

ABSTRACT
Epithelial-to-mesenchymal transition (EMT) confers cancer cells the ability of invasion and metastasis. However, how does EMT contribute to evasion of immune surveillance is unclear, especially in salivary adenoid cystic carcinoma (SACC). In this study, we investigated the molecular link between EGF-induced EMT and the immune checkpoint ligand programmed death-ligand 1 (PD-L1) by immunoprecipitation (IP) and Westernblot analysis. Cell migration and invasion activity was assayed by transwell assay. Immunohistochemical (IHC) staining analysis was performed for measurement of EMT markers and PD-L1 expression levels in tumor tissues. We found that EGF-induced EGFR activation stabilized Snail expression and induced EMT in SACC. Interestingly, EGFR activation induced simultaneously both EMT and PD-L1 in SACC. Importantly, knockdown of Snail greatly suppressed EGF-induced EMT, but not EGF-induced PD-L1 expression; whereas knockdown of c-Myc strongly repressed PD-L1 expression, but not snail expression and EMT. The molecular link is strongly supported by robust correlations between the EMT markers and PD-L1 expression in human cancer samples.These results suggest that EGFR activated EMT and PD-L1 via two distinct mechanisms. EGFR activation induced EMT and PD-L1 expression in SACC. Snail is required for EGF-induced EMT, but not PD-L1 expression; whereas c-Myc is required for EGFR-mediated PD-L1 upregulation but not EMT. Thus, targeting activated EGFR may inhibit both EMT and PD-L1, which may potentiate the therapeutic effect of PD-L1-based immunotherapy, especially in the malignant subgroups of SACC patients with activated EGFR.
KEYWORDS: SACC, EMT, PD-L, Snail, c-Myc
Introduction
Salivary adenoid cystic carcinoma (SACC) is a rare variant of adenocarcinomas that arise mostly in the salivary glands, and account for 25% of malignant tumors in the major salivary glands and 50% in the minor glands [1]. Clinical studies indicate that SACC is characterized by a protracted clinical course with local recurrences, hematogenous metastases, and poor response to classical chemotherapeutic approaches [2]. Despite a slow growing pattern, SACC shows intriguingly a notable ability to develop distant metastases in the very early phase [3]. Numerous studies have identified the factors related to the prognosis and outcome of SACC [4,5], but little is known regarding the molecular mechanisms that control SACC metastasis. Thus, understanding the biological process of SACC metastasis and revealing the molecular mechanisms of the process would be crucial to reduce both the morbidity and mortality in SACC patients.
Epithelial-mesenchymal transition (EMT) has been shown to play a critical role in tumor invasion and metastasis. The invasive ability of malignant tumor cells can be achieved by induction of EMT [6]. During EMT, cells loose epithelial characteristics such as cell polarity and cell-cell contact, and gain mesenchymal properties such as motility [7]. A hallmark of EMT is the loss of E-cadherin expression [8]. Several transcription factors are implicated in the transcriptional repression of E-cadherin, including the zinc finger proteins of Snail, Slug, Twist, δEF1/ZEB1, SIP1, and E12/E47 [9–13]. EMT is also a dynamic and reversible process, provoked by signals, such as EGF, TGF-β, Wnt and TNF-α, from the microenvironment [14–18].
Cancer cells have developed various strategies for evading host anti-cancer immunologic attacks, including the up-regulation of PD-L1 (encoded by CD274), which induces T cell anergy and apoptosis by interacting with programmed death-1 (PD-1) receptors [19,20]. The development of immune checkpoint inhibitors that block PD-1/PD-L1 interaction has been clinically successful, with a long response time noted [21,22]. However, more than half of patients evaluated were insensitive to these agents, it is therefore important to identify the mechanism distinguishing those who may be sensitive or resistant to PD-L1 inhibitors.
EMT induction has been linked to high PD-L1 expression [23]. SACC cancer cells often have activated EGFR pathway and may invade tissues through EMT [24]. But how SACC could evade immune surveillance is unclear. Previous reports suggest that there is a connection between immune suppression and EMT, such as that Snail-induced EMT induced regulatory T cells and impaired dendritic cells [25]. Aberrant activation of EGFR is involved in SACC. As reported, 85% of SACC are with overexpressed EGFR [26]. However, mutation and gene amplification of EGFR are rare in SACC [27]. Thus, autocrine and paracrine EGFR ligands are implicated in activation of EGFR, which participates in tumor proliferation, migration, invasion and metastasis. We hypothesize that, activated EGFR signaling induces EMT and PD-L1 in SACC, which promotes immune evasion, invasion and metastasis.
Results
EGF activates EGFR pathway and induces EMT in salivary adenoid cystic carcinoma
To determine the roles of EGF signaling in SACC cells, we examined the major downstream signaling proteins of EGFR pathway in SACC cells following EGF treatment. EGF treatment increased p-EGFR, p-STAT3, p-AKT, and p-Erk in different kinetic ways (Figure 1(a)). To determine the role of EGF on EMT induction in SACC, we examined the biological and biochemical properties of SACC cells in a 3D culture model following EGF treatment for 72 hours.SACC-83 cells appear with rounded/polygonal shape in 3D culture. However, EGF treatment resulted in a marked change in morphology of SACC-83 cells, with stellate projections shape (Figure 1(b)). Importantly, E-cadherin was suppressed while vimentin expression was increased, the hallmarks of EMT induction (Figure 1(c,d)). Moreover, we examined the migration and invasion capability in SACC-83 cells treated with EGF. We found thatSACC-83 cells were much more motile and invasive following treatment with EGF (Figure 1(e)). Taken together, these results suggest that EGF activates EGFR pathway and induces EMT in salivary adenoid cystic carcinoma.
Figure 1.
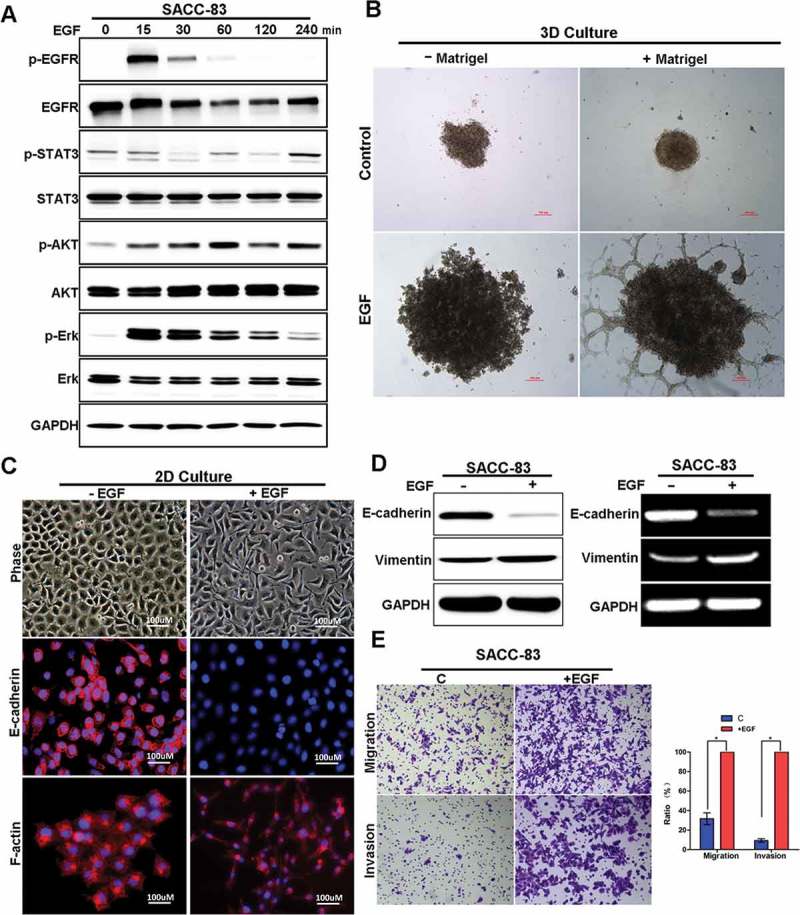
EGF activates EGFR pathway and induces EMT in SACC cells.
(A) Western blot analyzes for p-EGFR, EGFR, p-AKT, AKT, p-ERK, ERK, p-STAT3, STAT3 in SACC-83 cells treated with EGF for 0 to 4 hours.(B) 3D morphology of SACC-83 cells treated with or without EGF. Scale bar = 100 μm(C) Representative images of the 2D morphology and staining for E-cadherin and F-actin in SACC-83 cells treated with or without EGF for 72 hours. Scale bar = 100 μm.(D)Western blot and RT-PCR analysis of E-cadherin and Vimentin levels in SACC-83 cells treated with or without EGF for 72 hours.(E) The transwell migration and invasion assays established the migration and invasion capability of SACC-83 cells with representative images shown. And graphic representation of the percent of migrated cells from 3 separate experiments (mean ± SD). * indicates a p < 0.05.Scale bar = 200 μm.
Snail is crucial for EGF-induced EMT in salivary adenoid cystic carcinoma
Snail and Slug are two major transcription factors for induction of EMT and metastasis. To determine whether Snail and/or Slug were involved in EGF-induced EMT in SACC cells, we examined their expression levels in SACC-83 following EGF treatment. EGF treatment significantly increased Snail and slightly increased Slug (Figure 2(a)). For further characterization,we knocked down Snail or/and Slug expression in SACC-83 cells respectively, and treated cells with EGF for 72 hours. EGF treatment efficiently induced the morphological changes in cells that resemble mesenchymal phenotype (Figure 2(b)). However, silencing Snail, but not Slug, strongly inhibited EGF-induced EMT, suggesting that Snail is essential for EGF-induced EMT in SACC cells whereas Slug is negligible (Figure 2(b)). Consistently, immunoblot analysis showed that, while EGF treatment reduced E-cadherin in SACC cells, knockdown of Snail with siRNA restored E-cadherin in the same condition (Figure 2(c)). In addition, knockdown of Snail with siRNA dramatically decreased the numbers of migrated and invaded cells compared with those treated with siR-NC following EGF treatment (Figure 2(d,e)). Taken together, these results showed that Snail was essential for EGF-induced EMT in SACC cells.
Figure 2.
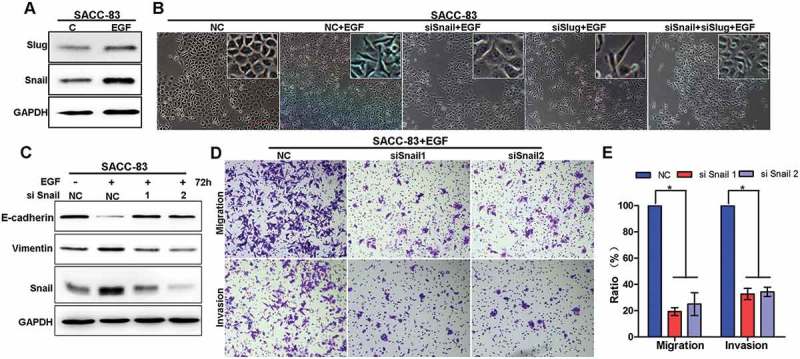
Snail is required for EGF-induced EMT in SACC cells.
(A)SACC-83 cells were treated with or without EGF for 72 h and then the expression of Snail and Slug was analyzed by Western blotting.(B) After 24-h pre-transfection with si-Snail, si-Slug, si-Snail+ Slug or si-NC siRNAs, cells were further treated with or without EGF for 72h and then the morphological changes of EMT in SACC-83 cell lines were detected by phase contrast microscopy. Scale bar = 100 μm.(C) After 24-h pre-transfection with si-Snail or si-NC short interfering RNAs (siRNAs), cells were further treated with or without EGF for 72 hand the expression of Snail, E-cadherin and Vimentin were detected by western blotting.(D) After 24-h pre-transfection with si-Snail or si-NC siRNAs, cells were further pre-treated with or without EGF for an additional 72 h. Then, cells were harvested to perform migration and invasion assays for 24 h. Cells that had migrated into the lower chamber or invaded through the matrix-gel into the under-side of the filter were fixed, stained and counted. The representative images was shown. Scale bar = 200 μm.(E) Graphic representation of the percent of migrated cells from 3 separate experiments (mean ± SD). * indicates a p < 0.05.
EGF induces Snail stabilization by inhibiting its ubiquitination
Next, we investigated how EGF regulates Snail expression for EMT induction in SACC cells. Firstly, we examined the kinetic features of Snail following EGF treatment. EGF treatment increased the level of Snail in SACC-83 (Figure 3(a,b)). Interestingly, the mRNA level of Snail did not show significant change after EGF treatment (Figure 3(c,d)). These results suggest that EGF-induced elevation of Snail is likely through protein stabilization, or in a transcription-independent manner.
Figure 3.
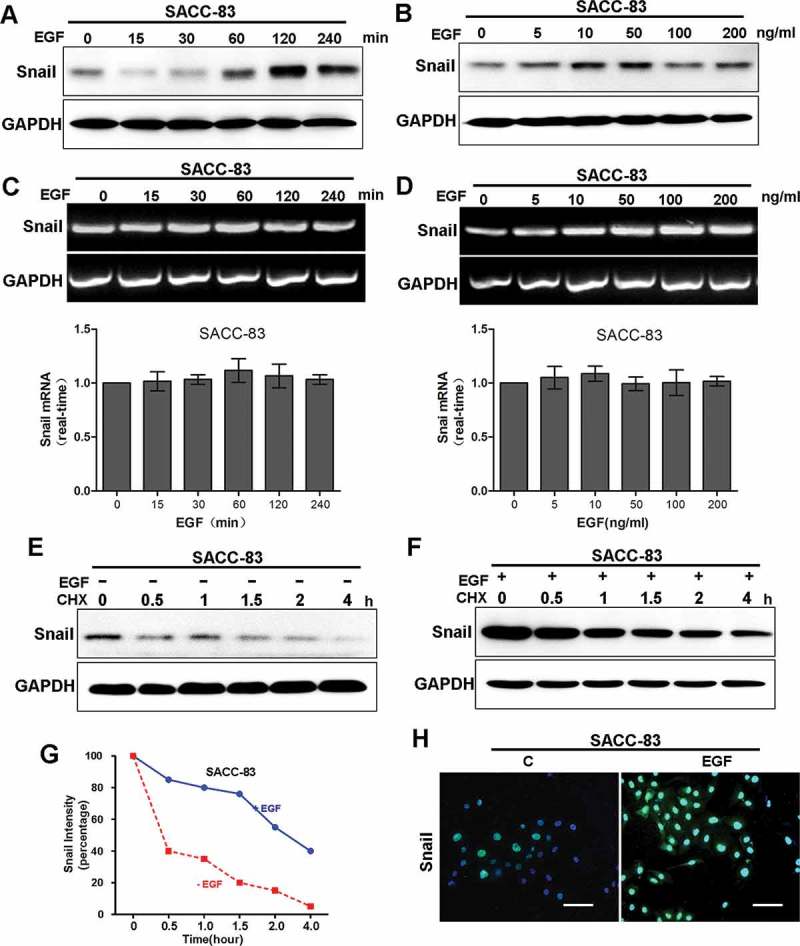
EGF induces protein stabilization of Snail.
(A) Western blot analyzes for Snail in SACC-83 cells treated with 50 ng/mL EGF for different time intervals for 4 hours.(B) Western blot analyzes for Snail from SACC-83 cells treated with 0 to 200 ng/mL EGF for 2 hours.(C) RT-PCR analyzes for mRNA levels of Snail for different time intervals.(D) RT-PCR analzses for mRNA levels of Snail from SACC-83 cells treated with 0 to 200 ng/mL EGF for 2 hours.(E-F) SACC-83 cells were treated with or without EGF for 2 hr, followed by incubation with cycloheximide (CHX; 10 μM) for an extended period of time. The levels of Snail were determined by western blot analysis.(G) Graphic representation of densitometry results for Snail after cycloheximide treatment (circle, with EGF; square, without EGF).(H) Immunofluorescence staining for Snail in SACC-83 cells treated with or without 50 ng/mL EGF for 2 hours.
Snail is a labile protein [18]. To determine the mechanism of EGF-induced Snail stabilization, we examined Snail protein levels in cells treated with or without EGF for various time (up to 4 hour) in presence or absence of protein translational inhibitor cycloheximide (CHX). The results showed that, Snail was more stable in cells treated with EGF than those without (Figure 3(e-g)). This observation was further confirmed via immunofluorescence (IF) microscopy, in which the staining intensity of Snail was dramatically increased in SACC-83 cells treated with EGF (Figure 3(h)). Taken together, these results suggest that EGF treatment induces stabilization of Snail.
We then asked whether EGF-induced Snail stabilization is through regulation of the ubiquitination pathway since EGF-induced stabilization of Snail occurs at protein level. Via immunoprecipitation (IP)-Westernblot analysis, we found that EGF treatment significantly reduced ubiquitination on exogenous Flag-tagged Snail expressed in transfected HEK293 cells (Figure 4(a)). Consistently, ubiquitination on endogenous Snail was also dramatically suppressed following EGF treatment (Figure 4(b)). Taken together, these results demonstrated that EGF stabilizes Snail by suppression of its ubiquitination.
Figure 4.
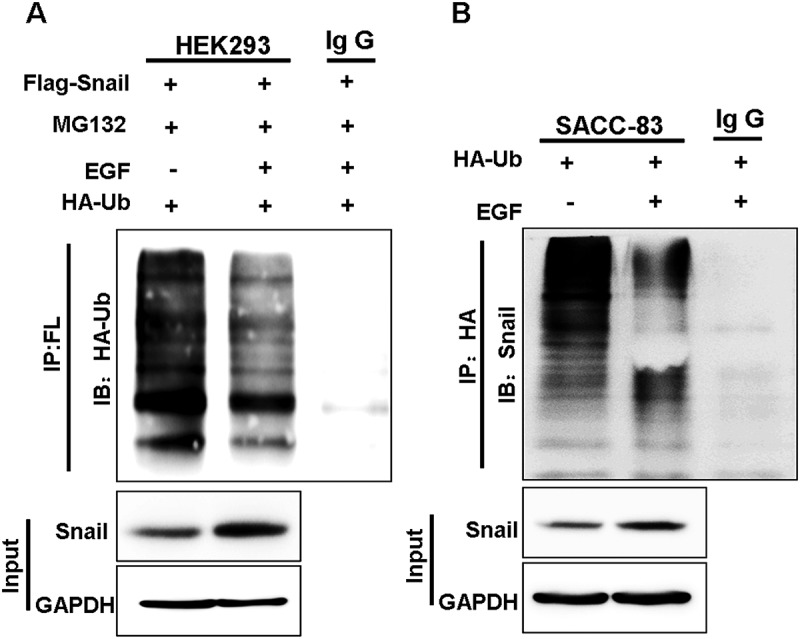
EGF inhibits the ubiquitination of Snail.
(A) HA-ubiquitin was expressed in Snail/HEK293 cells followed by treatment with or without EGFand MG-132 for 2 hr. Ubiquitin was immunoprecipitated from equal amount of lysates and the ubiquitinated Snail was examined by western blotting.(B) SACC-83 cells were either treated with or without EGF. Endogenous Snail was immunoprecipitated, and the bound of ubiquitin was detected by western blotting.
EGF-mediated Snail stabilization is dependent on EGFR activation
EGF triggers downstream signaling pathways via binding to its receptor, EGFR. To validate if EGFR activation is essential for EGF-induced stabilization of Snail, we examined the effects of different inhibitors of EGFR. The results showed that all EGFR inhibitors, including erlotinib, gefitinib and AG1478, completely blocked EGF-induced stabilization of Snail (Figure 5(a)). Consistently, while EGF induced the morphological changes of EMTinSACC-83 cells, from atypical polygonal morphology of epithelial cells to a disseminated, fibroblast-like morphology, erlotinib treatment abrogated EGF-induced EMT phenotype changes, remaining in the polygonal epithelial morphology (Figure 5(b)).Moreover, erlotinib treatment resulted in upregulation of E-cadherin (Figure 5(c)) and reduction of migration and invasion in the EGF-induced EMT model (Figure5(d)). Similarly, while EGF treatment induced large size colonies of SACC-83 cells in 3D culture model, erlotinib treatment resulted in a marked suppression the size of colonies and changed the morphology from aggressive shape back to benign one with rounded/polygonal features (Figure 5(e)). To determine which downstream signaling pathway of EGFR is essential for EGF-induced stabilization of Snail, we examined the effects of inhibitors of AKT, STAT3, and ERK pathways. The results showed that the STAT3 inhibitor stattic completely blocked EGF-induced stabilization of Snail, whereas the inhibitors of ERK and AKT showed only partial inhibition (Figure 5(f)). We also examined the effect of EGFR inhibitor in vivo. The result showed that, erlotinib treatment greatly reduced the number of metastatic nodules when compared to control group (Figure 5(g)). Taken together, these results strongly suggest that the activated EGFR pathway is crucial for EGF-induced stabilization of Snail, and STAT3 signaling plays a major role in mediating the effect.
Figure 5.
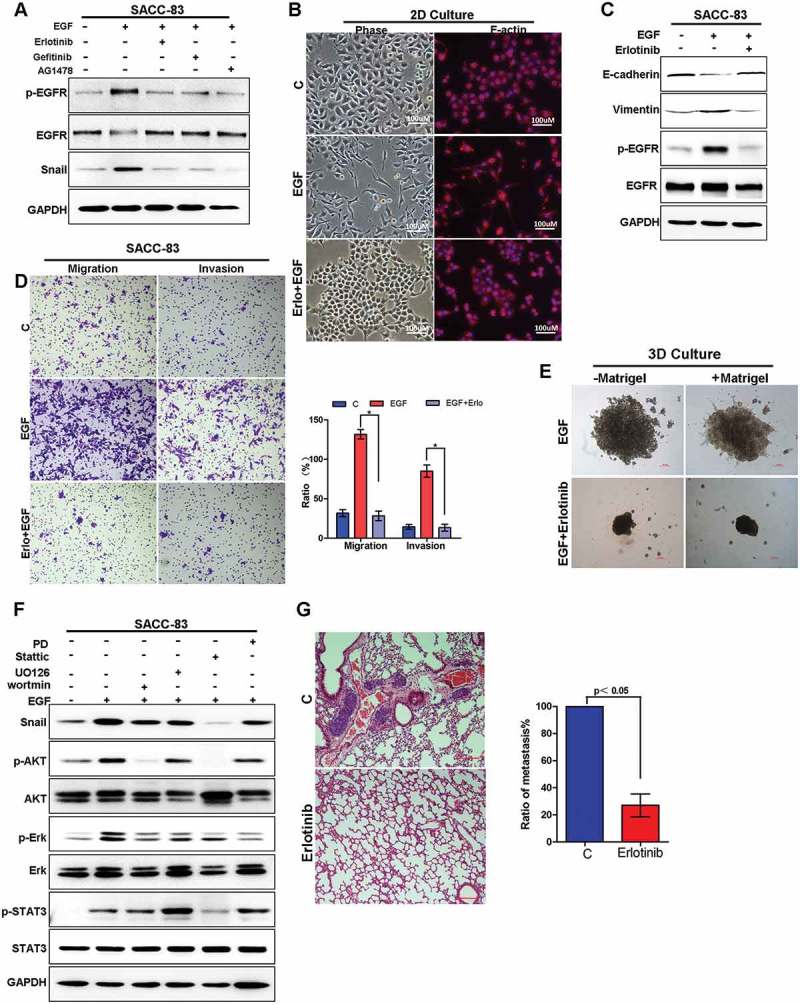
Targeting EGFR inhibits EGF-induced migration and invasion capabilities of SACC both in vitro and in vivo.
(A) SACC-83 cells were pre-treated with either erlotinib, geftinib or AG1478 for 1 h before stimulation with EGF for 2 h. The expression of Snail and the activation of EGFR were examined by western blotting.(B) The morphology change of SACC-83 cells treated with or without EGF and erlotinib visualized by phase-contrast microscopy, or the cells were stained with the F-actin.(C) Western blot analysis of E-cadherin, vimentin, p-EGFR and EGFR levels from SACC-83 cells treated with or without EGF and erlotinib for 72 hours.(D) Representative images of migration and invasion by SACC-83 cells treated with or without EGF and erlotinib for 72 hours. And graphic representation of percent migrated and invasion cells from 3 separate experiments with mean ± SD percent indicated. * indicates a p < 0.05.(E) 3D morphology of SACC-83 cells treated with or without EGF and erlotinib. Scale bar = 100 μm.(F) Western blot analysis of Snail, p-AKT, AKT, -ERK, ERK, p-STAT3, STAT3 from SACC-83 cells pretreated with various inhibitors for 1 hr followed by stimulation with EGF for 2 hr.(G) SACC-LM cells treated with or without erlotinib for 72 hours were injected into the tail vein of nude mice. Histopathologic analysis shows small metastatic nodules in lung tissues. Graphic representation of the area of lung metastases with mean ± SD; n = 5.
PD-L1 upregulationis associated with EGFR-mediated EMT in SACC
The immunomodulatory interaction of PD1 and PD-L1are emerged as the most promising targets for immunotherapy in malignant melanoma and non-small cell lung cancer[28].To determine if PD-L1 upregulation were involved in EGF-induced EMT status in salivary gland carcinoma, we examined the expression of PD-L1 in EGF-induced EMT in vitro. The analysis showed that EGF treatment increased the expression of PDL1 in both protein and mRNA levels (Figure 6(a)). The results is consistent with the report that EMT induction increases PD-L1 on breast cancer cells and hence promotesintratumoralCD8+ T cells immunosuppression and metastasis [29]. To determine whether EGF-induced PD-L1 is mediated by EGFR kinase activity, we treated SACC-83 cells with EGF in absence or presence of EGFR inhibitors erlotinib, or geftinib, or AG1478. The results showed that EGF-induced PD-L1 was greatly reduced in presence of EGFR inhibitors, suggesting induction of PD-L1 is dependent on the activated EGFR (Figure 6(b) left). Consistently, knockdown of EGFR with siRNA-EGFR dramatically suppressed EGF-induced expression of PD-L1 in SACC-83 cells (Figure 6(b) right). To determine which downstream signaling pathways of EGFR is essential for EGF-induced PD-L1 expression, we examined the effects of inhibitors of EGFR, Akt, STAT3, and ERK. The EGFR inhibitor erlotinib completely blocked EGF-induced PD-L1, whereas the inhibitors of STAT3, ERK and PI3K showed only partial inhibition (Figure 6(c)). Taken together, the activated EGFR pathway is crucial for EGF-induced expression of PDL1, and the downstream of EGFR, STAT3, MAPK and PI3K, all partially contribute to the effect.
Figure 6.
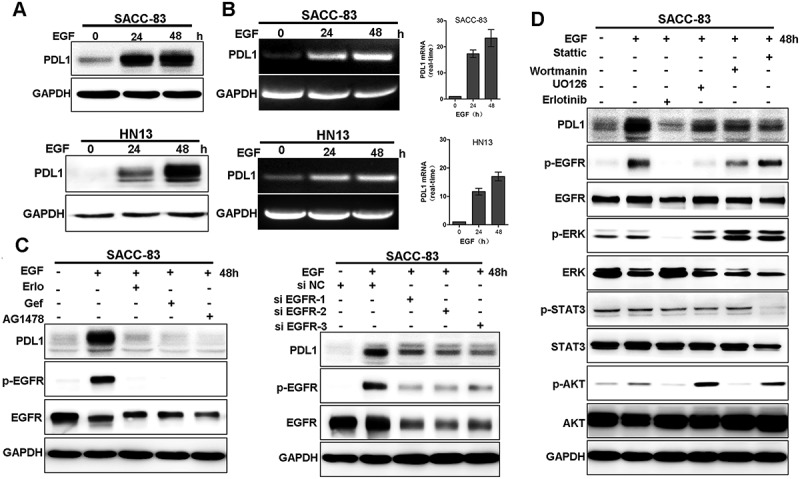
EGF induces PD-L1 expression through EGFR and downstream pathway.
(A) SACC-83 cells were treated with EGF for 24 and 48 hours and the expression of PD-L1 were examined by western blotting and RT-PCR.(B) SACC-83 cells were pre-treated with either erlotinib, or gefitinib or AG1478 for 1 h before stimulation with EGF for 48 h. Or after 24-h pre-transfection with si-EGFR or si-NC siRNAs, cells were further treated with EGF for an additional 48 h. The expression of PD-L1 and the activation of EGFR were examined by western blotting.(C) Western blot analysis of PD-L1, p-EGFR, EGFR, p-AKT, AKT, p-ERK, ERK, p-STAT3, STAT3 from SACC-83 cells pretreated with various inhibitors for 1 hr followed by stimulation with EGF for 48 hr.
c-Myc is required for EGF-induced PD-L1 expression in salivary adenoid cystic carcinoma
As PD-L1 upregulation was involved in EGFR-mediated EMT in SACC, we asked whether EMT status was required for PD-L1 induction. EGF-induced Snail stabilization results in EMT and cancer metastasis in SACC (Figures 2–5). To determine a putative role of Snail in regulation of PD-L1 expression, we silenced Snail in SACC-83 cells via siRNA. Interestingly, although Snail silencing strongly restored E-cadherin levels in SACC-83cells, EGF-induced PD-L1 was not affected (Figure 7(a)).This suggests that upregulation of PD-L1,although associated with, is actually independent of Snail and EMT.
Figure 7.
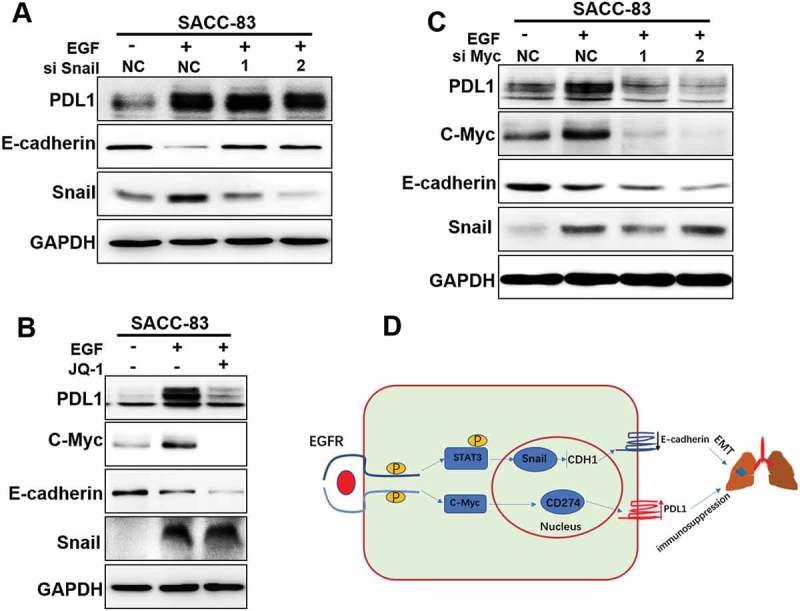
c-Myc is required for EGF-induced PD-L1 expression, but not EMT.
(A) After 24-h pre-transfection with si-Snail or si-NC short interfering RNAs (siRNAs), cells were further treated with or without EGF for 72 hand the expression of PD-L1, E-cadherin and Vimentin were detected by western blotting.(B) Western blot analysis of PD-L1 and c-Myc expression from SACC-83 cells pretreated with JQ-1 for 1 hr followed by stimulation with EGF for 48 hr.(C) After 24-h pre-transfection with si-c-Myc or si-NC short interfering RNAs (siRNAs), cells were further treated with EGF for 48 hand the expression of PD-L1 and C-Myc were detected by western blotting.(D) A proposed model to illustrate the crosstalk of EGF induced EMT and PD-L1 induction during SACC metastasis.
We then asked what mechanism is involved in upregulation of PD-L1 in EMT-activated SACC-83 cells. In SACC cells, we noted that the EGF-induced PD-L1upregulationis primarily at transcription level as EGF influence PD-L1 mRNA expression (Figure 6(a)), which is different from what reported in breast cancer, EGF-induced PD-L1is mainly via glycosylation at post transcription level [30]. Previous study suggests that c-myc is involved in regulation of PD-L1 and c-myc can bind to the promoters of the PD-L1 gene and induce its expression [31,32].To further determine the role of c-Myc in EGF-induced PD-L1, we treated cells with EGF, and EGF plus a Myc inhibitor JQ-1. The results showed that EGF induced c-myc as well as PD-L1. Importantly, although myc inhibitor JQ-1greatly suppressed EGF-induced PD-L1, the expression level of E-cadherin was not affected (Figure 7(b)).To further validate the result, we examined the effect of knocked down c-Myc by siRNA. And the results showed that knockdown c-Myc greatly suppressed EGF-induced PD-L1 in SACC-83 cells, whereas expression of snail and E-cadherin was not affected (Figure 7(c)). Thus, c-Myc is the critical mediator for EGF-induced PD-L1 expression, but not EMT in SACC.
Correlations of EMT, PD-L1, and p-EGFR expression in human tumor tissues
As reported before, primary ACC of the mandible is an extremely rare occurrence. And patients with primary ACC of the mandible have a poor prognosis due to local recurrence and distant metastases[33]. To search for the clinic relevance of our observations, we investigated whether there is a correlation between EGFR-mediated EMT and PD-L1 induction in the patients with mandible ACC. In one example, examination Patient #A revealed swelling of the left mandible, ranging from the medial of tooth #45 to the medial of tooth #48. Panoramic radiography and Maxillofacial CT suggested an odontogenic tumor in the left mandible (Figure 8(b)). The patient was diagnosed as primary mandible adenoid cystic carcinoma by the hematoxylin and eosin staining in histology pathology(Figure 8(a)). Moreover, compared with adjacent normal tissues, tumor tissues show relative low E-cadherin expression and high vimentin expression, which are EMT markers. Furthermore, tumor tissues also show high PD-L1 expression with activated EGFR and activated STAT3 (Figure 8(a)).
Figure 8.
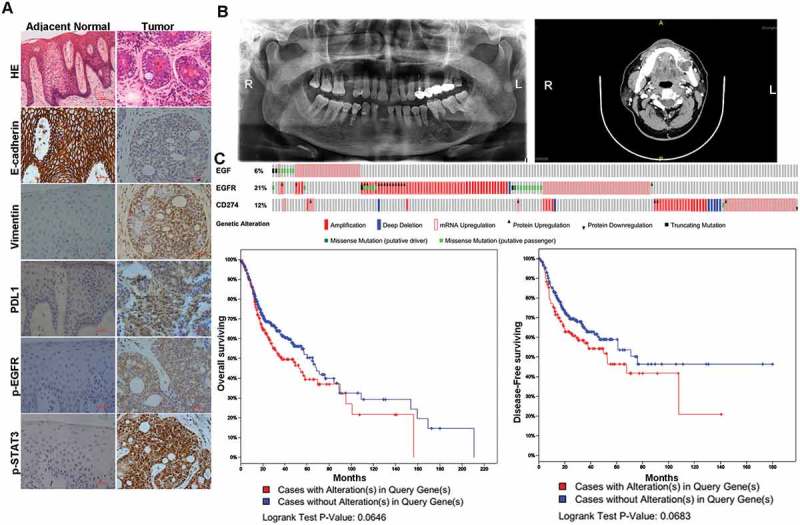
EGFR activation-mediated EMT is correlated with PD-L1 induction in human cancer simples.
(A) HE and IHC staining of E-cadherin, Vimentin, PD-L1, p-EGFR and p-STAT3 from a patient diagnosed as primary mandibular adenoid cystic carcinoma.(B) The panoramic radiography and Maxillofacial CT of the mandibular adenoid cystic carcinoma patient.(C) OncoPrint of EGF-EGFR-CD274 pathway alterations and survival analysis in head and neck cancer. Genomic alterations of different members in this pathway are mutually exclusive. Patients with EGF-EGFR-CD274 pathway alteration have worse overall survival and disease-free survival than cases without an alteration. Case with alteration(s) means patient with genetic alteration of either EGF, EGFR or CD274 and case without alteration(s) means patient has none genetic alteration of either EGF, EGFR or CD274.
To search for genomic alterations in the EGF-EGFR-PD-L1 pathway in the TCGA data, we performed analysis by using the cBio Cancer Genomics Portal (http://cbioportal.org) as described before [34]. The OncoPrint shows that alterations of genes within the EGF-EGFR-PD-L1 pathway tend to be mutually exclusive (Figure 8(c)), suggesting overexpression of these proteins have similar functional role. Moreover, survival analysis shows that cases with an EGF-EGFR-PD-L1 pathway alteration have worse overall survival and disease-free survival than cases without alteration(Figure 8(d)). Thus, activation of the EGFR–PD-L1 axis is associated with EMT, metastasis, which predicates with a poor prognosis in cancer patients.
Discussion
In this study, we provide evidence that EMT is linked to the evasion of anti-tumor immunity. We showed that activated EGFR induced both EMT and PD-L1 expression in SACC. We found that, EGFR-induced EMT is in a snail-dependent manner, whereas EGFR-induced PD-L1 is a Snail-independent but c-Myc-dependent manner.
Previously, we showed that the highly metastatic SACC cells exhibited the EMT-like characteristics compared to the low-metastatic cells. And activated EGFR signaling pathway promotes the EMT-like and metastatic features of SACC [24]. In this study, we confirm that EGF induces EMT and promotes SACC invasion and metastasis by activating EGFR pathway. Moreover we identify that Snail is critical for EGFR-mediated EMT in SACC. Previously studies showed that Snail is mainly regulated by GSK-3β-mediated phosphorylation and degradation [35,36]. Snail is also stabilized by inflammatory cytokines including IL-6 and TNF-α, in which activation of the NF-κB pathway is required to block the ubiquitination and degradation of Snail. In this study, we found that EGFR signaling stabilizes Snail and induce EMT.
The PD-1/PD-L1 pathway plays an important role in inhibition of T cell response and evasion of host immunity [22,37]. Blockade of the PD-L1/PD-1 pathway has emerged as a promising therapeutic strategy to reduces tumor aggressiveness and improves survival [38–40]. However, the mechanism of PD-L1 upregulation is not fully understood. Elevated tumor-associated PD-L1 levels could be caused by various mechanisms, including exposure to extracellular cytokines secreted by tumor bystander cells, or intrinsic cancerous changes linked to the EMT and carcinogenesis. Chen et al. recently demonstrated a molecular link between the epithelial-mesenchymal transition (EMT) and intra-tumoural CD8+ T cell suppression through PD-L1 regulation, in both animal models and human cell lines [41]. However, the mechanism underlying PD-L1 is unclear in salivary gland carcinoma. In this study, we provide a mechanism for EGFR-mediated EMT and EGFR-inducedPD-L1 upregulation in SACC, which may lead to develop effective therapeutic approach.
Previously, it was shown that EGF induced PD-L1 in breast cancer. In breast cancer cells, EGF stabilizes PD-L1 through glycosylation via GSK3β inactivation at post-translational level [30]. In our study, we found that knockdown c-Myc greatly suppressed EGF-induced PD-L1 in SACC-83 cells, suggesting that EGF-inducedPD-L1 is c-Myc-dependent in SACC. Our observation was consistent with several previous reports. It was shown that suppression of Myc in mouse tumors and human tumor cells caused a reduction in the levels ofPD-L1 messenger RNA and protein. And Myc was found to bind directly to the promoters of PD-L1 gene. Myc inactivation in mouse tumors down-regulated PD-L1expression and enhanced the antitumor immune response [32]. Thus, Myc was a direct regulator of PD-L1 expression, and therefore is the major target for PD-L1 based therapy.
Taken together, EGFR-mediated upregulation of PD-L1 was found in EMT-activated SACC cells driven by Snail and c-Myc. Therefore, the use of EMT inhibitors as adjuvant with immunotherapeutic strategies may be beneficial for immunotherapy against SACC with mesenchymal metastatic features.
Materials and methods
Cell cultures and reagents
SACC-83 and HN13 cells were obtained from Shanghai 9th People’s Hospital. The SACC-83 cell line with a low lung metastasis rate, was isolated from a patient diagnosed pathologically with SACC in the sublingual gland as describe before[42]. The HNSCC cell line HN13 was kindly provided by the University of Maryland School of Dentistry and cultured in Dulbecco’s modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum. The human embryonic kidney HEK293 was purchased from the American Type Culture Collection (Manassas, VA). SACC-83 cells were cultured in RPMI-1640 (Gibco BRL, Grand Island, NY) with 10% fetal bovine serum (Gibco) and incubated in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Recombinant Human EGF was obtained from R&D Systems (Minneapolis, MN, USA). Antibodies against GAPDH were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for E-cadherin, Vimentin, Snail, Slug, p-EGFR, p-AKT, p-ERK, p-SATAT3, EGFR, AKT, Erk, STAT3 and PD-L1 were from Cell Signaling Technology Inc. (Beverley, MA,). HRP-conjugated secondary antibodies were from eBioScience (San Diego, CA). Smartpool siRNA was from Biomics. The Snail siRNA target sequence was: F-CAGAUGUCAAGAAGUACCAdTdT; R-UGGUACUUCUUGACAUCUGdTdT. The Slug siRNA target sequence was: F-ACUACAGUCCAAGCUUUCAdTdT; R-UGAAAGCUUGGACUGUAGUdTdT. The Slug EGFR siRNA target sequence was: F-GUCGCUAUCAAGGAAUUAAdTdT; R-UUAAUUCCUUGAUAGCGACdTdT.
Western blotting and immunoprecipitation
The experimental protocol was performed as described previously[43]. Cells were lyzed with RIPA lysis buffer (Cell Signaling Technology). Cell lysates were separated by SDS-PAGE in a 10% acrylamide gel and transferred onto a nitrocellulose membrane for immunoblot analysis. Immunoprecipitation was performed with 2 ug of antibody against Snail, Ubiquitin or normal IgG (as a negative control) in 1.0-mg whole-cell lysate. Cell lysates and/or immunoprecipitation cellular proteins were separated by SDS-PAGE in a 10% acrylamide gel and transferred onto nitrocellulose membrane for immunoblot.
Immunofluorescence
The experimental protocol was performed as described previously[44]. Cultured cells rinsed three times with PBS and fixed with 3.7% formaldehyde were permeabilized with 0.1% Triton X-100. After blocking in 1% BSA for 1 hour, cells were incubated with the primary antibody in a moist, 4ºC chamber overnight, washed and then incubated for 1 hour with Alexa Fluor 488 (in the dark) donkey anti-rabbit IgG antibody (Invitrogen, Grand Island, NY) at room temperature . Washed cells (PBS containing 0.02% Tween 20), stained by mounting onto a slide with aqueous mounting medium containing 0.5 mg/ml 40–6-diamidino-2-phenylindole, were examined with a fluorescence microscope.
Reverse transcriptasepolymerase chain reaction (RT-PCR)
The experimental protocol was performed as described previously[6]. Total RNA samples were extracted with TriPure Isolation Reagent (Roche, Switzerland) and cDNA prepared from 1ug of total RNA using the SuperScript III System (Invitrogen Life Technologies). The mRNAs levels were determined by RT-PCR, using the following primers:
GAPDH (F: 5ʹ-TCCACCACCCTGTTGCTGTA −3ʹ and R: 5ʹ-ACCACAGTCCATGCCATCAC −3ʹ); Snail (F: 5ʹ-CGCGCTCTTTCCTCGTCAG-3ʹ and R: 5ʹ-TCCCAGATGAGCATTGGCAG-3ʹ); PDL1 (F: 5ʹ-GGTGGTGCCGACTACAA-3ʹ and R: 5ʹ-TAGCCCTCAGCCTGACAT-3ʹ).
Transwell assay
The experimental protocol was performed as described previously[6]. For the migration assay, cells (5x105) were seeded onto the upper chamber in 200 µL of serum-free medium; the lower compartment was filled with 0.6 mL of DMEM media supplemented with 10% of FBS. After 24 h incubation, migrated cells on the lower surface of the filter were fixed and stained using 1% crystal violet; cells on the upper side were removed using a rubber scraper. Reported data are counts of migrated cells with experiments performed in triplicate. The procedure used for invasion assay was similar to that of the cell migration assay, except that the transwell membrane was coated with Matrigel.
Experimental lung metastasis model
This study was approved by the ethics committee of our institution. The experimental protocol was performed as described previously [24].Cells (1 × 106/0.2 ml) were injected into the tail vein of BALB/c nude mice. Animals were sacrificed after 5 weeks, and the metastatic tumors in the lung assessed. No mice showed notable toxic effects or body weight loss during the experiment. Visible lung metastatic nodules were counted macroscopically or in paraffin embedded sections stained with H&E. Data were analyzed using Student’s t test; a P value less than 0.05 was considered significant.
Statistical analysis
Data analysis used SPSS (Statistic Package for Social Sciences) 13.0 for Windows (SPSS Inc., Chicago, IL, USA). Unpaired Student’s t-tests or U-Mann Whitney tests determined statistical significance between groups with P values < 0.05 considered significant.
Funding Statement
This work was supported by Shanghai Natural Science Foundation of China 17ZR1416300 (to YW), by grant of National Nature Science Foundation of China 8120213(to DXY), 81472516 (to JZH), 81620108022 (to JD), 81602370 (to DWZ), 81771127(to LZ), by Shanghai Summit & Plateau Disciplines, and by grants of Shanghai Municipal Planning Commission Clinical Center Project, grants of Shanghai Municipal Health Bureau 2012173(to DXY).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Laurie SA, Ho AL, Fury MG, et al. Systemic therapy in the management of metastatic or locally recurrent adenoid cystic carcinoma of the salivary glands: a systematic review. Lancet Oncol. 2011;12:815–824. [DOI] [PubMed] [Google Scholar]
- [2].Coca-Pelaz A, Rodrigo JP, Bradley PJ, et al. Adenoid cystic carcinoma of the head and neck–an update. Oral Oncol. 2015;51:652–661. [DOI] [PubMed] [Google Scholar]
- [3].Sung MW, Kim KH, Kim JW, et al. Clinicopathologic predictors and impact of distant metastasis from adenoid cystic carcinoma of the head and neck. Arch Otolaryngology–Head Neck Surg. 2003;129:1193–1197. [DOI] [PubMed] [Google Scholar]
- [4].He JF, Ge MH, Zhu X, et al. Expression of RUNX3 in salivary adenoid cystic carcinoma: implications for tumor progression and prognosis. Cancer Sci. 2008;99:1334–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vekony H, Raaphorst FM, Otte AP, et al. High expression of Polycomb group protein EZH2 predicts poor survival in salivary gland adenoid cystic carcinoma. J Clin Pathol. 2008;61:744–749. [DOI] [PubMed] [Google Scholar]
- [6].Liu S, Liu L, Ye W, et al. High vimentin expression associated with lymph node metastasis and predicated a poor prognosis in oral squamous cell carcinoma. Sci Rep. 2016;6:38834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liu S, Ye D, Guo W, et al. G9a is essential for EMT-mediated metastasis and maintenance of cancer stem cell-like characters in head and neck squamous cell carcinoma. Oncotarget. 2015;6:6887–6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kang Y, Massague J.. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118:277–279. [DOI] [PubMed] [Google Scholar]
- [9].Thiery JP, Morgan M.. Breast cancer progression with a Twist. Nat Med. 2004;10:777–778. [DOI] [PubMed] [Google Scholar]
- [10].Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. [DOI] [PubMed] [Google Scholar]
- [11].Bolos V, Peinado H, Perez-Moreno MA, et al. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116:499–511. [DOI] [PubMed] [Google Scholar]
- [12].Comijn J, Berx G, Vermassen P, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–1278. [DOI] [PubMed] [Google Scholar]
- [13].Vandewalle C, Comijn J, De Craene B, et al. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 2005;33:6566–6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Christofori G. New signals from the invasive front. Nature. 2006;441:444–450. [DOI] [PubMed] [Google Scholar]
- [15].Lopez-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med. 2009;1:303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wu Y, Zhou BP. Inflammation: a driving force speeds cancer metastasis. Cell Cycle (Georgetown, Tex). 2009;8:3267–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Miettinen PJ, Ebner R, Lopez AR, et al. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127:2021–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wu Y, Deng J, Rychahou PG, et al. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15:416–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. [DOI] [PubMed] [Google Scholar]
- [20].Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (New York, NY). 2011;331:1565–1570. [DOI] [PubMed] [Google Scholar]
- [21].Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–2532. [DOI] [PubMed] [Google Scholar]
- [22].Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ock CY, Kim S, Keam B, et al. PD-L1 expression is associated with epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Oncotarget. 2016;7:15901–15914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu S, Ye D, Xu D, et al. Autocrine epiregulin activates EGFR pathway for lung metastasis via EMT in salivary adenoid cystic carcinoma. Oncotarget. 2016;7:25251–25263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kudo-Saito C, Shirako H, Takeuchi T, et al. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell. 2009;15:195–206. [DOI] [PubMed] [Google Scholar]
- [26].Vered M, Braunstein E, Buchner A. Immunohistochemical study of epidermal growth factor receptor in adenoid cystic carcinoma of salivary gland origin. Head Neck. 2002;24:632–636. [DOI] [PubMed] [Google Scholar]
- [27].Williams MD, Roberts DB, Kies MS, et al. Genetic and expression analysis of HER-2 and EGFR genes in salivary duct carcinoma: empirical and therapeutic significance. Clin Cancer Res: an off J of the Am Assoc for Cancer Res. 2010;16:2266–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hughes PE, Caenepeel S, Wu LC. Targeted therapy and checkpoint immunotherapy combinations for the treatment of cancer. Trends Immunol. 2016;37:462–476. [DOI] [PubMed] [Google Scholar]
- [29].Noman MZ, Janji B, Abdou A, et al. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology. 2017;6:e1263412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li CW, Lim SO, Xia W, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen J, Jiang CC, Jin L, et al. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol: off J Eur Soc Med Oncol. 2016;27:409–416. [DOI] [PubMed] [Google Scholar]
- [32].Casey SC, Tong L, Li Y, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science (New York, NY). 2016;352:227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gillespie MB, Albergotti WG, Eisele DW. Recurrent salivary gland cancer. Curr Treat Options Oncol. 2012;13:58–70. [DOI] [PubMed] [Google Scholar]
- [34].Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yook JI, Li XY, Ota I, et al. Wnt-dependent regulation of the E-cadherin repressor snail. J Biol Chem. 2005;280:11740–11748. [DOI] [PubMed] [Google Scholar]
- [36].Zhou BP, Deng J, Xia W, et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. [DOI] [PubMed] [Google Scholar]
- [37].Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Strome SE, Dong H, Tamura H, et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003;63:6501–6505. [PubMed] [Google Scholar]
- [39].Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. [DOI] [PubMed] [Google Scholar]
- [40].Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17:133–144. [DOI] [PubMed] [Google Scholar]
- [41].Chen L, Gibbons DL, Goswami S, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dong L, Wang YX, Li SL, et al. TGF-beta1 promotes migration and invasion of salivary adenoid cystic carcinoma. J Dent Res. 2011;90:804–809. [DOI] [PubMed] [Google Scholar]
- [43].Liu SL, Zhong SS, Ye DX, et al. Repression of G protein-coupled receptor family C group 5 member A is associated with pathologic differentiation grade of oral squamous cell carcinoma. J Oral Pathol Med: off Publ Int Assoc Oral Pathol Am Acad Oral Pathol. 2013;42:761–768. [DOI] [PubMed] [Google Scholar]
- [44].Liu S, Shi L, Yang X, et al. Nuclear survivin promoted by acetylation is associated with the aggressive phenotype of oral squamous cell carcinoma. Cell Cycle (Georgetown, Tex). 2017;16:894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]