

Abstract
A majority of the world population is infected with herpes simplex viruses (HSV; human herpesvirus types 1 and 2). These viruses, perhaps best known for their manifestation in the genital or oral mucosa, can also cause herpes simplex encephalitis, a severe and often fatal disease of the central nervous system. Antiviral therapies for HSV are only partially effective since the virus can establish latent infections in neurons, and severe pathological sequelae in the brain are common. A better understanding of disease pathogenesis is required to develop new strategies against herpes simplex encephalitis, including the precise viral and host genetic determinants that promote virus invasion into the central nervous system and its associated immunopathology. Here we review the current understanding of herpes simplex encephalitis from the host genome perspective, which has been illuminated by groundbreaking work on rare herpes simplex encephalitis patients together with mechanistic insight from single-gene mouse models of disease. A complex picture has emerged, whereby innate type I interferon-mediated antiviral signaling is a central pathway to control viral replication, and the regulation of immunopathology and the balance between apoptosis and autophagy are critical to disease severity in the central nervous system. The lessons learned from mouse studies inform us on fundamental defense mechanisms at the interface of host–pathogen interactions within the central nervous system, as well as possible rationales for intervention against infections from severe neuropathogenic viruses.
Introduction
Herpes simplex virus types 1 and 2 (HSV-1 and HSV-2) are among the most common human pathogens. These viruses can establish life-long latency, such that an estimated 3.7 billion people under age 50 (67%) have been infected with HSV-1 and 417 million people aged 15–49 (11%) with HSV-2, worldwide (Looker et al. 2015a). There are important geographical differences however, where HSV-1 is universal in the developing world and in the United States, and HSV-2 is most prevalent in Africa (Looker et al. 2015b). HSV-2 disproportionally affects women and increases the risk of acquiring the human immunodeficiency virus (McQuillan et al. 2018). HSV infections are generally confined to the oro-labial and genital skin and mucosa, although the viruses are the most frequent cause of sporadic encephalitis (Looker et al. 2017; Steiner and Benninger 2013), a potentially deadly infection of the central nervous system (CNS). The characteristic clinical presentation of HSV encephalitis usually consists of fever, seizures, and often focal or generalized neurological deficits depending on the clinical form, namely neonatal HSV or herpes simplex encephalitis (HSE). In neonatal HSV, brain involvement is generalized, and the usual cause is HSV-2 (Long et al. 2011), which is acquired at the time of delivery in incident mothers. Post-partum infection is thought to be acquired by contact with HSV-1 shed by caregivers (10% of cases). HSV-1 is the predominant cause of HSE, with 10% of the cases caused by HSV-2 (Solomon et al. 2012). HSE typically affects the frontal and temporal lobes (Kaewpoowat et al. 2016), but in rare cases, the brainstem may be preferentially involved (Livorsi et al. 2010). The age-specific incidence is bimodal, with about 1/3 of cases observed in children between 3 months and 20 years, and the other in adults over 60, representing approximately 2/3 of the cases. Before the advent of acyclovir and other antiviral therapies, the mortality rate associated with central HSV infection was 70%. Treatment is effective if started promptly; in contrast, delays in treatment can be devastating. It is striking that in HSE, which still has a mortality approaching 30% and causes serious brain damage, there have been no significant enhancements of therapy in the last 30 years (Haubenberger and Clifford 2016). Improved tools are needed to treat, diminish the risk of, and prevent HSE based on a molecular understanding of HSE disease mechanisms.
Although there have been important advances in understanding disease neuropathogenesis [reviewed in Koyuncu et al. (2013), Kramer and Enquist (2013)], the precise mechanisms leading to HSE are not known. The virus enters the body by infecting epithelial or mucosal cells in peripheral tissues before entering sensory neurons. The virus genome reaches the soma of the sensory neuron as an episome that expresses a single transcript, LAT, and several microRNAs. The LAT RNA counteracts apoptosis of the neuronal cell and allows maintenance of the viral genome (Ahmed et al. 2002). The virus also has mechanisms (ICP34.5) that counteract autophagy, another antiviral mechanism in neurons (Orvedahl et al. 2007). During active replication, the virus expresses proteins (ICP0) that block innate responses mediated by type I interferon (IFN I), preventing the activation of the innate antiviral response (Lin et al. 2004). If the infection is not rapidly controlled, inflammatory responses are triggered and IFNs activate NK cell and T cell responses that are critical to clearance but also need to be tightly controlled to prevent inflammatory damage in the CNS (Menendez and Carr 2017). Pathological studies show that HSE is associated with virus replication, neuronal cell death, and the presence of inflammatory infiltrates; however, the relative contribution of each response to pathology or defense is not entirely clear (Michael et al. 2016; Wnek et al. 2016). Also outstanding is the question of which cellular and viral determinants can restrict replication at mucosal sites of infection, or permit the rare escape of virus to the CNS.
Host and viral genetic factors may interact to cause variability of herpesvirus neurovirulence. Genetic differences in isolates from HSE patients and oro-labial infections indicated that at least half of the cases of HSE are caused by a different viral strain than the one responsible for cold sores in the same individual, suggesting that HSE is due to primary infection rather than reactivation (Steiner 2011). Advancements in high-throughput sequencing have now made it possible to study the full extent of genetic variation in the viral population within an infected host, or that being transmitted between different hosts (Pandey et al. 2017; Parsons et al. 2015), and to provide the potential molecular basis for neurovirulence.
In recent years, the clinical genetic study of patients and families with HSE has revealed that single-gene inborn errors of innate or cell-intrinsic immunity can underlie enhanced susceptibility to specific viral infections in otherwise healthy individuals. Unique aspects of HSE molecular pathology have also been clarified with the advent of next-generation sequencing technologies. Thus, it was shown that HSE in two children was the consequence of an autosomal recessive deficiency in the intracellular protein UNC93B1 leading to impaired cellular IFN I responses (Casrouge et al. 2006). Subsequent work has confirmed that HSE in children may result from single-gene errors in Toll-like receptor 3 (TLR3)-IFN I pathways (Herman et al. 2012; Perez de Diego et al. 2010; Sancho-Shimizu et al. 2011; Zhang et al. 2007). These genetic defects all lead to reduced IFN I induction in patient cells, upon HSV-1 infection or ex vivo stimulation of the TLR3 pathway, in both fibroblasts and specific CNS cell lineages derived from induced pluripotent cells from the patient (Lafaille et al. 2012). Whereas defects in the TLR3 pathway are remarkably specific for manifestations of HSE caused by HSV-1, some patients have immunodeficiencies to various infectious phenotypes. Patients with susceptibility to herpes and mycobacterial infections have mutations in the transcription factor STAT1 and in NF-κB essential modulator NEMO (Audry et al. 2011; Dupuis et al. 2003). More recently, several patients with brainstem-localized HSV-1, norovirus, or influenza virus infections were found to lack intrinsic antiviral immunity due to mutations in the RNA binding protein, DBR1 (Zhang et al. 2018). Why such genetic determinants should manifest in childhood, but not in adult HSE, is unclear.
These groundbreaking discoveries (recently reviewed in (Casanova 2015; Zhang et al. 2013b)) have clarified the critical protective role of innate IFN responses that are non-redundant in the immune system. Specific mutations in components of the innate response account only for a minority of patients and fail to explain completely the different disease manifestations in the newborn HSV, childhood HSE, and the elderly population. Four decades ago, Lopez et al. reported that the infection of inbred mouse strains with HSV-1 mimicked dramatic differences in the presentation of encephalitis and survival observed in humans, justifying the search of genetic determinants of pathogenesis in mice (Lopez 1975). Whereas the phenotypes of human mutations have been recapitulated in mouse models for the most part, studies in mice have led to the discovery of new cell-intrinsic (Table 1) and immune cell-mediated (Table 2) disease mechanisms. This review will focus on how mouse models have contributed to our understanding of HSE and on the contributions of host genetics and of Trl3-dependent and independent mechanisms to HSV antiviral immunity. We also discuss future perspective on how these discoveries may lead to the development of improved therapies tailored to specific forms of human HSE.
Table 1.
Gene deficiencies affecting cell-intrinsic responses to HSV encephalitis in mice, and to other infections in humans
| Targeted allele(s) | Protein | Function | Survival phenotype† | Virus | Infection route | References | Human gene | Infectious agent or disease | OMIM number§ |
|---|---|---|---|---|---|---|---|---|---|
| Pvrl1 −/− | Nectin-1 | Cell-surface HSV entry receptor | Resistant | HSV-2 | Intracranial | Kopp et al. (2009) | |||
| Ifnar −/− | IFNα/βR1 | Type I IFN receptor | Susceptible | HSV-1 | Intracranial | Wang et al. (2012) | IFNAR2 | Disseminated vaccine measles, HHV-6 | 602376 |
| Susceptible | HSV-2 | Intravaginal | Lee et al. (2017), Reinert et al. (2012) | ||||||
| Tlr3 −/− | TLR3 | Endosomal pattern recognition receptor | Susceptible | HSV-2 | Intravaginal | Reinert et al. (2012) | TLR3 | HSV-1 | 603029 |
| Ticam1 −/− | TRIF | TLR3 cascade adaptor protein | Susceptible* | HSV-1 | Intranasal | Menasria et al. (2013) | TICAM1 | HSV-1, HSV-2 | 607601 |
| STING cascade adaptor protein | Susceptible* | HSV-1 | Corneal | Wang et al. (2016) | |||||
| Tlr4 −/− | TLR4 | Cell-surface pattern recognition receptor | As WT | HSV-1 | Intraperitoneal | Kurt-Jones et al. (2004) | |||
| Tlr2 −/− | TLR2 | Cell-surface pattern recognition receptor | As WT | HSV-1 | Intranasal | Lima et al. (2010) | |||
| Resistant | HSV-1 | Intraperitoneal | Kurt-Jones et al. (2004) | ||||||
| Resistant | HSV-1 | Intracranial | Wang et al. (2012) | ||||||
| Tlr9 −/− | TLR9 | Endosomal pattern recognition receptor | Susceptible* | HSV-1 | Intranasal | Lima et al. (2010) | |||
| As WT | HSV-1 | Intracranial | Wang et al. (2012) | ||||||
| Tlr2 −/− Tlr9 −/− | TLR2/9 | See above | Susceptible | HSV-1 | Intranasal | Lima et al. (2010) | |||
| As WT | HSV-1 | Intracranial | Wang et al. (2012) | ||||||
| Unc93b1 3 d | UNC93B1 | TLR cascade adaptor protein | As WT | HSV-1 | Intracranial | Wang et al. (2012) | UNC93B1 | HSV-1 | 608204 |
| Myd88 −/− | MYD88 | TLR cascade adaptor protein | Susceptible | HSV-1 | Intranasal | Mansur et al. (2005) | MYD88 | Bacterial (pyogenes) | 602170 |
| Resistant | HSV-1 | Intravenous | Honda et al. (2005) | ||||||
| Mb21d1 −/− | cGAS | Cytosolic DNA sensor | Susceptible | HSV-1 | Corneal | Reinert et al. (2016) | |||
| Sting gt/ gt | STING | Cytosolic DNA sensor | Susceptible | HSV-1 | Corneal | Reinert et al. (2016) | |||
| Sting −/− | STING | IFN I-dependent autophagy | As WT | HSV-1 | Corneal | Parker et al. (2015) | |||
| Susceptible | HSV-1 | Intravenous/intracranial | Parker et al. (2015) | ||||||
| Trim14 −/− | TRIM14 | Modifier of cGAS | Susceptible* | HSV-1 | Intravenous | Chen et al. (2016) | |||
| Usp13 −/− | USP13 | Modifier of STING | Resistant | HSV-1 | Intravenous | Sun et al. (2017a) | |||
| Usp21 fl/ fl Lyz2-cre | USP21 | Modifier of STING | Resistant | HSV-1 | Intravenous | Chen et al. (2017) | |||
| Rhbdf2 −/− | iRhom2 | STING signaling cascade | Susceptible | HSV-1 | Intravenous | Luo et al. (2016) | |||
| Mavs −/− | IPS-1 (MAVS) | RIG-I/MDA5 cytosolic RNA sensing pathway | As WT | HSV-1 | Intranasal | Menasria et al. (2013) | |||
| Irf3 −/− | IRF3 | IFN signaling transcription factor | Susceptible* | HSV-1 | Corneal | Murphy et al. (2013) | IRF3 | HSV-1 | 616532 |
| As WT | HSV-1 | Intravenous | Honda et al. (2005) | ||||||
| Irf7 −/− | IRF7 | IFN signaling transcription factor | Susceptible | HSV-1 | Corneal | Murphy et al. (2013) | IRF7 | Severe influenza disease | 605047 |
| Susceptible | HSV-1 | Intravenous | Honda et al. (2005) | ||||||
| Irf3 −/− Irf7 −/− | IRF3/7 | See above | Susceptible | HSV-1 | Corneal | Murphy et al. (2013) | |||
| Rnf128 −/− | RNF128 | Modifier of TBK1 | Susceptible | HSV-1 | Intravenous | Song et al. (2016) | |||
| Hcfc2 fls/ fls | HCFC2 | Facilitates IRF1/IRF2 binding to Tlr3 promoter | Susceptible | HSV-1 | Retro-orbital | Sun et al. (2017b) | |||
| Stat1 −/− | STAT1 | IFN signaling transcription factor | Susceptible | HSV-1 | Corneal | Katzenell et al. (2014) | STAT1 | Mycobacteria | 614892 |
| HSV-1, EBV, VZV | 613796 | ||||||||
| Candidiasis | 614162 | ||||||||
| Isg15 −/− | ISG15 | Interferon-stimulated gene | Susceptible* | HSV-1 | Intracranial/ corneal | Lenschow et al. (2007) | ISG15 | Mycobacteria | 147571 |
| Oasl1 −/− | OASL1 | Interferon-stimulated gene | Resistant | HSV-2 | Intravaginal | Oh et al. (2016) | |||
| Trp53 −/− | p53 | Regulator of cellular stress | Resistant | HSV-1 | Intracranial | Maruzuru et al. (2016) |
*Incomplete penetrance of survival phenotype. These gene-deficient animals, although more susceptible than WT controls, do not all succumb to HSV infection
†Respectively, “resistant” and “susceptible” denote reduced or increased survival to HSV infection, as compared to WT control mice. “As WT” describes gene-deficient animals that are equally susceptible or resistant to HSV infection as WT controls
§Reference to human gene deficiencies available on the Online Mendelian Inheritance in Man (OMIM) database (http://www.omim.org)
Table 2.
Gene deficiencies affecting cell-mediated responses to HSV encephalitis in mice, and to other infections in humans
| Targeted allele(s) | Protein | Function | Survival phenotype† | Virus | Infection route | References | Human genes | Infectious agent or disease | OMIM number§ |
|---|---|---|---|---|---|---|---|---|---|
| Ifnar −/− Ifngr −/− | IFN-αβγR | IFN I and IFN II receptors | Susceptible | HSV-1 | Corneal | Parker et al. (2016) | |||
| Irf9 −/− | IRF9 | IFN signaling transcription factor | Susceptible | HSV-2 | Intravaginal | Lee et al. (2017) | |||
| Il15 −/− | IL-15 | T and NK cell proliferation | Susceptible* | HSV-2 | Intravaginal | Ashkar and Rosenthal (2003) | |||
| Rag1 −/− | RAG-1 | B and T cell development | Susceptible | HSV-1 | Intranasal/cutaneous | Milora et al. (2017), Zolini et al. (2014) | RAG1 | T- B- NK + deficiency, HCMV | 609889 |
| Rag2 −/− | RAG-2 | B and T cell development | Susceptible | HSV-1 | Corneal | Ramakrishna and Cantin (2018) | RAG2 | T- B- NK + deficiency, respiratory infections | 233650 |
| Rag2 −/− Il2rg −/− | RAG-2, γc (CD132) | B, T, and NK cell development | Susceptible | HSV-2 | Intravaginal | Ashkar and Rosenthal (2003) | IL2RG | T- B + NK- deficiency, EBV | 308380 |
| Ighm −/− | Ig heavy chain μ | Component of B cell receptor | Susceptible | HSV-1 | Intraperitoneal | Beland et al. (1999) | IGHM | B- deficiency, severe bacterial infections | 147020 |
| B2m −/− | β2M | Component of MHC class I receptor | Susceptible | HSV-1, HSV-2 | Cutaneous | Holterman et al. (1999), Manickan and Rouse (1995) | B2M | CD8+ T- deficiency, impaired T/NK cytotoxicity, sinopulmonary infections | 109700 |
| Cd4 −/− | CD4 | CD4+ T cell development | Susceptible | HSV-1, HSV-2 | Cutaneous | Manickan and Rouse (1995) | |||
| Cd8 −/− | CD8 | CD8+ T cell development | Susceptible | HSV-1 | Intranasal | Zolini et al. (2014) | CD8 | CD8+ T deficiency, recurrent bacterial (H. influenzae) infections | 186910 |
| Ptprc L3 X | CD45 | Lymphoid cell development | Susceptible | HSV-1 | Intraperitoneal | Caignard et al. (2013) | PTPRC | T- NK- deficiency | 151460 |
| Lta −/− | LTα | TNF family cytokine | Susceptible | HSV-1 | Intramuscular | Kumaraguru et al. (2001) | |||
| Ifngr −/− | IFNγR | IFN II receptor | Susceptible* | HSV-1 | Corneal | Cantin et al. (1999) | IFNGR1 | Mycobacteria and Salmonella | 107470 |
| IFNGR2 | Mycobacteria and Salmonella | 147569 | |||||||
| Ifng −/− | IFNγ | Inflammatory cytokine | As WT | HSV-1 | Corneal | Cantin et al. (1999) | |||
| Susceptible* | HSV-1 | Corneal | Ramakrishna and Cantin (2018) | ||||||
| Susceptible* | HSV-1 | Intranasal | Mansur et al. (2005) | ||||||
| Susceptible* | HSV-2 | Intravaginal | Ashkar and Rosenthal (2003) | ||||||
| Socs2 −/− | SOCS2 | Suppressor of cytokine signaling | Resistant | HSV-1 | Intracranial | da Cunha Sousa et al. (2016) | |||
| Il10 −/− | IL-10 | Inflammatory cytokine | Susceptible* | HSV-1 | Corneal | Ramakrishna and Cantin (2018) | |||
| Il6 −/− | IL-6 | Inflammatory cytokine | Susceptible* | HSV-1 | Corneal | LeBlanc et al. (1999) | |||
| Tnf −/− | TNFα | Inflammatory cytokine | Susceptible* | HSV-1 | Intranasal/ corneal |
Lundberg et al. (2007), Sergerie et al. (2007) | |||
| Il1b −/− | IL-1β | Inflammatory cytokine | Susceptible | HSV-1 | Intranasal | Sergerie et al. (2007) | |||
| Tnf −/− Il1b −/− | TNFα IL-1β |
See above | Susceptible | HSV-1 | Intranasal | Sergerie et al. (2007) | |||
| Il36b −/− | IL-36β | Inflammatory cytokine | Susceptible* | HSV-1 | Cutaneous | Milora et al. (2017) | |||
| Tnfrsf1a −/− | p55 | TNF receptor subunit | As WT | HSV-1 | Corneal | Lundberg et al. (2007) | |||
| Susceptible* | HSV-1 | Corneal | Mohankrishnan et al. (2017) | ||||||
| Tnfrsf1a −/− Tnfrsf1b −/− | p55/p75 | TNF receptor subunits | As WT | HSV-1 | Corneal | Lundberg et al. (2007) | |||
| Nos2 −/− | iNOS | Synthesis of nitric oxide | Susceptible | HSV-1 | Intranasal | Zolini et al. (2014) | |||
| Ptafr −/− | PAFR | Platelet activating factor receptor | Resistant | HSV-1 | Intracranial | Vilela et al. (2016) | |||
| Cxcl10 −/− | CXCL10 | Chemokine | Susceptible* | HSV-1 | Corneal | Wuest and Carr (2008) | |||
| Cxcr3 −/− | CXCR3 | Chemokine receptor | Resistant | HSV-1 | Corneal | Wickham et al. (2005) | |||
| Ccr5 −/− | CCR5 | Chemokine receptor | As WT | HSV-1 | Intracranial | Vilela et al. (2013) |
CCR5
(32-BP DEL) |
Resistance to HIV | 601373 |
| Resistant | HSV-1 | Corneal | Carr et al. (2006) | ||||||
| Cx3cr1 −/− | CX3CR1 | Chemokine receptor | Susceptible* | HSV-1 | Intranasal | Menasria et al. (2017) |
*Incomplete penetrance of survival phenotype. These gene-deficient animals, although more susceptible than WT controls, do not all succumb to HSV infection
†Respectively, “resistant” and “susceptible” denote reduced or increased survival to HSV infection, as compared to WT control mice. “As WT” describes gene-deficient animals that are equally susceptible or resistant to HSV infection as WT controls
§Reference to human gene deficiencies available on the Online Mendelian Inheritance in Man (OMIM) database (http://www.omim.org)
Mouse experimental models of HSV infection and HSE
HSV infections can be modeled experimentally in mice. Although humans are the principal natural reservoir for HSV, mice share several cell-surface receptors that allow for both systemic and neurotropic infection with human herpes viruses. These receptors include Nectin-1 (Pvrl1) and herpes virus entry mediator HVEM (Tnfrsf14), both expressed on epithelial keratinocytes and fibroblasts (Petermann et al. 2015a, b). In addition, nectin-1 expression on neurons can facilitate the entry of HSV; intracranially infected Pvrl1−/− mice do not develop encephalitis, and lytic viral replication in severely limited in the CNS compared to WT mice (Haarr et al. 2001; Kopp et al. 2009). While the establishment of latent HSV infection in neurons of the trigeminal ganglia (TG) is well documented in mice, the spontaneous reactivation of latent virus is difficult to measure reliably in immunocompetent hosts, but can be triggered in vivo under stress, or ex vivo in explanted TG neuron cultures (Doll and Sawtell 2017; Matundan et al. 2016; Ramakrishna et al. 2015).
Different routes of infection recapitulate different aspects of HSV pathogenesis, with some routes better suited for investigating the peripheral host response. Cutaneous inoculation of the flank, footpad, or of the oral mucosa will often limit pathology and viral replication to the site of inoculation, and involves the early recruitment of neutrophils, NK cells, and antigen-presenting cells that will migrate to secondary lymphoid organs and prime the adaptive immune response (Milora et al. 2017). More commonly, intraperitoneal or intravenous inoculations are used to model systemic HSV infection, where viral replication may occur notably in the liver (Caignard et al. 2013; Chen et al. 2016; Parker et al. 2016). Intraperitoneal and intravenous models can also result in viral invasion of the CNS and lethal encephalitis, although many studies report lethality rather than HSV titers in brain tissue or infiltration of immune cells.
Other sites of delivery better approximate the natural course of primary infection observed in human patients. Intravaginal or intranasal inoculation, or intraocular delivery by corneal scarification, can each establish productive infection in the vaginal, nasal, and corneal epithelia, respectively (Menasria et al. 2017; Reinert et al. 2012, 2016). Through the contact of the nasal epithelium and eye with the sensory termini of TG neurons, HSV virions travel by retrograde axonal transport along the TG to invade the CNS (Cook and Stevens 1973). From here, the virus is typically localized to hindbrain (brainstem, cerebellum), and can be detected in the brain ependyma and lateral ventricles (Conrady et al. 2013; Kroll et al. 2014). However, mice rarely develop the temporal or frontal lobe-localized infected lesions that are characteristic of human childhood HSV encephalitis. The olfactory bulbs can also host lytic viral replication (Menasria et al. 2017), although the virus generally avoids them altogether in intranasal models (Shivkumar et al. 2013). Alternatively, intravaginal inoculation will lead to productive infection in the dorsal root ganglia, whereupon viral particles can reach the CNS via spinal cord neurons (Wang et al. 2013). While paracellular entry is also possible in most models—as pro-inflammatory cytokines and matrix metalloproteases weaken the tight junctions of the blood–brain barrier (BBB) (Sellner et al. 2006)—HSV cannot passively invade the CNS in the event of elevated systemic HSV replication (viremia), a common invasion mechanism in many kinds of arboviral encephalitis (Salimi et al. 2016). Finally, intracranial HSV inoculation is also frequently employed in mice, a route that bypasses retrograde axonal transport and often results in disseminated CNS viral replication (Wang et al. 2012).
Additional considerations for modeling HSE include the choice of HSV strain, which have different capacities for neurovirulence and CNS invasion. For HSV-1, highly neurovirulent strains including strain 17 and McKrae, and milder strains like KOS, are often used for experimental encephalitis in mice, whereas neurovirulent HSV-2 strains often result in meningitis (Bergstrom et al. 1990; Wang et al. 2013). Higher doses usually result in more severe pathology, but can be titrated in vivo to a level where susceptible controls succumb and resistant controls survive (Caignard et al. 2013). Other non-genetic factors include the age of mice at infection, where the severity and CNS viral invasion of HSV is greater in neonate mice than in adults (Wilcox et al. 2015).
As with human HSE, host genetics play an important role in mouse susceptibility to lethal encephalitis. Inbred strains of mice have shown differential susceptibility to HSV-1 infection, with C57BL/6 mice noted for their resistance compared to other strains such as A/J, BALB/c (Lopez 1975, 1980), or 129S6SvEv/Tac (Cantin et al. 1999), which are relatively susceptible to fatal infection. Forward genetic screens have been performed to identify new susceptibility loci or genes on resistant backgrounds (Caignard et al. 2013; Lundberg et al. 2003; Sun et al. 2017b), and significantly more reverse genetic studies have used single-gene knockout mice, or lymphopenic mice, to inform important mechanisms of host defense. Early targeted mutagenesis studies have made extensive use of embryonic stem cells obtained from 129S6SvEv/Tac mice, which as mentioned above are relatively susceptible to HSV-1 infection (Linder 2006). Hence to control for genetic background effects in these studies, the mutant allele has been serially back-crossed to HSV-resistant C57BL/6 to generate congenic mice. However, flanking gene differences between experimental animals, and between them and their controls, can present itself as variations in the degree of susceptibility (penetrance) of the phenotype. In establishing a hierarchy in the impact of the mutations, or for comparison between different laboratories, caution must be exerted in the interpretation of the results. To better guide the reader, we have thus indicated cases in which incomplete penetrance has been observed. Here, we will focus on groups of genes that together implicate cell-intrinsic and cell-mediated mechanisms, required in the periphery, in infiltrating hematopoietic immune cells, and in resident neural cells to assure a protective response to HSV-1 infection. Ultimately, these mouse models have helped to inform the genetics of HSE pathogenesis even beyond the TLR3/IFN I axis implicated in childhood human HSE.
Cell-intrinsic IFN I responses to HSV in mice
IFN I cytokines in mice consist of 13 isoforms for IFNα and one for IFNβ, and together ensure the control and restriction of viral replication in various cell types (Pestka et al. 2004). For DNA viruses like HSV, IFN I signaling can be initiated downstream of endosomal TLRs upon their recognition of foreign viral nucleotides (TLR3 recognizing double-stranded RNA, TLR8/7 recognizing single-stranded RNA, and TLR9 recognizing unmethylated cytosine-phosphate-guanine, or CpG, DNA). Furthermore, cytosolic viral DNA will be sensed by the cyclic GMP-AMP synthase/stimulator or interferon genes (cGAS/STING) pathway. On the other hand, RIG-I/MDA5-mediated recognition of cytosolic double-stranded RNA appears to have a more limited role in anti-HSV immunity (Liu et al. 2016; Menasria et al. 2013). IFNγ-inducible protein 16 (IFI16) is also a major viral DNA sensor in humans, with some evidence suggesting that IFI200-family genes (including Ifi204 and Ifi205) respond to IFN I or infection to fulfill a homologous role in mice (Ghosh et al. 2017; Hertel et al. 1999). The above signaling cascades converge notably on NF-κB and interferon regulatory factor (IRF) family transcription factors to modulate IFN I expression and other inflammatory cytokines. Upon binding to the IFN I receptor (IFNAR, including IFNAR1 and IFNAR2 subunits), IFN I stimulates the transcription of various interferon-stimulated genes (ISGs) that help to maintain a global antiviral program in the cell. Thus, Ifnar−/− mice are susceptible to lethal intracranial HSV-1 (Wang et al. 2012) and to intravaginal HSV-2 (Lee et al. 2017; Reinert et al. 2012). Mouse models that are deficient for discrete factors involved in IFN I signaling have further defined the role of TLR and cGAS/STING viral nucleotide sensing in the development of HSE (Table 1; Fig. 1).
Fig. 1.
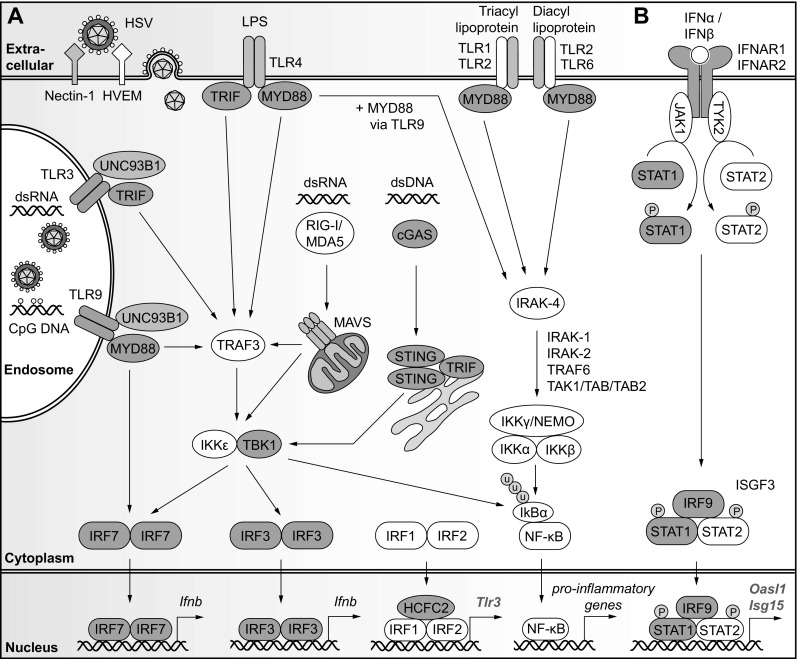
Selected genetic factors and pathways essential to the cell-intrinsic response to HSV encephalitis in mice. a Following infection and the entry of HSV via endosomes, or of HSV nucleocapsids into the cytoplasm, viral nucleic acids and other pathogen-associated molecular patterns are recognized by endosomal Toll-like receptors (including TLR3 and TLR9), TLR at the cell surface (including TLR1/2, TLR2/6 and TLR4), and by cytoplasmic DNA sensors cGAS/STING and RNA sensors RIG-I/MDA5. These pattern recognition receptors initiate signaling through factors including MYD88, TRIF, and TBK1 to promote the transcription of type I interferon (IFN I) and pro-inflammatory genes via transcription factors IFR3, IRF7, and NF-κB. b The subsequent production of IFNα and IFNβ and engagement of the type I interferon receptor (IFNAR1/2) initiates JAK/STAT signaling to promote the expression of antiviral interferon-stimulated genes (ISG) that establish a protective antiviral state in the infected cell. For each reported gene defect in mice, the encoded protein is color-coded above as follows: (1) red for gene defects that lead to HSV susceptibility in at least one mouse model, (2) blue for gene-deficient mice with equal or increased resistant compared to WT control mice, and (3) white for genes that have not been tested in mice. Further gene defects identified in mice that drive susceptibility or resistance to HSV infection including the cGAS modifier TRIM14, STING cascade modifiers USP13, USP21, and iRhom2, and TBK1 modifier RNF128 are detailed in Table 1. (Color figure online)
TLR signaling
Most genetic aberrations that drive HSE in children fit into the TLR3/IFN I axis, yet mice have broadened our understanding of innate viral sensing in the HSV-infected CNS to include additional TLRs. In glial cell cultures, murine astrocytes can upregulate TLR2, TLR3, and TLR4 after interacting with activated microglia, which themselves express most TLRs at steady-state and upon activation (Holm et al. 2012; Rosenberger et al. 2014). Following intravaginal HSV-2 infection, Tlr3−/− mice are highly susceptible to HSE, and show increased leukocyte infiltration, viral load, and infection of astrocytes and neurons in the CNS (Reinert et al. 2012). However, while global IFN I production was unaffected in the Tlr3−/− CNS, astrocytes from these animals failed to produce IFNβ in response to HSV-2 and were thus more readily infected ex vivo. Further, TRIF (Ticam1−/−) null mice, lacking the TLR3-specific intracellular adaptor TRIF, are partially susceptible to HSE, where decreased IFNβ production at day 5 post intranasal HSV-1 infection was associated with 60% of mice developing lethal encephalitis by day 8 (Menasria et al. 2013). Thus, the murine TLR3 cascade may contribute to protective CNS IFN I production, as observed in many human childhood HSE patients.
Other TLRs, including those that depend on downstream intracellular adaptor protein MYD88, are required for protective responses to HSV. Upon intranasal HSV-1 challenge, Tlr2−/− mice are resistant to HSE, and Tlr9−/− or double Trl2−/−Tlr9−/− knockout mice are partially and fully susceptible, respectively (Lima et al. 2010; Mansur et al. 2005). In this model, lesions and cellular infiltrate occur in the brain of HSE-susceptible mice. Yet, Tlr2 and Tlr9 gene expression is mostly upregulated in the TG after infection in resistant WT mice (Zolini et al. 2014), suggesting that the TG is a crucial checkpoint for viral recognition and control. Lower levels of CCL2 have also been reported in HSE-resistant Tlr2−/− mice, suggesting that the receptor may also play an auxiliary role in HSE pathology (Kurt-Jones et al. 2004). However, using a more acute intracranial HSV-1 infection model to bypass the TG altogether, Tlr9−/−, Trl2−/−Tlr9−/− and adaptor Unc93b1-null mutant mice are only as susceptible as C57BL/6 mice, and each with normal IFNβ expression in the brain (Wang et al. 2012); here Tlr2−/− mice are significantly more resistant than WT. Similarly, Myd88−/− mice are partially susceptible following intranasal inoculation (Mansur et al. 2005), but fully resistant to systemic HSV-1 infection (Honda et al. 2005). Overall, proper viral sensing via these MYD88-dependent TLRs is required in the TG to mount a protective innate response to CNS invasion of HSV, but may be dispensable when the virus is introduced directly in the brain parenchyma or in the periphery.
cGAS/STING signaling
Recent studies in mice have uncovered an important role for cytosolic viral DNA sensing in innate immunity to HSE. In infected mammalian cells, as HSV capsids are ubiquitinated and targeted to the proteasome for degradation, HSV genomic DNA may become available in the cytosol to bind cyclic GMP-AMP synthase (cGAS) (Horan et al. 2013). Newly produced cyclic GMP-AMP molecules can interact with and activate stimulator of interferon genes (STING), notably leading to downstream IFN I production. Accordingly, mice lacking either cGAS or STING are highly susceptible to HSE, and are characterized by elevated viral titers in the TG, brainstem, and further dissemination into the whole brain (Reinert et al. 2016). In particular, STING is required by infected microglia cells to produce IFN I and limit viral replication (Reinert et al. 2016), but has also been implicated in autophagy pathway-mediated viral clearance following systemic and intracranial HSV-1 infection (Parker et al. 2015).
Furthermore, modifiers of cGAS and STING function contribute to HSV-1 susceptibility in single-gene or tissue-specific knockout models. First, the stability and activation of cGAS is required to initiate effective antiviral responses. IFN I-induced expression of TRIM14 favors the recruitment of USP14, a deubiquitinase which in turn promotes further IFN I expression by modifying and protecting cGAS from autophagy-mediated degradation; thus, Trim14−/− mice are more susceptible to infection (Chen et al. 2016). Second, deubiquitination of STING on different residues can improve or exacerbate pathology. Both Usp13−/− mice (Sun et al. 2017a) and myeloid cell compartment-specific Usp21fl/fl null mice (Chen et al. 2017) are more resistant to HSV-1 infection due to heightened IFN I signaling occurring in the absence of these STING deubiquitinases. Yet, STING can also be stabilized by the site-specific removal of ubiquitin chains. As reported in Rhbdf2−/− mice, the failed iRhom2-dependent recruitment of deubiquitinase EIFS35 and adaptor TRAFβ to the STING complex, factors that normally stabilize STING and promote its transport to the endoplasmic reticulum, results in lower serum IFN I and increased HSV-1 susceptibility (Luo et al. 2016). Finally, it has been suggested that mice lacking TRIF, previously noted for their TLR3-dependent susceptibility to intranasal HSV-1 infection (Menasria et al. 2013), may otherwise produce lower IFN I due to a deficiency in STING signaling in a susceptible model of corneal infection (Wang et al. 2016). Here, an interaction with TRIF supports the activation and dimerization of STING, lowering viral replication in ex vivo infected primary Ticam1−/− cells. Thus, TRIF is an example of a factor that bridges multiple innate sensing pathways.
Transcription factors and IFN-stimulated genes
Viral molecular pattern sensors signal through downstream transcription factors that drive the expression of IFN I and the subsequent expression of ISGs. Among these transcription factors, the nuclear translocation of active IRF3 dimers leads to early IFNβ and IRF7 expression. In turn, active IRF7 promotes elevated IFNα, IFNβ, and ISG expression. Without both transcription factors, Irf3−/−Irf7−/− mice cannot mount an antiviral response and succumb fully to corneal HSV-1 infection (Murphy et al. 2013). In this model, Irf7 contributes most to the protective response, while Irf3−/− mice are partially susceptible to infection. Furthermore, IRF3 phosphorylation and downstream IFN production are dependent on TBK1 kinase activity; positive regulation of TBK1 by E3 ubiquitin ligase RNF128 confers some protection to systemic HSV-1 infection in Rnf128-sufficient mice (Song et al. 2016). Additionally, other IRF-family members including IRF1 and IRF2 are known to modulate the transcription of Tlr3. In this context, mice carrying a chemically induced null mutation in Hcfc2, a factor that facilitates the interaction of IRF1 and IRF2 with the Tlr3 promoter, show defects in TLR3-dependent IFN production and are susceptible to retro-orbital HSV-1 infection (Sun et al. 2017b).
Signal transducer and activator of transcription 1 (STAT1) is another key transcription factor in the response downstream of IFNAR signaling. Two Stat1 knockout mouse models are commonly used, one with an N-terminal deletion (Stat1−/− ΔNTD) that disrupts active complexes of phosphorylated STAT1 with phosphorylated STAT2 (Meraz et al. 1996), and another with a deletion in the DNA-binding domain (Stat1−/− ΔDBD) (Durbin et al. 1996). Although the DNA-binding domain plays a more prominent role in the mounting of an adequate response to IFNβ and control of ex vivo infection, both Stat1−/− models are highly sensitive to corneal HSV-1 infection and succumb by day 8 post-infection (Katzenell et al. 2014). Yet, HSV-1-infected Stat1−/− ΔDBD and ΔNTD mice distinguish themselves by their respective systemic or CNS viral tropisms (Pasieka et al. 2011b). Upon ocular infection of Stat1−/− ΔNTD mice that succumb to CNS-localized infection, active STAT3 and IL-6 drive inflammation in the corneal epithelium (Pasieka et al. 2009) before a heightened antiviral and pro-inflammatory response develops in the brainstem (Pasieka et al. 2011a). In these brainstems collected at the day 5 peak of HSV-1 replication, functional gene expression analysis revealed that IFN-I-dependent ISGs and IRF-family genes are upregulated in resistant 129S-background control animals; susceptible Stat1−/− mice show increased expression of inflammatory chemokines (Cxcl10, Ccl2), cytokines (Ifng, Il6, Il1b), markers of cell infiltration (Icam1, Il8rb), and matrix metalloproteases (Mmp3, Mmp8, Mmp9), all typical of acute HSE pathogenesis (Pasieka et al. 2011a). Thus, STAT1-dependent IFN I signaling is pivotal for the initiation of a protective innate antiviral response to HSV-1 in the mouse CNS.
Certain ISGs have also been found to contribute to a protective antiviral response to HSV infection in mice. Notably, the lack of ISG15—an IFN I-induced ubiquitin-like factor that modifies several protein targets that help to establish an antiviral state in infected cells—renders mice more sensitive to lethal HSV-1 following intracranial and intraocular infections (Lenschow et al. 2007). On the other hand, negative regulators of IFN I, including IRF7 antagonist OASL1, can exacerbate viral pathogenesis; Oasl1−/− mice are partially protected from lethal HSV-2 infection (Oh et al. 2016). Thus, genetic defects that target IFN I-responsive proteins, as well as components of viral pattern recognition pathways and their downstream transcription factor-dependent signaling, modify the innate capacity of mouse cells to reduce HSV-1 viral replication, and to ultimately clear the infection in vivo.
Cell-mediated responses to HSV infection in mice
In the context of neurotropic viral infection, immune cell populations contribute both to inflammation and to the recognition and targeted killing of infected cells. Spurred on by the production of antiviral factors, the protective cellular response to HSV-1 in mice progresses through three general phases. First, in the periphery, natural killer cells (NK), myeloid cells, and various dendritic cell (DC) subsets will produce IFN I, IFNγ, and other pro-inflammatory cytokines necessary for the further activation and proliferation of NK cells (Kassim et al. 2006; Swiecki et al. 2013). Antigen-presenting cells including DCs will migrate to the secondary lymphoid organs to expose B cells, CD4+ and CD8+ T cells to viral antigen. Next, these activated cytokine-producing lymphocytes and myeloid cells can invade the CNS by the lymphatics network or directly across an inflammation-weakened BBB. Ultimately, alongside activated glial cells, the CNS infiltration of immune cell can either benefit the host by eliminating infected cells and reducing viral titers, or drive overwhelming inflammation characteristic of HSE pathology. Mouse models have been especially useful to dissect the role of discrete genes in these three aspects of cell-mediated HSV-1 immunity (Table 2; Fig. 2).
Fig. 2.
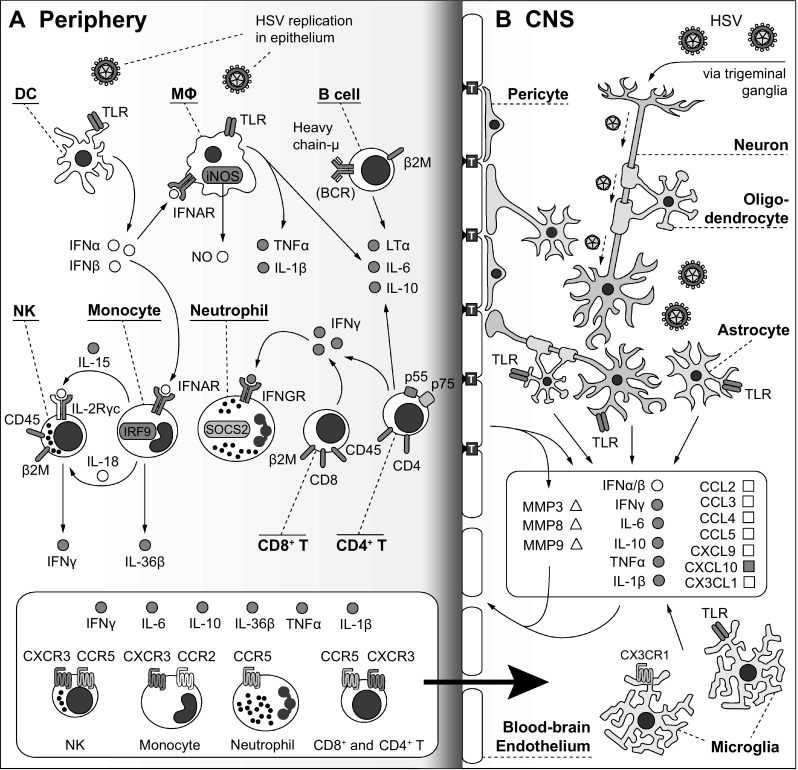
Selected genetic factors and pathways essential to the cell-mediated immune response to HSV encephalitis in mice. a HSV will first infect and replicate in epithelial cells and keratinocytes, activating chemokine pathways and triggering dendritic cells (DC) to produce type I interferon (IFN I). Responding to IFN I, macrophages (MΦ) and monocytes will produce inflammatory cytokines (TNFα, IL-1β), and contribute to the activation of cytotoxic IFNγ-producing natural killer cells (NK). Furthermore, activated B cells, and especially effector CD4+ T cells and cytotoxic CD8+ T cells, will help to maintain an adequate antiviral response including T cell-dependent production of cytokines like IFNγ, which will promote neutrophil expansion. While single-gene defects in factors including CD45, β2M, Heavy chain-μ, LTα, CD4, CD8, RAG-1, RAG-2, and IL-2Rγc have all been implicated in susceptibility to HSV encephalitis in mice, it remains unclear whether their essential function in immune cell development, or any specific effector functions, strictly drives susceptibility to infection. b In the CNS, the principal route of entry of HSV is via the axons of the trigeminal ganglia. HSV readily infects neurons, as well as glia including oligodendrocytes, astrocytes, and microglia. The TLR-dependent recognition of HSV by glia and neurons may drive expression of cytokines including IFN I, IFNγ, IL-6, TNFα, and IL-1β. Along with matrix metalloproteases (MMP3, 8, 9), these CNS cytokines and those produced in the periphery may disrupt the blood–brain barrier (BBB) by weakening the tight junctions (marked above by “T”-labeled blue squares) between BBB endothelial cells. Finally, the expression of various chemokines by CNS-resident cells will attract immune cells with cognate chemokine receptors (CCR2/CCL2; CCR5/CCL3, CCL4, CCL5; CXCR3/CXCL9, CXCL10; CX3CR1/CX3CL1) across the permeable BBB into the CNS (marked by the black arrow between panels a and b). In the CNS, these infiltrating immune cells may enhance virus clearance, but also contribute to HSE pathology. The pathways shown above have been directly involved in mouse HSE studies, but do not reflect the full production of factors by all immune, neuronal, and glial cells, or their complete downstream effects. For each reported gene defect in mice, the encoded protein is color-coded above as follows: (1) red for gene defects that lead to HSV susceptibility in at least one mouse model, (2) blue for gene-deficient mice with equal or increased resistant compared to WT control mice, and (3) white for genes that have not been tested in mice. (Color figure online)
Peripheral lymphoid and myeloid cell responses
Innate IFN production plays a fundamental role in the activation of the cellular immune response to early HSV-1 replication. Infected Ifnar−/−Ifngr−/− mice, lacking both type I and II IFN receptors, exhibit high viral loads in the liver due to a failure of the IFN signaling-deficient hematopoietic compartment to control the infection (Parker et al. 2016). For example, HSV-2 replication at the vaginal mucosa is controlled by the IFN I-dependent recruitment of cytotoxic Granzyme B+/IFNγ+ NK cells and CCL2-producing Ly6CHI inflammatory monocytes; Ifnar−/− mice recruit only CXCL1/CXCL2-producing Ly6CLOW monocytes that instead promote neutrophil invasion, and fail to initiate a protective antiviral response (Uyangaa et al. 2015). In a similar model, Ifnar−/− and Irf9−/− mice are both more sensitive to lethal HSV-2 infection, where few IL-18-producing inflammatory monocytes are recruited to the vaginal mucosa, resulting in low IL-18-dependent production of IFNγ in NK cells (Lee et al. 2017). Lower levels of IFNγ are also implicated in an HSV-2-susceptible Il15−/− model (Ashkar and Rosenthal 2003). As for the role of cytotoxic NK cells in the infected CNS, a forward genetics approach identified a locus on murine chromosome 6 that may underlie resistance to HSE in C57BL/6 mice (Kastrukoff et al. 2015). Susceptible BALB/c background mice, or NK-depleted C57BL/6 mice, exhibited viral spread to the cerebellum and augmented titers in the brainstem. Thus, NK-specific genes in this resistance locus, including Cd94, the Ly49 cluster, and NKG2 cluster, may contribute to the protective antiviral response to HSE.
Classical adaptive immune cells are also important in the control of acute HSV infection and the development of HSE in mice. Both Rag−/− (lacking mature B and T cells) and Rag2−/−Il2rg−/− mice (lacking mature B, T, and NK cells) are fully susceptible to lethal intranasal HSV-1 or intravaginal HSV-2 infection, respectively (Ashkar and Rosenthal 2003; Zolini et al. 2014). Furthermore, B cell (Ighm−/−)- , MHC I (B2m−/−)- , and T cell (Cd4−/− and Cd8−/−)-deficient animals are all more susceptible than WT mice in different models of HSV infection and HSE (Beland et al. 1999; Holterman et al. 1999; Manickan and Rouse 1995; Zolini et al. 2014). As with other viral infections, effector CD4+ T cells and cytotoxic CD8+ T cells play a major role in the elimination of replicating virus. A loss-of-function mutation in CD45 (PtprcL3X), identified in a chemical mutagenesis screen for HSV-1 susceptibility in mice, results in a lack of T and NK cells that drives susceptibility to HSE (Caignard et al. 2013). Here, CD8+ T cells, supported by IFNγ-producing CD4+ T effector cells, are required for effective viral clearance in the CNS. In response to infection, CD4+ T cell-specific Stat3−/− mice have increased HSV-1-specific CD8+ T cells that express lower levels of KLRG-1 and IFNγ, suggesting that antiviral CD8+ T cell function relies in part on their cooperation with STAT3-competent CD4+ cells (Yu et al. 2013). In addition, CD8+ T cell expression of Dok-1 and Dok-2 proteins, that each function in proliferation, activation, and migration of hematopoietic cells, has been shown to support and amplify HSV-1-specific CD8+ effector responses; the absence of Dok1 and Dok2 does not, however, impact viral titers in the cornea or TG during primary infection (Lahmidi et al. 2017). Other CD8+ T cell defects have also been found to increase susceptibility, notably in lymphotoxin-α-deficient (Lta−/−) mice that, without adequate cytotoxic IFNγ+CD8+ T cell responses, succumbed to lethal intramuscular HSV-1 infection (Kumaraguru et al. 2001).
Pro-inflammatory cytokines and immune cell invasion
The contribution of the immune system to HSE resistance is further supported by findings that single cytokine gene-knockout models are highly susceptible to infection. While the elevated and concerted expression of pro-inflammatory cytokines is a hallmark of pathological inflammation, individual cytokines are essential and non-redundant in a protective response. At high doses of corneal HSV-1, IFNγ receptor-null (Ifngr−/−) mice are more susceptible to HSE than both Ifng−/− and WT mice (Cantin et al. 1999). Yet, Ifng−/− mice are partially susceptible to HSV-1 and HSV-2 infection (Ashkar and Rosenthal 2003; Mansur et al. 2005), and approximately 75% of mice succumb to lower doses of corneal HSV-1 between days 10 to 16 post-infection, despite controlling virus as well as resistant WT mice by day 8 (Ramakrishna and Cantin 2018). This may suggest that IFNγ plays a role in limiting cell infiltration and inflammation, rather than as a direct antiviral mechanism (Ramakrishna and Cantin 2018). Yet in the context of HSV-1 infection, the proliferative and cytotoxic capacities of NK cells and of effector CD4+ and CD8+ T cells also rely on IFNγ expression to clear HSV-1-infected cells and reduce viral load in the CNS (Caignard et al. 2013).
Furthermore, susceptible Ifng−/− mice express elevated levels of granulocyte colony stimulating factor (G-CSF), promoting the excessive CNS recruitment of degranulating neutrophils (Ramakrishna and Cantin 2018). G-CSF also induces the expression of suppressor of cytokine signaling 2 (SOCS2). Validating this pathway, either depletion of G-CSF in Ifng−/− mice (Ramakrishna and Cantin 2018) or ablation of SOCS2 (Socs2−/− mice) (da Cunha Sousa et al. 2016), lowered neutrophil infiltration and thus increased HSE resistance. As for the repressive function of IL-10, Il10−/− mice are highly susceptible to lethal HSE (Ramakrishna and Cantin 2018). IL-6, which is known to be overexpressed in the CNS at the peak of HSE, also confers protection to HSE in IL-6-competent mice compared to Il6−/− mice (LeBlanc et al. 1999).
Tumor necrosis factor (TNFα) is another example of a key pro-inflammatory cytokine that is involved in neuroinflammation, and yet also acts to control infection and prevent encephalitis. Tnf−/− mice are susceptible to HSE, where HSV-1 disseminates past the brainstem and cerebellum (Lundberg et al. 2007; Sergerie et al. 2007). Tnf−/− mice share a similar susceptibility profile with Il1b−/− and Tnf−/−Il1b−/− double-knockout mice, which lack pro-inflammatory IL-1β processed downstream of the inflammasome complex and capable of activating lymphocytes and inducing IFN I and II production (Sergerie et al. 2007). IL-1-family cytokine IL-36β (Il36b gene) is also involved in protective immunity to lethal HSE (Milora et al. 2017). As with circulating immune cells, TNFα is produced by resident microglia in the brain; its cognate receptors p55 (Tnfrsf1a) and p75 (Tnfrsf1b), expressed on infiltrating and resident immune cells, play a more complex role in HSV immunity. One study found that a 3200 plaque-forming units (PFU) HSV-1 corneal infection of Tnfrsf1a−/− mice resulted in self-resolving viral replication in the brainstem and TG. WT and Tnfrsf1a−/−Tnfrsf1b−/− double-knockout mice were also resistant to HSE (Lundberg et al. 2007). In another study, Tnfrsf1a−/− mice were only slightly more sensitive at a higher dose (105 PFU) of corneal HSV-1 than WT mice (Mohankrishnan et al. 2017). Together, these studies suggest that a protective response to HSE depends on TNFα and not its receptors, or otherwise partly relies on p55-mediated inflammation and cytokine production in certain models of acute disease. A final cytokine that contributes to resistance is inducible nitric oxide synthase (iNOS; Nos2 gene), produced chiefly by myeloid cells. iNOS-deficient mice exhibit poor virus control following footpad HSV-1 infection (MacLean et al. 1998), and 100% mortality to intranasal infection and an overall increase in pro-inflammatory factors TNFα, CCL2, Rantes and CXCL10 in the TG characteristic of HSE (Zolini et al. 2014).
While protective cellular responses depend on the above cytokines, their elevated expression can compromise the integrity of the BBB and allow cells to infiltrate and further damage the infected CNS. In mouse models of neurotropic infection, the infiltration of immune cells is enabled by TNFα and IL-6, and various matrix metalloproteases are known to reduce the expression of endothelial and tight junction proteins at the barrier (Ashley et al. 2017; Li et al. 2015; Rochfort et al. 2016). CNS lymphatics can also facilitate infiltration (Louveau et al. 2015). Following HSV infection, the further development of blood and lymph vessels has been studied in the context of the cornea. TNFα accelerates HSV-1-driven lymphangiogenesis in the corneal epithelium, as does IL-6 in the absence of TNFα (Bryant-Hudson et al. 2014). Here, viral ICP4 transcripts act as enhancers of vascular endothelial growth factor A (VEGF-A) expression to increases vascularization and HSV-1-specific antiviral CD8+ T cells in the cornea and TG (Gurung et al. 2017). The role of lymphangiogenesis is less clear in the brain of HSE-susceptible mice. However, both vascular permeability and lymphoid or myeloid cell CNS infiltration is reduced in platelet activating factor receptor-deficient (Ptafr−/−) mice, resulting in delayed mortality upon intracranial HSV-1 (Vilela et al. 2016).
CNS-infiltrating immune cells and microglial activation
In the infected CNS, once thought to be off-limits to the immune system, the activation of resident cells and the infiltration of immune cells can either restrict viral replication to limit inflammation, or contribute directly to lethal encephalitis. Trafficking lymphocytes can be detected as early as day 5 post-HSV-1 infection in the TG, where TLR2- and TLR9-dependent pathways drive the production of granzyme B and perforin by cytotoxic NK and CD8+ T cells, of iNOS by macrophages, and of IL-1β by conventional DCs (Lucinda et al. 2017). The infected TG are also noted for their high expression of CCL2 in susceptible Tlr2−/−Tlr9−/− mice, which leads to further recruitment of monocytes and T cells to the CNS. In addition, infiltrating neutrophils and F4/80+ macrophages are known to contribute directly to HSE pathology in susceptible 129 background mice (Lundberg et al. 2008).
CXC-motif chemokines secreted by resident glial cells, blood–brain endothelial cells, or hematopoietic cells help to recruit inflammatory cells that express cognate chemokine receptors. CXCL10 expression is sharply upregulated upon infection and, like fellow chemokine CXCL9, binds receptor CXCR3 expressed on monocytes, NK cells, and T cells to promote their homing to the site of infection. Thus, Cxcl10−/− mice are more sensitive to lethal HSE than WT mice, and fail to control viral replication in the brain stem due to low recruitment of NK and CD8+ T cells (Wuest and Carr 2008). CXCL10 is also implicated the protective response to HSV-1-related pathology in the cornea, and in the absence of CXCL10, CXCL9 may play a compensatory role to limit herpes keratitis (Tajfirouz et al. 2017). Moreover, bone marrow chimeric mouse models have been very useful to limit monogenic defects in chemokine signaling to either the hematopoietic or tissue-resident cell compartments. The HSE-protective effect of CXCL10 is dependent on bone-marrow-derived cells that home to the CNS, and not on radio-resistant microglia or stromal cells of the CNS (Wuest et al. 2011). For example, it has been proposed that CXCR3+ NK cells, for which CXCL10 is thought to be the principle attractant, may be important effectors of the antiviral response in Cxcl10-competent mice (Wuest and Carr 2008).
Although CXCL10 has a non-redundant protective role in the CNS, mice that lack its receptor CXCR3 (Cxcr3−/−) are resistant to HSV-1 infection, and slightly more resistant than WT C57BL/6 mice (Wickham et al. 2005; Zimmermann et al. 2017), suggesting that some CXCR3+ immune cells may be the cause of lethal inflammation. Consistent with their survival, Cxcr3−/− mice maintain control of viral replication in the brain ependyma and hippocampus by relying on intact IFNβ signaling (Kroll et al. 2014; Zimmermann et al. 2017). However, low cytokine (TNFα, IFNγ) and chemokine (CXCL9, CXCL10, CCL2) expression in resistant animals limits leukocyte infiltration (Zimmermann et al. 2017). CXCR3-deficiency also completely excludes activated CD8+ T cells from the CNS, cells that otherwise upregulate CXCR3 in WT hosts. Thus, the infiltration of CXCR3+ monocytes and T cells from the periphery drives HSE pathology in mice (Menasria et al. 2015; Zimmermann et al. 2017).
Of note, other models of defective chemokine receptor signaling show altered recruitment of immune cells to the CNS without necessarily affecting survival or outcome. For example, Ccr5−/− and WT mice are both completely susceptible to intracranial HSV-1, yet knockouts display increased neutrophil homing to the CNS (Vilela et al. 2013). Alternatively, Ccr5−/− mice feature higher numbers of T cells in the TG, but are only slightly more resistant to corneal HSV-1 infection compared to WT (Carr et al. 2006). CCR5 receptor is expressed on most immune cells, neurons, and glial cells, and engages many chemokines including CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES); the expressions of these and other chemokines are all increased in Ccr5−/− mice in both infection models, hinting at an intricate compensatory mechanism. Thus, while the role of individual chemokine signaling pathways in protective or detrimental host responses is complex and not fully understood, the excessive infiltration of cytotoxic and inflammatory cells to the CNS is most often auxiliary to HSE pathogenesis.
Finally, CNS-resident microglia can play an important role in the development of HSE, especially in the context of innate antiviral immunity. Abundant macrophage-like glial cells, microglia are activated in the HSV-1-infected brain and can produce a number of chemokines and cytokines including IFN I that can help control viral replication, or in the long-term promote HSE pathology (Conrady et al. 2013; Marques et al. 2008; Wuest and Carr 2008). Expression of CX3CR1 on microglia, which recognizes inhibitory CX3CL1 produced by healthy CNS cells, helps maintain a protective, anti-inflammatory environment in the brain. Therefore, Cx3cr1-deficiency in the radio-resistant compartment of chimeric mice is sufficient to render them susceptible to lethal intranasal HSV-1 infection (Menasria et al. 2017). Here, Cx3cr1−/− microglia fail to contain HSV-1 spread in the CNS, which recruits elevated numbers of neutrophils and inflammatory monocytes to the site of infection. Furthermore, the ablation of CD118 (IFNAR1) in radio-resistant microglia has been shown to drive HSV-1 viral load in the CNS, implicating TRIF-dependent production of IFN I by microglia in the protective response to HSE (Conrady et al. 2013). Ultimately, microglia are required for proper IFN I-mediated control of HSV-1 in infected neurons and glial cells of the CNS, and to avoid a general shift towards lytic cell death and neuroinflammation that underlie HSE.
Cell death and autophagy in mouse HSE pathology
Recent studies have also relied on mouse models and primary cells to investigate cell death pathways and their role in the pathology of the infected CNS. In the brain, homeostasis is maintained by patrolling microglia that detect pathogens and damage signals, while the more numerous astrocytes promote neuronal survival. In primary murine cultures, HSV-1 infects both these glial cell types, and both secrete pro-inflammatory factors and IFN I (Aravalli et al. 2005). Virus replication is controlled and self-limited in microglia that preferentially undergo early caspase 3- and TNF-dependent apoptosis; infected astrocytes yield higher viral titers and die at later time-points from Fas-dependent apoptosis (Aravalli et al. 2006).
In the context of the CNS, apoptotic cell death can function as a cell-intrinsic antiviral mechanism by depriving the virus of host cells within which to replicate, but can also be a consequence of infection that drives pathology and inflammation. For example, cells lacking X-box binding protein 1 (Xpb1−/−), a key player in the unfolded protein response that relieves stress at the endoplasmic reticulum, are unable to initiate apoptosis upon HSV-1 infection, resulting in more elevated viral titers (Fink et al. 2017). More generally, various cellular stress responses, including infection, can signal through p53 to promote cell death and apoptosis. Following in vitro HSV-1 infection, p53 can enhance or depress the expression of viral proteins ICP22 and ICP0, respectively, to modulate viral replication in a human epithelial cell line (Maruzuru et al. 2013). In vivo, p53-null (Trp53−/−) mice are significantly more resistant to intracranial HSV-1 infection than susceptible WT mice (Maruzuru et al. 2016). Further, the p53-deficient CNS exhibits no important cell death or HSE pathology, and reduced infiltration of immune cells, suggesting that virus is controlled independently of cell death; p53-dependent gene signatures are associated with HSE in susceptible Trp53+/+ control mice. Thus, apoptosis of infected glial cells, and likely including infected neurons, appears to be detrimental and central to HSE pathogenesis in mice.
Neurons are especially vulnerable to acute neurotropic viral infections. Damage caused by lytic neuronal cell death can have severe impact on the host. Consequently, neurons respond very differently from glial cells to IFN I signals, and aim to avoid IFN I-induced apoptosis. Upon infection in vivo, neuron-specific STAT1-deficiency (Stat1N−/−) leads to high viral titers in the TG and brain stem and the development of HSE (Rosato et al. 2016). Murine neurons also do not upregulate apoptosis in response to IFN unlike fibroblasts (Rosato et al. 2016; Yordy et al. 2012). Rather, STAT1-dependent IFNβ signaling only managed to restrict the replication of a mutant Δγ34.5 HSV-1 virus lacking viral protein ICP34.5 (Rosato and Leib 2014). While ICP34.5 expression is known to reduce the activity of host IFN I (Manivanh et al. 2017), these data also support a role for IFN I signaling in initiating antiviral mechanisms alternate to apoptosis, namely, IFN I-induced autophagy.
Viral ICP34.5 has been extensively studied in the context of its Beclin-binding domain, which can interact with Beclin 1 to prevent autophagy in infected neurons (Orvedahl et al. 2007). Thus, neurotropic HSV viruses are equipped to counter autophagy, a well-established recycling mechanism that is preferentially induced in neurons to reduce viral replication while avoiding pathological cell death and ensuring survival (Yakoub and Shukla 2015). Neurons lacking key autophagy factor ATG5 (Atg5−/−) yield higher viral titers in culture, following infection with both a Beclin-binding domain-deficient and functional HSV-1 virus, suggesting that while ICP34.5 certainly reduces autophagy, autophagy can also be initiated at early time-points before ICP34.5 is expressed to help reduce viral load (Yordy et al. 2012). Alternatively, Sting−/− mice are also susceptible to intracranial and corneal infection with Beclin-binding domain-deficient HSV-1 compared to resistant control mice, highlighting the role of STING-dependent autophagy in in vivo antiviral control (Parker et al. 2015). Finally, if not to induce apoptotic cell death or traditional antiviral responses, IFN I does induce the formation of unique autophagosome clusters in treated neurons that are absent in IFN signaling-deficient cells (Irf3−/−, Ifnar−/−Ifngr−/−, Stat1−/−) (Katzenell and Leib 2016). As autophagy is less impeded in infections with Beclin-binding domain-deficient HSV-1, the mutant virus also spreads differently in the BALB/c brain, where increased autophagy and NLRP3-dependent inflammasome activation correlated with a reduction in viral burden in the brain and lower HSE incidence (Zhang et al. 2013a). Thus, these data suggest that neurons can rely on the IFN I/autophagy axis to control viral replication without resorting to apoptotic cells death, but the efficient blocking of these pathways by viral proteins further complicates our understanding of HSE development in vivo.
Perspectives
Altogether, mouse models have refined our understanding of HSV infection and invasion of the CNS. Reverse genetic approaches have not only confirmed several childhood HSE-protective genes (Tlr3, Trif, Stat1), or revealed others belonging to overarching TLR pathways (Myd88, Irf3, Irf7, Tlr9), but have also implicated many innate and adaptive immunity genes that better define mechanisms underlying HSE. Further, genetic mouse models that are differentially susceptible, based on the route of viral entry to the CNS, highlight the importance of early antiviral control by different viral pattern recognition mechanisms at mucosal surfaces, in the TG or in the CNS proper. Accordingly, antiviral treatment with acyclovir is the first-line treatment for HSE in humans, and works in mice to improve survival (Long et al. 2011; Quenelle et al. 2018). IFNα is also capable of limiting HSV infection in human neuronal culture (Pourchet et al. 2017), and is sometimes used in combination therapy to treat other viral infection (Sagnelli et al. 2017). Treatment of mice with cyclic dinucleotide agonists of the cGAS/STING pathway protected animals from HSV-2 replication, while TLR agonist treatment in mice also limited HSE development (Boivin et al. 2008; Skouboe et al. 2018); topical TLR7 agonist imiquimod has been shown to be ineffective in human HSV-2-infected patients (Schacker et al. 2002). In mice under the Tlr3-independent IFN I pathway, drug-targetable genes might also include deubiquitinases (TRIM14, USP13, USP21, iRhom2) that modify factors in the cGAS/STING cascade to effect IFN I production and HSE outcome.
These mouse studies have also highlighted an important contribution of trafficking and infiltrating immune cells to HSE pathogenesis. For example, peripheral and CNS-invading NK cells are involved in several gene-knockout models (Irf9, Ifng, Ifngr, Il15, Cxcl10); the protective antiviral function of NK cells in murine HSE may warrant further study into using NK cells as a targeted antiviral therapy. In general, cytokines appear to be protective in mouse HSE, despite being involved in late-stage neuroinflammation. Chemokine dynamics play complex roles in limiting or promoting CNS invasion, or even activating resident glial cells. Understanding the balance between inflammation and cell death in the CNS might help to reduce the risk of long-term sequelae that may develop in many cases of acyclovir-treated encephalitis.
Furthermore, these aspects of mouse HSE might help inform mechanisms that underlie different types of viral encephalitis in humans, which are challenging to model appropriately in mice. While neonate and adult mice react differently to CNS infection, current mouse models do not capture the individual variation in human HSE pathologies, which may depend on the neurovirulence of the viral strain, on the primary site of infection, or on the reactivation of HSV from latency in sensory neurons. Other types of encephalitis, including autoimmune anti-NMDAR (N-methyl-D-aspartate receptor) encephalitis, have been reported in older adults that have recovered from HSE earlier in life, but later develop neuroinflammation due to anti-NMDAR antibodies in the cerebrospinal fluid that may stem for exposure to HSV antigens (Armangue et al. 2014; Omae et al. 2018). Viral escape from latency can also be triggered by certain drugs, most strikingly by natalizumab, approved for treatment of relapsing-remitting multiple sclerosis and Crohn’s disease, which has been reported to drive John Cunningham virus (JCV) reactivation from latency in the brain and lead to progressive multifocal leukoencephalopathy in some patients (Bloomgren et al. 2012).
Cases of HSE worldwide are usually reported at 2 to 4 per million individuals per year (Jorgensen et al. 2017). While HSV-1 and HSV-2 prevalence are decreasing in the United States (McQuillan et al. 2018), the ubiquity of these viruses has prompted many studies on the involvement of herpes viruses in complex CNS neurodegenerative or chronic inflammatory diseases. Most notably, HSV-1 infection has been linked to Alzheimer’s disease and to the expression of the apolipoprotein 4 (APOE-ε4) allele isoform (Itzhaki et al. 1997; Steel and Eslick 2015). Besides host genetics, HSV infection is known to increase as people age, and women are twice as likely to be infected with HSV-2; both age and gender are known to affect risk for certain neurodegenerative and chronic inflammatory diseases (McQuillan et al. 2018). Given these important genetic, environmental and pathogen factors, mouse models will continue be useful to dissect the cells and pathways that are implicated in HSV infections of the CNS. As novel techniques are applied to study human HSE in rare patients, including induced pluripotent stem cell differentiation of patient cells to CNS cells (Lafaille et al. 2012; Pourchet et al. 2017; Zhang et al. 2018), we expect that these essential genes identified in animal models and human patients will together help to define mechanisms of pathogenesis and viral control, and better clarify how a widespread and successful viral pathogen only seldom results in lethal encephalitis.
Acknowledgements
M.M. was supported by the Fonds de recherche santé - Québec (FRQS). S.M.V. was supported by the Canada Research Chairs Program and the Canadian Institutes of Health Research (CIHR).
Compliance with ethical standards
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Ahmed M, Lock M, Miller CG, Fraser NW. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J Virol. 2002;76:717–729. doi: 10.1128/JVI.76.2.717-729.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravalli RN, Hu S, Rowen TN, Palmquist JM, Lokensgard JR. Cutting edge: TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus. J Immunol (Baltimore, Md: 1950) 2005;175:4189–4193. doi: 10.4049/jimmunol.175.7.4189. [DOI] [PubMed] [Google Scholar]
- Aravalli RN, Hu S, Rowen TN, Gekker G, Lokensgard JR. Differential apoptotic signaling in primary glial cells infected with herpes simplex virus 1. J Neurovirol. 2006;12:501–510. doi: 10.1080/13550280601064921. [DOI] [PubMed] [Google Scholar]
- Armangue T, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol. 2014;75:317–323. doi: 10.1002/ana.24083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkar AA, Rosenthal KL. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J Virol. 2003;77:10168–10171. doi: 10.1128/JVI.77.18.10168-10171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley SL, et al. Matrix metalloproteinase activity in infections by an encephalitic virus, mouse adenovirus type 1. J Virol. 2017 doi: 10.1128/JVI.01412-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audry M, et al. NEMO is a key component of NF-kappaB- and IRF-3-dependent TLR3-mediated immunity to herpes simplex virus. J Allergy Clin Immunol. 2011;128:610–617, e611. doi: 10.1016/j.jaci.2011.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beland JL, Sobel RA, Adler H, Del-Pan NC, Rimm IJ. B cell-deficient mice have increased susceptibility to HSV-1 encephalomyelitis and mortality. J Neuroimmunol. 1999;94:122–126. doi: 10.1016/s0165-5728(98)00238-0. [DOI] [PubMed] [Google Scholar]
- Bergstrom T, Alestig K, Svennerholm B, Horal P, Skoldenberg B, Vahlne A. Neurovirulence of herpes simplex virus types 1 and 2 isolates in diseases of the central nervous system. Eur J Clin Microbiol Infect Dis. 1990;9:751–757. doi: 10.1007/BF02184688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomgren G, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366:1870–1880. doi: 10.1056/NEJMoa1107829. [DOI] [PubMed] [Google Scholar]
- Boivin N, Sergerie Y, Rivest S, Boivin G. Effect of pretreatment with toll-like receptor agonists in a mouse model of herpes simplex virus type 1 encephalitis. J Infect Dis. 2008;198:664–672. doi: 10.1086/590671. [DOI] [PubMed] [Google Scholar]
- Bryant-Hudson KM, Gurung HR, Zheng M, Carr DJ. Tumor necrosis factor alpha and interleukin-6 facilitate corneal lymphangiogenesis in response to herpes simplex virus 1 infection. J Virol. 2014;88:14451–14457. doi: 10.1128/JVI.01841-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caignard G, et al. Genome-wide mouse mutagenesis reveals CD45-mediated T cell function as critical in protective immunity to HSV-1. PLoS Pathog. 2013;9:e1003637. doi: 10.1371/journal.ppat.1003637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin E, Tanamachi B, Openshaw H, Mann J, Clarke K. Gamma interferon (IFN-gamma) receptor null-mutant mice are more susceptible to herpes simplex virus type 1 infection than IFN-gamma ligand null-mutant mice. J Virol. 1999;73:5196–5200. doi: 10.1128/jvi.73.6.5196-5200.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DJ, Ash J, Lane TE, Kuziel WA. Abnormal immune response of CCR5-deficient mice to ocular infection with herpes simplex virus type 1. J General Virol. 2006;87:489–499. doi: 10.1099/vir.0.81339-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova JL. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci USA. 2015;112:E7128–E7137. doi: 10.1073/pnas.1521651112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casrouge A, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- Chen M, et al. TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol Cell. 2016;64:105–119. doi: 10.1016/j.molcel.2016.08.025. [DOI] [PubMed] [Google Scholar]
- Chen Y, et al. p38 inhibition provides anti-DNA virus immunity by regulation of USP21 phosphorylation and STING activation. J Exp Med. 2017;214:991–1010. doi: 10.1084/jem.20161387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrady CD, Zheng M, van Rooijen N, Drevets DA, Royer D, Alleman A, Carr DJ. Microglia and a functional type I IFN pathway are required to counter HSV-1-driven brain lateral ventricle enlargement and encephalitis. J Immunol (Baltimore Md: 1950) 2013;190:2807–2817. doi: 10.4049/jimmunol.1203265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook ML, Stevens JG. Pathogenesis of herpetic neuritis and ganglionitis in mice: evidence for intra-axonal transport of infection. Infect Immun. 1973;7:272–288. doi: 10.1128/iai.7.2.272-288.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cunha Sousa LF, et al. Suppressor of cytokine signaling 2 (SOCS2) contributes to encephalitis in a model of Herpes infection in mice. Brain Res Bull. 2016;127:164–170. doi: 10.1016/j.brainresbull.2016.09.011. [DOI] [PubMed] [Google Scholar]
- Doll JR, Sawtell NM. Analysis of herpes simplex virus reactivation in explant reveals a method-dependent difference in measured timing of reactivation. J Virol. 2017 doi: 10.1128/JVI.00848-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33:388–391. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- Fink SL, Jayewickreme TR, Molony RD, Iwawaki T, Landis CS, Lindenbach BD, Iwasaki A. IRE1alpha promotes viral infection by conferring resistance to apoptosis. Sci Signal. 2017 doi: 10.1126/scisignal.aai7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Wallerath C, Covarrubias S, Hornung V, Carpenter S, Fitzgerald KA. The PYHIN Protein p205 Regulates the Inflammasome by controlling Asc expression. J Immunol (Baltimore Md 1950) 2017;199:3249–3260. doi: 10.4049/jimmunol.1700823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung HR, Carr MM, Carr DJ. Cornea lymphatics drive the CD8(+) T-cell response to herpes simplex virus-1. Immunol Cell Biol. 2017;95:87–98. doi: 10.1038/icb.2016.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarr L, Shukla D, Rodahl E, Dal Canto MC, Spear PG. Transcription from the gene encoding the herpesvirus entry receptor nectin-1 (HveC) in nervous tissue of adult mouse. Virology. 2001;287:301–309. doi: 10.1006/viro.2001.1041. [DOI] [PubMed] [Google Scholar]
- Haubenberger D, Clifford DB. Clinical trials in neurovirology: successes, challenges, and pitfalls. Neurotherapeutics. 2016;13:571–581. doi: 10.1007/s13311-016-0440-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman M, et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med. 2012;209:1567–1582. doi: 10.1084/jem.20111316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel L, De Andrea M, Azzimonti B, Rolle A, Gariglio M, Landolfo S. The interferon-inducible 204 gene, a member of the Ifi 200 family, is not involved in the antiviral state induction by IFN-alpha, but is required by the mouse cytomegalovirus for its replication. Virology. 1999;262:1–8. doi: 10.1006/viro.1999.9885. [DOI] [PubMed] [Google Scholar]
- Holm TH, Draeby D, Owens T. Microglia are required for astroglial Toll-like receptor 4 response and for optimal TLR2 and TLR3 response. Glia. 2012;60:630–638. doi: 10.1002/glia.22296. [DOI] [PubMed] [Google Scholar]
- Holterman AX, Rogers K, Edelmann K, Koelle DM, Corey L, Wilson CB. An important role for major histocompatibility complex class I-restricted T cells, and a limited role for gamma interferon, in protection of mice against lethal herpes simplex virus infection. J Virol. 1999;73:2058–2063. doi: 10.1128/jvi.73.3.2058-2063.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Horan KA, et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol (Baltimore Md 1950) 2013;190:2311–2319. doi: 10.4049/jimmunol.1202749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet. 1997;349:241–244. doi: 10.1016/S0140-6736(96)10149-5. [DOI] [PubMed] [Google Scholar]
- Jorgensen LK, Dalgaard LS, Ostergaard LJ, Norgaard M, Mogensen TH. Incidence and mortality of herpes simplex encephalitis in Denmark: a nationwide registry-based cohort study. J Infect. 2017;74:42–49. doi: 10.1016/j.jinf.2016.09.004. [DOI] [PubMed] [Google Scholar]
- Kaewpoowat Q, Salazar L, Aguilera E, Wootton SH, Hasbun R. Herpes simplex and varicella zoster CNS infections: clinical presentations, treatments and outcomes. Infection. 2016;44:337–345. doi: 10.1007/s15010-015-0867-6. [DOI] [PubMed] [Google Scholar]
- Kassim SH, Rajasagi NK, Zhao X, Chervenak R, Jennings SR. In vivo ablation of CD11c-positive dendritic cells increases susceptibility to herpes simplex virus type 1 infection and diminishes NK and T-cell responses. J Virol. 2006;80:3985–3993. doi: 10.1128/JVI.80.8.3985-3993.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastrukoff LF, Lau AS, Takei F, Carbone FR, Scalzo AA. A NK complex-linked locus restricts the spread of herpes simplex virus type 1 in the brains of C57BL/6 mice. Immunol Cell Biol. 2015;93:877–884. doi: 10.1038/icb.2015.54. [DOI] [PubMed] [Google Scholar]
- Katzenell S, Leib DA. Herpes simplex virus and interferon signaling induce novel autophagic clusters in sensory neurons. J Virol. 2016;90:4706–4719. doi: 10.1128/JVI.02908-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenell S, Chen Y, Parker ZM, Leib DA. The differential interferon responses of two strains of Stat1-deficient mice do not alter susceptibility to HSV-1 and VSV in vivo. Virology. 2014;450–451:350–354. doi: 10.1016/j.virol.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp SJ, et al. Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc Natl Acad Sci USA. 2009;106:17916–17920. doi: 10.1073/pnas.0908892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyuncu OO, Hogue IB, Enquist LW. Virus infections in the nervous system. Cell Host Microbe. 2013;13:379–393. doi: 10.1016/j.chom.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer T, Enquist LW. Directional spread of alphaherpesviruses in the nervous system. Viruses. 2013;5:678–707. doi: 10.3390/v5020678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll CM, Zheng M, Carr DJ. Enhanced resistance of CXCR3 deficient mice to ocular HSV-1 infection is due to control of replication in the brain ependyma. J Neuroimmunol. 2014;276:219–223. doi: 10.1016/j.jneuroim.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaraguru U, Davis IA, Deshpande S, Tevethia SS, Rouse BT. Lymphotoxin alpha-/- mice develop functionally impaired CD8+ T cell responses and fail to contain virus infection of the central nervous system. J Immunol (Baltimore, Md: 1950) 2001;166:1066–1074. doi: 10.4049/jimmunol.166.2.1066. [DOI] [PubMed] [Google Scholar]
- Kurt-Jones EA, et al. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci USA. 2004;101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafaille FG, et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature. 2012;491:769–773. doi: 10.1038/nature11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahmidi S, Yousefi M, Dridi S, Duplay P, Pearson A. Dok-1 and Dok-2 are required to maintain herpes simplex virus 1-specific CD8(+) T cells in a murine model of ocular infection. J Virol. 2017 doi: 10.1128/JVI.02297-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc RA, Pesnicak L, Cabral ES, Godleski M, Straus SE. Lack of interleukin-6 (IL-6) enhances susceptibility to infection but does not alter latency or reactivation of herpes simplex virus type 1 in IL-6 knockout mice. J Virol. 1999;73:8145–8151. doi: 10.1128/jvi.73.10.8145-8151.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AJ, et al. Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J Exp Med. 2017;214:1153–1167. doi: 10.1084/jem.20160880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenschow DJ, et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Natl Acad Sci USA. 2007;104:1371–1376. doi: 10.1073/pnas.0607038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, et al. Viral infection of the Central Nervous System and neuroinflammation precede blood-brain barrier disruption during Japanese Encephalitis. Virus Infect J Virol. 2015;89:5602–5614. doi: 10.1128/JVI.00143-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima GK, et al. Toll-like receptor (TLR) 2 and TLR9 expressed in trigeminal ganglia are critical to viral control during herpes simplex virus 1 infection. Am J Pathol. 2010;177:2433–2445. doi: 10.2353/ajpath.2010.100121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J Virol. 2004;78:1675–1684. doi: 10.1128/JVI.78.4.1675-1684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder CC. Genetic variables that influence phenotype. ILAR J. 2006;47:132–140. doi: 10.1093/ilar.47.2.132. [DOI] [PubMed] [Google Scholar]
- Liu Y, et al. RIG-I-mediated STING upregulation restricts herpes simplex virus 1 infection. J Virol. 2016;90:9406–9419. doi: 10.1128/JVI.00748-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livorsi D, Anderson E, Qureshi S, Howard M, Wang YF, Franco-Paredes C. Brainstem encephalitis: an unusual presentation of herpes simplex virus infection. J Neurol. 2010;257:1432–1437. doi: 10.1007/s00415-010-5600-x. [DOI] [PubMed] [Google Scholar]
- Long SS, Pool TE, Vodzak J, Daskalaki I, Gould JM. Herpes simplex virus infection in young infants during 2 decades of empiric acyclovir therapy. Pediatr Infect Dis J. 2011;30:556–561. doi: 10.1097/INF.0b013e31820e3398. [DOI] [PubMed] [Google Scholar]
- Looker KJ, Magaret AS, May MT, Turner KM, Vickerman P, Gottlieb SL, Newman LM. Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS ONE. 2015;10:e0140765. doi: 10.1371/journal.pone.0140765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looker KJ, Magaret AS, Turner KM, Vickerman P, Gottlieb SL, Newman LM. Correction: global estimates of prevalent and incident herpes simplex virus type 2 infections in 2012. PLoS ONE. 2015;10:e0128615. doi: 10.1371/journal.pone.0128615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looker KJ, Magaret AS, May MT, Turner KME, Vickerman P, Newman LM, Gottlieb SL. First estimates of the global and regional incidence of neonatal herpes infection. Lancet Global Health. 2017;5:e300–e309. doi: 10.1016/S2214-109X(16)30362-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez C. Genetics of natural resistance to herpesvirus infections in mice. Nature. 1975;258:152–153. doi: 10.1038/258152a0. [DOI] [PubMed] [Google Scholar]
- Lopez C. Resistance to HSV-1 in the mouse is governed by two major, independently segregating, non-H-2 loci. Immunogenetics. 1980;11:87–92. doi: 10.1007/BF01567772. [DOI] [PubMed] [Google Scholar]
- Louveau A, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucinda N, et al. Dendritic cells, macrophages, NK and CD8(+) T lymphocytes play pivotal roles in controlling HSV-1 in the trigeminal ganglia by producing IL1-beta, iNOS and granzyme B. Virol J. 2017;14:37. doi: 10.1186/s12985-017-0692-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg P, Welander P, Openshaw H, Nalbandian C, Edwards C, Moldawer L, Cantin E. A locus on mouse chromosome 6 that determines resistance to herpes simplex virus also influences reactivation, while an unlinked locus augments resistance of female mice. J Virol. 2003;77:11661–11673. doi: 10.1128/JVI.77.21.11661-11673.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg P, Welander PV, Edwards CK, 3rd, van Rooijen N, Cantin E. Tumor necrosis factor (TNF) protects resistant C57BL/6 mice against herpes simplex virus-induced encephalitis independently of signaling via TNF receptor 1 or 2. J Virol. 2007;81:1451–1460. doi: 10.1128/JVI.02243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg P, et al. The immune response to herpes simplex virus type 1 infection in susceptible mice is a major cause of central nervous system pathology resulting in fatal encephalitis. J Virol. 2008;82:7078–7088. doi: 10.1128/JVI.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo WW, Li S, Li C, Lian H, Yang Q, Zhong B, Shu HB. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat Immunol. 2016;17:1057–1066. doi: 10.1038/ni.3510. [DOI] [PubMed] [Google Scholar]
- MacLean A, Wei XQ, Huang FP, Al-Alem UA, Chan WL, Liew FY. Mice lacking inducible nitric-oxide synthase are more susceptible to herpes simplex virus infection despite enhanced Th1 cell responses. J General Virol. 1998;79(Pt 4):825–830. doi: 10.1099/0022-1317-79-4-825. [DOI] [PubMed] [Google Scholar]
- Manickan E, Rouse BT. Roles of different T-cell subsets in control of herpes simplex virus infection determined by using T-cell-deficient mouse-models. J Virol. 1995;69:8178–8179. doi: 10.1128/jvi.69.12.8178-8179.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manivanh R, Mehrbach J, Knipe DM, Leib DA (2017) Role of herpes simplex virus 1 gamma34.5 in the regulation of IRF3 signaling. J Virol. 10.1128/jvi.01156-17 [DOI] [PMC free article] [PubMed]
- Mansur DS, et al. Lethal encephalitis in myeloid differentiation factor 88-deficient mice infected with herpes simplex virus 1. Am J Pathol. 2005;166:1419–1426. doi: 10.1016/S0002-9440(10)62359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques CP, Cheeran MC, Palmquist JM, Hu S, Urban SL, Lokensgard JR. Prolonged microglial cell activation and lymphocyte infiltration following experimental herpes encephalitis. J Immunol (Baltimore, Md: 1950) 2008;181:6417–6426. doi: 10.4049/jimmunol.181.9.6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruzuru Y, Fujii H, Oyama M, Kozuka-Hata H, Kato A, Kawaguchi Y. Roles of p53 in herpes simplex virus 1 replication. J Virol. 2013;87:9323–9332. doi: 10.1128/JVI.01581-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruzuru Y, et al. p53 is a host cell regulator during herpes simplex encephalitis. J Virol. 2016;90:6738–6745. doi: 10.1128/JVI.00846-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matundan HH, et al. Interrelationship of primary virus replication, level of latency, and time to reactivation in the trigeminal ganglia of latently infected mice. J Virol. 2016;90:9533–9542. doi: 10.1128/JVI.01373-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuillan G, Kruszon-Moran D, Flagg EW, Paulose-Ram R. Prevalence of herpes simplex virus type 1 and type 2 in persons aged 14–49: United States, 2015–2016. NCHS Data Brief. 2018;304:1–8. [PubMed] [Google Scholar]
- Menasria R, Boivin N, Lebel M, Piret J, Gosselin J, Boivin G. Both TRIF and IPS-1 adaptor proteins contribute to the cerebral innate immune response against herpes simplex virus 1 infection. J Virol. 2013;87:7301–7308. doi: 10.1128/JVI.00591-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menasria R, Canivet C, Piret J, Boivin G. Infiltration pattern of blood monocytes into the Central Nervous System during experimental herpes simplex virus encephalitis. PLoS ONE. 2015;10:e0145773. doi: 10.1371/journal.pone.0145773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menasria R, Canivet C, Piret J, Gosselin J, Boivin G. Protective role of CX3CR1 signalling in resident cells of the central nervous system during experimental herpes simplex virus encephalitis. J General Virol. 2017;98:447–460. doi: 10.1099/jgv.0.000667. [DOI] [PubMed] [Google Scholar]
- Menendez CM, Carr DJJ. Defining nervous system susceptibility during acute and latent herpes simplex virus-1 infection. J Neuroimmunol. 2017;308:43–49. doi: 10.1016/j.jneuroim.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz MA, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Michael BD, et al. The interleukin-1 balance during encephalitis is associated with clinical severity, blood-brain barrier permeability, neuroimaging changes, and disease outcome. J Infect Dis. 2016;213:1651–1660. doi: 10.1093/infdis/jiv771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milora KA, Uppalapati SR, Sanmiguel JC, Zou W, Jensen LE. Interleukin-36beta provides protection against HSV-1 infection, but does not modulate initiation of adaptive immune responses. Sci Rep. 2017;7:5799. doi: 10.1038/s41598-017-05363-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohankrishnan A, Parmar R, Bhurani V, Dalai SK. Lack of TNF-alpha signaling through p55 makes the mice more susceptible to acute infection but does not alter state of latency and reactivation of HSV-1. Virus Res. 2017;244:1–5. doi: 10.1016/j.virusres.2017.11.004. [DOI] [PubMed] [Google Scholar]
- Murphy AA, Rosato PC, Parker ZM, Khalenkov A, Leib DA. Synergistic control of herpes simplex virus pathogenesis by IRF-3, and IRF-7 revealed through non-invasive bioluminescence imaging. Virology. 2013;444:71–79. doi: 10.1016/j.virol.2013.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JE, Lee MS, Kim YJ, Lee HK. OASL1 deficiency promotes antiviral protection against genital herpes simplex virus type 2 infection by enhancing type I interferon production. Sci Rep. 2016;6:19089. doi: 10.1038/srep19089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omae T, Saito Y, Tsuchie H, Ohno K, Maegaki Y, Sakuma H. Cytokine/chemokine elevation during the transition phase from HSV encephalitis to autoimmune anti-NMDA receptor encephalitis. Brain Dev. 2018;40:361–365. doi: 10.1016/j.braindev.2017.12.007. [DOI] [PubMed] [Google Scholar]
- Orvedahl A, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Pandey U, Renner DW, Thompson RL, Szpara ML, Sawtell NM. Inferred father-to-son transmission of herpes simplex virus results in near-perfect preservation of viral genome identity and in vivo phenotypes. Sci Rep. 2017;7:13666. doi: 10.1038/s41598-017-13936-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker ZM, Murphy AA, Leib DA. Role of the DNA sensor STING in protection from lethal infection following corneal and intracerebral challenge with herpes simplex virus 1. J Virol. 2015;89:11080–11091. doi: 10.1128/JVI.00954-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker ZM, Pasieka TJ, Parker GA, Leib DA. Immune- and nonimmune-compartment-specific interferon responses are critical determinants of herpes simplex virus-induced generalized infections and acute liver failure. J Virol. 2016;90:10789–10799. doi: 10.1128/JVI.01473-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons LR, Tafuri YR, Shreve JT, Bowen CD, Shipley MM, Enquist LW, Szpara ML. Rapid genome assembly and comparison decode intrastrain variation in human alphaherpesviruses. MBio. 2015 doi: 10.1128/mBio.02213-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasieka TJ, Cilloniz C, Lu B, Teal TH, Proll SC, Katze MG, Leib DA. Host responses to wild-type and attenuated herpes simplex virus infection in the absence of Stat1. J Virol. 2009;83:2075–2087. doi: 10.1128/JVI.02007-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasieka TJ, Cilloniz C, Carter VS, Rosato P, Katze MG, Leib DA. Functional genomics reveals an essential and specific role for Stat1 in protection of the central nervous system following herpes simplex virus corneal infection. J Virol. 2011;85:12972–12981. doi: 10.1128/JVI.06032-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasieka TJ, et al. Bioluminescent imaging reveals divergent viral pathogenesis in two strains of Stat1-deficient mice, and in alphassgamma interferon receptor-deficient mice. PLoS ONE. 2011;6:e24018. doi: 10.1371/journal.pone.0024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez de Diego R, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. 2010;33:400–411. doi: 10.1016/j.immuni.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- Petermann P, Rahn E, Thier K, Hsu MJ, Rixon FJ, Kopp SJ, Knebel-Morsdorf D. Role of nectin-1 and herpesvirus entry mediator as cellular receptors for herpes simplex virus 1 on primary murine dermal fibroblasts. J Virol. 2015;89:9407–9416. doi: 10.1128/JVI.01415-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann P, et al. Entry mechanisms of herpes simplex virus 1 into murine epidermis: involvement of nectin-1 and herpesvirus entry mediator as cellular receptors. J Virol. 2015;89:262–274. doi: 10.1128/JVI.02917-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourchet A, Modrek AS, Placantonakis DG, Mohr I, Wilson AC. Modeling HSV-1 latency in human embryonic stem cell-derived neurons. Pathogens. 2017 doi: 10.3390/pathogens6020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle DC, et al. Efficacy of pritelivir and acyclovir in the treatment of herpes simplex virus infections in a mouse model of herpes simplex encephalitis. Antiviral Res. 2018;149:1–6. doi: 10.1016/j.antiviral.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishna C, Cantin EM. IFNgamma inhibits G-CSF induced neutrophil expansion and invasion of the CNS to prevent viral encephalitis. PLoS Pathog. 2018;14:e1006822. doi: 10.1371/journal.ppat.1006822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishna C, et al. Establishment of HSV1 latency in immunodeficient mice facilitates efficient in vivo reactivation. PLoS Pathog. 2015;11:e1004730. doi: 10.1371/journal.ppat.1004730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinert LS, et al. TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J Clin Investig. 2012;122:1368–1376. doi: 10.1172/JCI60893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinert LS, et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun. 2016;7:13348. doi: 10.1038/ncomms13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochfort KD, Collins LE, McLoughlin A, Cummins PM. Tumour necrosis factor-alpha-mediated disruption of cerebrovascular endothelial barrier integrity in vitro involves the production of proinflammatory interleukin-6. J Neurochem. 2016;136:564–572. doi: 10.1111/jnc.13408. [DOI] [PubMed] [Google Scholar]
- Rosato PC, Leib DA. Intrinsic innate immunity fails to control herpes simplex virus and vesicular stomatitis virus replication in sensory neurons and fibroblasts. J Virol. 2014;88:9991–10001. doi: 10.1128/JVI.01462-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato PC, Katzenell S, Pesola JM, North B, Coen DM, Leib DA. Neuronal IFN signaling is dispensable for the establishment of HSV-1 latency. Virology. 2016;497:323–327. doi: 10.1016/j.virol.2016.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger K, Derkow K, Dembny P, Kruger C, Schott E, Lehnardt S. The impact of single and pairwise Toll-like receptor activation on neuroinflammation and neurodegeneration. J Neuroinflamm. 2014;11:166. doi: 10.1186/s12974-014-0166-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagnelli E, Sagnelli C, Macera M, Pisaturo M, Coppola N. An update on the treatment options for HBV/HCV coinfection. Expert Opin Pharmacother. 2017;18:1691–1702. doi: 10.1080/14656566.2017.1398233. [DOI] [PubMed] [Google Scholar]
- Salimi H, Cain MD, Klein RS. Encephalitic arboviruses: emergence, clinical presentation, and neuropathogenesis. Neurotherapeutics. 2016;13:514–534. doi: 10.1007/s13311-016-0443-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho-Shimizu V, et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Investig. 2011;121:4889–4902. doi: 10.1172/JCI59259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacker TW, Conant M, Thoming C, Stanczak T, Wang Z, Smith M. Imiquimod 5-percent cream does not alter the natural history of recurrent herpes genitalis: a phase II, randomized, double-blind, placebo-controlled study. Antimicrob Agents Chemother. 2002;46:3243–3248. doi: 10.1128/AAC.46.10.3243-3248.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellner J, Simon F, Meyding-Lamade U, Leib SL. Herpes-simplex virus encephalitis is characterized by an early MMP-9 increase and collagen type IV degradation. Brain Res. 2006;1125:155–162. doi: 10.1016/j.brainres.2006.09.093. [DOI] [PubMed] [Google Scholar]
- Sergerie Y, Rivest S, Boivin G. Tumor necrosis factor-alpha and interleukin-1 beta play a critical role in the resistance against lethal herpes simplex virus encephalitis. J Infect Dis. 2007;196:853–860. doi: 10.1086/520094. [DOI] [PubMed] [Google Scholar]
- Shivkumar M, Milho R, May JS, Nicoll MP, Efstathiou S, Stevenson PG. Herpes simplex virus 1 targets the murine olfactory neuroepithelium for host entry. J Virol. 2013;87:10477–10488. doi: 10.1128/JVI.01748-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouboe MK, et al. STING agonists enable antiviral cross-talk between human cells and confer protection against genital herpes in mice. PLoS Pathog. 2018;14:e1006976. doi: 10.1371/journal.ppat.1006976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon T, et al. Management of suspected viral encephalitis in adults—association of British Neurologists and British Infection Association National Guidelines. J Infect. 2012;64:347–373. doi: 10.1016/j.jinf.2011.11.014. [DOI] [PubMed] [Google Scholar]
- Song G, et al. E3 ubiquitin ligase RNF128 promotes innate antiviral immunity through K63-linked ubiquitination of TBK1. Nat Immunol. 2016;17:1342–1351. doi: 10.1038/ni.3588. [DOI] [PubMed] [Google Scholar]
- Steel AJ, Eslick GD. Herpes viruses increase the risk of alzheimer’s disease: a meta-analysis. J Alzheimers Dis. 2015;47:351–364. doi: 10.3233/JAD-140822. [DOI] [PubMed] [Google Scholar]
- Steiner I. Herpes simplex virus encephalitis: new infection or reactivation? Curr Opin Neurol. 2011;24:268–274. doi: 10.1097/WCO.0b013e328346be6f. [DOI] [PubMed] [Google Scholar]
- Steiner I, Benninger F. Update on herpes virus infections of the nervous system. Curr Neurol Neurosci Rep. 2013;13:414. doi: 10.1007/s11910-013-0414-8. [DOI] [PubMed] [Google Scholar]
- Sun H, et al. USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat Commun. 2017;8:15534. doi: 10.1038/ncomms15534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, et al. HCFC2 is needed for IRF1- and IRF2-dependent Tlr3 transcription and for survival during viral infections. J Exp Med. 2017;214:3263–3277. doi: 10.1084/jem.20161630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M, Wang Y, Gilfillan S, Colonna M. Plasmacytoid dendritic cells contribute to systemic but not local antiviral responses to HSV infections. PLoS Pathog. 2013;9:e1003728. doi: 10.1371/journal.ppat.1003728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajfirouz D, West DM, Yin XT, Potter CA, Klein R, Stuart PM. CXCL9 compensates for the absence of CXCL10 during recurrent Herpetic stromal keratitis. Virology. 2017;506:7–13. doi: 10.1016/j.virol.2017.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyangaa E, Kim JH, Patil AM, Choi JY, Kim SB, Eo SK. Distinct upstream role of type I IFN signaling in hematopoietic stem cell-derived and epithelial resident cells for concerted recruitment of Ly-6Chi monocytes and NK cells via CCL2-CCL3 cascade. PLoS Pathog. 2015;11:e1005256. doi: 10.1371/journal.ppat.1005256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilela MC, et al. Absence of CCR5 increases neutrophil recruitment in severe herpetic encephalitis. BMC Neurosci. 2013;14:19. doi: 10.1186/1471-2202-14-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilela MC, et al. Platelet activating factor (PAF) receptor deletion or antagonism attenuates severe HSV-1 meningoencephalitis. J Neuroimmune Pharmacol. 2016;11:613–621. doi: 10.1007/s11481-016-9684-7. [DOI] [PubMed] [Google Scholar]
- Wang JP, et al. Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. J Virol. 2012;86:2273–2281. doi: 10.1128/JVI.06010-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Davido DJ, Morrison LA. HSV-1 strain McKrae is more neuroinvasive than HSV-1 KOS after corneal or vaginal inoculation in mice. Virus Res. 2013;173:436–440. doi: 10.1016/j.virusres.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. STING requires the adaptor TRIF to trigger innate immune responses to microbial infection. Cell Host Microbe. 2016;20:329–341. doi: 10.1016/j.chom.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham S, Lu B, Ash J, Carr DJ. Chemokine receptor deficiency is associated with increased chemokine expression in the peripheral and central nervous systems and increased resistance to herpetic encephalitis. J Neuroimmunol. 2005;162:51–59. doi: 10.1016/j.jneuroim.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Wilcox DR, Wadhwani NR, Longnecker R, Muller WJ. Differential reliance on autophagy for protection from HSV encephalitis between newborns and adults. PLoS Pathog. 2015;11:e1004580. doi: 10.1371/journal.ppat.1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wnek M, et al. Herpes simplex encephalitis is linked with selective mitochondrial damage; a post-mortem and in vitro study. Acta Neuropathol. 2016;132:433–451. doi: 10.1007/s00401-016-1597-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuest TR, Carr DJ. Dysregulation of CXCR3 signaling due to CXCL10 deficiency impairs the antiviral response to herpes simplex virus 1 infection. J Immunol (Baltimore, Md: 1950) 2008;181:7985–7993. doi: 10.4049/jimmunol.181.11.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuest TR, Thapa M, Zheng M, Carr DJ. CXCL10 expressing hematopoietic-derived cells are requisite in defense against HSV-1 infection in the nervous system of CXCL10 deficient mice. J Neuroimmunol. 2011;234:103–108. doi: 10.1016/j.jneuroim.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakoub AM, Shukla D. Autophagy stimulation abrogates herpes simplex virus-1 infection. Sci Rep. 2015;5:9730. doi: 10.1038/srep09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yordy B, Iijima N, Huttner A, Leib D, Iwasaki A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe. 2012;12:334–345. doi: 10.1016/j.chom.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CR, Dambuza IM, Lee YJ, Frank GM, Egwuagu CE. STAT3 regulates proliferation and survival of CD8+ T cells: enhances effector responses to HSV-1 infection, and inhibits IL-10+ regulatory CD8+ T cells in autoimmune uveitis. Mediat Inflamm. 2013;2013:359674. doi: 10.1155/2013/359674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SY, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- Zhang M, Covar J, Zhang NY, Chen W, Marshall B, Mo J, Atherton SS. Virus spread and immune response following anterior chamber inoculation of HSV-1 lacking the Beclin-binding domain (BBD) J Neuroimmunol. 2013;260:82–91. doi: 10.1016/j.jneuroim.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SY, Abel L, Casanova JL. Mendelian predisposition to herpes simplex encephalitis. Handb Clin Neurol. 2013;112:1091–1097. doi: 10.1016/B978-0-444-52910-7.00027-1. [DOI] [PubMed] [Google Scholar]
- Zhang SY, et al. Inborn errors of RNA lariat metabolism in humans with brainstem viral infection. Cell. 2018;172:952–965.e918. doi: 10.1016/j.cell.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann J, et al. Enhanced viral clearance and reduced leukocyte infiltration in experimental herpes encephalitis after intranasal infection of CXCR3-deficient mice. J Neurovirol. 2017;23:394–403. doi: 10.1007/s13365-016-0508-6. [DOI] [PubMed] [Google Scholar]
- Zolini GP, et al. Defense against HSV-1 in a murine model is mediated by iNOS and orchestrated by the activation of TLR2 and TLR9 in trigeminal ganglia. J Neuroinflamm. 2014;11:20. doi: 10.1186/1742-2094-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]