

Summary
In 2008 the Ludwig Boltzmann Cluster Oncology (LBC ONC) was established on the basis of two previous Ludwig Boltzmann Institutes working in the field of hematology and cancer research. The general aim of the LBC ONC is to improve treatment of hematopoietic neoplasms by eradicating cancer-initiating and disease-propagating cells, also known as leukemic stem cells (LSC) in the context of leukemia. In a first phase, the LBC ONC characterized the phenotype and molecular aberration profiles of LSC in various malignancies. The LSC phenotypes were established in acute and chronic myeloid leukemia, in acute lymphoblastic leukemia and in chronic lymphocytic leukemia. In addition, the concept of preleukemic (premalignant) neoplastic stem cells (pre-L-NSC) was coined by the LBC ONC and was tested in myelodysplastic syndromes and myeloproliferative neoplasms. Phenotypic characterization of LSC provided a solid basis for their purification and for the characterization of specific target expression profiles. In a second phase, molecular markers and targets were validated. This second phase is ongoing and should result in the development of new diagnostics parameters and novel, more effective, LSC-eradicating, treatment strategies; however, many issues still remain to be solved, such as sub-clonal evolution, LSC niche interactions, immunologic control of LSC, and LSC resistance. In the forthcoming years, the LBC ONC will concentrate on developing LSC-eradicating strategies, with special focus on LSC resistance, precision medicine and translation of LSC-eradicating concepts into clinical application.
Keywords: Cancer stem cells, Leukemic stem cells, Targeted therapy, Precision medicine, Immunotherapy
Introduction and historical overview
During the past 20 years, cancer-initiating stem cells (CSC) have been extensively analyzed in basic science and in translational medicine [1–7]. In contrast to their more mature daughter cells, CSC have the capacity to propagate a neoplasm (cancer) in vivo for unlimited time periods. In line with this definition, CSC exhibit self-renewal and long-term disease-propagating capabilities. In addition, CSC have the ability to undergo asymmetrical cell division and subsequent differentiation to form the bulk of “more mature” cells in a malignancy. The concept of CSC was first established in myeloid leukemia where CSC are also termed leukemic stem cells (LSC) [8–10]. In certain disease models, such as chronic myeloid leukemia (CML), the clonal hierarchy and LSC dependence are obvious predominant features [11–13].
Over the years, the CSC hypothesis has also been tested in diverse forms of solid tumors and in melanoma patients [6, 7, 14–22]. To a degree, clonal evolution and stem cell hierarchies are also demonstrable in these malignancies [14–22]; however, in advanced (metastatic) cancer and melanoma as well as in most forms of acute leukemia, the stem cell hierarchy in (most of) the dominant clones is gradually (and often rapidly) diminishing [23–28]. Moreover, in advanced neoplasms, neoplastic stem cells are heterogeneous populations of cells, and depending on the stage and type of neoplasm, “stemness” may be or may become a reversible or newly acquired feature of (certain subsets of) neoplastic precursor cells [20–28].
Clinically relevant questions and issues in CSC research are the discovery of more or less specific (defining, diagnostic and/or prognostic) markers and oncogenic pathways in CSC/LSC, the identification and characterization of molecular targets in these cells, and the stem cell-eliminating capacity of various targeted drugs and drug combinations [4, 5, 7, 29–34]; however, whereas a number of different markers, targets, and target pathways have been identified in CSC and LSC, numerous questions remain, such as the origin and clonal evolution of these cells, mechanisms contributing to the multiple forms of stem cell resistance, and the interactions of neoplastic stem cells with the specific microenvironment (stem cell niche) and the immune system (Table 1; [35–40]).
Table 1.
Major issues in cancer/leukemic stem cell research
| Origin of neoplastic/leukemic stem cells (NSC/LSC) |
| Definition of NCS/LSC (biology and function) |
| Terminology/nomenclature: e.g., pre-L-NSC |
| Classification of NSC: premalignant versus malignant |
| Comparing various disease models: NSC, LSC, CSC. |
| Selecting the optimal stem cell assays and models |
| Optimal mouse xenograft model to study NSC engraftment |
| Selection of optimal purification protocols |
| Impact of the stem cell niche |
| Impact of the immune system |
| Markers and targets of NCS (LSC and CSC) |
| Effects of targeted drugs on NSC, LSC, and CSC |
| Definition of eradication and cure in NSC context |
| Eradication of CSC/LSC versus eradication of all NSC |
| Mechanisms of drug resistance of NSC, LSC and CSC |
| How to overcome drug resistance of NSC/LSC/CSC |
CSC cancer stem cell(s); LSC leukemic stem cell(s); pre-L-NSC preleukemic neoplastic stem cell(s)
An important aspect is that during malignant progression, proliferation of CSC/LSC gradually shifts from asymmetrical to (more and more) symmetrical cell divisions, resulting in a huge accumulation of cells possessing stem cell features. During this process of stem cell expansion, more and more somatic aberrations and epigenetic changes are acquired by the dominant sub-clone(s) (Fig. 1; [38–46]). In advanced neoplasms and acute leukemia, CSC/LSC indeed exhibit diverse combinations of molecular lesions and oncogenic pathways that lead to an increasing tumorigenic potential [43–50]. These processes are triggered by the genomic instability and plasticity. Sub-clone formation, expansion and diversification of clonogenic cells may also be regulated by the disease-specific microenvironment (niche) and the immune system (Fig. 1). There is also growing evidence that extensive sub-clone formation and diversification essentially contributes to acquired drug resistance of CSC/LSC, which occurs during disease evolution [32–34, 42–50].
Fig. 1.
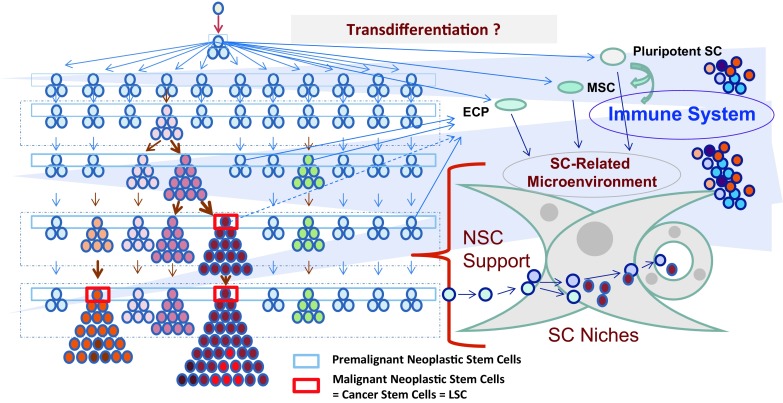
Evolution of neoplastic disorders and impact of the microenvironment. In an early phase of cancer evolution, normal stem cells acquire early lesions and thereby transform into self-renewing neoplastic stem cells (blue rectangle boxes). These premalignant neoplastic stem cells are slowly cycling or dormant cells that persist and slowly acquire additional pro-oncogenic lesions and hits (color change) in distinct sub-clones. The resulting diversification into many different sub-clones is a constantly ongoing process. In a next step, the stem cells of one or more of these premalignant sub-clones acquire critical combinations of oncogenic hits and thereby an enhanced proliferative potential and the capacity to produce an overt neoplasm. Finally, these stem cells and their sub-clones acquire additional pro-oncogenic lesions and convert into fully malignant neoplastic stem cells (red rectangle boxes) producing an overt cancer. In the context of a solid tumor, these cells are then called cancer stem cells (CSC), and in the context of leukemia, these cells are termed leukemic stem cells (LSC). Cancer/leukemia evolution is triggered by the genetic background, a number of different exogenous factors (viruses, toxins, etc.), epigenetic events, the microenvironment and the immune system. The stem cell-related microenvironment, also called stem cell (SC) niche, has been implicated in stem cell survival, stem cell homing and stem cell resistance. In certain neoplasms, some of the micro-environmental cells may be derived from clonal cells through transdifferentiation. The immune system has been implicated in immunosurveillance; however, in an overt malignancy, neoplastic stem cells have the capacity to escape immunosurveillance
Research on LSC developed rapidly in various countries in the Western world over the past two decades. In Austria, first attempts were made to establish robust xenotransplantation models for primary, patient-derived cells between 2000 and 2005. An essential step forward was the introduction of more permissive immunodeficient mice, including the NSG mouse model that has been used successfully in various leukemia models over the past 15 years.
The Ludwig Boltzmann Cluster Oncology (LBC ONC) was established in 2008 by merging two institutes working in the field of hematology and cancer research, the Ludwig Boltzmann Institute for Leukemia Research and Hematology and the Ludwig Boltzmann Institute for Clinical and Experimental Oncology. The newly formed LBC ONC was soon able to attract a group of capable scientists and to establish a number of fruitful collaborations with local and international experts. During the past 10 years, the LBC ONC expanded substantially and launched a series of projects on LSC in various disease models. In these projects, members of the LBC ONC were able to delineate the phenotype and biology of LSC and to develop theoretical concepts around these cells. A major concept propagated by the LBC ONC is that neoplastic stem cells (NSC) can be divided into premalignant (preleukemic) neoplastic stem cells (pre-L-NSC) and fully malignant (leukemic/cancerous) neoplastic stem cells [32, 43–46]. In the context of leukemia, these malignant NSC are called LSC.
Members of the LBC ONC also organized a series of international meetings and conferences in the past 10 years. In the current article, the aims, achievements and future perspectives of the LBC ONC are summarized.
Major aims of the LBC ONC
The general aim of the LBC ONC is to identify and characterize LSC in myeloid and lymphoid neoplasms, to identify clinically relevant (diagnostic and/or prognostic) markers and therapeutic targets (target expression profiles) in these cells, and to examine the effects of various targeted drugs and drug combinations on growth and survival of LSC. In the past 10 years, the LBC ONC has made considerable progress in the phenotyping and characterization of LSC and pre-L-NSC in various leukemia models and preleukemic conditions and has started to validate a number of promising markers and therapeutic targets in these cells. The ultimate aim of the LBC ONC is to establish a solid basis for the development of novel LSC-eradicating treatment concepts. The related long-term vision of the research program of the LBC ONC is to establish improved, potentially curative approaches for human leukemias by applying drugs and drug combinations or immunotherapies that can eliminate LSC and pre-L-NSC in these malignancies, either directly or by targeting the LSC-supporting niche and/or by modulating immunologic mechanisms, such as checkpoint expression, and other processes contributing to LSC resistance.
A general strategic aim of the LBC ONC is to provide a multidisciplinary platform for interactive, cooperative research on NSC/LSC in Vienna and Austria. A related specific aim is to assist in the initiation of specific collaborations in the field of LSC research and to intensify and assist ongoing collaborations with national and international partners and networks working in the field of LSC. To reach these goals, the LBC ONC has established a series of collaboration meetings and platforms, with the aim to discuss novel developments in the field and new results in ongoing projects and collaborations. Moreover, the LBC ONC organized a series of international meetings and conferences on CSC/LSC in the past 10 years. Another important aim of the LBC ONC is to educate young scientists interested in LSC research, to promote junior group leaders in the field, and to motivate clinical colleagues to translate LSC concepts and results into clinical application. With respect to clinical trials and translational projects, the LBC ONC exploits a strong local network of clinical groups working in the field of leukemia research and several different interactions between the cluster core group, the Medical University of Vienna and external partners, such as the Hanusch Hospital, where clinical hematology is a well-established focus, several national and international study groups, and various industrial partners. Moreover, the LBC ONC cooperates with the Comprehensive Cancer Center (CCC) of the Medical University of Vienna.
Major topics and project lines
At the time of initiation in 2008, four project lines were launched: one focusing on myeloid neoplasms, one on lymphoid neoplasms, one on solid tumors, and one on skin cancer models and mastocytosis. Over the years, the cluster focused primarily on myeloid, lymphoid and mast cell neoplasms, following the recommendations of their scientific advisors and members of the evaluation board. Nevertheless, the LBC ONC also published results on putative CSC in solid tumors (colon adenoma and colon cancer) and putative melanoma stem cells [7, 22, 38, 51–54].
Each project line is based on 2 or more research groups and a series of interconnected projects on LSC. Based on evaluation reports and recommendations of the scientific advisory board (SAB) and depending on the developments in the field and available infrastructure, the LBC ONC decided to focus on certain disease models in each group of neoplasms. Among myeloid neoplasms, the LBC ONC is focusing on acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), and chronic myeloid leukemia (CML). More recently, the LBC ONC has also started to work in the field of Ph-negative myeloproliferative neoplasms (MPN) and on chronic myelomonocytic leukemia (CMML). Within lymphoid neoplasms, the LBC ONC started to focus on chronic lymphocytic leukemia (CLL) and acute lymphoblastic leukemia (ALL). Later, the LBC ONC also examined putative stem cells and their response to targeted drugs in multiple myeloma [55]. Within the group of solid tumors, the LBC ONC started to focus on colon adenomas and colon carcinomas [51]; however, over time most of these projects had to be discontinued in the LBC ONC. Similarly, although the cluster published on melanoma-initiating cells [54], melanoma projects were discontinued over time. Within mast cell neoplasms, the LBC ONC started to characterize stem cells in aggressive systemic mastocytosis (ASM) and mast cell leukemia (MCL). More recently, the LBC ONC also started to characterize pre-L-NSC in indolent SM (ISM) and smoldering SM (SSM).
Overall, the LBC ONC characterized disease-initiating and propagating cells in diverse forms of acute (aggressive) malignancies in a first research phase. More recently, the LBC ONC also started to characterize preleukemic neoplastic stem cells (pre-L-NSC) in various indolent neoplasms, including MDS, MPN, CMML and ISM/SSM. In addition, the LBC ONC has started to characterize pre-L-NSC in prediagnostic stages of a malignancy and residual pre-L-NSC and LSC in patients who are in complete remission after drug therapy (e.g., chemotherapy or targeted drugs).
In each project line, the LBC ONC was able to organize constant access to primary patient-derived samples and established a robust biobank which was only possible to accomplish through the constant support of the Medical University of Vienna. In addition, the LBC ONC was able to establish robust xenotransplantation models through a strong essential collaboration with the University of Veterinary Medicine in Vienna. Based on this fruitful collaboration, the LBC ONC invited the University of Veterinary Medicine Vienna to join the cluster, and in 2017 this important institution indeed joined and became an official partner of the LBC ONC.
Management, internal collaborations and core facility platforms
The LBC ONC was established in 2008 by merging two previous Ludwig Boltzmann Institutes, one located at Hanusch Hospital and the other at the Medical University of Vienna. In 2017, the University of Veterinary Medicine Vienna joined as a third partner institution. The three academic partners are interconnected via a number of well-established and fruitful scientific collaborations in the field of leukemia research, translational hematology and clinical hematology. In addition, based on fruitful collaborations on mast cell neoplasms between the University of Veterinary Medicine Vienna and the Medical University of Vienna, the new emerging field of comparative oncology has become a new focus in the LBC ONC.
The LBC ONC is based on a robust communication system that promotes continuous networking and scientific collaborations and discussion. Internal communication in the LBC ONC and interactions with external partners are coordinated and guided by the administration team of the cluster. This team is instrumental for the management of the LBC ONC and is coordinated by the administrative coordinator, Emir Hadzijusufovic and his team. Members of the LBC ONC meet regularly in weekly progress report meetings at the Medical University of Vienna. These meetings are incorporated in the Vienna Cancer Stem Cell Club (VCSCC) [56] and represent a suitable (permanent) platform for fruitful scientific discussion and collaborations. New projects are introduced and ongoing projects are discussed on a regular basis (at least monthly) in these meetings. In collaboration with the VCSCC, the LBC ONC also hosts a weekly journal club and a seminar series where international experts are invited to Vienna. Finally, the LBC ONC organized a number of smaller and larger international scientific meetings on CSC/LSC, including several major working conferences (Table 2).
Table 2.
International conferences and meetings organized by the LBC ONC (2008–2018)
| Title of conference/workshopa | Place/year | Publicationsb |
|---|---|---|
| Second Vienna Cancer Stem Cell Meeting | Vienna 2008 | – |
| Year 2011 Working Conference on Cancer Stem Cells | Vienna 2011 | [43, 46] |
| 10 Year Jubilee Meeting of the VCSCC, Billroth Haus, Vienna | Vienna 2012 | [56] |
| Ph+ CML: from LSC eradication to cure | Vienna 2014 | – |
| From cellular basis and targets to targeted therapies | Vienna 2015 | [95] |
| Classification and nomenclature of clonal conditions | Vienna 2015 | [96] |
| Workshop on mast cells and mastocytosis | Vienna 2015 | [87] |
| Biology, classification and therapy of basophil disorders | Vienna 2016 | [97] |
| Workshop on MDS and pre-MDS conditions | Vienna 2016 | [98] |
| Workshop comparative oncology—Mast cell neoplasms | Vienna 2017 | Submitted |
| Workshop on erythropoiesis and erythroid disorders | Vienna 2017 | Submitted |
| 10 Year Jubilee Meeting of the LBC ONC | Vienna 2018 | This manuscript |
| Working Conference on Chronic Myelomonocytic Leukemia | Vienna 2018 | Planned |
aIn each conference, the first day included an education session open to the public and free of registration (no registration fee)
bPosition papers were prepared to summarize the event and the most important conference outcomes in form of an overview and/or in form of consensus statements. VCSCC, Vienna Cancer Stem Cell Club
In 2017 an updated contract that regulates the interactions between partners and their financial supporters in the LBC ONC was signed. Based on this new contract, a new board of partners assembling major representatives of all partner institutions was established. The board of the LBC ONC meets two times per year to discuss the general strategies and conceptual issues in the LBC ONC and to report about achievements and plans as well as the budget of the cluster. The LBC ONC has also established a scientific advisory board (SAB) consisting of 3 international experts in the field. The SAB members visit the LBC ONC once per year and provide critical input and important scientific advice to cluster coordinators as well as to postdocs and students working on LBC ONC projects.
During the first 10 years, the LBC ONC has established seven core facility platforms (CF-PF) in collaboration with their academic partners. These CF-PF include a management and administration CF-PF, a flow cytometry and LSC-sorting CF-PF, an omics CF-PF, a mouse (NSG) xenotransplantation CF-PF, an education CF-PF, a clinical study CF-PF and a CF-PF for viral-mediated gene delivery and the establishment of related cell lines. The CF-PF are interconnected with each other and with all ongoing cluster projects and provide an essential technology network and basis for the successful research in the three project lines of the LBC ONC.
Established collaborations and networking
The LBC ONC established numerous collaborations with local, national and international groups and institutes working in the field of hematology and cancer research and with various academic organizations and societies. Important local collaborating institutions include the Children’s Cancer Research Institute (CCRI), the Institute for Molecular Pathology (IMP), the Center for Molecular Medicine (CeMM) of the Austrian Academy of Science, the Ludwig Boltzmann Institute for Cancer Research (LBI-CR), the Ludwig Boltzmann Cluster for Cardiovascular Research, and the Ludwig Boltzmann Institute for Osteology. Several of these collaborations are embedded in strong scientific networks that were formed on the basis of the VCSCC and the LBC ONC. One good example is a special research program (SFB) on MPN that has been initiated by the Medical University of Vienna and is coordinated by members of the LBC ONC. Another good example is the international networking on MDS where members of the LBC ONC from the Hanusch Hospital and the Medical University of Vienna worked together with their international partners and essentially contributed to numerous studies [57–60]. The LBC ONC has also established important collaborations with certain industrial partners interested in translational and clinical hematology and LSC research. Based on mutual interest, several of these collaborations have been intensified in the recent past. Whereas the LBC ONC is able to test new innovative drugs against LSC, the industrial partners can learn whether their drugs may indeed have a curative (LSC-eliminating) potential.
Major events and conferences
In 2008, members of the LBC ONC and the VCSCC co-organized the second Cancer Stem Cell Meeting in Vienna (Table 2). Several pioneers in the field of CSC/LSC research participated and the LBC ONC was able to present key researchers, postdocs and students as well as the strategic research plan to interested visitors and to the international community. In 2011, the LBC ONC and the VCSCC organized the Year 2011 Working Conference on CSC (Table 2). In this meeting, a panel of authorities in the field discussed various aspects of CSC/LSC research, including terminologies, the biology and heterogeneity of NSC (pre-C-NSC/pre-L-NSC vs. CSC/LSC) as well as limitations of these concepts, and the assays that have to be applied to detect and quantify CSC/LSC [43]. As a result a new classification of NSC was proposed by the faculty. Based on this proposal, neoplastic stem cells (the umbrella term) are divided into premalignant (in leukemia context preleukemic) neoplastic stem cells (pre-L-NSC or pre-C-NSC) and malignant (leukemic) neoplastic stem cells (Table 3; [43]). Premalignant NSC can produce a premalignant/preleukemic neoplastic condition (e.g., MDS or MPN) but not a fully transformed malignancy (e.g. AML). Over time, pre-L-NCS can transform into fully malignant LSC, depending on additional lesions and hits that are acquired by these cells [43, 46]. Stem cell evolution is a multistep process that takes years or even decades, and is propagated (by driver and passenger mutations) in one, more, or many different (separately developing) sub-clones (Fig. 1). This new concept and the related classification of neoplastic stem cells may explain a number of different clinical phenomena, such as the extensive plasticity and heterogeneity of primary and secondary cancer lesions, lineage switches of cancer cells, or the different biology and resistance of relapsing disease. The faculty delivered consensus statements and the proposed classification in 2012 [43]. In October 2012, the VCSCC celebrated its 10 Year Jubilee in the form of a Scientific Workshop on CSC/LSC in the “Billroth Haus” in Vienna (Table 2). Members of the LBC ONC co-organized this event and in April 2018, members of the LBC ONC celebrated the 10 Year Jubilee of the cluster in a research meeting in Vienna.
Table 3.
Classification of stem cells in preleukemia and leukemia patients and clinical implications
| Stem cell class | Major clinical implications |
|---|---|
| Normal stem cells | Can be detected in patients with leukemia and preleukemic conditionsa Basis of curative drug therapies (regeneration of normal BM) |
| Pre-L-NSC | (1) Can produce a preleukemic neoplastic condition, such as MDS or MPN |
| (2) Can transform into LSC and thereby trigger a full-blown leukemia, such as AML | |
| (3) Often escapes interventional therapy, including poly-chemotherapy and targeted therapies | |
| (4) Can cause a late relapse after therapyb | |
| (5) Can increase the risk of thromboembolic events (when ARCH mutations are expressed) | |
| LSC | Produce an overt leukemia; are often resistant against conventional chemotherapy and can cause early relapse or drug-resistant disease |
| These cells need to be eradicated to achieve a complete remission (CR) after chemotherapy. In many patients long-term CR is achievedb |
BM bone marrow Pre-L-NCS pre-leukemic neoplastic stem cells, MDS myelodysplastic syndrome, MPN myeloproliferative neoplasms, AML acute myeloid leukemia, ARCH age-related clonal hematopoiesis
aPreleukemic conditions are all clonal processes that may precede a leukemia; preleukemic neoplasms are all overt hematopoietic neoplasms that can precede a leukemia
bWhen all Pre-L-NSC and LSC can be eradicated therapy is curative and no relapse can occur. When all LSC but only some or most Pre-L-NSC can be eradicated, the patient may or may not enter long-term CR, depending on the progression potential of the residual Pre-L-NSC
Major concepts around LSC and pre-L-NSC
Cancer evolution is an extremely complex and long-lasting process that can essentially be divided into prediagnostic, premalignant and malignant phases (stages) of the disease. The same holds true for leukemic disorders, where preleukemic neoplasms and overt leukemia have to be distinguished. As mentioned before, NSC can also be divided into premalignant neoplastic stem cells (pre-C-NCS or pre-L-NSC) and fully malignant stem cells (CSC/LSC) [43, 46]. Whereas pre-L-NSC can produce a preleukemic neoplasm, such as MDS, only LSC can produce an overt leukemia (Table 3). The concept of pre-L-NSC and LSC has been confirmed in various preclinical models and clinical studies and has major implications for the development of novel diagnostic and prognostic concepts and novel therapies.
A major discovery has been that pre-L-NSC are frequently detectable in older, apparently healthy individuals. In fact, age-related somatic mutations (ARCH), also known as clonal hematopoiesis of indeterminate potential (CHIP) [61–64], are considered to be produced by pre-L-NCS, provided that the somatic clone persists. Moreover, it has been shown that these early clones can acquire additional mutations over time and that these additional mutations can contribute to progression to an overt neoplasm or a relapse [65, 66]. It has also been described that after successful chemotherapy or successful targeted therapy, when the dominant clones are no longer detectable, early pre-L-NSC may persist and may be detectable by testing blood cells for the presence of CHIP/ARCH mutations (Table 3; [67–69]). Finally, CHIP/ARCH mutations have been associated with a high risk to develop atherosclerosis and related (life-threatening) thromboembolic events [63, 69, 70]. This unexpected observation may explain why many (if not most) blood cell disorders are associated with an increased risk of thromboembolic events.
Phenotypic characterization of LSC and LSC resistance
During the past 10 years, the LBC ONC has been able to characterize the phenotype of putative NSC/LSC in various hematopoietic neoplasms. The LSC were identified in patients with Ph+ CML, AML, CLL, Ph+ ALL, Ph− ALL and MCL [71–80]. Putative NSC (pre-L-NSC) were identified in patients with MDS, systemic mastocytosis and Ph− MPN [77, 79]. As assessed by gene array and antibody-based screening, a number of cell surface antigens, such as roundabout-4 (ROBO4), the IL-2 receptor alpha chain (CD25), dipeptidyl-peptidase IV (DPPIV = CD26), Siglec-3 (CD33) or Campath-1 (CD52), were identified on neoplastic stem cells. An interesting observation was that in all overt leukemias, including even CLL, the NSG mouse-engrafting NSC/LSC apparently resided in a CD34+ sub-fraction of the neoplastic clone (Table 4). Another interesting observation was that several markers identified on LSC are also detectable on stem cell populations in preleukemic conditions (pre-L-NSC) and even also in non-hematologic (solid) tumors. For example, CD26 and CD133 are expressed on putative colon CSC as well as on CML LSC [53, 72]. Finally, several of the newly identified surface markers on NSC/LSC may serve as diagnostic indicators or as molecular targets. A summary of surface markers and targets detected on pre-L-NSC and LSC in hematologic neoplasms is shown in Table 4. In Ph+ CML, the NSG mouse-engrafting LSC reside within a sub-fraction of CD34+/CD38− cells that aberrantly co-express CD25, CD26 and IL-1RAP (Table 4; [72]). In addition, these cells co-express increased amounts of CD33, CD44, CD56 and CD90 when compared to normal bone marrow stem cells. In AML, the putative CD34+/CD38− LSC display, among other antigens, CD25, CD33, CD44, IL-1RAP and CD371 (CLL-1; Table 4). In Ph+ ALL, CD34+/CD38− LSC often express CD25, CD26 and IL-1RAP, whereas in Ph− ALL, LSC usually lack CD25, CD26 and IL-1RAP [80]. In patients with MDS, the putative CD34+/CD38− pre-L-NSC often express CD25, CD33, and CD44 as well as CD52 [77, 81].
Table 4.
Phenotype of leukemic stem cells (LSC) and identified targetsa
| Disease/neoplasm | Primary phenotype | Molecular targetsa |
|---|---|---|
| AML (CD34+)b | CD34+/CD38−; CD34+/CD38+ | CD33, CD44, CD52, KIT, |
| CD34+/CD38−/CD123+ | FLT3, HO-1, BRD4, MYC | |
| CD34+/CD38−/CD25+ | ||
| CD34+/CD38−/CLL-1+ | ||
| Ph+ CML | CD34+/CD38−/CD25+/CD26+/ | CD33, CD44, CD52, KIT, |
| CD56+/CD93+/L-1RAP+ | HO-1, MCL-1, AURKA/B | |
| Ph+ ALL | CD34+/CD38−; CD34+/CD38+ | CD19, CD22, CD33, CD52 |
| CD34+/CD38−/CD25+/CD26+ | HO-1, BLC-2, MCL-2 | |
| Ph− ALL | CD34+/CD38−; CD34+/CD38+ | CD19, CD22, CD33, CD52 |
| CD34+/CD38−/CD25+ | HO-1, BCL-2, MLC-1 | |
| CLL | CD34+/CD19+/CD5+ | CD20, CD23, CD44, CD52 |
| Notch1/2, PTEN, PI3K | ||
| ASM/MCL | CD34+/CD38−/CD123+ | CD33, CD44, CD52, KIT |
| HO-1, FES, AURKA/B, |
Cell surface expression of markers and targets on LSC was determined by multi-color flow cytometry
AML acute myeloid leukemia, HO-1 heme oxygenase-1, CML chronic myeloid leukemia, ALL acute lymphoblastic leukemia, CLL chronic lymphocytic leukemia, PI3K phosphoinositide-3-kinase, ASM aggressive systemic mastocytosis, MCL mast cell leukemia, AURK aurora kinase
aThe Table includes a few selected molecular targets identified in LSC by members of the LBC ONC in the past 10 years
bIn AML, both the CD34+/CD38− and CD34+/CD38+ subsets of CD34+ cells contain LSC
During validation, members of the LBC ONC were interested to learn (i) about the mechanism(s) underlying abnormal or even selective aberrant expression (compared to normal stem cells) of markers and targets on LSC, (ii) about the functional pathogenetic role of these antigens, and (iii) about possibilities to employ these antigens as suitable diagnostic or prognostic markers or as molecular targets of therapy. For example, CD25 was found to be a STAT5-dependent negative regulator of growth of CML LSC [74]. The antigen CD26, also known as DPPIV, was identified as an enzyme that is involved in the mobilization of CML LSC out of the stem cell niche which may explain the huge spread of LSC into extramedullary organs in CML [72]. Another example is CD52. Expression of this antigen is triggered by the RAS pathway and often upregulated on LSC in MDS and AML patients exhibiting the 5q-anomaly [77].
During the past 5 years, members of the LBC ONC have explored the mechanisms underlying the interactions between LSC and the organ and disease-specific microenvironment, the so-called stem cell niche. As mentioned before, CD26 is an important regulator of LSC-niche interaction. Another observation has been that certain LSC-niche interactions contribute to drug resistance. Therefore, the LBC ONC has also explored the mechanisms of various targeted drugs on niche cells. For example, certain tyrosine kinase inhibitors directed against BCR-ABL1 and other (vascular) targets, like nilotinib, suppress endothelial cell proliferation and angiogenesis and may thus impact on the vascular stem cell niche [69].
Finally, certain cell surface antigens on pre-L-NSC and/or LSC may be associated with or even trigger, drug resistance. Among these are the multidrug-resistance gene product MDR-1 (CD243), “the don’t eat me” checkpoint receptor CD47, and the T cell-PD1-interacting checkpoint molecule PD-L1 (CD274). These antigens are frequently detectable on LSC in AML, CML and other hematopoietic neoplasms, and are regulated by specific pathways. For example, expression of PD-L1 on LSC depends on a BET-MYC pathway that can be induced by the oncogenic machinery and/or the cytokine storm involving interferon-gamma [82]. Currently, members of the LBC ONC are exploring the mechanisms underlying the well-known resistance of LSC against various targeted and conventional drugs and how LSC-niche and LSC-immune cell interactions contribute to the intrinsic and acquired forms of resistance of LSC.
Clinical translation of LSC concepts: examples and status 2018
During the past few years, members of the LBC ONC were able to translate several of the concepts that were developed in their cluster projects into clinical application. These include novel diagnostic approaches, prognostication, and therapies. In the following paragraphs, a few highlighting examples are provided.
1. Diagnostic LSC phenotyping: during the past 5 years members of the LBC ONC have established LSC expression profiles in myeloid and lymphoid leukemias by testing antibody combinations in larger cohorts of patients. During validation, aberrantly expressed markers were also examined for their specificity and sensitivity. As a result, several markers have been introduced into application in clinical practice. In CML, the recommended LSC markers are CD25, CD26 and IL-1RAP [72–74]. In most patients with chronic phase CML, all three antigens are detectable on CD34+/CD38− stem cells; however, in advanced CML one or more of these antigens may not be detectable on LSC. Even in chronic phase CML, LSC may sometimes lack one of these antigens. Therefore, all three markers should be applied in diagnostic phenotyping. The CCL-1 (CD371), a marker of AML LSC, is usually not expressed on CML LSC. In AML, CD34+/CD38− LSC often display CD25 (in about 40% of patients) and/or CCL-1 (CD371; in about 30% of patients). In addition, IL-1RAP may be detected on AML LSC. By contrast, AML LSC usually stain negative for CD26. Normal bone marrow stem cells usually lack all these LSC-related surface antigens; however, some of these antigens, such as CD25, may be expressed on early preleukemic neoplastic CD34+/CD38− stem cells (pre-L-NSC). For example, in patients with MDS, the putative CD34+/CD38− pre-L-NSC often display CD25. Despite these encouraging results, diagnostic LSC phenotyping needs further validation in forthcoming studies.
2. Linking pre-L-NSC to age-related clonal hematopoiesis (ARCH): recent data suggest that ARCH is a persistent (and thus stem cell-derived) molecular signature of somatic mutations predisposing (a) for the development of clonal hematopoietic neoplasms and (b) for the occurrence of life-threatening cardiovascular events [63, 69, 70]. The name ARCH implies that the prevalence of these mutations increases with age. The alternative name for ARCH is clonal hematopoiesis with indeterminate potential (CHIP) which implies that the final outcome is uncertain; however, it is well known that several of these patients develop a myeloid neoplasm, and it is also established that in patients with myeloid neoplasms the rate of cardiovascular events is relatively high. Therefore, analysis of patients with myeloid neoplasms for the presence of ARCH/CHIP mutations will soon be regarded as the standard. Even after successful debulking of all dominant sub-clones, ARCH/CHIP mutations may still be detected [66–69]. Based on concepts developed in the LBC ONC, these residual clones may be derived from pre-L-NSC and recent data suggest that carriers of ARCH/CHIP after therapy may indeed bear an increased risk to develop vascular events [69].
3. Clinical translation of KIT-targeting drugs in advanced mast cell neoplasms: KIT acts as a major and largely selective driver in systemic mastocytosis (SM). This neoplasm is defined by uncontrolled expansion and accumulation of neoplastic mast cells, certain clinical and molecular characteristics, and the transforming KIT mutation D816V. Whereas in indolent SM (ISM) the prognosis is excellent (normal life expectancy), patients with advanced SM, including aggressive SM (ASM) or MCL have a poor prognosis. Based on the specificity of KIT for SM, the LBC ONC focused research on this molecular target. In earlier studies, members of the LBC ONC were able to show that the KIT mutation D816V confers resistance against imatinib, and that this resistance can be overcome by a few novel TKIs. Among these, midostaurin was found to exert profound effects on the autonomous growth of KIT-mutated neoplastic mast cells [83]. In addition, members of the LBC ONC were able to show that midostaurin blocks mediator secretion in mast cells [84, 85]. Based on these observations and first pilot case reports, a global trial examining the effects of midostaurin in patients with advanced SM was conducted. In this study, the overall response rate was 60% despite the poor risk of the study group. Median overall survival was 28.7 months and the median progression-free survival amounted to 14.1 months [86, 87]. Among the 16 patients with MCL, the median survival time was 9.4 months and in some of the patients, long-lasting major responses were obtained [86, 87]. These results are superior compared to previous treatment options in advanced SM and MCL and have recently resulted in the approval of midostaurin for use in advanced SM by major health authorities [87]. Moreover, based on this study, several companies have recently started to develop novel more potent and more specific interventional drugs targeting KIT and various KIT mutant forms.
4. Revival of antibody-based therapy in AML: whereas in lymphoproliferative neoplasms the addition of targeted antibodies to induction poly-chemotherapy has clearly improved the overall treatment outcome and survival in these patients, no comparable antibody therapy is available in myeloid neoplasms. This is mainly due to the fact that the antibodies used thus far in myeloid leukemias, especially the CD33-directed antibody-conjugate gemtuzumab-ozogamicin (GO, mylotarg®) produced substantial myelotoxicity in clinical trials, which in turn can be explained by the stem cell-targeting effect of this drug. Notably, as mentioned before, CD33 is also expressed on normal stem cells. In addition, these antibodies, such as alemtuzumab (anti-CD52 antibody) can induce immunosuppressive effects by eliminating lymphocytes and monocytes. As a result, some of these agents, although very effective as anti-leukemic agents, such as GO, have been labeled as being “too toxic” and GO has even been (transiently) removed from the market as an AML therapy. During the past 10 years, members of the LBC ONC have shown that CD33 is indeed a robust target and that the levels of CD33 detectable on LSC in AML and CML are much higher (10–1000-fold) compared to expression levels on normal stem cells [71, 88, 89]. In addition, the in vitro effects of GO on LSC are much more pronounced than the effects on normal stem cells, providing a clear therapeutic window. More recently, several studies have shown that the myelotoxic effects of GO can be minimized by reducing the dose of GO and/or the numbers of applications. In addition, most of the larger studies and metanalyses have shown higher remission rates and a survival benefit for AML patients treated with GO [90, 91]. Based on these observations, mylotarg® (GO) is again available for the treatment of AML [91].
5. Treatment of older patients AML patients with intensive (high dose) chemotherapy: a special challenge in clinical hematology is the optimal choice of therapy for older patients (aged over 60 years) with AML. Many of these patients cannot tolerate intensive chemotherapy and most of them cannot be referred to hematopoietic stem cell transplantation (HSCT). On the other hand, less intensive therapy or therapy with hypomethylating agents often fails or is only effective for a certain time period. Even when these patients tolerate induction chemotherapy and reach complete remission, the (predicted) outcome is often poor, as many of these patients have secondary forms of AML or AML variants characterized by an unfavorable molecular or cytogenetic signature. Therefore, it is of utmost importance to develop new, improved treatment strategies for these patients. A straightforward approach would be to intensify the overall chemotherapy in these patients; however, due to the risk of severe infections and other side effects this strategy is questionable. Therefore, members of the LBC ONC and the Medical University of Vienna conducted a clinical trial exploring the clinical tolerability and efficacy of a special form of intensified consolidation after remission induction in older patients with AML [92]. The consolidation regimen was developed on the basis of a previous study showing that intermediate ARA‑C (cytosine arabinoside) is not only a highly effective but also a less toxic and extremely well-tolerated type of consolidation chemotherapy [93, 94]. In the trial conducted recently, AML patients ≥ 60 years in complete remission after induction therapy received fludarabine and ARA-C in consolidation cycle 1 and intermediate-dose ARA-C (IDAC) in cycles 2 through 4. In addition, pegfilgrastim (G-CSF) was applied: to examine the effect of pegfilgrastim on neutropenia and the time of hospitalization, the study compared cycles 2 and 4 where pegfilgrastim was given routinely from day 6 (IDAC-P) with cycle 3 where pegfilgrastim was only administered in case of severe infections and/or prolonged neutropenia. This regimen was not only found to be feasible but also to be less toxic and highly effective with superior response and survival data compared to (all known) previous trials conducted in older AML patients so far. The median duration of severe neutropenia was 7 days in cycle 2 (IDAC-P), 11.5 days in cycle 3 (IDAC), and 7.5 days in cycle 4 (IDAC-P; p < 0.05) [92]. The probability of survival after 5 years was 32%. Together, intensified consolidation can be administered in AML patients ≥ 60 years, and those who are <75 years were found to benefit from this therapy. Routine administration of pegfilgrastim seems important to shorten the time of neutropenia and hospitalization in AML. Whether this therapy approach can be regarded as a new standard of therapy in AML patients over 60 years, remains to be investigated in forthcoming, randomized clinical studies.
Summary and future perspectives
The LBC ONC was established in 2008 with the aim to define phenotypic properties and target expression profiles in disease-initiating and propagating cells in various hematopoietic neoplasms and to improve anti-cancer therapy by eliminating these cells with more or less specific drugs and suitable (most effective) drug combinations. In addition, the LBC ONC has developed novel diagnostic tools and identified new prognostic stem cell markers. Several of these concepts have been translated from a preclinical stage into clinical application. The LBC ONC has also identified premalignant phases and cells in various myeloid malignancies. Finally, the LBC ONC has deciphered mechanisms underlying stem cell resistance and developed strategies to overcome resistance. In the project phase, the LBC ONC will continue to define the function and druggable vulnerabilities of neoplastic stem cells and will try to translate diagnostic, prognostic and stem cell-eradicating concepts into clinical application. In the context of new emerging therapeutic concepts, tools, and immunotherapies, there is hope that these translational approaches can lead to improved (curative) anti-neoplastic therapies in myeloid malignancies in the foreseeable future.
Acknowledgments
This research is supported by the Austrian Science Fund (FWF) under Grant No F4701 and F4704.
Funding
Open access funding provided by Austrian Science Fund (FWF).
Conflict of interest
P. Valent, E. Hadzijusufovic, T. Grunt, H. Karlic, B. Peter, H. Herrmann, G. Eisenwort, G. Hoermann, A. Schulenburg, M. Willmann, R. Hubmann, M. Shehata, E. Selzer, K.V. Gleixner, T. Rülicke, W.R. Sperr, B. Marian, M. Pfeilstöcker, H. Pehamberger, F. Keil, U. Jäger and C. Zielinski declare that they have no competing interests.
References
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 3.Polyak K, Hahn WC. Roots and stems: stem cells in cancer. Nat Med. 2006;12:296–300. doi: 10.1038/nm1379. [DOI] [PubMed] [Google Scholar]
- 4.Guzman ML, Jordan CT. Considerations for targeting malignant stem cells in leukemia. Cancer Control. 2004;11:97–104. doi: 10.1177/107327480401100216. [DOI] [PubMed] [Google Scholar]
- 5.Sperr WR, Hauswirth AW, Florian S, Ohler L, Geissler K, Valent P. Human leukaemic stem cells: a novel target of therapy. Eur J Clin Invest. 2004;34(S2):31–40. doi: 10.1111/j.0960-135X.2004.01368.x. [DOI] [PubMed] [Google Scholar]
- 6.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schulenburg A, Ulrich-Pur H, Thurnher D, Erovic B, Florian S, Sperr WR, et al. Neoplastic stem cells: a novel therapeutic target in clinical oncology. Cancer. 2006;107:2512–2520. doi: 10.1002/cncr.22277. [DOI] [PubMed] [Google Scholar]
- 8.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 9.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 10.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–743. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 11.Kavalerchik E, Goff D, Jamieson CH. Chronic myeloid leukemia stem cells. J Clin Oncol. 2008;26:2911–2915. doi: 10.1200/JCO.2008.17.5745. [DOI] [PubMed] [Google Scholar]
- 12.Copland M. Chronic myelogenous leukemia stem cells: What’s new? Curr Hematol Malig Rep. 2009;4:66–73. doi: 10.1007/s11899-009-0010-9. [DOI] [PubMed] [Google Scholar]
- 13.Sloma I, Jiang X, Eaves AC, Eaves CJ. Insights into the stem cells of chronic myeloid leukemia. Leukemia. 2010;24(11):1823–1833. doi: 10.1038/leu.2010.159. [DOI] [PubMed] [Google Scholar]
- 14.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 15.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 16.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 17.Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. U.S.A. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, Zhan Q, Jordan S, Duncan LM, Weishaupt C, Fuhlbrigge RC, Kupper TS, Sayegh MH, Frank MH. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zabierowski SE, Herlyn M. Melanoma stem cells: the dark seed of melanoma. J Clin Oncol. 2008;26:2890–2894. doi: 10.1200/JCO.2007.15.5465. [DOI] [PubMed] [Google Scholar]
- 20.Huff CA, Matsui W. Multiple myeloma cancer stem cells. J Clin Oncol. 2008;26:2895–2900. doi: 10.1200/JCO.2007.15.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulenburg A, Brämswig K, Herrmann H, Karlic H, Mirkina I, Hubmann R, Laffer S, Marian B, Shehata M, Krepler C, Pehamberger H, Grunt T, Jäger U, Zielinski CC, Valent P. Neoplastic stem cells: current concepts and clinical perspectives. Crit Rev Oncol Hematol. 2010;76(2):79–98. doi: 10.1016/j.critrevonc.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 23.Eaves CJ. Cancer stem cells: here, there, everywhere? Nature. 2008;456:581–582. doi: 10.1038/456581a. [DOI] [PubMed] [Google Scholar]
- 24.Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, Johnson TM, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2010;18:510–523. doi: 10.1016/j.ccr.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- 26.Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 28.Goardon N, Marchi E, Atzberger A, Quek L, Schuh A, Soneji S, et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19:138–152. doi: 10.1016/j.ccr.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 29.Al-Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev. 2004;14:43–47. doi: 10.1016/j.gde.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Trumpp A, Wiestler OD. Mechanisms of disease: cancer stem cells-targeting the evil twin. Nat Clin Pract Oncol. 2008;5:337–347. doi: 10.1038/ncponc1110. [DOI] [PubMed] [Google Scholar]
- 31.Helgason GV, Young GA, Holyoake TL. Targeting chronic myeloid leukemia stem cells. Curr Hematol Malig Rep. 2010;5:81–87. doi: 10.1007/s11899-010-0043-0. [DOI] [PubMed] [Google Scholar]
- 32.Valent P. Targeting of leukemia-initiating cells to develop curative drug therapies: straightforward but nontrivial concept. Curr Cancer Drug Targets. 2011;11:56–71. doi: 10.2174/156800911793743655. [DOI] [PubMed] [Google Scholar]
- 33.McCubrey JA, Steelman LS, Abrams SL, Misaghian N, Chappell WH, Basecke J, et al. Targeting the cancer initiating cell: the ultimate target for cancer therapy. Curr Pharm Des. 2012;18:1784–1795. doi: 10.2174/138161212799859701. [DOI] [PubMed] [Google Scholar]
- 34.Barnes DJ, Melo JV. Primitive, quiescent and difficult to kill: the role of non-proliferating stem cells in chronic myeloid leukemia. Cell Cycle. 2006;5:2862–2866. doi: 10.4161/cc.5.24.3573. [DOI] [PubMed] [Google Scholar]
- 35.Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21:926–935. doi: 10.1038/sj.leu.2404609. [DOI] [PubMed] [Google Scholar]
- 36.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol. 2011;29:591–599. doi: 10.1200/JCO.2010.31.0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schulenburg A, Blatt K, Cerny-Reiterer S, Sadovnik I, Herrmann H, Marian B, Grunt TW, Zielinski CC, Valent P. Cancer stem cells in basic science and in translational oncology: can we translate into clinical application? J Hematol Oncol. 2015;8:16. doi: 10.1186/s13045-015-0113-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou HS, Carter BZ, Andreeff M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: yin and yang. Cancer Biol Med. 2016;13:248–259. doi: 10.20892/j.issn.2095-3941.2016.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holyoake TL, Vetrie D. The chronic myeloid leukemia stem cell: stemming the tide of persistence. Blood. 2017;129(12):1595–1606. doi: 10.1182/blood-2016-09-696013. [DOI] [PubMed] [Google Scholar]
- 41.Greaves M. Cancer stem cells: back to Darwin? Semin Cancer Biol. 2010;20:65–70. doi: 10.1016/j.semcancer.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 42.Clappier E, Gerby B, Sigaux F, Delord M, Touzri F, Hernandez L, et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med. 2011;208:653–661. doi: 10.1084/jem.20110105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer. 2012;12:767–775. doi: 10.1038/nrc3368. [DOI] [PubMed] [Google Scholar]
- 44.Horton SJ, Huntly BJ. Recent advances in acute myeloid leukemia stem cell biology. Haematologica. 2012;97:966–974. doi: 10.3324/haematol.2011.054734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valent P, Bonnet D, Wöhrer S, Andreeff M, Copland M, Chomienne C, Eaves C. Heterogeneity of neoplastic stem cells: theoretical, functional, and clinical implications. Cancer Res. 2013;73(3):1037–1045. doi: 10.1158/0008-5472.CAN-12-3678. [DOI] [PubMed] [Google Scholar]
- 47.Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 48.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366:1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulenburg A, Cech P, Herbacek I, Marian B, Wrba F, Valent P, et al. CD44-positive colorectal adenoma cells express the potential stem cell markers musashi antigen (msi1) and ephrin B2 receptor (EphB2) J Pathol. 2007;213:152–160. doi: 10.1002/path.2220. [DOI] [PubMed] [Google Scholar]
- 52.Karlic H, Herrmann H, Schulenburg A, Grunt TW, Laffer S, Mirkina I, et al. Tumor stem cell research—basis and challenge for diagnosis and therapy. Wien Klin Wochenschr. 2010;122:423–436. doi: 10.1007/s00508-010-1408-z. [DOI] [PubMed] [Google Scholar]
- 53.Grunt TW, Hebar A, Laffer S, Wagner R, Peter B, Herrmann H, et al. Prominin-1 (CD133, AC133) and dipeptidyl-peptidase IV (CD26) are indicators of infinitive growth in colon cancer cells. Am J Cancer Res. 2015;5:560–574. [PMC free article] [PubMed] [Google Scholar]
- 54.Mirkina I, Hadzijusufovic E, Krepler C, Mikula M, Mechtcheriakova D, Strommer S, et al. Phenotyping of human melanoma cells reveals a unique composition of receptor targets and a subpopulation co-expressing ErbB4, EPO-R and NGF-R. PLoS ONE. 2014;9:e84417. doi: 10.1371/journal.pone.0084417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blatt K, Herrmann H, Stefanzl G, Sperr WR, Valent P. Evaluation of in vitro effects of various targeted drugs on plasma cells and putative neoplastic stem cells in patients with multiple myeloma. Oncotarget. 2016;7:65627–65642. doi: 10.18632/oncotarget.11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valent P, Grunt T, Karlic H, Herrmann H, Marian B, Schulenburg A, et al. The Vienna Cancer Stem Cell Club (VCSCC): first 10 years and future perspectives. J Life Sci. 2013;7:577–585. [Google Scholar]
- 57.Valent P, Horny HP, Bennett JM, Fonatsch C, Germing U, Greenberg P, et al. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: consensus statements and report from a working conference. Leuk Res. 2007;31:727–736. doi: 10.1016/j.leukres.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 58.Schanz J, Tüchler H, Solé F, Mallo M, Luño E, Cervera J, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30:820–829. doi: 10.1200/JCO.2011.35.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–2465. doi: 10.1182/blood-2012-03-420489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pfeilstöcker M, Tuechler H, Sanz G, Schanz J, Garcia-Manero G, Solé F, et al. Time-dependent changes in mortality and transformation risk in MDS. Blood. 2016;128:902–910. doi: 10.1182/blood-2016-02-700054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert BL. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Malcovati L, Cazzola M. The shadowlands of MDS: idiopathic cytopenias of undetermined significance (ICUS) and clonal hematopoiesis of indeterminate potential (CHIP) Hematology Am Soc Hematol Educ Program. 2015;2015:299–307. doi: 10.1182/asheducation-2015.1.299. [DOI] [PubMed] [Google Scholar]
- 63.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shlush LI. Age-related clonal hematopoiesis. Blood. 2018;131:496–504. doi: 10.1182/blood-2017-07-746453. [DOI] [PubMed] [Google Scholar]
- 65.Gibson CJ, Lindsley RC, Tchekmedyian V, Mar BG, Shi J, Jaiswal S, et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol. 2017;35:1598–1605. doi: 10.1200/JCO.2016.71.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takahashi K, Wang F, Kantarjian H, Doss D, Khanna K, Thompson E, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol. 2017;18:100–111. doi: 10.1016/S1470-2045(16)30626-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gaidzik VI, Weber D, Paschka P, Kaumanns A, Krieger S, Corbacioglu A, et al. German-Austrian Acute Myeloid Leukemia Study Group (AMLSG). DNMT3A mutant transcript levels persist in remission and do not predict outcome in patients with acute myeloid leukemia. Leukemia. 2018;32:30–37. doi: 10.1038/leu.2017.200. [DOI] [PubMed] [Google Scholar]
- 68.Jongen-Lavrencic M, Grob T, Hanekamp D, Kavelaars FG, Al Hinai A, Zeilemaker A, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378:1189–1199. doi: 10.1056/NEJMoa1716863. [DOI] [PubMed] [Google Scholar]
- 69.Hadzijusufovic E, Albrecht-Schgoer K, Huber K, Hoermann G, Grebien F, Eisenwort G, et al. Nilotinib-induced vasculopathy: identification of vascular endothelial cells as a primary target site. Leukemia. 2017;31:2388–2397. doi: 10.1038/leu.2017.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Herrmann H, Cerny-Reiterer S, Gleixner KV, Blatt K, Herndlhofer S, Rabitsch W, et al. CD34(+)/CD38(−) stem cells in chronic myeloid leukemia express Siglec-3 (CD33) and are responsive to the CD33-targeting drug gemtuzumab/ozogamicin. Haematologica. 2012;97:219–226. doi: 10.3324/haematol.2010.035006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Herrmann H, Sadovnik I, Cerny-Reiterer S, Rülicke T, Stefanzl G, Willmann M, et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood. 2014;123:3951–3962. doi: 10.1182/blood-2013-10-536078. [DOI] [PubMed] [Google Scholar]
- 73.Valent P, Sadovnik I, Ráčil Z, Herrmann H, Blatt K, Cerny-Reiterer S, et al. DPPIV (CD26) as a novel stem cell marker in Ph+ chronic myeloid leukaemia. Eur J Clin Invest. 2014;44:1239–1245. doi: 10.1111/eci.12368. [DOI] [PubMed] [Google Scholar]
- 74.Sadovnik I, Hoelbl-Kovacic A, Herrmann H, Eisenwort G, Cerny-Reiterer S, Warsch W, et al. Identification of CD25 as STAT5-dependent growth regulator of leukemic stem cells in Ph+ CML. Clin Cancer Res. 2016;22:2051–2061. doi: 10.1158/1078-0432.CCR-15-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Herrmann H, Kneidinger M, Cerny-Reiterer S, Rülicke T, Willmann M, Gleixner KV, et al. The Hsp32 inhibitors SMA-ZnPP and PEG-ZnPP exert major growth-inhibitory effects on CD34+/CD38+ and CD34+/CD38− AML progenitor cells. Curr Cancer Drug Targets. 2012;12(1):51–63. doi: 10.2174/156800912798888992. [DOI] [PubMed] [Google Scholar]
- 76.Herrmann H, Blatt K, Shi J, Gleixner KV, Cerny-Reiterer S, Müllauer L, et al. Small-molecule inhibition of BRD4 as a new potent approach to eliminate leukemic stem- and progenitor cells in acute myeloid leukemia AML. Oncotarget. 2012;3(12):1588–1599. doi: 10.18632/oncotarget.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blatt K, Herrmann H, Hoermann G, Willmann M, Cerny-Reiterer S, Sadovnik I, et al. Identification of campath-1 (CD52) as novel drug target in neoplastic stem cells in 5q-patients with MDS and AML. Clin Cancer Res. 2014;20:3589–3602. doi: 10.1158/1078-0432.CCR-13-2811. [DOI] [PubMed] [Google Scholar]
- 78.Shehata M, Hubmann R, Hilgarth M, Schnabl S, Demirtas D, Ponath E, et al. Partial characterization and in vitro expansion of putative CLL precursor/stem cells which are dependent on bone marrow microenvironment for survival. Blood. 2010;116:2433. doi: 10.1182/blood-2009-10-248054. [DOI] [Google Scholar]
- 79.Eisenwort G, Peter B, Blatt K, Cerny-Reiterer S, Hoermann G, Sadovnik I, et al. Identification of a neoplastic stem cell in human mast cell leukemia. Blood. 2014;124:817. [Google Scholar]
- 80.Blatt K, Cerny-Reiterer S, Herrmann H, Herndlhofer S, Eisenwort G, Rabitsch W, et al. Phenotypic characterization and target expression profiles in CD34+/CD38− and CD34+/CD38+ stem- and progenitor cells in acute lymphoblastic leukemia (ALL) Neoplasia. 2018 doi: 10.1016/j.neo.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Loeffler-Ragg J, Germing U, Sperr WR, Herrmann H, Zwierzina H, Valent P, et al. Serum CD44 levels predict survival in patients with low-risk myelodysplastic syndromes. Crit Rev Oncol Hematol. 2011;78:150–161. doi: 10.1016/j.critrevonc.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 82.Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 2017;18:2162–2174. doi: 10.1016/j.celrep.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gleixner KV, Mayerhofer M, Aichberger KJ, Derdak S, Sonneck K, Böhm A, et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood. 2006;107:752–759. doi: 10.1182/blood-2005-07-3022. [DOI] [PubMed] [Google Scholar]
- 84.Krauth MT, Mirkina I, Herrmann H, Baumgartner C, Kneidinger M, Valent P. Midostaurin (PKC412) inhibits immunoglobulin E‑dependent activation and mediator release in human blood basophils and mast cells. Clin Exp Allergy. 2009;39:1711–1720. doi: 10.1111/j.1365-2222.2009.03353.x. [DOI] [PubMed] [Google Scholar]
- 85.Peter B, Winter GE, Blatt K, Bennett KL, Stefanzl G, Rix U, et al. Target interaction profiling of midostaurin and its metabolites in neoplastic mast cells predicts distinct effects on activation and growth. Leukemia. 2016;30:464–472. doi: 10.1038/leu.2015.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gotlib J, Kluin-Nelemans HC, George TI, Akin C, Sotlar K, Hermine O, et al. Efficacy and safety of Midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374:2530–2541. doi: 10.1056/NEJMoa1513098. [DOI] [PubMed] [Google Scholar]
- 87.Valent P, Akin C, Hartmann K, George TI, Sotlar K, Peter B, et al. Midostaurin: a magic bullet that blocks mast cell expansion and activation. Ann Oncol. 2017;28:2367–2376. doi: 10.1093/annonc/mdx290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Florian S, Sonneck K, Hauswirth AW, Krauth MT, Schernthaner GH, Sperr WR, et al. Detection of molecular targets on the surface of CD34+/CD38−-stem cells in various myeloid malignancies. Leuk Lymphoma. 2006;47:207–222. doi: 10.1080/10428190500272507. [DOI] [PubMed] [Google Scholar]
- 89.Hauswirth AW, Florian S, Printz D, Sotlar K, Krauth MT, Fritsch G, et al. Expression of the target receptor CD33 in CD34+/CD38−/CD123+ AML stem cells. Eur J Clin Invest. 2007;37:73–82. doi: 10.1111/j.1365-2362.2007.01746.x. [DOI] [PubMed] [Google Scholar]
- 90.Loke J, Khan JN, Wilson JS, Craddock C, Wheatley K. Mylotarg has potent anti-leukaemic effect: a systematic review and meta-analysis of anti-CD33 antibody treatment in acute myeloid leukaemia. Ann Hematol. 2015;94:361–373. doi: 10.1007/s00277-014-2218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Appelbaum FR, Bernstein ID. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood. 2017;130:2373–2376. doi: 10.1182/blood-2017-09-797712. [DOI] [PubMed] [Google Scholar]
- 92.Sperr WR, Herndlhofer S, Gleixner K, Girschikofsky M, Weltermann A, Machherndl-Spandl S, et al. Intensive consolidation with G‑CSF support: tolerability, safety, reduced hospitalization, and efficacy in acute myeloid leukemia patients ≥60 years. Am J Hematol. 2017;92:E567–E574. doi: 10.1002/ajh.24847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sperr WR, Piribauer M, Wimazal F, Fonatsch C, Thalhammer-Scherrer R, Schwarzinger I, et al. A novel effective and safe consolidation for patients over 60 years with acute myeloid leukemia: intermediate dose cytarabine (2 x 1 g/m2 on days 1, 3, and 5) Clin Cancer Res. 2004;10(12 Pt 1):3965–3971. doi: 10.1158/1078-0432.CCR-04-0185. [DOI] [PubMed] [Google Scholar]
- 94.Krauth MT, Florian S, Böhm A, Sonneck K, Agis H, Samorapoompichit P, et al. Immunological characterization and antibacterial function of persisting granulocytes in leukemic patients receiving pulse cytosine arabinoside-consolidation chemotherapy on days 1, 3, and 5. J Immunol. 2006;176:1759–1768. doi: 10.4049/jimmunol.176.3.1759. [DOI] [PubMed] [Google Scholar]
- 95.Valent P, Groner B, Schumacher U, Superti-Furga G, Busslinger M, Kralovics R, et al. Paul Ehrlich (1854–1915) and his contributions to the foundation and birth of translational medicine. J Innate Immun. 2016;8:111–120. doi: 10.1159/000443526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Valent P, Akin C, Arock M, Bock C, George TI, Galli SJ, et al. Proposed terminology and classification of pre-malignant neoplastic conditions: a consensus proposal. EBioMedicine. 2017;26:17–24. doi: 10.1016/j.ebiom.2017.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Valent P, Sotlar K, Blatt K, Hartmann K, Reiter A, Sadovnik I, et al. Proposed diagnostic criteria and classification of basophilic leukemias and related disorders. Leukemia. 2017;31(4):788–797. doi: 10.1038/leu.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Valent P, Orazi A, Steensma DP, Ebert BL, Haase D, Malcovati L, et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget. 2017;8:73483–73500. doi: 10.18632/oncotarget.19008. [DOI] [PMC free article] [PubMed] [Google Scholar]