‐BR) in the gastrointestinal tract41 and liver42 during the first pass. We also confirmed high in vitro formation of AED (46%) from testosterone using primary human hepatocyte, but it was marginally higher than TG levels (42%). Because 17β‐HSDs are also expressed in the intestine,41 testosterone to AED formation can occur in the enterocyte. Thus, inhibition of 17β‐HSD2 in the intestine and the liver can significantly increase testosterone circulating levels and could be used as a potential therapeutic target for augmenting the effect of oral TRT. This is important as the development of 17β‐HSDs inhibitors is in the early stage for treatment of osteoporosis (17β‐HSD1/2) and prostate cancer (17β‐HSD3/5).43
Although relative to AED, TG formation was observed to be a minor pathway in overall testosterone metabolism, it is noteworthy that testosterone to TG conversion is mediated by a highly variable enzyme, UGT2B17 (Figure 5). Because ∼50% population has a very low expression (nondetectable in the liver,36 testosterone‐glucuronidation will be important only in the high UGT2B17 expressers (e.g., in 10% population), perhaps not captured in this study. This is supported with controversial association of UGT2B17 with testosterone‐related pathophysiological conditions, prostate cancer.44 That is, whereas some studies support association of UGT2B17 gene deletion with prostate cancer,18 a recent large study failed to confirm the same,40 perhaps due to high variability in UGT2B17.17, 45 Particularly, we recently demonstrated that UGT2B17 is associated with single nucleotide polymorphism (rs7436962, rs9996186, rs28374627, and rs4860305), age, and gender besides gene deletion.36
In our study, a fourfold increase in dose (200 to 800 mg) only resulted in twofold increase in circulating testosterone concentrations. We report here for the first time that testosterone metabolism to TG and AED increases more than proportionally with increasing dose. Higher TG formation after 800 mg dose could be because of the cumulative effect of: (a) the dose‐limiting testosterone solubility resulting in longer gastrointestinal retention of testosterone; and (b) higher UGT2B17 abundance in lower part of gastrointestinal tract.36 Interestingly, AG and EtioG formation seems to saturate at the higher dose (800 mg). The molecular mechanisms underlying these observations will require further research. Nevertheless, because metabolite ratios are used to create individual passports, these findings could be considered when analyzing doping test results.
As discussed above, this study was a retrospective analysis of previously collected samples,25 in which 5‐AR inhibitor, dutasteride, was also co‐administered to minimize increase in DHT levels, which is relevant to long‐term effects of elevated DHT. Therefore, we also tested the effect of dutasteride co‐administration on testosterone metabolome. We observed that dutasteride resulted twofold higher serum testosterone concentration after 800 mg oral testosterone dose. This increase in serum testosterone levels are due to dual inhibition of 5‐AR types 1 and 2, and would likely not be evident with a specific inhibitor for Type‐2 5‐AR (e.g., finasteride).46 Interestingly, there was no significant increase in serum testosterone levels at 200 mg testosterone dose with dutasteride, perhaps due to low levels of AED in T200 as compared with T800, which seemed necessary to drive the back‐conversion (Figure 4, 15). Upon inhibiting 5‐AR, testosterone metabolism is shifted toward AED, Etio, and EtioG pathways, albeit TG formation was not altered significantly (Figure 4 a). A moderate reduction in TG levels was observed with dutasteride at 200 and 800 mg testosterone. However, inhibition of UGT2B17 or induction of 17‐HSD2 do not explain these observations as indicated by in vitro data (Supplementary Figure S6).
Our study has several limitations, including a small number of subjects, and the nonavailability of urine samples and DNA for genotyping. The sulfate conjugates could not be detected in this study due to analytical challenges of these low‐level metabolites or perhaps due to rapid elimination of sulfates in urine. Nevertheless, the data presented here are novel with respect to levels of circulating glucuronides, nonlinear PK, and effect of dutasteride on testosterone metabolite profiles. In particular, the relative prominence of the AG vs. the TG pathway for the metabolism of oral testosterone was unexpected and may be useful in the development of novel formulation strategies of oral testosterone. In addition, the nonlinear PK of oral testosterone observed in this study should be taken into consideration while developing antidoping strategies. Multiple peaking phenomenons in the PK profiles suggest a potential role of microbiome in deconjugation of glucuronides, indicating another potential reasons of interindividual variability. This is consistent with the Markle et al.47 data, which revealed that transfer of cecal contents from male mice to female mice was associated with increased circulating testosterone in female mice. Further investigations are warranted to elucidate quantitative role of glucuronidation in the overall testosterone‐metabolism in the UGT2B17 high expressers. In addition, to better interpret results of this study, future studies are needed to delineate kinetic parameters (substrate affinity (Km) and maximal rate of metabolism (Vmax)) of testosterone and its metabolites for individual enzymes and the absolute protein abundance of these enzymes in liver and intestines. Finally, mechanisms of transport of testosterone and its metabolites across cells could explain our observations.
Conflict of Interest
The authors declared no competing interests for this work.
Supporting information
Supplemental Material
Source of Funding
This study was primarily supported by the start‐up funds of Dr Prasad (Department of Pharmaceutics, University of Washington, Seattle, WA) and in parts by Dr Prasad's grant R01HD081299 from the Eunice Kennedy Shriver National Institutes of Child Health and Human Development (NICHD), a branch of the National Institutes of Health. Dr Amory is supported, in part, by grant K24HD082231 from the NICHD.
Author Contributions
A.B., J.K.A., and B.P. wrote the manuscript. A.B., J.K.A., and B.P. designed the research. A.B., J.K.A., and B.P. performed the research. A.B. and B.P. analyzed the data. J.K.A. contributed new reagents/tools.
References
- 1. Kaufman, J.M. & Vermeulen, A . The decline of androgen levels in elderly men and its clinical and therapeutic implications. Endocr. Rev. 26, 833–876 (2005). [DOI] [PubMed] [Google Scholar]
- 2. Bremner, W.J . Testosterone deficiency and replacement in older men. N. Engl. J. Med. 363, 189–191 (2010). [DOI] [PubMed] [Google Scholar]
- 3. Golds, G. , Houdek, D. & Arnason, T. Male hypogonadism and osteoporosis: the effects, clinical consequences, and treatment of testosterone deficiency in bone health. Int. J. Endocrinol. 2017, 4602129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang, C. et al Low testosterone associated with obesity and the metabolic syndrome contributes to sexual dysfunction and cardiovascular disease risk in men with type 2 diabetes. Diabetes Care. 34, 1669–1675 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bassil, N. , Alkaade, S. & Morley, J.E. The benefits and risks of testosterone replacement therapy: a review. Ther. Clin. Risk Manag. 5, 427–448 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Snyder, P.J . et al Effects of testosterone treatment in older men. N. Engl. J. Med. 374, 611–624 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pastuszak, A.W. , Gomez, L.P. , Scovell, J.M. , Khera, M. , Lamb, D.J. & Lipshultz, L.I. Comparison of the effects of testosterone gels, injections, and pellets on serum hormones, erythrocytosis, lipids, and prostate‐specific antigen. Sex. Med. 3, 165–173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mazer, N. et al Comparison of the steady‐state pharmacokinetics, metabolism, and variability of a transdermal testosterone patch versus a transdermal testosterone gel in hypogonadal men. J. Sex. Med. 2, 213–226 (2005). [DOI] [PubMed] [Google Scholar]
- 9. Schedl, H.P. Absorption of steroid hormones from the human small intestine. J. Clin. Endocrinol. Metab. 25, 1309–1316 (1965). [DOI] [PubMed] [Google Scholar]
- 10. Farthing, M.J. , Vinson, G.P. , Edwards, C.R. & Dawson, A.M. Testosterone metabolism by the rat gastrointestinal tract, in vitro and in vivo. Gut. 23, 226–234 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rao, G.S. , Rao, M.L. & Breuer, H . Studies on a testosterone glucuronyltransferase from the cytosol fraction of human liver. Biochem. J. 119, 635–642 (1970). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tax, L . Absolute bioavailability of testosterone after oral administration of testosterone‐undecanoate and testosterone. Eur. J. Drug Metab. Pharmacokinet. 12, 225–226 (1987). [DOI] [PubMed] [Google Scholar]
- 13. Tauber, U. , Schroder, K. , Dusterberg, B. & Matthes, H . Absolute bioavailability of testosterone after oral administration of testosterone‐undecanoate and testosterone. Eur. J. Drug Metab. Pharmacokinet. 11, 145–149 (1986). [DOI] [PubMed] [Google Scholar]
- 14. Frey, H. , Aakvaag, A. , Saanum, D. & Falch, J . Bioavailability of oral testosterone in males. Eur. J. Clin. Pharmacol. 16, 345–349 (1979). [DOI] [PubMed] [Google Scholar]
- 15. Ghayee, H.K. & Auchus, R.J . Basic concepts and recent developments in human steroid hormone biosynthesis. Rev. Endocr. Metab. Disord. 8, 289–300 (2007). [DOI] [PubMed] [Google Scholar]
- 16. Wells, P.G . et al Glucuronidation and the UDP‐glucuronosyltransferases in health and disease. Drug Metab. Dispos. 32, 281–290 (2004). [DOI] [PubMed] [Google Scholar]
- 17. Zhu, A.Z . et al Genetic and phenotypic variation in UGT2B17, a testosterone‐metabolizing enzyme, is associated with BMI in males. Pharmacogenet. Genomics 25, 263–269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karypidis, A.H. , Olsson, M. , Andersson, S.O. , Rane, A. & Ekstrom, L . Deletion polymorphism of the UGT2B17 gene is associated with increased risk for prostate cancer and correlated to gene expression in the prostate. Pharmacogenomics J. 8, 147–151 (2008). [DOI] [PubMed] [Google Scholar]
- 19. Yang, T.L . et al Genome‐wide copy‐number‐variation study identified a susceptibility gene, UGT2B17, for osteoporosis. Am. J. Hum. Genet. 83, 663–674 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Uddin, M . et al UGT2B17 copy number gain in a large ankylosing spondylitis multiplex family. BMC Genet. 14, 67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hirata, H . et al Function of UDP‐glucuronosyltransferase 2B17 (UGT2B17) is involved in endometrial cancer. Carcinogenesis 31, 1620–1626 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shin, H.C . et al Comparative gene expression of intestinal metabolizing enzymes. Biopharm. Drug Dispos. 30, 411–421 (2009). [DOI] [PubMed] [Google Scholar]
- 23. Wang, Y.H . et al UGT2B17 genetic polymorphisms dramatically affect the pharmacokinetics of MK‐7246 in healthy subjects in a first‐in‐human study. Clin. Pharmacol. Ther. 92, 96–102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Herbst, K.L. , Coviello, A.D. , Page, S. , Amory, J.K. , Anawalt, B.D. & Bremner, W.J . A single dose of the potent gonadotropin‐releasing hormone antagonist acyline suppresses gonadotropins and testosterone for 2 weeks in healthy young men. J. Clin. Endocrinol. Metab. 89, 5959–5965 (2004). [DOI] [PubMed] [Google Scholar]
- 25. Amory, J.K. & Bremner, W.J. Oral testosterone in oil plus dutasteride in men: a pharmacokinetic study. J. Clin. Endocrinol. Metab. 90, 2610–2617 (2005). [DOI] [PubMed] [Google Scholar]
- 26. Martin‐Escudero, P . et al Impact of UGT2B17 gene deletion on the steroid profile of an athlete. Physiol. Rep. 3, e12645 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Strahm, E . et al Dose‐dependent testosterone sensitivity of the steroidal passport and GC‐C‐IRMS analysis in relation to the UGT2B17 deletion polymorphism. Drug Test. Anal. 7, 1063–1070 (2015). [DOI] [PubMed] [Google Scholar]
- 28. Sten, T. , Bichlmaier, I. , Kuuranne, T. , Leinonen, A. , Yli‐Kauhaluoma, J. & Finel, M. UDP‐glucuronosyltransferases (UGTs) 2B7 and UGT2B17 display converse specificity in testosterone and epitestosterone glucuronidation, whereas UGT2A1 conjugates both androgens similarly. Drug Metab. Dispos. 37, 417–423 (2009). [DOI] [PubMed] [Google Scholar]
- 29. Tria, A. , Hiort, O. & Sinnecker, G.H . Steroid 5alpha‐reductase 1 polymorphisms and testosterone/dihydrotestosterone ratio in male patients with hypospadias. Horm. Res. 61, 180–183 (2004). [DOI] [PubMed] [Google Scholar]
- 30. Usmani, K.A. & Tang, J. Human cytochrome P450: metabolism of testosterone by CYP3A4 and inhibition by ketoconazole. Curr. Protoc. Toxicol. Chapter 4, Unit 4.13 (2004). [DOI] [PubMed] [Google Scholar]
- 31. Bhasin, S . et al Effect of testosterone supplementation with and without a dual 5alpha‐reductase inhibitor on fat‐free mass in men with suppressed testosterone production: a randomized controlled trial. JAMA. 307, 931–939 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wurzel, R. , Ray, P. , Major‐Walker, K. , Shannon, J. & Rittmaster, R . The effect of dutasteride on intraprostatic dihydrotestosterone concentrations in men with benign prostatic hyperplasia. Prostate Cancer Prostatic. Dis. 10, 149–154 (2007). [DOI] [PubMed] [Google Scholar]
- 33. Vrana, M. , Whittington, D. , Nautiyal, V. & Prasad, B. Database of optimized proteomic quantitative methods for human drug disposition‐related proteins for applications in physiologically based pharmacokinetic modeling. CPT Pharmacometrics Syst. Pharmacol. 6, 267–276 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Groer, C . et al Absolute protein quantification of clinically relevant cytochrome P450 enzymes and UDP‐glucuronosyltransferases by mass spectrometry‐based targeted proteomics. J. Pharm. Biomed. Anal. 100, 393–401 (2014). [DOI] [PubMed] [Google Scholar]
- 35. Kahma, H . et al Clopidogrel carboxylic acid glucuronidation is mediated mainly by UGT2B7, UGT2B4, and UGT2B17: Implications for pharmacogenetics and drug‐drug interactions. Drug Metab. Dispos. 46, 141–150 (2018). [DOI] [PubMed] [Google Scholar]
- 36. Bhatt, D.K . et al Hepatic abundance and activity of androgen‐ and drug‐metabolizing enzyme, UGT2B17, are associated with genotype, age, and sex. Drug Metab. Dispos. 46, 888–896 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Araujo, A.B . et al Prevalence and incidence of androgen deficiency in middle‐aged and older men: estimates from the Massachusetts male aging study. J. Clin. Endocrinol. Metab. 89, 5920–5926 (2004). [DOI] [PubMed] [Google Scholar]
- 38. Wu, F.C . et al Identification of late‐onset hypogonadism in middle‐aged and elderly men. N. Engl. J. Med. 363, 123–135 (2010). [DOI] [PubMed] [Google Scholar]
- 39. Auchus, M.L. & Auchus, R.J . Human steroid biosynthesis for the oncologist. J. Investig. Med. 60, 495–503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bhasin, S . et al Contributors to the substantial variation in on‐treatment testosterone levels in men receiving transdermal testosterone gels in randomized trials. Andrology 6, 151–157 (2018). [DOI] [PubMed] [Google Scholar]
- 41. Sano, T . et al 17 beta‐Hydroxysteroid dehydrogenase type 2 expression and enzyme activity in the human gastrointestinal tract. Clin. Sci. (Lond.) 101, 485–491 (2001). [PubMed] [Google Scholar]
- 42. Labrie, F . et al The key role of 17 beta‐hydroxysteroid dehydrogenases in sex steroid biology. Steroids 62, 148–158 (1997). [DOI] [PubMed] [Google Scholar]
- 43. Day, J.M. , Tutill, H.J. , Purohit, A. & Reed, M.J . Design and validation of specific inhibitors of 17beta‐hydroxysteroid dehydrogenases for therapeutic application in breast and prostate cancer, and in endometriosis. Endocr. Relat. Cancer 15, 665–692 (2008). [DOI] [PubMed] [Google Scholar]
- 44. Gallagher, C.J. , Kadlubar, F.F. , Muscat, J.E. , Ambrosone, C.B. , Lang, N.P. & Lazarus, P . The UGT2B17 gene deletion polymorphism and risk of prostate cancer. A case‐control study in Caucasians. Cancer Detect. Prev. 31, 310–315 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vidal, A.C . et al Novel associations of UDP‐glucuronosyltransferase 2B gene variants with prostate cancer risk in a multiethnic study. BMC Cancer 13, 556 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vaughan, E.D. Long‐term experience with 5‐alpha‐reductase inhibitors. Rev. Urol. 5(Suppl 4), S28–S33 (2003). [PMC free article] [PubMed] [Google Scholar]
- 47. Markle, J.G . et al Sex differences in the gut microbiome drive hormone‐dependent regulation of autoimmunity. Science 339, 1084–1088 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material