

Abstract
Abstract
A new series of 2-alkylthio-N-(quinazolin-2-yl)benzenesulfonamide derivatives have been synthesized and evaluated in vitro for their antiproliferative activity by MTT assay against cancer cell lines HCT-116, MCF-7, and HeLa as well as the NCI-60 human tumor cell lines screen. In NCI screen, three compounds inhibited approximately 50% growth of RPMI-8226 and A549/ATCC cell lines. The mean of IC50 calculated in MTT assays for three tested cell lines was about 45 μM for four compounds. The QSAR allowed finding statistically significant OPLS models for HeLa cell line. Metabolic stability in vitro studies indicated favorable and unfavorable structural elements. The good metabolic stability, with t1/2 higher than 40 min, was observed for three derivatives, which together with their antiproliferative activity and good ADMET profile, makes them good leading structures for further research.
Graphical abstract
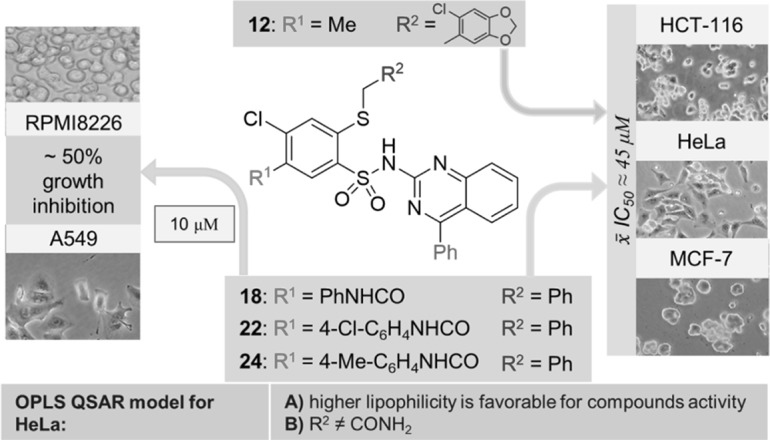
Keywords: 2-Alkylthiobenzenesulfonamide, Quinazoline, Anti-tumor agents, QSAR, Metabolic stability, ADMET
Introduction
Cancer diseases are the second leading cause of death in developed countries and are expected to surpass heart diseases as the leading cause of death in the next few years [1]. Furthermore, finding new antineoplastic agents constitutes a great challenge for medicinal chemistry as their molecular mechanisms of action needed to be based on subtle differences in biochemical processes of healthy and cancerous cells.
Aryl sulfonamide derivatives are a group of compounds widely used in medicine, and show an interesting spectrum of anti-tumor activity [2, 3]. Among them, one of the most important and the largest is a group of N-arylsulfonamides in which the aryl/heteroaryl substituent is connected directly to the sulfonamide nitrogen atom [4–8]. Our team, carrying research on a multidirectional activity of various 2-mercaptobenzenosulfonamide (MBSA) derivatives, have found high anticancer potential in this class of compounds [9–12]. Thus, aiming to obtain a synergistic effect by combining MBSA scaffold and an aryl substituent with proven biological activity, we decided to synthesize and examined the anti-tumor properties of the 4-chloro-2-thio-N-(quinazolin-2-yl)benzenesulfonamides 9–24 (Scheme 1) in which the quinazoline ring is attached to the 2-mercaptobenzenesulfonamide core. The choice of quinazoline as N-aryl substituent was prompted by the fact that its derivatives are being examined for the enormous spectrum of biological activity [13–15], including anti-HIV, anti-inflammatory, anti-microbial, and anti-tumor activity as well as occurs in several approved drugs such as gefitinib, erlotinib, vandetanib, or raltitrexed [16]. The structures of some quinazoline-bearing compounds with anticancer activity are presented in Fig. 1.

Fig. 1.
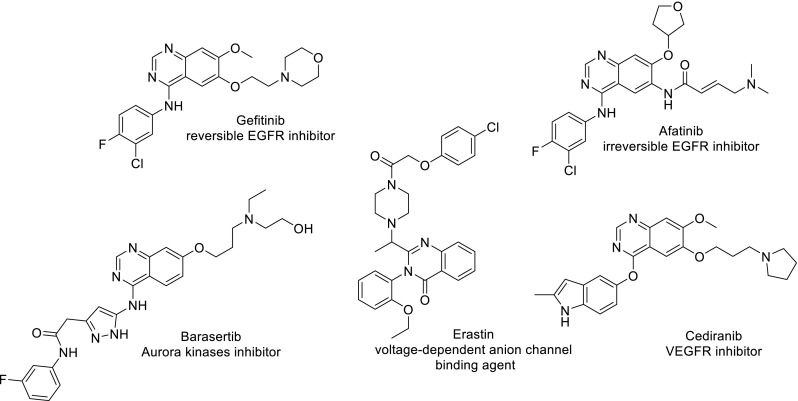
Anti-cancer quinazoline derivatives
The mechanism of action of the most important clinically approved anticancer quinazolines is reversible (lapatinib, erlotinib, gefitinib) [17–20] or irreversible (afatinib) [17, 21] inhibition of EGFR tyrosine kinase receptors. Furthermore, in clinical and pre-clinical trials, there are several other quinazoline derivatives with unique anticancer molecular mechanisms such as cediranib—anti-angiogenic VEGFR inhibitor [22, 23], barasertib aurora kinases inhibitor [24, 25], or erastin altering mitochondrial voltage-dependent anion channels leading to induction of ferroptosis [26, 27]. Despite the facts about the anti-tumor activity of quinazolines and N-arylsulfonamides, there are only a few reports on the anti-tumor activity of N-(quinazolin)sulfonamide derivatives [28–30] against several human cancer cell lines, i.e., SCLC cell lines NCIH889, NCI-H1963, and NCI-H146, lung cancer cell line (A549), cervical (HeLa) cancer cell line, colorectal cell line (LoVo), and breast cancer cell line (MDA-MB-231). However, there is no such research on N-(quinazolin-2-yl)sulfonamides. Therefore, we hope that presented research on the synthesis and the in vitro anticancer activity of 2-alkylthio-4-chloro-N-(quinazolin-2-yl)benzenesulfonamide derivatives will provide new information and will lead to better understanding of the structure–activity relationships of N-(quinazolin)sulfonamides.
Results and discussion
Chemistry
As presented in Scheme 1, the desired 2-alkylthio-N-(quinazolin-2-yl)benzenesulfonamide derivatives 9–24 have been obtained by reacting the appropriate N-(2-alkylthio-4-chloro-5-methylbenzenesulfonyl)cyanamide potassium salts 1–8 with 2′-aminoacetophenone or 2-aminobenzophenone. The syntheses were carried out in glacial acetic acid by refluxing for 3.5–5 h and the reactions progression were monitored using TLC method. According to Scheme 1, at the initial step the N-(benzenesulfonyl)cyanamide potassium salts 1–8 react with amine group yielding an intermediate of type A, which is then cyclized to the desired final products.
The structures of new compounds 9–24 were confirmed by spectroscopic methods IR, 1H NMR, and 13C NMR as well as HRMS spectrometry and elemental analyses. IR spectra of compounds 9–24 showed absorption bands derived from NH group in the range 3396–3159 and 1670–1618 cm−1. The bands at range of 1379–1309 and 1165–1141 cm−1 were due to SO2 group. For derivatives 13 and 14 and 17–24, the characteristic absorption bands for C=O amide groups from 1700 to 1641 cm−1 were observed. The ester C=O absorption bands in compounds 15 and 16 were detected at 1729 and 1723 cm−1, respectively.
The 1H NMR spectra of the series of N-(quinazolin-2-yl)benzenesulfonamides 9–24 revealed singlet signals at range 3.66–4.39 ppm for 2 protons of the methylthio group. The odd-numbered compounds gave singlet signals at around 2.5 ppm, which correspond to three protons from methyl group (R3 = Me). In turn, the spectra of compounds 17–24 having unsubstituted phenylcarbamoyl- or 4-substituted phenylcarbamoyl moiety, showed singlet signals in the range of 10.48–10.77 ppm attributable to NH proton of the amide function (CONH–Ar). Additionally, the 1H NMR spectra of all compounds showed singlet or broad singlet signal of the SO2NH proton in the downfield region δ = 12.50–13.75 ppm.
Cytotoxic activity
All of the newly synthesized compounds 9–24 were evaluated for their effects on cell viability in three human cancer cell lines: HCT-116 (colon cancer), HeLa (cervical cancer), and MCF-7 (breast cancer). Cisplatin was used as reference drug. The results expressed as the concentration required for 50% inhibition of cell viability IC50 have been shown in Table 1. Among studied compounds, derivatives 12, 18, 22, and 24 displayed the most potent cytotoxic effects against all tested cell lines. The mean IC50 values were 44.67 µM for 12, 18, and 22 while IC50 of 24 was 45.67 µM. On the other hand, compounds 13–15, 19, and 20 showed no antiproliferative effects. This suggests that amide group (R2 = CONH2) is undesirable, regardless of the kind of both R1 and R3, and aromatic ring in R2 position seem to be necessary for anticancer activity. Considering the activity of derivatives 9–12, 15, 17, 18, and 21–24, it can be noticed that phenyl group as R3 bring an increase of cytotoxicity in comparison with methyl in place of R3. However, this impact seems to be the most important for compounds with the bulky group in R1 and generally, it is less for a set of derivatives with methyl group substituted in R1 (9–12).
Table 1.
IC50 values for compounds 9–24
| Compd | IC50/µM | HaCaT | ||
|---|---|---|---|---|
| HCT-116 | HeLa | MCF-7 | ||
| 9 | 61 ± 1 | 75 ± 5 | 100 ± 2 | – |
| 10 | 44 ± 1 | 61 ± 2 | 57 ± 1 | – |
| 11 | 46 ± 1 | 58 ± 2 | 57 ± 2 | – |
| 12 | 37 ± 1 | 46 ± 1 | 51 ± 2 | 64 ± 2 |
| 13 | 260 ± 5 | 320 ± 22 | 440 ± 4 | – |
| 14 | 85 ± 3 | 120 ± 1 | 155 ± 9 | – |
| 15 | 110 ± 2 | 230 ± 7 | 160 ± 6 | – |
| 16 | 62 ± 1 | 74 ± 4 | 105 ± 3 | – |
| 17 | 84 ± 3 | 72 ± 1 | 175 ± 10 | – |
| 18 | 42 ± 1 | 46 ± 2 | 46 ± 1 | 68 ± 2 |
| 19 | 100 ± 4 | 117 ± 5 | 105 ± 3 | – |
| 20 | 140 ± 4 | 110 ± 7 | 92 ± 1 | – |
| 21 | 54 ± 1 | 71 ± 2 | 115 ± 2 | – |
| 22 | 39 ± 1 | 42 ± 0.5 | 53 ± 1 | 59 ± 1 |
| 23 | 70 ± 1 | 75 ± 2 | 97 ± 2 | – |
| 24 | 41 ± 1 | 44 ± 3 | 52 ± 3 | 58 ± 1 |
| Cisplatin | 3.8 ± 0.2 | 2.2 ± 0.1 | 3.0 ± 0.1 | |
– Not tested
For the compounds with the strongest cytotoxicity (12, 18, 22, and 24), an investigation of cytotoxic effect against non-carcinogenic cell line HaCaT was done. All compounds displayed promising selectivity toward cancer cells, especially HCT-116 and HeLa lines (Table 1). The activity against HCT-116 was 1.7-, 1.6-, 1.5-, and 1.4-fold higher when compared with HaCaT cell line, for compounds 12, 18, 22, and 24, respectively. In turn, the inhibition of the growth of HaCaT cells was 1.4-, 1.5-, 1.4-, and 1.3-fold weaker than HeLa cells, for compounds 12, 18, 22, and 24, respectively.
Aside from above, compounds 9–14 and 17–24 were also submitted to National Cancer Institute and evaluated for the cytotoxic effects toward 60 cell lines at a single dose of 10 μM (Table 2). These compounds exhibited preferential growth inhibition effects toward either leukemia or non-small cell lung cancer cell lines. As with MTT assays against HCT-116, HeLa and MCF-7, compounds 12, 18, 22, and 24 showed the strongest antiproliferative effect while derivatives 13, 14, 19, and 20 exhibited very weak cytotoxic activity. As it was summarized in Table 2, leukemia RPMI-8226 and NSCLC A549/ATCC were most sensitive to compounds 18, 22, and 24. These derivatives in 10 μM concentration inhibited the growth of approximately 50% cells belonging to the above mention cell lines.
Table 2.
The inhibition growth percent of selected NCI-60 cancer cells (IGP) at a single concentration of 10−5 M of compounds 9–14 and 17–24
| Panel | Cell line | IGP/% of compound | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 9 | 10 | 11 | 12 | 13 | 14 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | ||
| Leukemia | MOLT-4 | 12 | 25 | 20 | 12 | 11 | 9 | 9 | 36 | 3 | 5 | 7 | 52 | 5 | 21 |
| RPMI-8226 | 15 | 25 | 32 | 2 | a | a | 22 | 56 | 3 | 10 | 12 | 52 | 17 | 59 | |
| SR | 6 | 14 | 3 | a | 12 | 17 | 12 | 28 | 4 | 6 | 3 | 40 | 8 | 23 | |
| NSCLC | A549/ATCC | a | N | 34 | 14 | a | a | 11 | 47 | a | a | 8 | 51 | 6 | 48 |
| EKVX | 3 | 6 | 17 | a | 5 | a | 18 | 35 | a | a | 8 | 28 | 15 | 45 | |
| HOP-92 | 3 | 17 | N | N | a | a | 20 | 26 | 8 | 4 | 8 | 34 | a | 26 | |
| NCI-H522 | 9 | a | 19 | 16 | 4 | 4 | 12 | 25 | 8 | 11 | 20 | 35 | a | 28 | |
| Colon cancer | HCT-116 | a | 8 | 30 | 6 | a | a | 5 | 31 | 4 | a | 4 | 39 | a | 44 |
| Melanoma | UACC-62 | 29 | 29 | 23 | 33 | a | a | 14 | 39 | a | 8 | 6 | 40 | a | 28 |
| Renal cancer | UO-31 | 27 | 5 | 15 | 25 | 8 | 2 | 24 | 3 | 7 | 3 | 22 | 35 | 21 | 25 |
| Prostate cancer | PC-3 | 2 | N | 23 | 14 | a | a | 3 | 24 | 4 | 2 | 8 | 44 | 18 | 49 |
| Breast cancer | MCF-7 | 2 | 9 | 4 | 5 | 6 | a | a | 14 | 4 | 3 | 5 | 20 | 8 | 23 |
N not tested
aGrowth percent ≥ 100%
QSAR studies
QSAR analysis was performed to extract information regarding possible structure–activity relationship (SAR), especially to point out the most important parameters controlling pharmacological effects [31, 32].
The three-dimensional structure of the all studied compounds was built and optimized using the Gaussian software (Gaussian Inc.) [33] by density functional theory (DFT) method and B3LYP/6-31G(d) basis set. Subsequently, structures with optimized geometry were submitted to molecular descriptors calculation using Dragon 7.0 software (Talete, Milano, Italy) [34]. Among over 5200 descriptors only those easily interpretable were selected, namely constitutional indices, ring descriptors, functional group counts, atom-centered fragments, atom-type E-state indices, CATS 2D, 2D Atom Pairs, molecular properties and charge descriptors [35].
Next, a multiple linear regression technique (MLR) was used to find QSAR equations correlating the cytotoxic activity expressed as IC50 with values of selected molecular descriptors used as independent variables. This basic approach led to statistically insignificant equations, so it was decided to apply multivariate PLS and OPLS techniques using SIMCA software [36, 37]. Such an approach enabled to achieve valuable information in one of our previous reports [38]. Before regression analysis compounds with outlying cytotoxicity values were excluded. For HeLa and HCT-116 cell lines, we established statistically significant OPLS models. Figure 2 shows the relation between observed and predicted IC50 values as well as some statistical parameters describing the models.
Fig. 2.

Plot of experimental versus predicted by OPLS model cytostatic activity of tested compounds towards HCT-116 (a) and HeLa (b) cell lines
The model for HeLa cell line is able to describe over 99% of activity and predict over 87% of the variability in IC50 with cross-validated root mean squared error 8.14 µM.
The main advantage of OPLS is the possibility to point out relative influence of variables on the predictive model. The Variable Influence on Projection (VIP) values is used for such a comparison Table 3.
Table 3.
List of molecular descriptors characterized by the highest VIP values in OPLS model built for cytostatic activity towards cervical cancer HeLa cell line
| Descriptor | VIP | Full name of descriptor | Block of descriptors |
|---|---|---|---|
| ALOGP | 4.83 | Ghose–Crippen octanol–water partition coeff. (logP) | Molecular properties |
| ALOGP2 | 4.77 | Squared Ghose–Crippen octanol–water partition coeff. (logP2) | Molecular properties |
| N% | 4.70 | Percentage of N atoms | Monstitutional indices |
| F04[C-S] | 4.63 | The frequency of C–S at topological distance 4 | 2D atom pairs |
| MCD | 4.60 | Molecular cyclized degree | Ring descriptors |
| nRCONH2 | 4.59 | Number of primary amides (aliphatic) | Functional group counts |
The two most important descriptors are logP and its square obtained from the Ghose–Crippen algorithm for ALOGP calculation [39]. Table 4 shows values of descriptors and uses shades of green color for easy visual interpretation. Obviously, higher lipophilicity is favorable for compounds activity. The third highest VIP value stands for N% descriptor that corresponds to the percent of N atoms in relation to all atoms in the molecule. It seems to be reversely proportional to logP as N-containing functional groups usually increase the polarity of a molecule. A more detailed analysis indicates that N% values in case of the present group of compounds relate specifically to unsubstituted amide in position R2 (compounds 19 and 20). The presence of this functional group clearly changes values of nRCONH2 but also F04[C-S] into the less favorable level. MCD, the last among the most important descriptors, calculates molecular cyclized degree as a ratio between number of atoms belonging to any ring system and all atoms in the molecule [40]. There is an evidence that higher percent of the ring system is preferable for higher cytotoxic activity in this group of compounds.
Table 4.
Values of descriptors selected as the most influential for OPLS model describing IC50 against HeLa cell line

Table is sorted with descending activity of compounds. Color intensity reflects descriptor values—the more intense color, the higher value
Metabolic stability
Selected derivatives were submitted to metabolic stability study to assess their ability to remain unchanged in the presence of human metabolic enzymes. Human liver microsomes were used as they are a rich source of all common CYP isoenzymes. In vitro incubations were performed in the presence of NADPH as a cofactor and the disappearance of a derivative was followed using LC–MS technique. The in vitro metabolic half-life values are gathered in Fig. 3.
Fig. 3.

The in vitro metabolic half-life values, obtained in the presence of human liver microsomes and NADPH
The selected group of derivatives varies significantly in the in vitro metabolic half-life values. The most stable and the least stable compounds are marked green and red, respectively (Fig. 3). High metabolic stability is a desirable property. Derivatives 13 and 14 are characterized by the best t1/2: 75.4 and 90.8 min, respectively. Interestingly, they both possess unsubstituted amide moiety in R2 position. Possibly, the amide is less reactive against metabolic enzymes than phenyl (9 and 10) or 6-chloro-2H-1,3-benzodioxole (11 and 12) substituents. On the other hand, compounds 17 and 18 are the least stable with half-life values 16.0 and 11.2 min, respectively. These derivatives also share the same chemical group-N-phenyl substituted amide in R1 position. The results show that additional substituent (either 4-chloro or 4-methyl) in R1 enable to increase metabolic stability twofold (21 and 23 versus 17) to four- to sixfold (20, 22, 24 versus 18). Regarding R3 position, there is no clear difference between methyl and phenyl substituents.
The most informative approach is to compare metabolic stability within one chemical group of derivatives. This way, we can assess which derivative is the most promising in particular group of new compounds. The comparison between different series of compounds is difficult due to the lack of standard experiment, which is widely used worldwide. For this reason, it is more advisable to compare metabolic stability between our previous reports, because we have used the same test to determine metabolic stability and calculate in vitro metabolic half-life. Our report [38] concerning sulfonamide derivatives with 1,3,4-oxadiazole ring revealed t1/2 between ca 5 and over 60 min, whereas another study [41] showed metabolic stability in a range 13–38 min. In this report, the most stable compounds 13 and 14 are thus one of the most stable among all tested sulfonamide derivatives. In the presented research, the substituent R2, amide group, is replaced by phenyl, ethyl ester, or 6-Cl-benzo[d] [1, 3] dioxol-5-yl substituent. The amide group is reported as a stable one. It is not vulnerable to oxidation mediated by CYP enzymes. However, substituted amides can be cleaved by enzymatic hydrolysis [42]. On the other hand, phenyl (as well as benzodioxole) ring can be easily hydroxylated in I phase metabolism [43]. Our findings confirm previous reports and show that incorporation of unsubstituted amide is beneficial for metabolic stability.
ADMET in silico prediction
Based on the cytotoxicity studies, the compounds 12, 18, 22, and 24 were selected for in silico predictions of absorption, distribution, metabolism, elimination, and toxicity (ADMET) processes. The ADMET properties of 12, 18, 22, and 24 are detailed in Table 5.
Table 5.
In silico ADMET parameters of compounds 12, 18, 22, and 24 predicted by pkCSM approach [44]
| Descriptor | Predicted value | |||
|---|---|---|---|---|
| 12 | 18 | 22 | 24 | |
| Absorption | ||||
| logPapp | 0.911 | − 0.167 | 0.555 | 0.563 |
| P-gp substrate | + | + | + | + |
| P-gp I inhibitor | + | + | + | + |
| P-gp II inhibitor | + | + | + | + |
| Distribution | ||||
| logVDss (human) | − 0.946 | − 1.209 | − 0.92 | − 0.929 |
| log BB | − 1.049 | − 0.578 | − 0.471 | − 0.296 |
| Metabolism | ||||
| CYP2D6 substrate | − | − | − | − |
| CYP3A4 substrate | + | + | + | + |
| CYP1A2 inhibitor | − | − | − | − |
| CYP2C19 inhibitor | + | − | + | + |
| CYP2C9 inhibitor | + | − | − | − |
| CYP2D6 inhibitor | − | − | − | − |
| CYP3A4 inhibitor | + | − | − | − |
| Toxicity | ||||
| Max. tolerated dose (log mg/kg/day) | 0.426 | 0.436 | 0.443 | 0.444 |
| hERG I inhibitor | − | − | − | − |
| hERG II inhibitor | + | + | + | + |
In a drug discovery process, the Lipinski’s rule is the standard for the development of orally available drugs. However, an experience in drug development shows that highly promising drug candidate is often not accepted for further research because it did not comply the rule-of-five. Furthermore, the Lipinski’s rule seems to overemphasize since only 51% of all FDA-approved drugs comply with this protocol [45]. In the literature, there is a lot of in silico models to predict if drug candidate will be orally bioavailable. The Caco-2 monolayer is an in vitro model of the human intestinal mucosa. It is widely used to predict the absorption of orally administrated drugs. A compound has a high Caco-2 permeability if Papp > 8 × 10−6 cm/s (logPapp > 0.9). Although none of the studied compounds do comply with Lipinski’s rule, results in Table 5 showed that compound 12 is considered to have a high Caco-2 permeability. Furthermore, the results of the calculated property for absorption of compounds 23 and 24 revealed that these can be absorbed throughout the intestine but to a lower degree than 12.
A distribution of compounds 12, 18, 22, and 24 were determined as free parameters, namely volume of distribution (logVDss) and blood–brain barrier permeability (logBB). The VDss defines the drug distribution between plasma and the rest of the body. It is considered low if below 0.71 L/kg (log VDss < − 0.15) and high if above 2.81 L/kg (log VDss > 0.45). A compound is able to readily cross the blood–brain barrier if logBB > 0.3 while a molecule with logBB < − 1 is poorly distributed to the brain. The VDss of all studied derivatives was lower than − 0.15. A low volume of distribution indicates that there is a low probability of compounds accumulation in body tissues. On the other hand, the logBB was lower than − 1 only for compound 12. However, derivatives 18 and 22 with logBB − 0.578 and − 0.471, respectively, seem to display limited brain penetration. The highest logBB = − 0.296 was observed for 24 but still, this is a low value. The obtained logBB values suggest that compounds display rather a low brain toxicity, if at all.
Compounds transport and metabolism were expressed as a modulation of the activity of P-glycoprotein (P-gp) and various cytochrome P450 enzymes. Modulation of P-glycoprotein transport is essential for compounds pharmacokinetic properties. A number of drugs, including drugs in cancer chemotherapy, are substrates of P-gp transporter [46]. The data in Table 5 indicate that all compounds are potential P-gp substrates. On the other hand, all of them are also P-gp inhibitors. The inhibition of P-gp activity is considered as a strategy in cancer therapy to avoid P-gp-mediated drug resistance [47]. However, an efficient P-gp inhibitor should not inhibit the activity of cytochrome P450 enzymes involved in drug metabolism, particularly CYP3A. The simultaneous inhibition of both P-gp transport and CYP3A metabolism may result in an increase plasma drug concentration and leads to higher toxicity [48]. The obtained predictions indicate that only compound 12 could potentially inhibit the activity of both P-gp and CYP3A4. Derivatives 18, 22, and 24 were not considered to have the capability for inhibiting CYP3A4. On the other hand, compounds 18, 22, and 24 are substrates of P-gp and CYP3A4. Owing to the complementary function of these proteins [46], this may result in a greater than expected reduction in systematic exposure to 18, 22, and 24. Furthermore, these effects may allow for the maintenance of drug elimination when either CYP3A4 or P-gp activity is inhibited [46].
To consider the compounds toxicity, maximum tolerated dose and hERG inhibition were calculated in silico. The maximum dose of all compounds is significantly higher than their cytotoxic concentration. On the other hand, the discussed derivatives, as a considered hERG inhibitors, may cause drug-induced (acquired) QT interval prolongation associated with an increase in the incidence of sudden unexplained deaths. The hERG inhibition is considered as the main cause of it. However, the predicted ADMET properties indicated that only hERG II may be inhibited by 2-alkylthio-N-(quinazolin-2-yl)benzenesulfonamide derivatives. Thus, further studies with in vitro models are needed for compounds safety related with hERG inhibition.
Conclusion
We have developed methods for the synthesis of novel series of 2-alkylthio-N-(quinazolin-2-yl)benzenesulfonamide derivatives. The compounds were tested in vitro by MTT assay for their cytotoxic activity against three cancer cell lines: colon HCT-116, cervical HeLa, and breast MCF-7. Furthermore, compounds 9–14 and 17–24 were evaluated by NCI as potential growth inhibitors of 60 human cancer cell lines. We have found that the studied compounds display moderate cytotoxic activity and the best antiproliferative effect was observed for compounds 18, 22, and 24 which showed good results in MTT assays as well as NCI studies. Aside from HCT-116, HeLa, and MCF-7, noticeable growth inhibition of RPMI-8226 and A549/ATCC were also observed. Quite good results of MTT assays were also noticed for compound 12. The mean IC50 value against three tested cell lines was 44.67 µM (identical with IC50 obtained for 18 and 22). What important, the activity of compounds 12, 18, 22, and 24 was lower against non-cancerogenic HaCaT cell line than susceptible cancer cells. Structure–activity relationship revealed that amide group (R2 = CONH2) is undesirable, while aromatic ring in R2 position is important for anticancer effect. Furthermore, phenyl as R3 is more favorable than methyl and this impact is probably stronger in compounds with the bulky group in R1.
QSAR studies showed that higher lipophilicity is important for better compound activity against HeLa cell line. Furthermore, unsubstituted amide in position R2 is inadvisable for HeLa growth inhibition.
All but two (17, 18) of the 2-alkylthio-N-(quinazolin-2-yl)benzenesulfonamide derivatives displayed good metabolic stability with t1/2 in the range of 35–90.8 min. Compounds 12, 22, and 24 were characterized by t1/2 higher than 40 min, which together with the outstanding activity, makes them good leaders for further research.
The predicted in silico ADMET properties of compounds 12, 18, 22, and 24 indicated that new derivatives display rather low probability of side effects. However, further studies with in vitro models are needed to determine their complete safety profile.
Experimental
Melting points were measured using Boethius PHMK apparatus. IR spectra were measured on Thermo Mattson Satellite FTIR spectrometer in KBr pellets; an absorption range was 400–4000 cm−1. 1H NMR and 13C NMR spectra were recorded on a Varian Gemini 200 apparatus or Varian Unity Plus 500 apparatus. Chemical shifts are expressed at δ values relative to Me4Si (TMS) as an internal standard. The apparent resonance multiplicity is described as: s (singlet), d (doublet), dd (doublet of doublets), t (triplet), m (multiplet), and br (broad) signal. The addition of equimolar TFA was necessary to obtain 13C NMR spectra. Due to a poor solubility of compounds 21 and 23, the obtained 13C NMR spectra were not sufficient. HRMS analyses were performed on a TripleTOF 5600 + System (AB SCIEX, USA) in positive ion mode. Elemental analyses were performed on PerkinElmer 2400 Series II CHN Elemental Analyzer and the results were within ± 0.4% of the theoretical values. Thin-layer chromatography (TLC) was performed on Merck Kieselgel 60 F254 plates and visualized with UV. The commercially unavailable monopotassium salts were obtained according to the following methods described previously: 1, 4 [49], 2 [50], 3–5, 8 [51], and 7 [52].
N-[4-Chloro-5-(4-chlorophenylcarbamoyl)-2-carbamoylthiobenzenesulfonyl]cyanamide potassium salt (6, C16H11Cl2KN4O4S2)
The mixture of 4.020 g 3-amino-6-chloro-N-(4-chlorophenyl)-1,4,2-benzodithiazine-7-carboxamide 1,1-dioxide (10 mmol), 1.026 g 2-chloroacetamide (11 mmol), and 8.26 g anhydrous K2CO3 (59.8 mmol) in 67 cm3 dry tetrahydrofuran was stirred for 7.5 h at reflux. After cooling the solid was filtered off, dried, treated with 58 cm3 water and stirred for 20 min. Crude product was filtered off and crystallized from ethanol, giving 3.460 g (70%) 6. M.p.: 245–248 °C (dec.); 1H NMR (500 MHz, DMSO-d6): δ = 3.76 (s, 2H, CH2), 7.29 (s, 1H, NH2), 7.41 (d, 2H, arom), 7.64 (s, 1H, NH2), 7.65 (s, 1H, H-3), 7.72 (d, 2H, arom), 7.86 (s, 1H, H-6), 10.77 (s, 1H, NH) ppm; IR (KBr): = 3462, 3357 (NH), 2923, 2853 (CH), 2176 (C≡N), 1671 (NH def), 1314, 1143 (SO2) cm−1.
General procedure for the preparation of 2-alkylthio-N-(4-R3-quinazolin-2-yl)benzenesulfonamides 9–24
A mixture of the appropriate N-(benzenesulfonyl)cyanamide potassium salts 1–8 (2 mmol) and 2′-aminoacetophenone or 2-aminobenzophenone (2.2 mmol) in 6 cm3 glacial acetic acid was refluxed with stirring for 3.5–5 h. Then the mixture was cooled in an ice bath. The solid was collected by filtration, washed with glacial acetic acid (2 × 1 cm3) and dried. The final products 9–24 were purified as described below.
2-Benzylthio-4-chloro-5-methyl-N-(4-methylquinazolin-2-yl)benzenesulfonamide (9, C23H20ClN3O2S2)
Starting from 0.780 g 1 to 0.299 g 2′-aminoacetophenone for 5 h, the crude product was obtained. Crystallization from 10 cm3 ethanol gave 0.400 g (43%) 9. M.p.: 183–186 °C; TLC: Rf = 0.75 (benzene-EtOH 4:1); 1H NMR (200 MHz, DMSO-d6): δ = 2.39 (s, 3H, CH3), 2.58 (s, 3H, CH3), 4.25 (s, 2H, CH2), 7.04–7.06 (m, 3H, arom), 7.25–7.27 (m, 2H, arom), 7.40–7.50 (m, 3H, arom), 7.82–7.89 (t, 1H, arom), 8.02–8.12 (m, 2H, arom), 13.30 (br s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 19.1, 22.1, 36.4, 118.3, 120.3, 124.9, 127.0, 127.3, 127.4, 128.5, 128.6, 129.3, 131.7, 133.9, 135.7, 136.1, 136.6, 137.2, 138.8, 143.7, 153.1 ppm; IR (KBr): = 3256 (NH), 1623 (NH def), 1580, 1524 (C=C, C=N), 1379, 1141 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C23H20ClN3O2S2 ([M+H]+) 470.0764, found 470.0764.
2-Benzylthio-4-chloro-5-methyl-N-(4-phenylquinazolin-2-yl)benzenesulfonamide (10, C28H22ClN3O2S2)
Starting from 0.780 g 1 to 0.430 g 2-aminobenzophenone for 3.5 h, the crude product was obtained. Extraction with 7 cm3 boiling ethanol gave 0.590 g (55%) 10. M.p.: 192–198 °C; TLC: Rf = 0.75 (benzene-EtOH 4:1); 1H NMR (200 MHz, DMSO-d6): δ = 2.20 (s, 3H, CH3), 4.26 (s, 2H, CH2), 6.86–6.87 (m, 3H, arom), 7.11–7.30 (m, 4H, arom), 7.42–7.53 (m, 3H, arom), 7.59–7.65 (m, 3H, arom), 7.82–7.97 (m, 3H, arom), 13.50 (br s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 19.1, 36.3, 117.2, 120.8, 125.3, 127.1, 127.9, 128.2, 128.4, 128.6, 129.1, 130.1, 131.1, 132.1, 133.6, 135.0, 135.8, 136.3, 136.7, 137.1, 139.4, 145.8, 153.1 ppm; IR (KBr): = 3238 (NH), 2922 (CH), 1620 (NH def), 1582, 1567 (C=C, C=N), 1358, 1138 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C28H22ClN3O2S2 ([M+H]+) 532.0920, found 532.0910.
4-Chloro-2-(6-chlorobenzo[1,3]dioxol-5-ylmethylthio)-5-methyl-N-(4-methylquinazolin-2-yl)benzenesulfonamide (11, C24H19Cl2N3O4S2)
Starting from 0.940 g 2 to 0.299 g 2′-aminoacetophenone for 5 h, the crude product was obtained. Crystallization from 40 cm3 acetonitrile gave 0.470 g (43%) 11. M.p.: 198–200 °C; TLC: Rf = 0.78 (benzene-EtOH 4:1); 1H NMR (200 MHz, DMSO-d6): δ = 2.41 (s, 3H, CH3), 2.58 (s, 3H, CH3), 4.19 (s, 2H, SCH2), 5.90 (s, 2H, OCH2O), 6.83 (s, 1H, arom), 6.92 (s, 1H, arom), 7.42–7.48 (m, 3H, arom), 7.81–7.85 (t, 1H, arom), 8.01–8.05 (m, 2H, arom), 13.3 (br s, 1H, SO2NH) ppm; 13C NMR (50 MHz, DMSO-d6): δ = 19.19, 22.21, 34.65, 102.23, 109.63, 110.46, 124.78, 125.36, 126.83, 126.96, 127.71, 131.91, 135.22, 146.65, 147.61, 152.90 ppm; IR (KBr): = 3279 (NH), 2920 (CH), 1618 (NH def), 1582, 1475 (C=C, C=N), 1363, 1165 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C24H19Cl2N3O4S2 ([M+H]+) 548.0272, found 548.0268.
4-Chloro-2-(6-chlorobenzo[d][1,3]dioxol-5-ylmethylthio)-5-methyl-N-(4-phenylquinazolin-2-yl)benzenesulfonamide (12, C29H21Cl2N3O4S2)
Starting from 0.940 g 2 to 0.430 g 2-aminobenzophenone for 5 h, the crude product was obtained. Crystallization from 34 cm3 acetonitrile gave 0.500 g (43%) 12. M.p.: 112–116 °C; TLC: Rf = 0.77 (benzene-EtOH 4:1); 1H NMR (200 MHz, DMSO-d6): δ = 2.23 (s, 3H, CH3), 4.21 (s, 2H, SCH2), 5.64 (s, 2H, OCH2O), 6.30 (br s, 1H), 6.89 (s, 1H, arom), 7.05–7.10 (m, 2H, arom), 7.40–7.51 (m, 3H, arom), 7.62 (m, 3H, arom), 7.79–7.96 (m, 3H, arom), 13.5 (br s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 19.0, 34.6, 102.0, 109.5, 110.6, 116.8, 120.1, 125.0, 125.3, 127.2, 128.4, 128.5, 128.6, 130.1, 131.1, 132.6, 133.4, 134.2, 135.6, 136.2, 137.0, 140.5, 145.2, 146.5, 147.4, 152.9 ppm; IR (KBr): = 3239 (NH), 2919 (CH), 1619 (NH def), 1582, 1570, 1478 (C=C, C=N), 1357, 1141 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C29H21Cl2N3O4S2 ([M+H]+) 610.0429, found 610.0219.
2-Carbamoylmethylthio-4-chloro-5-methyl-N-(4-methylquinazolin-2-yl)benzenesulfonamide (13, C18H17ClN4O3S2)
Starting from 0.720 g 3 to 0.299 g 2′-aminoacetophenone for 5 h, the crude product was obtained. Crystallization from 50 cm3 ethanol gave 0.360 g (40%) 13. M.p.: 232–235 °C (dec); TLC: Rf = 0.43 (benzene-EtOH 4:1); 1H NMR (200 MHz, DMSO-d6): δ = 2.40 (s, 3H, CH3), 2.67 (s, 3H, CH3), 3.66 (s, 2H, CH2), 7.20 (s, 1H, CONHA), 7.39–7.63 (m, 4H, arom, CONHB), 7.81–7.88 (t, 1H, arom), 8.03–8.07 (m, 2H, arom), 13.35 (br s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 19.1, 22.3, 36.6, 118.3, 120.2, 125.0, 127.0, 127.2, 131.8, 133.9, 136.1, 136.2, 137.3, 138.5, 143.6, 153.3, 170.0 ppm; IR (KBr): = 3393, 3176 (NH), 2918 (CH), 1659 (CO), 1631 (NH def), 1587, 1524, 1493 (C=C, C=N), 1362, 1145 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C18H17ClN4O3S2 ([M+H]+) 437.0509, found 437.0516.
2-Carbamoylmethylthio-4-chloro-5-methyl-N-(4-phenylquinazolin-2-yl)benzenesulfonamide (14, C23H19ClN4O3S2)
Starting from 0.720 g 3 to 0.430 g 2-aminobenzophenone for 5 h, the crude product was obtained. Crystallization from 59 cm3 DMSO/methanol (v/v 29:30) gave 0.350 g (36%) 14. M.p.: 275–279 °C (dec.); TLC: Rf = 0.54 (benzene-EtOH 4:1); 1H NMR (200 MHz, DMSO-d6): δ = 2.22 (s, 3H, CH3), 3.67 (s, 2H, CH2), 7.19 (s, 1H, CONHA), 7.40–7.66 (m, 9H, arom, CONHB), 7.85–7.99 (m, 3H, arom), 13.58 (br s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 18.8, 36.5, 117.2, 120.7, 125.1, 127.5, 128.2, 128.6, 130.1, 131.1, 132.2, 133.8, 135.1, 135.5, 136.0, 136.2, 137.4, 138.8, 145.7, 153.3, 170.0 ppm; IR (KBr): = 3441 (NH), 2921 (CH), 1650 (CO), 1629 (NH def), 1584, 1453 (C=C, C=N), 1363, 1139 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C23H19ClN4O3S2 ([M+H]+) 499.0665, found 499.0518.
4-Chloro-2-(2-ethoxy-2-oxoethylthio)-5-methyl-N-(4-methylquinazolin-2-yl)benzenesulfonamide (15, C20H20ClN3O4S2)
Starting from 0.775 g 4 to 0.299 g 2′-aminoacetophenone for 5 h, the reaction mixture was evaporated under the diminished pressure and the residue was crystallized from 2 cm3 acetonitrile. The crude product (0.640 g, 69%) was filtered and dried. Crystallization from 31 cm3 ethanol gave 0.280 g (30%) 15. M.p.: 192–194 °C; TLC: Rf = 0.71 (benzene-EtOH 4:1); 1H NMR (200 MHz, DMSO-d6): δ = 0.95–1.09 (m, 3H, CH3), 2.41 (s, 3H, CH3), 2.65 (s, 3H, CH3), 3.87–3.98 (m, 4H, CH2, SCH2), 7.32–7.52 (m, 3H, arom), 7.84 (t, 1H, arom), 8.04 (d, 1H, arom), 8.14 (s, 1H, arom), 13.30 (br s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 13.8, 18.9, 22.0, 34.5, 61.2, 118.2, 120.0, 124.8, 126.8, 127.4, 132.1, 134.0, 134.6, 136.0, 137.2, 143.4, 153.2, 169.2 ppm; IR (KBr): = 3258 (NH), 2976, 2918 (CH), 1729 (C=O), 1623 (NH def), 1579, 1524, 1497 (C=C, C=N), 1358, 1140 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C20H20ClN3O4S2 ([M+H]+) 466.0662, found 466.0517.
4-Chloro-2-(2-ethoxy-2-oxoethylthio)-5-methyl-N-(4-phenyl-quinazolin-2-yl)benzenesulfonamide (16, C25H22ClN3O4S2)
Starting from 0.775 g 4 to 0.430 g 2-aminobenzophenone for 5 h, the reaction mixture was evaporated under the diminished pressure and the residue was crystallized from 2 cm3 acetonitrile. Solid was filtered out. The filtrate was evaporated to dryness and crystallized from 2 cm3 ethanol giving 0.350 g (33%) 16. M.p.: 162–165 °C; TLC: Rf = 0.69 (benzene-EtOH 4:1); 1H NMR (500 MHz, DMSO-d6): δ = 0.92 (t, 3H, CH3), 2.24 (s, 3H, CH3), 3.83 (q, 2H, CH2), 3.97 (s, 2H, SCH2), 7.38–7.44 (m, 4H, arom), 7.54–7.57 (m, 2H, arom), 7.60–7.67 (m, 2H, arom), 7.86 (d, 1H, arom), 7.91 (t, 1H, arom), 7.97 (br s, 1H, arom), 13.65 (br s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 14.0, 19.1, 34.4, 61.2, 112.0, 117.0, 120.3, 125.3, 127.6, 128.4, 128.8, 130.2, 131.3, 132.4, 133.7, 134.3, 135.9, 136.4, 137.2, 139.4, 145.3, 153.3, 169.2 ppm; IR (KBr): = 3244 (NH), 2978 (CH), 1723 (C=O), 1620 (NH def), 1567, 1514, 1493 (C=C, C=N), 1357, 1139 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C25H22ClN3O4S2 ([M+H]+) 528.0819, found 528.0717.
2-Benzylthio-4-chloro-5-phenylcarbamoyl-N-(4-methylquinazolin-2-yl)benzenesulfonamide (17, C29H23ClN4O3S2)
Starting from 0.990 g 5 to 0.299 g 2′-aminoacetophenone for 5 h, the crude product was obtained. Extraction with 12 cm3 boiling ethanol gave 0.490 g (43%) 17. M.p.: 228–230 °C; TLC: Rf = 0.70 (benzene-EtOH 4:1); 1H NMR (500 MHz, DMSO-d6): δ = 2.59 (s, 3H, CH3), 4.36 (s, 2H, CH2), 7.10–7.16 (m, 4H, arom), 7.34 (d, 2H, arom), 7.39 (t, 2H, arom), 7.46–7.51 (m, 2H, arom), 7.58 (s, 1H, arom), 7.73 (d, 2H, arom), 7.88 (t, 1H, arom), 8.08 (d, 1H, arom), 8.22 (br s, 1H, arom), 10.60 (s, 1H, NHCO), 13.46 (br s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 22.3, 36.0, 118.0, 119.3, 120.2, 124.3, 125.0, 127.2, 127.3, 127.5, 128.6, 129.2, 129.3, 132.2, 132.3, 133.5, 136.4, 136.5, 138.7, 139.2, 140.5, 153.2, 164.3 ppm; IR (KBr): = 3417, 3318 (NH), 2968 (CH), 1673 (C=O), 1626 (NH def), 1585, 1526, 1497 (C=C, C=N), 1360, 1143 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C29H23ClN4O3S2 ([M+H]+) 575.0978, found 575.0973.
2-Benzylthio-4-chloro-5-phenylcarbamoyl-N-(4-phenylquinazolin-2-yl)benzenesulfonamide (18, C34H25ClN4O3S2)
Starting from 0.990 g 5 to 0.430 g 2-aminobenzophenone for 3 h 45 min, the crude product was obtained. Extraction of byproducts with 7 cm3 boiling ethanol allowed to obtain 0.450 g (36%) 18. M.p.: 243–245 °C; TLC: Rf = 0.70 (benzene-EtOH 4:1); 1H NMR (500 MHz, DMSO-d6): δ = 4.39 (s, 2H, CH2), 6.98–7.06 (m, 3H, arom), 7.16 (t, 1H, arom), 7.24–7.36 (m, 3H, arom), 7.40 (t, 2H, arom), 7.46–7.58 (m, 4H, arom), 7.60–7.68 (m, 2H, arom), 7.73 (d, 2H, arom), 7.87–7.97 (m, 2H, arom), 8.15 (br s, 1H, arom), 10.57 (s, 1H, NHCO), 13.70 (br s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 36.1, 117.0, 120.1, 124.2, 125.2, 127.4, 127.9, 128.4, 128.5, 128.9, 129.0, 129.1, 130.2, 131.2, 131.5, 132.3, 134.2, 135.8, 136.2, 136.3, 138.8, 139.2, 140.4, 153.3, 163.9 ppm; IR (KBr): = 3396, 3290 (NH), 2926 (CH), 1660 (C=O), 1621 (NH def), 1582, 1551, 1494 (C=C, C=N), 1316, 1146 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C34H25ClN4O3S2 ([M+H]+) 637.1135, found 637.1133.
2-Carbamoylmethylthio-4-chloro-5-(4-chlorophenylcarbamoyl)-N-(4-methylquinazolin-2-yl)benzenesulfonamide (19, C24H19Cl2N5O4S2)
Starting from 0.995 g 6 to 0.299 g 2′-aminoacetophenone for 5 h 15 min, the crude product was obtained. Extraction with 250 cm3 boiling ethanol gave 0.690 g (61%) 19. M.p.: 288–292 °C (dec.); TLC: Rf = 0.42 (benzene-EtOH 4:1); 1H NMR (500 MHz, DMSO-d6): δ = 2.68 (s, 3H, CH3), 3.76 (s, 2H, CH2), 7.26 (s, 1H, CONHA), 7.45 (d, J = 8.3 Hz, 2H, 4-ClPh), 7.48–7.56 (m, 2H, arom), 7.58–7.62 (m, 2H, arom, CONHB), 7.76 (d, J = 8.3 Hz, 2H, 4-ClPh), 7.87 (br s, 1H, arom), 8.09 (d, 1H, arom), 8.26 (br s, 1H, arom), 10.77 (s, 1H, NHCO), 13.48 (s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 22.4, 36.1, 118.1, 119.3, 121.6, 125.0, 127.1, 128.1, 129.1, 132.0, 132.2, 133.6, 136.4, 138.0, 138.5, 141.1, 142.5, 153.4, 164.4, 169.7 ppm; IR (KBr): = 3436, 3291, 3159 (NH), 2995, 2913, 2678 (CH), 1657, 1641 (C=O), 1591, 1512 (C=C, C=N), 1310, 1143 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C24H19Cl2N5O4S2 ([M+H]+) 576.0334, found 576.0340.
2-Carbamoylmethylthio-4-chloro-5-(4-chlorophenylcarbamoyl)-N-(4-phenylquinazolin-2-yl)benzenesulfonamide (20, C29H21Cl2N5O4S2)
Starting from 0.995 g 6 to 0.430 g 2-aminobenzophenone for 4 h 45 min, the crude product was obtained. Extraction with 12 cm3 boiling ethanol gave 0.550 g (43%) 20. M.p.: 310–313 °C (dec.); TLC: Rf = 0.52 (benzene-EtOH 4:1); 1H NMR (500 MHz, DMSO-d6): δ = 3.80 (s, 2H, CH2), 7.26–7.36 (m, 2H, arom, CONHA), 7.47–7.52 (m, 7H, arom), 7.58–7.61 (m, 3H, arom, CONHB), 7.77 (d, J = 8.3 Hz, 2H, 4-ClPh), 7.90–7.92 (m, 2H, arom), 8.19 (br s, 1H), 10.71 (s, 1H, NHCO), 13.75 (br s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 36.2, 117.0, 121.5, 125.3, 127.6, 128.1, 128.5, 128.9, 129.0, 130.3, 131.3, 131.5, 132.0, 134.2, 135.9, 136.4, 138.1, 138.6, 141.2, 153.5, 163.9, 169.6 ppm; IR (KBr): = 3425, 3286 (NH), 2925 (CH), 1660, 1645 (C=O), 1582, 1556, 1493 (C=C, C=N), 1364, 1160 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C29H21Cl2N5O4S2 ([M+H]+) 638.0490, found 638.0489.
2-Benzylthio-4-chloro-5-(4-chlorophenylcarbamoyl)-N-(4-methylquinazolin-2-yl)benzenesulfonamide (21, C29H22Cl2N4O3S2)
Starting from 1.060 g 7 to 0.299 g 2′-aminoacetophenone for 5 h, the crude product was obtained. Crystallization from 7 cm3 ethanol gave 0.620 g (51%) 21. M.p.: 278–281 °C; TLC: Rf = 0.67 (benzene-EtOH 4:1); 1H NMR (500 MHz, DMSO-d6): δ = 2.61 (s, 3H, CH3), 4.37 (s, 2H, CH2), 7.07–7.12 (m, 3H, arom), 7.32 (d, 2H, arom), 7.44–7.48 (m, 3H, arom), 7.51 (d, 1H, arom), 7.59 (s, 1H, H-3), 7.76 (d, 2H, arom), 7.88 (t, 1H, arom), 8.07 (d, 1H, arom), 8.25 (s, 1H, H-6), 10.75 (s, 1H, NHCO), 12.50 (br s,1H, SO2NH) ppm; IR (KBr): = 3321 (NH), 2924 (CH), 1681 (C=O), 1634 (NH def), 1586, 1537, 1493 (C=C, C=N), 1310, 1145 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C29H22Cl2N4O3S2 ([M+H]+) 609.0589, found 609.0593.
2-Benzylthio-4-chloro-5-(4-chlorophenylcarbamoyl)-N-(4-phenylquinazolin-2-yl)benzenesulfonamide (22, C34H24Cl2N4O3S2)
Starting from 1.060 g 7 to 0.430 g 2-aminobenzophenone for 5 h, the crude product was obtained. Extraction with 9 cm3 boiling ethanol gave 0.620 g (46%) 22. M.p.: 283–286 °C (dec.); TLC: Rf = 0.72 (benzene-EtOH 4:1); 1H NMR (500 MHz, DMSO-d6): δ = 4.39 (s, 2H, CH2), 6.98–7.08 (m, 3H, arom), 7.23–7.34 (m, 4H, arom), 7.46 (d, J = 8.8 Hz, 2H, 4-ClPh), 7.50–7.55 (m, 4H, arom), 7.60–7.68 (m, 2H, arom), 7.76 (d, J = 8.8 Hz, 2H, 4-ClPh), 7.90 (d, 1H, arom), 7.94–7.99 (m, 1H, arom), 8.13 (br s, 1H), 10.70 (s, 1H, NHCO), 13.75 (br s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 36.1, 117.0, 121.5, 125.2, 127.3, 127.9, 128.1, 128.4, 128.5, 128.8, 128.9, 129.1, 130.1, 131.2, 131.5, 131.9, 134.1, 135.7, 136.2, 136.3, 138.1, 138.9, 140.6, 145.0, 153.3, 163.9 ppm; IR (KBr): = 3394, 3275 (NH), 2927, 2853 (CH), 1657 (C=O), 1622 (NH def), 1582, 1554, 1493 (C=C, C=N), 1309, 1147 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C34H24Cl2N4O3S2 ([M+H]+) 671.0745, found 671.0746.
2-Benzylthio-4-chloro-5-(4-methylphenylcarbamoyl)-N-(4-methylquinazolin-2-yl)benzenesulfonamide (23, C30H25ClN4O3S2)
Starting from 1.020 g 8 to 0.299 g 2′-aminoacetophenone for 5 h, the crude product was obtained. Extraction of byproducts with 4 cm3 boiling ethanol gave 0.630 g (54%) 23. M.p.: 273–277 °C (dec.); TLC: Rf = 0.68 (benzene-EtOH 4:1); 1H NMR (500 MHz, DMSO-d6): δ = 2.30 (s, 3H, CH3), 2.62 (s, 3H, CH3), 4.36 (s, 2H, SCH2), 7.06–7.12 (m, 3H, arom), 7.19 (d, J = 8.3 Hz, 2H, 4-MePh), 7.35 (d, 2H, arom), 7.46 (t, 1H, arom), 7.51 (d, 1H, arom), 7.58 (s, 1H, H-3), 7.61 (d, J = 8.3 Hz, 2H, 4-MePh), 7.89 (t, 1H, arom), 8.08 (d, 1H, arom), 8.24 (s, 1H, H-6), 10.51 (s, 1H, NHCO) ppm; IR (KBr): = 3326 (NH), 2921 (CH), 1700 (C=O), 1670 (NH def), 1633, 1587, 1540 (C=C, C=N), 1316, 1150 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C30H25ClN4O3S2 ([M+H]+) 589.1135, found 589.1132.
2-Benzylthio-4-chloro-5-(4-methylphenylcarbamoyl)-N-(4-phenylquinazolin-2-yl)benzenesulfonamide (24, C35H27ClN4O3S2)
Starting from 1.020 g 8 to 0.430 g 2-aminobenzophenone for 3.5 h, the crude product was obtained. Extraction of byproducts with 7 cm3 boiling ethanol gave 0.610 (47%) 24. M.p.: 257–260 °C (dec.); TLC: Rf = 0.70 (benzene-EtOH 4:1); 1H NMR (500 MHz, DMSO-d6): δ = 2.31 (s, 3H, CH3), 4.38 (s, 2H, CH2), 6.98–7.08 (m, 3H, arom), 7.20 (d, J = 8.3 Hz, 2H, 4-MePh), 7.27–7.33 (m, 3H, arom), 7.48–7.54 (m, 5H, arom), 7.62 (d, J = 8.3 Hz, 2H, 4-MePh), 7.62–7.67 (m, 2H, arom), 7.90–7.95 (m, 2H, arom), 8.11 (br s, 1H, H-6), 10.48 (s, 1H, NHCO), 13.7 (br s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6/TFA): δ = 20.1, 36.5, 117.3, 120.3, 120.7, 125.0, 127.3, 128.0, 128.2, 128.3, 128.7, 129.0, 129.2, 130.0, 130.9, 131.6, 132.6, 133.6, 134.3, 135.8, 136.0, 136.4, 138.8, 140.0, 145.7, 153.3, 163.6 ppm; IR (KBr): = 3394, 3276 (NH), 2922 (CH), 1656 (C=O), 1621 (NH def), 1582, 1519, 1494 (C=C, C=N), 1362, 1147 (SO2) cm−1; HRMS (ESI-TOF): m/z calcd. for C35H27ClN4O3S2 ([M+H]+) 651.1291, found 651.1103.
Cell culture and cell viability assay
All chemicals, if not stated otherwise, were obtained from Sigma-Aldrich (St. Louis, MO, USA). The MCF-7 and HeLa cell lines were purchased from Cell Lines Services (Eppelheim, Germany), the HCT-116 cell line was purchased from ATCC (ATCC-No: CCL-247). Cells were cultured in in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 units/cm3 penicillin, and 100 μg/cm3 streptomycin. Cultures were maintained in a humidified atmosphere with 5% CO2 at 37 °C in an incubator (Heraceus, HeraCell).
Cell viability was determined using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Cells were seeded in 96-well plates at a density of 5 × 103 cells/well and treated for 72 h with the examined compounds in the concentration range 1–100 μM (1, 10, 25, 50, and 100 μM). Following treatment, MTT (0.5 mg/cm3) was added to the medium and cells were further incubated for 2 h at 37 °C. Cells were lysed with DMSO and the absorbance of the formazan solution was measured at 550 nm with a plate reader (Victor, 1420 multilabel counter). The optical density of the formazan solution was measured at 550 nm with a plate reader (Victor, 1420 multilabel counter). The experiment was performed in triplicate. Values are expressed as the mean ± SD of at least three independent experiments.
NCI screening
Cytotoxicity evaluation of compounds 9–14 and 17–24 was performed at the National Cancer Institute according to NCI-60 DTP human tumor cell line screen procedure [53–56].
QSAR study
Three-dimensional models of studied compounds were built in Gaussian (Gaussian Inc.) software using DFT geometry optimization and B3LYP/6-31G(d) basis set. The optimized structures were imported to Dragon software (Talete, Milano, Italy) to calculate molecular descriptors. Descriptors with constant values or variance less than 0.0001 were discarded. Multiple linear regression along with forward stepwise algorithm and model validation was performed in Statistica software (Statsoft, Tulsa, USA). PLS, OPLS model and its leave one out cross-validation as well as VIP calculations were made in SIMCA (Umetrics, Umea, Sweden).
Metabolic stability
Stock solutions of studied compounds were prepared at concentration of 10 mM in DMSO. Working solutions were prepared daily by dilution of stock with reaction buffer or acetonitrile, final concentration of organic solvent did not exceed 1%. Incubation mixture contained 10 μM of a studied derivative, 1 mM of NADPH (Sigma-Aldrich) and 0.5 mg/cm3 of pooled human liver microsomes (HLM, Sigma-Aldrich) in potassium phosphate buffer (0.1 M, pH 7.4). Incubation was carried out in thermostat at 37 °C and started by addition of studied compound. 50 mm3 samples were taken at starting point and after 5, 15, 30, 45, and 60 min. Enzymatic reaction was terminated by the addition of the equal volume of ice-cold acetonitrile. Incubations were performed in triplicates, the average t1/2 and standard deviation was calculated. Control incubations were performed without NADPH to assess chemical instability. After collection, samples were immediately centrifuged (10 min, 10,000 rpm) and resulted supernatant was directly analyzed or kept in − 80 °C until LC–MS analysis. Natural logarithm of a compound over IS peak area ratio was plotted versus incubation time. Metabolic half-time (t1/2) was calculated from the slope of the linear regression.
LC–MS analysis was performed on an Agilent 1260 system coupled to SingleQuad 6120 mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). Poroshell EC-C18 (2.1 mm × 150 mm, 2.7 μm, Agilent Technologies, Santa Clara, CA, USA) was used in reversed-phase mode with gradient elution starting with 5% of phase A (10 mM ammonium formate in water) and 95% of phase B (10 mM ammonium formate in acetonitrile–water mixture, 95:5 v/v). The amount of phase B was linearly increased to 100% in 10 min. Total analysis time was 15 min at 40 °C, flow rate was 1 cm3/min and the injection volume was 5 mm3. The mass spectrometer was equipped with electrospray ion source and operated in positive ionization. Mass analyzer was set individually to each compound to detect [M+H]+ protonated molecule. MSD parameters of the ESI source were as follows: nebulizer pressure 35 psig (N2), drying gas 10 cm3/min (N2), drying gas temperature 300 °C, capillary voltage 3 kV, fragmentor voltage 150 V.
In silico prediction of ADMET properties
Pharmacokinetic and toxicity properties (ADMET) of compounds were determined by an ADMET descriptors algorithm protocol of pkCSM approach (http://biosig.unimelb.edu.au/pkcsm/prediction) [44].
Acknowledgements
This project was financed by National Science Centre based on the decision number DEC-2013/09/B/NZ7/00048.
Contributor Information
Aneta Pogorzelska, Email: aneta.pogorzelska@gumed.edu.pl.
Jarosław Sławiński, Email: jaroslaw.slawinski@gumed.edu.pl.
References
- 1.Siegel R, Miller K, Jemal A. Ca-Cancer J Clin. 2015;65:5. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Scozzafava A, Owa T, Mastrolorenzo A, Supuran CT. Curr Med Chem. 2003;10:925. doi: 10.2174/0929867033457647. [DOI] [PubMed] [Google Scholar]
- 3.Casini A, Scozzafava A, Mastrolorenzo A, Supuran CT. Curr Cancer Drug Targets. 2002;2:55. doi: 10.2174/1568009023334060. [DOI] [PubMed] [Google Scholar]
- 4.Reddy MVR, Mallireddigari MR, Pallela VR, Cosenza SC, Billa VK, Akula B, Subbaiah DRCV, Bharathi EV, Padgaonkart A, Lv H, Gallo JM, Reddy EP. J Med Chem. 2013;56:5562. doi: 10.1021/jm400575x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy NS, Mallireddigari MR, Cosenza S, Gumireddy K, Bell SC, Reddy EP, Reddy MVR. Bioorg Med Chem Lett. 2004;14:4093. doi: 10.1016/j.bmcl.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 6.Motavallizadeh S, Fallah-Tafti A, Maleki S, Shirazi AN, Pordeli M, Safavi M, Ardestani SK, Asd S, Tiwari R, Oh D, Shafiee A, Foroumadi A, Parang K, Akbarzadeh T. Tetrahedron Lett. 2014;55:373. doi: 10.1016/j.tetlet.2013.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Liu T, Wu R, Lou J, Cao J, Dong X, Yang B, He Q, Hu Y. Bioorg Med Chem. 2010;18:8478. doi: 10.1016/j.bmc.2010.10.047. [DOI] [PubMed] [Google Scholar]
- 8.Ghorab MM, Ragab FA, Heiba HI, El-Gazzar MG, Zahran SS. Eur J Med Chem. 2015;92:682. doi: 10.1016/j.ejmech.2015.01.036. [DOI] [PubMed] [Google Scholar]
- 9.Żołnowska B, Sławiński J, Belka M, Bączek T, Kawiak A, Chojnacki J, Pogorzelska A, Szafrański K. Molecules. 2015;20:19101. doi: 10.3390/molecules201019101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sławiński J, Żołnowska B, Brzozowski Z, Kawiak A, Belka M, Bączek T. Molecules. 2015;20:5754. doi: 10.3390/molecules20045754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pogorzelska A, Sławiński J, Brożewicz K, Ulenberg S, Bączek T. Molecules. 2015;20:21960. doi: 10.3390/molecules201219821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brożewicz K, Sławiński J. Eur J Med Chem. 2012;55:384. doi: 10.1016/j.ejmech.2012.07.042. [DOI] [PubMed] [Google Scholar]
- 13.Vijaychand A, Manjula SN, Bharath EN, Divya B. Int J Pharma Bio Sci. 2011;2:780. [Google Scholar]
- 14.Sciences A. Int J Pharm Pharm Sci. 2015;7:1. [Google Scholar]
- 15.Rajput R, Mishra AP. Int J Pharm Pharm Sci. 2012;4:2. [Google Scholar]
- 16.Selvam TP, Kumar PV. Res Pharm. 2011;1:1. [Google Scholar]
- 17.Ravez S, Castillo-Aguilera O, Depreux P, Goossens L. Expert Opin Ther Pat. 2015;25:789. doi: 10.1517/13543776.2015.1039512. [DOI] [PubMed] [Google Scholar]
- 18.Medina PJ, Goodin S. Clin Ther. 2008;30:1426. doi: 10.1016/j.clinthera.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Schmid-Bindert G, Zhou C. Ther Adv Med Oncol. 2011;4:19. doi: 10.1177/1758834011427927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haringhuizen A, van Tinteren H, Vaessen HFR, Baas P, van Zandwijk N. Ann Oncol. 2004;15:786. doi: 10.1093/annonc/mdh177. [DOI] [PubMed] [Google Scholar]
- 21.Keating GM. Drugs. 2014;74:207. doi: 10.1007/s40265-013-0170-8. [DOI] [PubMed] [Google Scholar]
- 22.Siemann DW, Brazelle WD, Jürgensmeier JM. Int J Radiat Oncol Biol Phys. 2009;73:897. doi: 10.1016/j.ijrobp.2008.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ledermann JA, Embleton AC, Raja F, Perren TJ, Jayson GC, Rustin GJS, Kaye SB, Hirte H, Eisenhauer E, Vaughan M, Friedlander M, González-Martín A, Stark D, Clark E, Farrelly L, Swart AM, Cook A, Kaplan RS, Parmar MKB. Lancet. 2016;387:1066. doi: 10.1016/S0140-6736(15)01167-8. [DOI] [PubMed] [Google Scholar]
- 24.Helfrich BA, Kim J, Gao D, Chan DC, Zhang Z, Tan AC, Bunn PA. Mol Cancer Ther. 2016;15:2314. doi: 10.1158/1535-7163.MCT-16-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamauchi T, Uzui K, Shigemi H, Negoro E, Yoshida A, Ueda T. Cancer Sci. 2013;104:926. doi: 10.1111/cas.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR. Nature. 2007;447:865. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huo H, Zhou Z, Qin J, Liu W, Wang B, Gu Y. PLoS ONE. 2016;11:e0154605. doi: 10.1371/journal.pone.0154605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sleebs BE, Czabotar PE, Fairbrother WJ, Fairlie WD, Flygare JA, Huang DCS, Kersten WJA, Koehler MFT, Lessene G, Lowes K, Parisot JP, Smith BJ, Smith ML, Souers AJ, Street IP, Yang H, Baell JB. J Med Chem. 2011;54:1914. doi: 10.1021/jm101596e. [DOI] [PubMed] [Google Scholar]
- 29.Ghorab MM, Alsaid MS, Al-Dosari MS, El-Gazzar MG, Parvez MK. Molecules. 2016;21:189. doi: 10.3390/molecules21020189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghorab M, Alsaid M, Al-Dosary M, El-Gazzar M. Med Chem. 2016;12:448. doi: 10.2174/1573406412666160201120921. [DOI] [PubMed] [Google Scholar]
- 31.Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, Dearden J, Gramatica P, Martin YC, Todeschini R, Consonni V, Kuz’min VE, Cramer R, Benigni R, Yang C, Rathman J, Terfloth L, Gasteiger J, Richard A, Tropsha A. J Med Chem. 2014;57:4977. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tropsha A. Mol Inf. 2010;29:476. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- 33.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich A, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ. Gausian 09, Revision A.02. Wallingford: Gaussian Inc; 2016. [Google Scholar]
- 34.Mauri A, Consonni V, Pavan M, Todeschini R. MATCH. 2006;56:237. [Google Scholar]
- 35.Todeschini R, Consonni V. Handbook of molecular descriptors. Hoboken: Wiley; 2008. [Google Scholar]
- 36.Wu Z, Li D, Meng J, Wang H. Introduction to SIMCA-P and its application. In: Esposito Vinzi V, Chin W, Henseler J, Wang H, editors. Handbook of partial least squares. Springer handbooks of computational statistics. Berlin: Springer; 2010. [Google Scholar]
- 37.UMETRI AB (1996) UMETRI AB: SIMCA-P for Windows: Maltivariate Modeling, Analysis and SPC of Process Data
- 38.Sławiński J, Szafrański K, Pogorzelska A, Żołnowska B, Kawiak A, Macur K, Belka M, Bączek T. Eur J Med Chem. 2017;132:236. doi: 10.1016/j.ejmech.2017.03.039. [DOI] [PubMed] [Google Scholar]
- 39.Viswanadhan VN, Reddy MR, Bacquet RJ, Erion MD. J Comput Chem. 1993;14:1019. doi: 10.1002/jcc.540140903. [DOI] [Google Scholar]
- 40.Lin TH, Li HT, Tsai KC. J Chem Inf Comput Sci. 2004;44:76. doi: 10.1021/ci030295a. [DOI] [PubMed] [Google Scholar]
- 41.Żołnowska B, Sławiński J, Pogorzelska A, Szafrański K, Kawiak A, Stasiłojć G, Belka M, Zielińska J, Bączek T. Chem Biol Drug Des. 2017;90:380. doi: 10.1111/cbdd.12955. [DOI] [PubMed] [Google Scholar]
- 42.Nassar AEF, Kamel AM, Clarimont C. Drug Discov Today. 2004;9:1020. doi: 10.1016/S1359-6446(04)03280-5. [DOI] [PubMed] [Google Scholar]
- 43.Guengerich FP. Chem Res Toxicol. 2001;14:611. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 44.Pires DEV, Blundell TL, Ascher DB. J Med Chem. 2015;58:4066. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang MQ, Wilkinson B. Curr Opin Biotechnol. 2007;18:478. doi: 10.1016/j.copbio.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 46.Kim RB. Drug Met Rev. 2002;34:47. doi: 10.1081/DMR-120001389. [DOI] [PubMed] [Google Scholar]
- 47.Amin L. Drug Target Insights. 2013;7:27. doi: 10.4137/DTI.S12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wandel C, Kim RB, Guengerich FP, Wood AJJ. Drug Metab Dispos. 2000;28:895. [PubMed] [Google Scholar]
- 49.Sławiński J. Pol J Chem. 2001;75:1309. [Google Scholar]
- 50.Żołnowska B, Sławiński J, Pogorzelska A, Chojnacki J, Vullo D, Supuran CT. Eur J Med Chem. 2014;71:135. doi: 10.1016/j.ejmech.2013.10.081. [DOI] [PubMed] [Google Scholar]
- 51.Sławiński J, Gdaniec M. Eur J Med Chem. 2005;40:377. doi: 10.1016/j.ejmech.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 52.Sławiński J, Bednarski P, Grünert R, Reszka P. Pol J Chem. 2003;77:53. [Google Scholar]
- 53.Alley MC, Scudiero DA, Monks PA, Hursey ML, Czerwinski MJ, Fine DL, Abbtt BJ, Mayo JG, Shoemaker RH, Boyd MR. Cancer Res. 1988;48:589. [PubMed] [Google Scholar]
- 54.Grever MR, Schepartz SA, Chabner BA. Semin Oncol. 1992;19:622. [PubMed] [Google Scholar]
- 55.Boyd MR, Paull KD. Drug Dev Res. 1995;34:91. doi: 10.1002/ddr.430340203. [DOI] [Google Scholar]
- 56.Shoemaker RH. Nat Rev. 2006;6:813. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]