

Supplemental Digital Content is available in the text.
Key Words: immunotherapy, immunomodulatory antibodies, cisplatin, tumor microenvironment
Abstract
While immunomodulatory monoclonal antibodies (mAbs) have therapeutic efficacy against many tumors, few patients are cured. Attempting to improve their therapeutic efficacy we have applied the TC1 mouse lung carcinoma model and injected established subcutaneous tumors intratumorally with 3 weekly doses of various combinations of mAbs. Combinations of mAbs to CTLA4/PD1/CD137 (the 3 mAb combination) and to CTLA4/PD1/CD137/CD19 (the 4 mAb combination) were most efficacious to induce complete regression of both the injected tumor and an untreated tumor in the same mouse. Tumor cure was consistently associated with shifting a Th2 to a Th1 response in tumor-draining lymph nodes and spleen and it involved epitope specific and long-lived memory T cells as well as M1 macrophages. This shift and accompanying tumor rejection was harder to achieve as the treated tumors increased in size. Relapse of tumors which had initially regressed following treatment with immunomodulatory mAbs was associated with return of a Th2 microenvironment in tumors, tumor-draining lymph nodes and spleens rather than the emergence of immune-resistant tumor cells. While mAbs to CTLA4 plus PD-1 were therapeutically ineffective, combining the 2 of them with intraperitoneal cisplatin, 10 mg/kg, induced long-term complete tumor regression in most mice with small TC1 tumors and the therapeutic efficacy against larger tumors improved by administrating cisplatin together with the 3 or 4 mAb combination.
It has been known since the 1960s that tumor-bearing subjects, humans as well as mice, form a cell-mediated antitumor immune response that is detectable in vitro also when the tumor burden is large.1 More recently, promising approaches have been introduced to make tumor immunity therapeutically effective both in mice2–7 and human patients.8–18 Most clinical success has been obtained by administering immunomodulatory monoclonal antibodies (mAbs) to counteract the immunosuppressive tumor microenvironment. Although relatively few patients have been cured, such mAbs are becoming key contributors to the treatment of cancer. Trying to extend their ability to cure established tumors we apply mouse tumor models and presently focus on the TC1 lung carcinoma whose human papillomavirus (HPV) epitopes facilitate analyses with tetramers.
A Th2 type inflammation at the tumor site facilitates carcinogenesis and tumor progression.19–21 The cervix mucosa from women infected with high-risk HPV provides a good model to investigate this, since the causative agent is known and the stages characterizing the progression from premalignant carcinoma in situ (CIN1) to invasive cancer are well defined. Already at CIN1 the mucosa is infiltrated by lymphocytes expressing B-cell or plasma cell markers, and cellular infiltration characteristic of a Th2 type response increases during progression to invasive carcinoma.22,23 Likewise, increased numbers of lymphocytes expressing the B-cell marker CD19 and tumor-associated Th2 lymphokines can be detected in tumor-draining lymph nodes (TLN) shortly after mice have been transplanted with cells from the TC1 lung carcinoma.6,23
In view of the dominant role of a Th2 response during carcinogenesis and tumor progression we hypothesized that shifting the tumor microenvironment to Th1 can induce regression and have published results with the SW1 and B16 melanoma, TC1 lung carcinoma, and 1D8 ovarian carcinoma which support this hypothesis.5,6 Long-term regression of established mouse tumors was demonstrated with mAbs targeting CD137/PD-1/CTLA4/CD19 (the “4 mAb combination”) or CD137/PD-1/CTLA4 (the “3 mAb combination”) and intratumoral mAb delivery was found to be more therapeutically effective than systemic administration.6 We now show that a sustained immunologic shift from a Th2 to a Th1 milieu is fundamental for treatment to be curative, that this shift is harder to achieve with a larger tumor load, and that recurrence of treated tumors is associated with a return of a Th2 tumor microenvironment. We also extend our previous demonstration that cisplatin acts synergistically with immunomodulatory mAbs.24 A combination of anti-CTLA4, and anti-PD-1 mAbs together with cisplatin, all approved for clinical application in humans, cured ∼50% of mice with small TC1 tumors and the therapeutic efficacy against larger TC1 tumors improved by also including mAb to CD137 or to CD137 plus CD19.
MATERIALS AND METHODS
Tumor Lines and Mice
TC125 is a C57BL/6 lung carcinoma line transfected with HPV-16 E6 and E7, SW1 is a clone derived from the K1735 melanoma of C3H origin,26 and B16 is a clone derived from a spontaneous melanoma in a C57BL/6 mouse.27 Tumor cells were cultured in IMEM supplemented with 10% fetal bovine serum (Atlanta Biological, Norcross, GA), penicillin, and streptomycin before cell suspensions were prepared and transplanted to syngeneic mice.
Six to 8-week female C57BL and C3H mice were purchased (Charles River Laboratories, Wilmington, MA). The animal facilities are certified by the Association for Assessment and Accreditation of Laboratory Animal Care, and our protocols are approved by the institution (University of Washington).
mAbs for Immunotherapy
The following mAbs were purchased from BioXcell: anti-CD137 (Rat IgG1; LOB12.3; Cat. #BE0169), anti-PD-1 (Rat IgG2a; RMP1-14; Cat. #BE0146), anti-CTLA4 (Mouse IgG2b; 9D9; Cat. #BE0164), anti-CD19 (Mouse IgG2b; 1D3; Cat. #BE0150), anti-PD-L1 (Rat IgG2b; 10F.9G2, Cat. #BE0101), and control (Rat IgG2a; 2A3; Cat. #BE0089). In most experiments we applied mAbs to CD137/CTLA4/PD-1 or to CD137/CTLA4/PD-1/CD19, referred to as the 3 and 4 mAb combinations, respectively.
Animal Studies
In most experiments, 5×105 tumor cells were transplanted subcutaneously (SC) on the right flank. When the mice had tumors of about 30 (or 60 when indicated under the Results section) mm2 surface area, they were randomized into treatment groups and injected with mAb combinations, 0.5 mg of each mAb; the mAbs (200 μL) were injected intratumorally (IT) at weekly intervals for a total of 3 times; as control, mice were injected IT with the same dose of irrelevant IgG mAb. In many experiments (see the Results section) mice were also injected with cisplatin (10 mg/kg), which was given intraperitoneally (IP). Mice were monitored daily for evidence of toxicity, and 2 perpendicular tumor diameters were measured twice/week and tumor surfaces were calculated. Overall survival was recorded.
We also performed experiments in which mice were transplanted with 5×105 TC1 cells SC on their right and 1.5×105 TC1 cells on their left flank. When the right tumors had ∼5 to 6 mm and those on the left side 2 to 3 mm mean diameter, we commenced treatment by injecting IT, a combination of mAbs into the right tumors while leaving the left tumors untreated; this treatment was repeated twice at weekly intervals, as for the other experiments.
For in vitro mechanistic studies, mice were euthanized and samples were obtained of spleens and TLN and in some experiments also from tumors as tumor-infiltrating lymphoid cells (TIL). Samples were prepared for flow cytometry and quantitative polymerase chain reaction (qPCR).
In order to investigate the different responses to treatment of big and small tumors, 5×105 TC1 cells were transplanted SC on the right side of the back. When the mice had a tumor of either about 8 or 4 mm in diameter they were injected IT with the 3 mAb combination given either alone or together with cisplatin IP; the mAb combination was also injected, IT, on days 7 and 21. For comparison, one group of tumor-bearing mice only received control mAb and another group comprised naive mice which were injected SC with the 3 mAb combination and also given cisplatin IP. The mice were euthanized after 3 and 24 days when TLNs and spleens were collected and their cells were analyzed by fluorescence-activated cell sorting (FACS) and qPCR.
We also investigated the immune responses in mice which had sustained long-term tumor regression by IT injection of the 3 mAb combination either 3 or 6 months before they were euthanized; for comparison we investigated cells from naive and from tumor-bearing controls. Lymph node and spleen cells were analyzed by FACS and qPCR.
Flow Cytometry
Single cell suspensions from spleens and lymph nodes were prepared as described.6 TIL were isolated from pooled tumors as described.26 All flow cytometry experiments were performed at least 3 times. Single cell suspensions were incubated with mouse Fc receptor binding inhibitor for 10 minutes before staining with antibodies to CD45 (clone 30-F11), CD3 (clone 145-2C11), CD4 (clone GK1.5), CD8 (clone 53-6.7), CD19 (clone eBio1D3), CD86 (clone GL1), CD11b (clone M1/70), Gr-1 (clone RB6-8C5), CD69 (clone H1.2F3), CD44 (clone IM9), CD62L (clone MEL-14), and CD11c (clone N418; all from eBioscience, San Diego, CA) for 30 minutes. Flow cytometry was performed using FACS Calibur (BD Biosciences) and the lymphocyte population was selected by gating CD45+ cells. The data were analyzed using Flow Jo software (Tree Star, Ashland, OR). For tetramer staining, 10 μL of PE labeled HLA-A*02:01 Human HPV16 E7 tetramer (NIH Tetramer Core Facility) was added to 200 μL mouse lymphocyte suspension (1×106 cells per tube). After incubation for 30 minutes, cells were centrifuged and resuspended in phosphate-buffered saline with 1% paraformaldehyde and then analyzed by flow cytometry. PE labeled HLA-A*02:01 human mesothelin tetramer (NIH Tetramer Core Facility) was used as control.
qPCR
qPCR assays were performed on 4 mice per treatment group to assess the transcription levels of genes of Tbx21, IFNγ, Il12, TNFα, Gata3, IL4, TIM3, IDO1, Ahr, and p27. Total RNA was extracted from tumors and TLNs and reverse transcribed to cDNA using the First Strand cDNA synthesis kit (Fischer Scientific, IL). Subsequently, cDNA was measured by qPCR assays on ABI Prism 7900 Sequence Detector System (Applied Biosystems, Foster City, CA). The relative quantification was performed using the ΔΔCt method described by the manufacturer.
Statistics
Results were expressed as mean±SEM. Student t test was used to compare the statistical difference between 2 groups and 1-way analysis of variance was used to compare 3 or more groups. Kaplan-Meier survival analyses were performed using GraphPad Prism 5, and the Gehan-Breslow-Wilcoxon test was used to determine significance. P<0.05 was considered to be statistically significant.
RESULTS
Immunomodulatory mAb Combinations Can Cure Mice With Established TC1 Tumors and Cisplatin Dramatically Improves the Therapeutic Efficacy
As shown in Figure 1, the 3 and 4 mAb combinations (anti-CTLA4/PD-1/CD137 and anti-CTLA4/PD-1/CD137/CD19, respectively) induced complete tumor regression in 6 of 15 (40%) and 9 of 20 (45%) mice, respectively, which had TC1 tumors of ∼30 mm2 surface area when first treated. In contrast, mAbs to CTLA4, CD137, PD-1 or PDL1 were ineffective as single agents, and mAbs to CD137 plus PD-1 or CTLA4 plus PD1 failed to cure any of the mice. However, together with 1 IP injection of cisplatin (10 mg/kg) on the first day of mAb administration, the anti-CTLA4/PD1 combination induced complete regression in 6 of 10 treated mice while cisplatin by itself had only a small effect (Fig. 1A).
FIGURE 1.
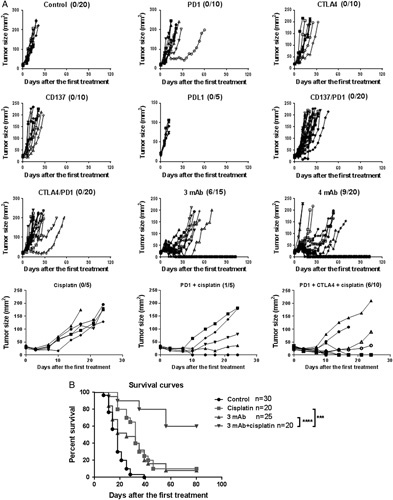
A, Intratumorally administration of mAbs and mAb combinations to mice with subcutaneous TC1 tumors; cured mice/mice per group is shown within parentheses. Control mice received equivalent dose of irrelevant IgG mAb (Rat IgG2a). B, Cisplatin plus the 3 mAb combination produces 60% tumor-free survival of mice with TC1 tumors on both sides of the back. The right side tumors were injected intratumorally with the mAbs which were given weekly 3 times; on the first day of treatment cisplatin was injected intraperitoneally. mAbs indicates monoclonal antibodies.
We next performed a similar experiment with mice that had TC1 tumors on both sides of the back, injecting the tumors on the right side with the mAbs. As shown in Figure 1B, cisplatin plus the 3mAb combination dramatically prolonged survival while cisplatin or the mAbs by themselves had <10% efficacy. The reason why the mAb combination alone was less efficacious than in Figure 1A is that the tumor load (2 per mouse) was larger when treatment commenced. There was a close relationship between the growth of the injected and untreated tumors in the same mice, demonstrating that the IT injection of mAb combination induced a systemic effect (Fig. 2).
FIGURE 2.
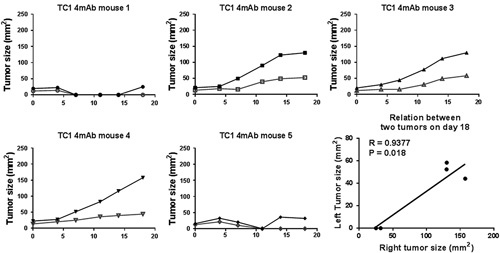
Growth curves for TC1 tumors on both sides of the back. The right side tumors (solid points) were injected with the 4 mAb combination (without cisplatin) while the left side tumor (hollow points) were not injected. The not-injected tumors regressed in mice showing strong therapeutic effect against the injected tumor. mAb indicates monoclonal antibody.
Immunomodulatory mAb Combinations More Effectively Induce a Th1 Response and Regressions When the Tumors are Small
Mice which had SW1, B16 or TC1 tumors with a surface area of 30 (smaller) or 64 (larger) mm2 were injected IT with the 4 mAb combination, which was repeated weekly for 2 more times. As shown in Figure 3A, complete regression of SW1 and B16 tumors was significantly more frequent with the smaller tumors and treatment of larger TC1 tumors did not prolong survival at all.
FIGURE 3.
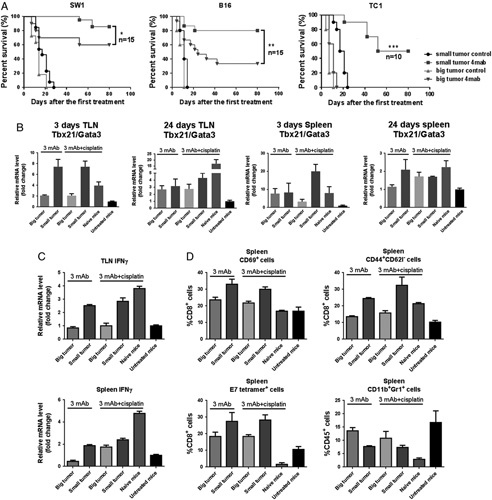
A, Survival of mice with SW1, B16 or TC1 tumors which had 4 (small tumor) or 8 mm (big tumor) diameters on the right side of the back when the 4 mAb combination was first injected; 3 weekly treatments. B, Tbx/Gata3 ratios for TLN and spleen cells 3 and 24 days after the first injection of 3 mAb with or without cisplatin. Naive (ie, not tumor-bearing) mice were injected subcutaneous with the 3 mAb combination and given cisplatin intraperitoneally; untreated tumor-bearing mice received control mAbs. C, Expression level of IFNγ for TLN and spleen cells 24 days after the first treatment. D, Percentages of CD69+, CD44+CD62l−, E7 tetramer+ and CD11b+Gr1+ spleen cells on day 24. IFN indicates interferon; mAb, monoclonal antibody; TLN, tumor-draining lymph nodes.
To relate the therapeutic response to the in vitro findings, a similar experiment was performed with mice which had TC1 tumors on one side of the back with a surface area of either 30 (smaller) or 64 (larger) mm2. The mice received the mAb combination weekly, given alone or together with one administration of cisplatin IP on the first day of mAb injection. Groups were also included with untreated tumor-bearing mice and with naive mice which received the mAb combination SC and cisplatin IP. The mice were euthanized 3 or 24 days after the first mAb injection and their immune status investigated. As shown in Figure 3B, the mAb treatment generated high Tbx21/Gata 3 ratios in TLN and spleens in mice with small tumors but not in mice with large tumors, and it increased expression of INFγ in mice with small tumors (Fig. 3C). Furthermore, it more effectively generated CD69+ activated T cells, long-term memory (CD44+CD62L−) T cells and epitope specific T cells (Fig. 3D) in mice with small tumors and fewer CD11b+Gr1+ myeloid derived suppressor cells. The findings were similar for TLNs and spleens. An immunologic profile analogous to that in responding mice was seen in naive mice injected with the 3 mAb combination plus cisplatin.
We also investigated the immune status of TLNs and spleens from mice whose tumors remained regressed 3 or 6 months after the first administration of the 3 mAb combination plus cisplatin; in parallel, the lymph nodes and spleens were tested from naive (ie, not tumor transplanted) mice which were injected with the 3 mAbs SC plus cisplatin IP. The reactivity of TLN from cured mice and from treated naive mice was similar (data not shown), while spleens from cured mice had a stronger Th1 response than spleens from the naive mice. The Tbet/Gata3 ratio with lymphoid cells from the cured mice was higher than from tumor-bearers, and the ratio was higher 3 months after the first treatment as compared with 6 months (Fig. 4) indicating that the mAb-induced Th1 response decreases with time.
FIGURE 4.
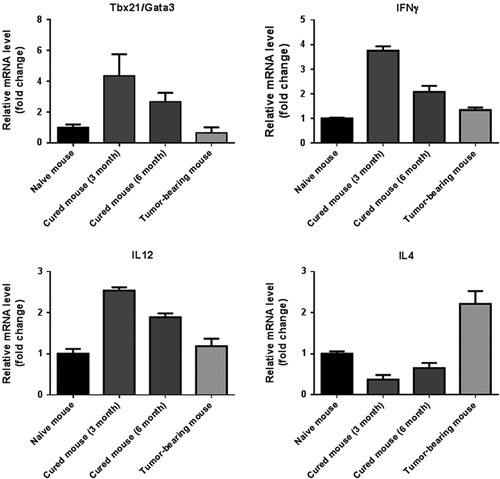
Quantitative polymerase chain reaction data for spleen cells from mice whose TC1 tumor regressed and did not recur after treatment with the 3 mAb combination intratumorally plus cisplatin intraperitoneally. Naive mice received the 3 mAb combination subcutaneous and cisplatin intraperitoneally; data are included from tumor-bearing mice which received control mAbs and no cisplatin. The data indicate that a shift from Th2 to Th1 immune status accompanies the cure of TC1 tumors. IFN indicates interferon; IL, interleukin; mAb, monoclonal antibody.
Relapse of Treated Tumors is Associated With a Th2 Tumor Microenvironment and Not With Selection of Immunotherapy Resistant Cells
Eight of 45 (18%) mice which macroscopically rejected their TC1 tumors relapsed 20 to 40 days after the last mAb administration, while the other 37 regressors remained tumor free for an observation period >100 days. Two types of experiments were performed to investigate the mechanisms responsible for the relapse.
First, cells were isolated from a relapsing TC1 tumor and cultured in vitro after which they were transplanted to mice whose resulting tumors were treated with the 3 mAb combination plus cisplatin. As shown in Figure 5 the therapeutic efficacy was similar to the original tumors that were not derived from recurrent tumors.
FIGURE 5.
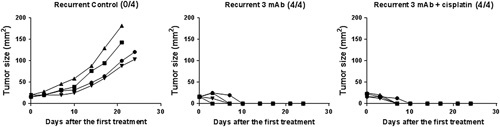
Tumor cells were collected from a treated tumor which recurred and cultured in vitro. Subsequently, the cultured tumor cells were transplanted to C57 mice and 3 times weekly tumor injection with the 3 mAb combination, with or without an initial dose of cisplatin, was started on day 7. mAb indicates monoclonal antibody.
Second, we tested cells from TLN and spleens of mice whose treated tumors had relapsed after 2 to 3 weeks of temporary regression and compared the data with those from mice whose tumors remained regressed. As shown in Figure 6, lymphoid cells from mice whose tumors recurred were consistently of the Th2 type and thus similar to lymphoid cells from nonresponders. In contrast, mice whose tumors remained regressed had decreased numbers of CD19 cells (Fig. 6A) and expanded numbers of CD3 (Fig. 6A), CD4 (Fig. 6E), and CD8 cells (Fig. 6E), including CD3 cells with increased expression of the activation marker CD69 (Fig. 6B), and there was a higher percentage of T cells which were positive for the E7 tetramers (Fig. 6D) in mice whose tumors remained regressed. In the relapsing mice, the number of CD44+CD62l− effector memory T cells was decreased (Fig. 6C) as compared with regressors. PCR data supported the notion that mice with recurrent tumors had a Th2 response (Fig. 6F) in TLN and spleens. In contrast, mice whose tumors remained regressed had significantly increased expression of Th1-related genes such as Tbx21, IL12, IFNγ, TNFα and a decreased expression of Th2 genes such as Gata3, IL4, TIM3.
FIGURE 6.
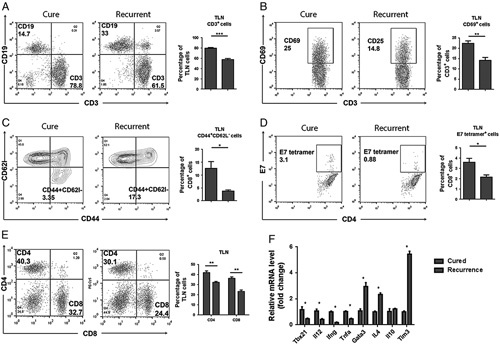
A–E, Flow cytometry data for lymphoid cells from TLN from mice whose TC1 tumors either remained regressed or recurred after treatment with the 3 mAb combination plus cisplatin. F, Upregulation of Th1 and downregulation of Th2 related genes expression in the cured mice compared with mice with recurrent tumors. mAb indicates monoclonal antibody; TLN, tumor-draining lymph nodes.
In some mice the treated tumor remained stable for a period of time and then recurred while other tumors gradually regressed and did not come back within an observation period over 6 months, that is, they were most likely cured. In order to clarify the underlying mechanism, we compared by flow cytometry and qPCR TLN and spleen cells from mice whose treated tumors either regressed or remained stable. As shown in Figure 7, more CD3, CD4, CD8 and CD11c+CD86+ cells and fewer CD11b+Gr1+ cells were found in the regressing mice and their Th2-related genes such as GATA3, Il4 and TGFβ were dramatically increased, supporting the view that a shift from Th2 immune response to Th1 response plays a key role for tumor regression.
FIGURE 7.
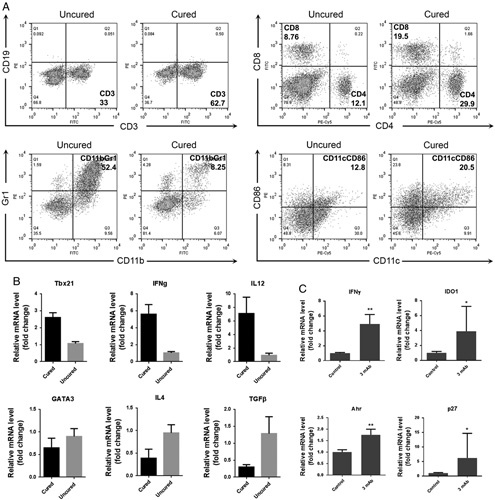
A and B, Flow cytometry and quantitative polymerase chain reaction data for lymphoid cells of spleen from mice whose TC1 tumors were either completely regressed (cured) or not regressed (uncured) 3 days after the last treatment with the 4 mAb combination plus cisplatin. C, Quantitative polymerase chain reaction data for tumors from mice whose tumors were stable 3 days after the last treatment of 3 mAb combination as compared to untreated tumor-bearing control mice. IFN indicates interferon; IL, interleukin; mAb, monoclonal antibody.
As reported by Liu et al,28 although IFNγ signalling triggers tumor cell apoptosis, it can induce tumor cell dormancy when IDO1 and AhR are highly expressed at the tumor site. As shown in this and previous studies, treatment with “our” mAb combinations significantly increases the produce of IFNγ, and we observed that sometimes TC1 growth was inhibited for a period of time after which it resumed. We investigated, therefore, tumors whose growth was stabilized after treatment. Our data indicated a high level of IFNγ in the “dormant” tumors and involvement of the IDO1-AhR-p27 signal pathway (Fig. 7C). We speculate that an IDO or Ahr inhibitor could abrogate the IFNγ-induced tumor dormancy and result in enhanced therapeutic efficacy.
TILs from responding and nonresponding mice were also analyzed. As shown in Figure 8A, a much higher level of CD3 cells was found in responding tumors and many more CD11b+Gr1+ cells in nonresponding ones (Fig. 8B). Relapsing mice had larger spleens than mice which remained tumor-free, the number of CD11b+Gr1+ cells in their spleens was about 5-fold higher (Fig. 8B), and these cells expressed higher level of IDO (Fig. 8B). According to qPCR, expression of arginase29 was much higher in the splenocytes from mice with recurrent tumors, indicating that they were of the M2 phenotype while splenocytes from cured mice had the M1 phenotype (Fig. 8C).
FIGURE 8.
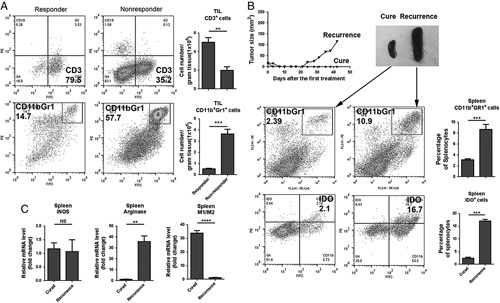
A, Flow cytometry for TIL from mice whose TC1 tumors either responded (partial regression) or did not respond (progressive growth) after treatment with the 3 mAb combination plus cisplatin. B, Enlarged spleen and increased CD11b+Gr1+ and IDO+ cells in spleen from a mouse with recurrent tumor compared with a cured mouse. C, Increased expression of arginase and reversed M1/M2 response in the spleen of a mouse with recurrent tumor. mAb indicates monoclonal antibody; TIL, tumor-infiltrating lymphoid cells.
Treatment With the 3 or 4 mAb Combinations, Alone or Together With Cisplatin, has Low Toxicity
Except for 5 deaths in >300 treated mice, there were few side effects in 3 different tumor models when the mice were treated with 3 or 4 mAb combinations given alone or together with cisplatin except modest hair loss and depigmentation in <5% of the treated animals There is no significant weight loss after the injection of 3 mAb or 4 mAb combination and there is no difference between the treated groups and control groups (Supplemental Figure, Supplemental Digital Content 1, http://links.lww.com/JIT/A505).
DISCUSSION
The promise of immunotherapy for cancer is now widely recognized.30,31 One of its advantages is the ability to destroy cancer cells in several ways, including antigen-specific and nonspecific cellular mechanisms, antibody-dependent cellular cytotoxicity and molecules such as TNFα. Furthermore, the high mutability of cancer cells32 creates new T-cell epitopes which can be recognized for tumor destruction33 together with various shared tumor epitopes while a single therapeutic target is often lost during during tumor progression. Remarkable clinical successes have been seen with many tumors after administration of mAbs to anti-PD-1/PDL-1 and CTLA4 and further improved therapeutic efficacy is likely to result from combination with chemotherapy, additional immunomodulators, cancer vaccines, adoptive transfer of engineered cells, etc.
Our previous studies demonstrate that combinations of immunomodulatory mAbs to CTLA4/PD-1/CD137 or CTLA4/PD-1/CD137/CD19 (referred to as the 3 and 4 mAb combinations, respectively) can produce complete regression of established mouse tumors5,6 and that addition of cisplatin improves the therapeutic efficacy.24 The present data expand these findings and show long-term tumor-free remission after treatment with immunomodulatory mAbs plus cisplatin when neither the mAbs alone nor the drug alone was curative. Our data also confirm the ability of cisplatin to counteract the immunosuppressive tumor microenvironment,34 including a population of IDO positive MDSC which is tolerogenic.35 Besides these immunosuppressive effects, a long-term tumor dormancy has recently been demonstrated, induced in a minor “stem-like tumor subpopulation” by IFNγ via an IDO-kynurenine-aryl-hydrocarbon-receptor (AhR)-p27-dependent mechanism which protects from IFNγ-induced apoptosis and temporarily slows down proliferation28 followed by recurrent tumor growth.
Importantly, complete regression was generated in approximately 50% of mice with established TC1 tumors by combining anti-CTLA4 plus anti-PD-1 mAbs with cisplatin, that is, by applying agents that are clinically approved for human patients. We hypothesize that the therapeutic efficacy can be further improved by drugs that counteract the induction of IDO by IFNγ whose level is increased by the immunomodulating mAbs.28 It is noteworthy that combination of radiotherapy with immunomodulatory mAbs can also act synergistically.36–38
As in our previous studies5,6 IT administration of immunomodulatory mAbs induces a systemic effect which can destroy a second, not-injected TC1 tumor in the same mouse and involves T lymphocytes, M1 macrophages, and natural killer cells, that is it involves both antigen specific and nonspecific mechanisms. The latter compensates for the fact that tumor antigens, and the ability of major histocompatibility complex (MHC) to present them, are commonly lost during tumor progression.39 While intraveneous administration is clinically preferable, it is noteworthy that intratumoral delivery also of other therapeutic agents can generate a more effective systemic response.40
The ability of immunomodulatory mAbs to shift the tumor microenvironment from Th2 to Th1 decreases with tumor size as does the therapeutic efficacy. We speculate that immunosuppressive molecules contributed by both the tumor cells and the tumor-bearing host such as PDL, IDO, TGF-β, IL10, tumor antigens, etc., are responsible to maintain a peripheral immunologic tolerance to the tumor, but this needs to be studied further.
The mAb induced Th1 response declines within weeks after mAb administration. This probably explains why several macroscopically regressed TC1 tumors relapsed, as the relapsed tumors were sensitive to the original treatment protocol. Importantly, lymphoid cells from recurrent tumors as well as from TLN and spleens from relapsing mice had the characteristics of a Th2 type inflammation, that is, the relapses were caused by failure of the immune system to retain a tumor-destructive Th1 response. Studies are needed to show whether prolonged treatment with the mAbs, cisplatin, and/or a therapeutic tumor vaccine can overcome this problem and we expect that monitoring Th1 and Th2 antitumor responses will be informative.
Supplementary Material
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.
ACKNOWLEDGMENT
The authors thank Dr P. Abrams for valuable suggestions.
Conflicts of Interest/Financial Disclosures
None reported. All authors have declared there are no financial conflicts.
Footnotes
K.E.H., M.D., H.O.S., and I.H.: conception and design. M.D. and Y.Y.Y.: acquisition of data. M.D., K.E.H., I.H., and H.O.S.: analysis and interpretation of data. K.E.H. and M.D.: writing. K.E.H. and I.H.: administrative, technical or material support.
REFERENCES
- 1.Hellstrom KE, Hellstrom I. Cellular immunity against tumor specific antigens. Adv Cancer Res. 1969;12:167–223. [DOI] [PubMed] [Google Scholar]
- 2.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. [DOI] [PubMed] [Google Scholar]
- 3.Melero I, Shuford W, Newby SA, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3:682–685. [DOI] [PubMed] [Google Scholar]
- 4.Pardoll D. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai M, Wei H, Yip YY, et al. Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation. J Immunother. 2013;36:248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai M, Yip YY, Hellstrom I, et al. Curing mice with large tumors by locally delivering combinations of immunomodulatory antibodies. Clin Cancer Res. 2014;21:1127–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen LP. Lymphocyte co-signal molecules in cancer immunotherapy. J Immunother. 2006;29:664. [Google Scholar]
- 8.Hodi F, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl J Med. 2010;363:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ascierto P, Simeone E, Sznol M, et al. Clinical experiences with anti-CD137 and anti-PD1 therapeutic antibodies. Semin Oncol. 2010;37:508–516. [DOI] [PubMed] [Google Scholar]
- 10.Balachandran VP, Cavnar M, Zeng S, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17:1094–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galon J, Angell HK, Bedognetti D, et al. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39:11–26. [DOI] [PubMed] [Google Scholar]
- 15.Couzin-Frankel J. Cancer immunotherapy. Science. 2013;342:1432–1433. [DOI] [PubMed] [Google Scholar]
- 16.Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol. 2011;29:4828–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. [DOI] [PubMed] [Google Scholar]
- 20.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ovestad IT, Gudlaugsson E, Skaland I, et al. Local immune response in the microenvironment of CIN2-3 with and without spontaneous regression. Mod Pathol. 2010;23:1231–1240. [DOI] [PubMed] [Google Scholar]
- 23.Feng Q, Wei H, Morihara J, et al. Th2 type inflammation promotes the gradual progression of HPV-infected cervical cells to cervical carcinoma. Gynecol Oncol. 2012;127:412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei H, Zhao L, Li W, et al. Combinatorial PD-1 blockade and CD137 activation has therapeutic efficacy in murine cancer models and synergizes with cisplatin. PLOS One. 2013;8:e84927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tuve S, Liu Y, Tragoolpua K, et al. In situ adenovirus vaccination engages T effector cells against cancer. Vaccine. 2009;27:4225–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang SC, Yang Y, Raycraft J, et al. Melanoma cells transfected to express CD83 induce anti-tumor immunity that can be increased by also engaging CD137. Proc Natl Acad Sci. 2004;101:4990–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fidler IJ, Gersten DM, Budmen MB. Characterization in vivo and in vitro of tumor cells selected for resistance to syngeneic lymphocyte-mediated cytotoxicity. Cancer Res. 1976;36:3160–3165. [PubMed] [Google Scholar]
- 28.Liu Y, Liang X, Yin X, et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-gamma-induced immunologic dormancy of tumor-repopulating cells. Nat Commun. 2017;8:15207–15222. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Briken V, Mosser DM. Editorial: switching on arginase in M2 macrophages. J Leukoc Biol. 2011;90:839–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prendergast GC, Jaffee EM. Cancer immunologists and cancer biologists: why we didn’t talk then but need to now. Cancer Res. 2007;67:3500–3504. [DOI] [PubMed] [Google Scholar]
- 31.Chen D, Mellman I. Oncology meets immunology: the cancer immunity cycle. Immunity. 2013;39:1–10. [DOI] [PubMed] [Google Scholar]
- 32.Bielas JH, Loeb KR, Rubin BP, et al. Human cancers express a mutator phenotype. ProcNatl Acad Sci. 2006;103:18238–18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. [DOI] [PubMed] [Google Scholar]
- 34.de Biasi AR, Villena-Vargas J, Adusumilli PS. Cisplatin-induced antitumor immunomodulation: a review of preclinical and clinical evidence. Clin Cancer Res. 2014;20:5384–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baban B, Hansen AM, Chandler PR, et al. A minor population of splenic dendritic cells expressing CD19 mediates IDO-dependent T cell supppression via type I IFN signaling following B7 ligation. Int Immunol. 2005;17:909–919. [DOI] [PubMed] [Google Scholar]
- 36.Oweida A, Lennon S, Calame D, et al. Ionizing radiation sensitizes tumors to PD-L1 immune checkpoint blockade in orthotopic murine head and neck squamous cell carcinoma. Oncoimmunology. 2017;6:e1356153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hwang WL, Niemierko A, Hwang KL, et al. Clinical outcomes in patients with metastatic lung cancer treated with PD-1/PD-L1 inhibitors and thoracic radiotherapy. JAMA Oncol. 2017;4:253–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sundahl N, De Wolf K, Rottey S, et al. A phase I/II trial of fixed-dose stereotactic body radiotherapy with sequential or concurrent pembrolizumab in metastatic urothelial carcinoma: evaluation of safety and clinical and immunologic response. J Transl Med. 2017;15:150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrone S, Marincola FM. Loss of HLA class I antigens by melanoma cells: molecular mechanisms, functional significance and clinical relevance. Immunol Today. 1995;16:487–494. [DOI] [PubMed] [Google Scholar]
- 40.Sagiv-Barfi I, Czerwinski DK, Levy S, et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med. 2018;10:426–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.