

Abstract
Gcn4 is a yeast transcriptional activator induced by amino acid starvation. ChIP-seq analysis revealed 546 genomic sites occupied by Gcn4 in starved cells, representing ~30% of Gcn4 binding-motifs. Surprisingly, only ~40% of the bound sites are in promoters, of which only ~60% activate transcription, indicating extensive negative control over Gcn4 function. Most of the remaining ~300 Gcn4-bound sites are within coding sequences (CDS), with ~75 representing the only bound sites near Gcn4-induced genes. Many such unconventional sites map between divergent antisense and sub-genic sense transcripts induced within CDS, adjacent to induced TBP peaks—consistent with Gcn4 activation of cryptic, bidirectional internal promoters. Mutational analysis confirms that Gcn4 sites within CDS can activate sub-genic and full-length transcripts from the same or adjacent genes, showing that functional Gcn4 binding is not confined to promoters. Our results show that internal promoters can be regulated by an activator that functions at conventional 5′-positioned promoters.
Keywords: Gcn4, transcription, activation, nucleosomes, cryptic promoters
In Brief
Rawal et al. showed that yeast transcription factor Gcn4 frequently binds within nucleosome-occupied protein coding regions in yeast cells, and that a substantial fraction of these events confer activation of cryptic internal promoters as well as canonical 5′-positioned promoters at the same or adjacent genes.
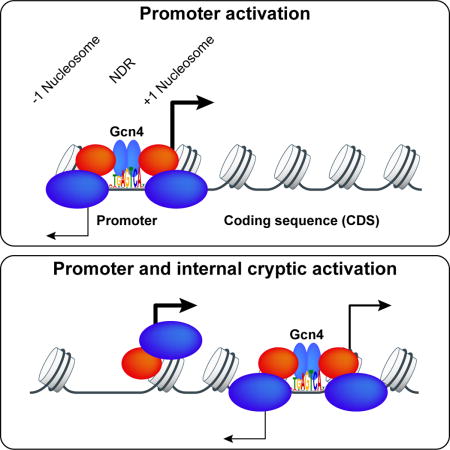
INTRODUCTION
Promoters for RNA Polymerase II (Pol II) in Saccharomyces cerevisiae harbor upstream activation sequences (UASs) that bind transcriptional activators, typically found in nucleosome-depleted regions (NDRs) 5′ of the transcription start site (TSS). UASs can function bidirectionally at variable distances upstream from the TSS (Rando and Winston, 2012), but appear to function poorly from downstream of the TSS (Struhl, 1984) (Guarente and Hoar, 1984). This restriction might reflect occlusion by nucleosomes, as UAS elements normally reside within NDRs, while CDSs are covered by nucleosomes (Jiang and Pugh, 2009). Indeed, general regulatory factors (GRFs) bind in NDRs and exclude nucleosomes (Bai et al., 2011) (Rando and Winston, 2012), which can facilitate activator binding (Devlin et al., 1991; Yu and Morse, 1999) (Levo et al., 2017).
Gcn4 is a transcriptional activator responsible for induction of >500 genes in response to amino acid limitation (Jia et al., 2000; Natarajan et al., 2001), which inducesGcn4 synthesis (Hinnebusch, 2005). ChIP-chip analysis of myc-tagged Gcn4 binding to intergenic regions in cells starved for isoleucine and valine (ILV) (Harbison et al., 2004), and filtering of the data for evolutionary conservation (MacIsaac et al., 2006), revealed 100 high-confidence, conserved Gcn4 binding sites upstream of 126 genes. While these include many amino acid biosynthetic genes induced by Gcn4 (Natarajan et al., 2001), it appears that most of the ~500 genes induced by Gcn4 are activated indirectly (Natarajan et al., 2001). In addition, ~1/4th of the 126 genes (MacIsaac et al., 2006) were not induced in cells starved for histidine or ILV (Natarajan et al., 2001; Saint et al., 2014), suggesting that Gcn4 binding at many promoters does not activate transcription. GCN4-dependent repression of hundreds of genes also occurs in starved cells, including most genes encoding ribosomal proteins (RPGs) (Natarajan et al., 2001), and there is evidence that Gcn4 acts as a repressor at RPG promoters (Joo et al., 2010).
To increase understanding of genes directly activated or repressed by Gcn4, we conducted ChIP-seq analysis of native Gcn4 in cells starved for ILV using the inhibitor of acetolactate synthase, sulfometuron methyl (SM) (Jia et al., 2000). By not limiting the analysis to intergenic sequences, we discovered that ~60% of Gcn4 occupancy peaks (~300) are not in promoters, occurring instead within CDS or 3′ non-coding regions; and 74 of these unconventional Gcn4 peaks are the only occupied binding sites detected nearby a Gcn4-induced gene. We present multiple lines of evidence that Gcn4 binding within CDS frequently activates cryptic, bidirectional internal promoters, and can also stimulate transcription from the canonical 5′-positioned promoter of the same or adjacent gene.
RESULTS
Similarity to the consensus motif and low nucleosome occupancy are major determinants of Gcn4 binding in vivo
To identify direct target genes of Gcn4, we conducted ChIP-seq analysis of native Gcn4 in wild-type (WT) cells treated with SM to induce Gcn4 expression (WT_I cells). Uninduced WT (WT_U) and SM-treated gcn4Δ cells (gcn4Δ_I) were analyzed as controls. Gcn4 peaks were identified from 2 or more biological replicates of WT_I and WT_U cultures using the program MACS2. After eliminating “hyper-ChIP-able” regions where signals were observed in gcn4Δ cells (Fig. S1A), we identified 546 Gcn4 peaks whose occupancies are much greater in WT_ I versus WT_U chromatin, and very low in gcn4Δ_I chromatin (‘All Gcn4 sites’ of Data S1). These encompass Gcn4 peaks in 101 of 126 promoters identified previously as containing Gcn4-myc binding sites (MacIsaac et al., 2006) (‘Potential target scoring’, Data S1), including known Gcn4 target genes ARG1, ARG4, HIS4, and CPA2, which exhibit strong induction of the occupancy of Pol II subunit Rpb3 across the CDSs (Cole et al., 2011; Qiu et al., 2015) (Fig. 1A). Hence, our ChIP-seq analysis captured a large proportion of Gcn4 binding sites in the genome.
Figure 1. Most Gcn4 occupancy peaks are centered on consensus Gcn4 binding motifs that tend to occur distal to nucleosome dyads.
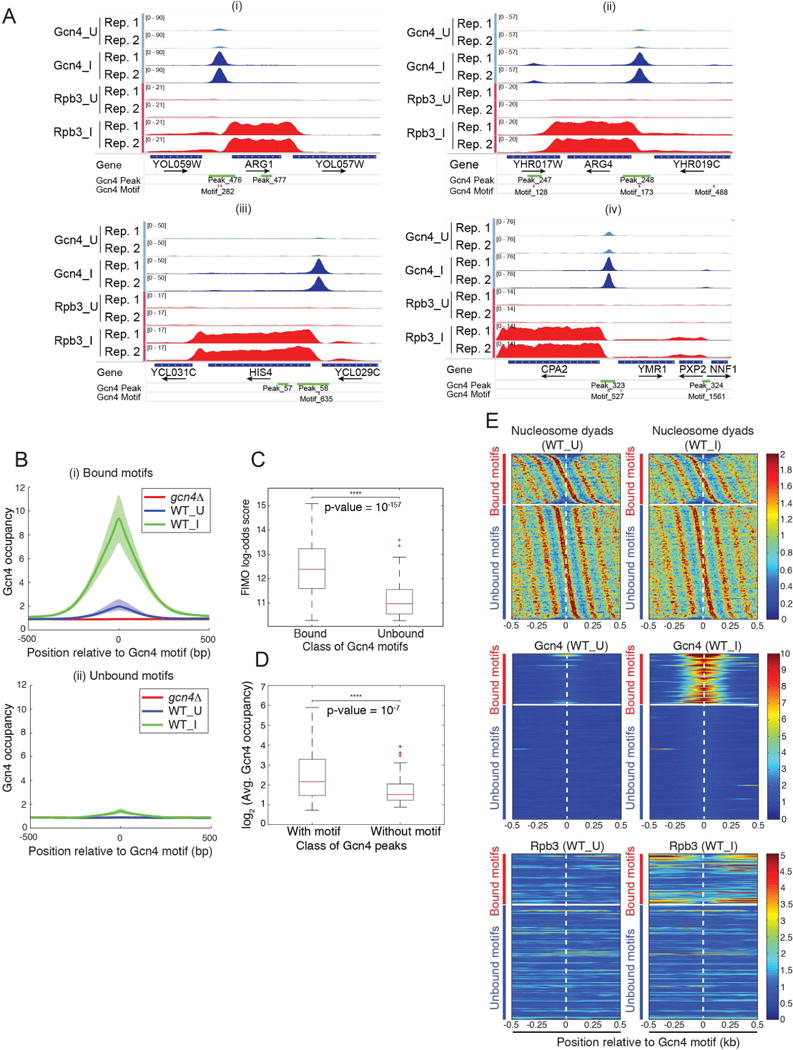
(A) Gcn4 occupancy peaks 5′ of canonical Gcn4 target genes. Gcn4 occupancies from WT SM-induced (Gcn4_I) or uninduced cells (Gcn4_U) for two biological replicates (Rep.1, Rep.2) (tracks 1-4), and Rpb3 occupancies from the corresponding cells (tracks 5-8), plotted with the Integrated Genomics Viewer (IGV). Tracks 1-4 and 5-8 were autoscaled as groups, and the range of values is indicated on the left side of each track. Occupancies were normalized such that the average occupancy of Gcn4 or Rpb3 for each chromosome is unity. Positions of Gcn4 peaks identified by MACS2 and consensus Gcn4 motifs found by FIMO are shown in the bottom tracks. (B) SM-induced Gcn4 occupancies center around canonical Gcn4 consensus motifs. Average Gcn4 occupancy surrounding the consensus motifs in Fig. S1B that are (i) bound or (ii) unbound by Gcn4 in WT_U, WT_I, or gcn4Δ_I cells. The solid lines show the averages of ≥3 replicates; shaded areas show the ranges of values for individual replicates. (C) Comparison of the log-odds scores reported by FIMO for the two classes of Gcn4 consensus motifs. Paired-sample t-test p-value = 10−157; null hypothesis: pairwise difference between FIMO scores has a mean equal to zero). (D) Gcn4 occupancies (log2) for 471 Gcn4 occupancy peaks containing consensus motifs versus 75 peaks lacking significant matches (FIMO match p-value ≥ 10−4) to the consensus motifs in Fig. S1B, from ‘All Gcn4 sites’ in Data S1. Paired-sample t-test p-value = 10−7; null hypothesis: pairwise difference between occupancies has a mean equal to zero. (E) Gcn4 motifs in bound occupancy peaks are depleted near nucleosome dyads. Nucleosome dyad distribution (top), Gcn4 occupancy (middle), and Rpb3 occupancy (bottom) near the Gcn4 motifs in WT_U (left) or WT_I cells (right). Motifs are split into two classes, Gcn4-bound (upper group) and Gcn4-unbound (lower group); in each group, Gcn4 motifs are sorted according to the relative position of the nearest nucleosome in WT_U cells. See also Fig. S1.
We identified a consensus Gcn4 binding site present in sequences covered by the 546 Gcn4 occupancy peaks using MEME; and this motif and its reverse complement (Fig. S1B) are essentially identical to consensus sequences determined from the ChIP-chip analysis of Gcn4-myc binding sites (MacIsaac et al., 2006) and by in vitro binding of Gcn4 to synthetic DNA arrays (Zhu et al., 2009). Most of the observed 546 Gcn4 peaks (471 peaks; 86%) contain at least one significant match to the consensus motif and 56 contain two or more such motifs, whereas 75 peaks have no consensus site (‘Gcn4 peak motifs’, Data S1). Gcn4 occupancies averaged over all 471 peaks with consensus motifs are unimodally distributed about the motifs, as expected if the latter are Gcn4 binding sites (Fig. 1B, (i)). The average occupancy in these peaks increased ~5-fold on SM-induction, whereas no binding was observed in SM-treated gcn4Δ cells (Fig. 1B, (i)). A much smaller average occupancy peak centered over consensus motifs was also observed for the 1217 motifs that did not exhibit statistically significant Gcn4 binding (Fig. 1B, (ii)), indicating that a small fraction of these motifs are bound by Gcn4 with occupancies below the threshold for statistical significance. The “Find Individual Motif Occurrences” (FIMO) scores, quantifying the similarity of each motif to the consensus (Grant et al., 2011), are much greater for the 537 motifs bound by Gcn4 versus the 1217 unbound motifs (Fig. 1C), indicating that similarity to the consensus sequence is an important determinant of detectable Gcn4 occupancy.
The 75 Gcn4 peaks lacking a strong match to the consensus exhibit Gcn4 occupancies significantly lower than the 471 Gcn4 peaks containing consensus motifs (Fig. 1D). MEME analysis of sequences within 100bp of the centers of these peaks revealed a degenerate version of the consensus motif (Fig. S1C) in 40 of 75 peaks, coinciding with the mode of Gcn4 occupancy (Fig. S1D), suggesting that the degenerate motifs are the Gcn4 binding sites in these peaks. Thus, ~92% of 546 identified Gcn4 peaks contain a strong match to the consensus motif (~86%) or a centrally positioned degenerate version of this sequence (~6%).
Among the 1754 statistically significant matches to the consensus motif, only ~31% (537) fall within the 546 Gcn4 occupancy peaks. Previous studies indicated that nucleosomal occlusion of binding sites can limit transcription factor binding in vivo (Liu et al., 2006). To determine whether Gcn4 binding is impeded by nucleosomes, we determined distances to the nearest nucleosome dyad for each of the 1754 motifs in a high-resolution nucleosome map determined for SM-treated cells (Qiu et al., 2015); and arranged the motif-to-dyad distances in a heat-map to reveal the presence of surrounding nucleosomes (Fig. 1E, top panels). Gcn4 occupancies in WT_I cells for the same order of motifs revealed an “hourglass” pattern for the bound motifs (Fig. 1E, middle panels, right), indicating a tendency for greater Gcn4 binding to motifs between nucleosomes versus motifs closer to dyads. Gcn4 binding at sites furthest from dyads was associated with the highest levels of induced Rpb3 occupancy in WT_I cells, determined previously (Qiu et al., 2015) (Fig. 1E, bottom panels, right). Both trends are consistent with the most potent Gcn4 binding sites residing within NDRs where nucleosome occupancies are lowest.
As summarized in Fig. S1E, we also used supervised machine learning to obtain evidence that a strong match to the consensus motif is the most important determinant of Gcn4 binding in vivo, but that motifs with inferior matches can still bind Gcn4 if located in regions depleted of nucleosomes. Accessibility to DNase I and occupancies of Abf1, histone variant H2A.Z, TATA-binding protein (TBP), or subunits of chromatin remodelers were found to make only marginal contributions to the probability of Gcn4 occupancy.
Only a subset of 5′ Gcn4 occupancy peaks activate transcription
Unexpectedly, only 42% (227) of the identified Gcn4 peaks are located upstream of the nearest annotated TSS, in the canonical location of UAS elements (5′ sites; Figs. 2A–B, dark blue sector). Taking into account divergently transcribed genes, the 227 peaks in 5′ non-coding (NC) regions could potentially activate 303 genes (NC/5′ genes in ‘Potential target scoring’, Data S1). To evaluate whether a 5′ peak is functional, we interrogated previous mRNA expression data that identified 512 genes induced by starvation for histidine (using the inhibitor 3-aminotriazole (3-AT) or ILV in a Gcn4-dependent manner (i.e. greater mRNA induction in WT versus gcn4Δ cells), or by a constitutively activated GCN4c allele (Natarajan et al., 2001) (‘Natarajan 2001 targets’, Data S2). We also examined a compilation of SM-induced mRNAs (Saint et al., 2014) (‘Saint et al_microarray’, Data S2); and our own ChIP-seq analysis of Rpb3 in SM-treated WT cells (Qiu et al., 2015), which we complemented with Rpb3 ChIP-seq analysis in isogenic gcn4Δ cells and thereby identified 294 genes with greater Rpb3 occupancies in SM-treated WT versus gcn4Δ cells (‘Rpb3 ChIP-seq’, Data S2). About 2/3rd of these 294 genes belong to the group of 512 genes mentioned above (Natarajan et al., 2001), demonstrating extensive overlap between these two groups of Gcn4-induced genes (P-value of 8.5 × 10−140 in the hypergeometric test).
Figure 2. Most Gcn4 peaks are not in 5′ non-coding regions and a fraction of UC peaks appear to activate transcription.
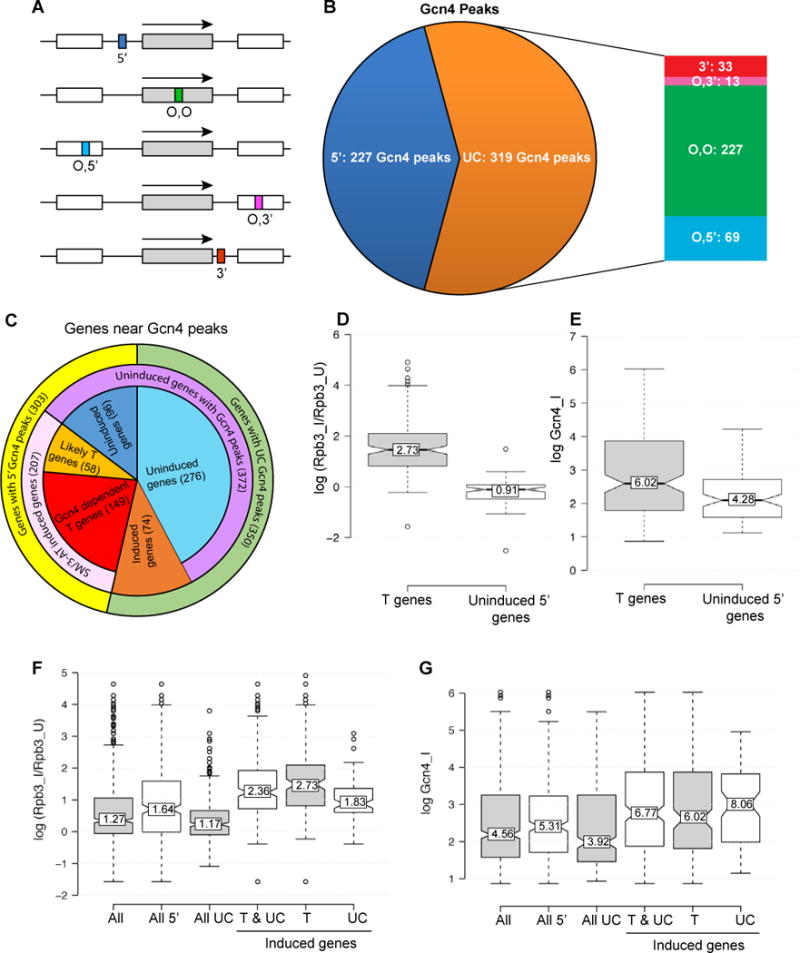
(A-B) Proportion of 546 Gcn4 peaks found in different locations relative to TSSs (from ‘All Gcn4 sites’ in Data S1). 5′ peaks depicted as a dark blue box (A) or sector (B). UC peaks (orange sector in (B)) occur in the ORF of the same gene (O,O green box/sector), the ORF of the upstream gene (O,5′, blue), the ORF of the downstream gene (O,3′, dark pink), or 3′ non-coding region of the same gene (3′, red); arrows indicate direction of transcription. (C) Pie chart showing proportions of genes with 5′ Gcn4 peaks (yellow sector) or UC peaks (green sector) that exhibit mRNA or Rpb3 induction in response to 3-AT, SM, or a GCN4c allele (pink sector), in a manner shown to be Gcn4-dependent (red or dark orange sectors). As explained in Methods, 97% of the 223 T and induced UC target genes exhibit induction of mRNA or Rpb3, or reduced expression in gcn4Δ versus WT cells, on SM treatment. (D-E) Rpb3 induction ratios (D) and induced Gcn4 occupancies (E) of 149 T genes and 96 uninduced genes with 5′ Gcn4 peaks. (F-G) Rpb3 induction ratios (F) and induced Gcn4 occupancies (G) of 647 genes with all arrangements of proximal Gcn4 peaks (All); all 303 genes with 5′ peaks (All 5′); all 344 genes with UC Gcn4 peaks (All UC); all 223 induced T and UC target genes (T & UC); 149 T genes (T); and 74 UC target genes.
Among the 303 genes with 5′ Gcn4 peaks, 2/3rd show induction of mRNA or Rpb3 occupancy on starvation induced by 3-AT or SM (Fig. 2C, pink). Of these 207 genes, ~72% also showed Gcn4-dependence for transcriptional induction by the aforementioned criteria, and were classified as direct Gcn4 targets (Fig. 2C, red) (149 “T” genes in ‘T and UC targets’, Data S1). The remaining 58 induced genes are likely direct targets, as Gcn4 binds in their promoters (Fig. 2C, light orange) (‘Induced 5prime likely-T genes’, Data S1), but we confined our analysis to the 149 T genes.
Gene ontology analysis of the T genes revealed the expected enrichment in genes encoding amino acid biosynthetic enzymes (45 genes, P<1×10−14), enzymes of vitamin/co-factor biosynthesis (9 genes), amino acid/vitamin transporters (5 genes), mitochondrial carrier proteins (4 genes), autophagy-related proteins (4 genes), and four transcription factors activating genes in one of these GO categories (4 genes) (‘T gene GO summary’, Data S1). As all of these genes can be viewed as instrumental in replenishing amino acids (Natarajan et al., 2001), the T genes are highly represented in canonical Gcn4 targets that mediate an adaptive response to amino acid limitation.
Interestingly, 96 of the 303 genes with 5′ Gcn4 peaks showed no significant SM-induction of mRNA expression or Rpb3 occupancies (Fig. 2C, dark blue) (‘Uninduced 5′ genes’, Data S1), exhibiting a median SM-induction ratio for Rpb3 of 0.91, compared to 2.73 for the 149 T genes (Fig. 2D). However, these uninduced genes have a median Gcn4 occupancy only ~30% lower than that of T genes (Fig. 2E), suggesting that appreciable Gcn4 binding in many promoters evokes little transcription. In fact, 27 of these uninduced 5′ genes have a median Gcn4 occupancy (7.44) even greater than that of T genes (6.02), but exhibit ~10% repression in SM rather than induction of Rpb3 (’27 high-Gcn4 Unind 5′ genes’, Data S1).
We considered different possibilities to account for the uninducibility of the 27 genes with high-occupancy 5′ Gcn4 peaks. For YFR057W, the Gcn4 peak maps ~1.5kb upstream from the CDS—possibly too distant for activation—and also within ~10kb of a telomere in a transcriptionally silent region (Fig. 3A). Ten genes contain Gcn4 peaks nearby tRNA genes (e.g. YPL112C/YPL111W, Fig. 3B), which can silence Pol II promoters (Good et al., 2013). Other mechanisms are required to account for the latent, high-occupancy Gcn4 peaks upstream of the remaining 16 genes (e.g. YKL016C-YKL015W, Fig. 3C), such as repressor binding or promoter structures or chromatin organization incompatible with activation.
Figure 3. Examples of non-functional 5′ Gcn4 peaks or UC peaks associated with uninduced or SM-induced genes.
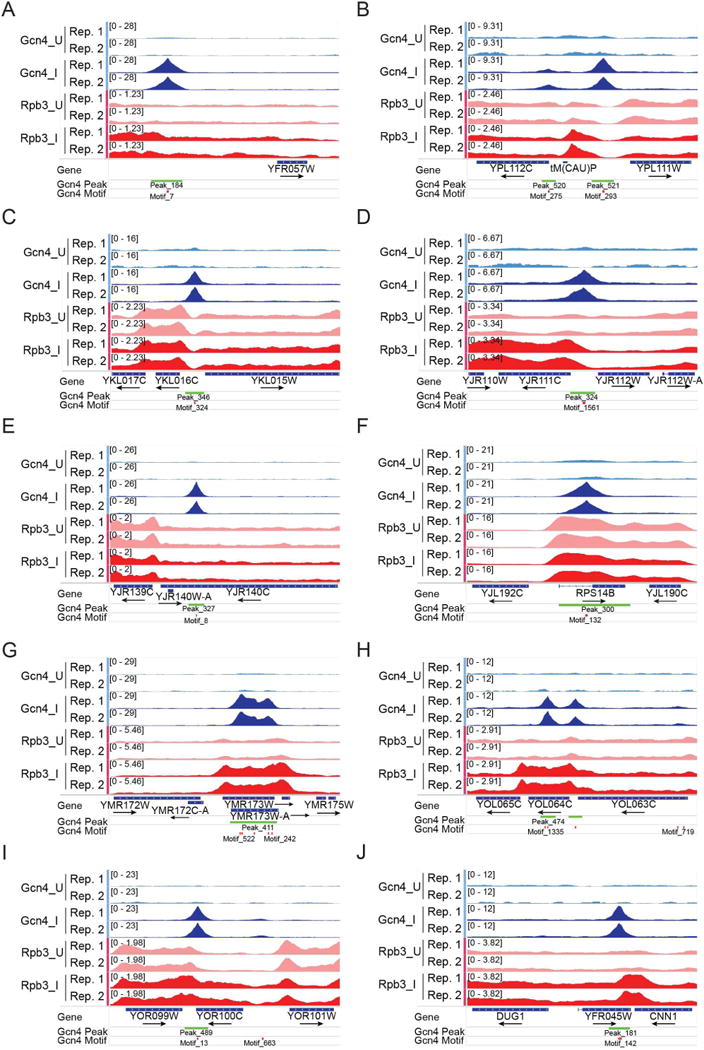
(A-D) Non-functional 5′ peaks. (E-F) Non-functional UC peaks. (G-J) UC peaks associated with SM-induction of the same or nearby gene. All displayed as in Fig. 1A.
Other evidence for the inability of Gcn4 to activate certain promoters came from analyzing 15 pairs of divergently transcribed genes that share a Gcn4 5′ peak, where only one gene is induced (e.g. YJR111C-YJR112W, Fig. 3D) (‘T-StrictUnind Diverg pairs’, Data S1); and four divergent pairs of T genes where induction of one gene is >4-fold higher than the other (‘Divergent T genes’, Data S1), as exemplified by CPA2-YMR1 (Fig. 1A(iv)). The differential activation of divergently arranged genes by Gcn4 supports findings that bidirectional transcriptional activation is an intrinsic property of yeast UASs, but the extent of activation in each direction is highly tuned in evolution (Jin et al., 2017).
Prevalent unconventional Gcn4 occupancy peaks in coding sequences and 3′ non-coding regions
Remarkably, ~60% of the 546 Gcn4 peaks do not reside in 5′ non-coding sequences (Fig. 2B, orange), and ~90% of these 319 unconventional (UC) peaks map within CDS (Fig. 2A–B, “O” for ORF peaks). For 71% of these peaks, Gcn4 binds in the ORF belonging to the nearest annotated TSS, where it could activate its own promoter from downstream of the TSS (Figs. 2A–B, O,O peaks, green); 22% lie in the CDS of the gene upstream of the nearest TSS (O,5′, blue); and ~7% reside in the CDS of the gene downstream (O,3′, pink). The remaining ~10% of UC sites occur in 3′ non-coding sequences of the gene belonging to the nearest TSS (Fig. 2A–B, 3′, red).
Most (276 of 319) UC peaks appear to be functionally inactive, as they are not associated with Gcn4-dependent SM-induction of Rpb3 or mRNA abundance of the nearby gene (Fig. 2C, light blue). Thus, the 350 genes in proximity to a UC peak have a median Rpb3 induction ratio of only 1.17, lower than observed for all genes with 5′ peaks, or for T genes (Fig. 2F, All UC vs. All 5′ and T genes). On the whole, UC peaks also display lower Gcn4 occupancies compared to all 5′ peaks, or those at T genes (Fig. 2G), helping to explain their relative inactivity. However, there are 55 genes associated with UC peaks with Gcn4 occupancies above the median value for T genes that show no SM-induction of Rpb3 (‘Uninduced UC genes’, Data S1) (e.g. YJR139C-YJR140C and RPS14B, Fig. 3E–F). Thus, UC peaks on the whole are less likely than 5′-positioned peaks to be associated with transcriptional activation, even at comparable Gcn4 occupancies.
It is intriguing that 74 UC genes are induced by Gcn4 and contain only a UC Gcn4 peak in proximity to the TSS (Fig. 2C, dark orange) (‘T and UC Targets’, Data S1). These include classical Gcn4 targets involved in amino acid and vitamin biosynthesis (TYR1, HOM2, HIS2, LYS12, LEU3, BIO5, BIO4, BIO3, CAB1, POS5, PDX1, NRK1), suggesting that certain UC peaks might function in transcriptional activation. Consistent with this, the median Gcn4 occupancy of these 74 peaks is much higher than that of all UC peaks, and even greater than that of 5′ peaks at T genes (Fig. 2G, induced UC genes vs. All UC genes and T genes). Moreover, 16/74 induced UC genes exceed the median Rpb3 induction ratio of T genes, and one member of the group, YMR173W, contains high-level Gcn4 occupancy peaks spanning the CDS in the absence of a statistically significant 5′ peak (Fig. 3G). YOR100C and DUG1 illustrate other induced genes where the only proximal Gcn4 peak is located in the 3′ non-coding sequences (Fig. 3I) or the adjacent downstream gene (Fig. 3J). YOL064C represents a separate group of genes with strong 5′ peaks but also containing extra UC binding sites in the CDS (Fig. 3H).
Gcn4 binding in CDSs can activate internal bidirectional promoters
Despite the numerous Gcn4-activated genes containing only UC Gcn4 peaks in their vicinity, these genes could be activated by Gcn4 binding to 5′ sites below the detection limit, or by other transcription factors induced by Gcn4. If however UC peaks are functional, we reasoned that they might activate cryptic promoters within the CDS or 3′ non-coding regions of the resident genes. Indeed, RNA-seq data from WT cells starved for histidine by 3-AT treatment for different periods of time revealed revealed many instances of bidirectional transcription originating within the CDS of induced genes harboring UC Gcn4 peaks. For example, 3-AT induced both antisense (AS) and sub-genic sense (SGS) transcripts within POS5, SNX41, SPO21, and COG1 CDS, which appear to initiate within ~100 bp upstream or downstream, respectively, of the internal Gcn4 peaks (Fig. 4A–D). The full-length (FL) sense transcripts of POS5 and COG1 also were induced by 3-AT.
Figure 4. Exemplar Gcn4 peaks in CDS that appear to recruit TBP and drive bi-directional sub-genic transcription.
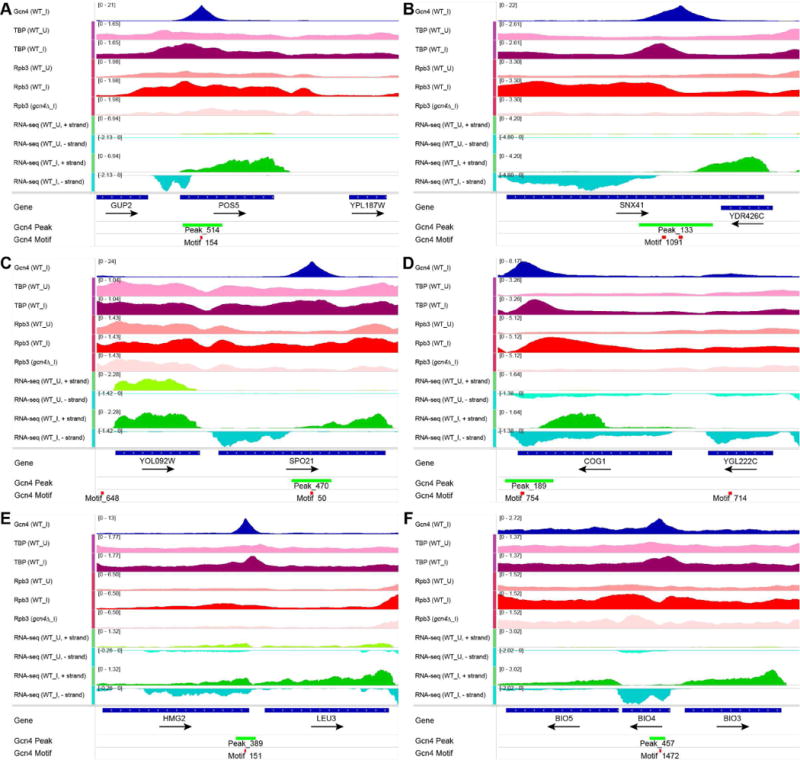
(A-F) Tracks 1-6: Occupancies of Gcn4, Rpb3, or TBP in uninduced (_U) or SM-induced (_I) cells; tracks 7-10: RNA read densities complementary to the Crick (+) or Watson (-) strand from WT cells uninduced (_U) or induced with 3-AT for 40 min (WT_I); plotted with IGV, as in Fig. 1A. RNA read densities were normalized such that the average density of the combined reads, aligned either to forward or reverse strand, is unity.
To examine more broadly 3-AT activation of internal promoters, we plotted the average RNA read densities of sense and AS transcripts relative to the positions of internal Gcn4 peaks for the subset of 62 induced “O” genes containing a single Gcn4 consensus motif coinciding with the peak of Gcn4 occupancy (‘O UC targets’, Data S3). The results indicated 3-AT induction of AS and SGS transcripts initiating ~100-150 bp upstream or downstream, respectively, of the Gcn4 motifs (Fig. 5A), with the SGS transcripts accumulating to higher levels than the induced FL transcripts of these genes (Fig. 5B). That the induced SGS and FL sense transcripts are more abundant than the induced AS transcripts (Fig. 5A–B) could indicate that Gcn4 activation in the sense direction is more efficient, or that sense transcripts are more stable than AS transcripts. A heat-map of AS RNA read densities reveals that most of the induced AS transcription derives from 20-25% of the genes (Fig. 5C, right), as quantified in Fig. 5E. Nevertheless, internal induced AS transcripts were detectable at nearly all of these genes (Fig. 5D).
Figure 5. Gcn4 binding to “O” target genes induces bidirectional transcription within ORFs.
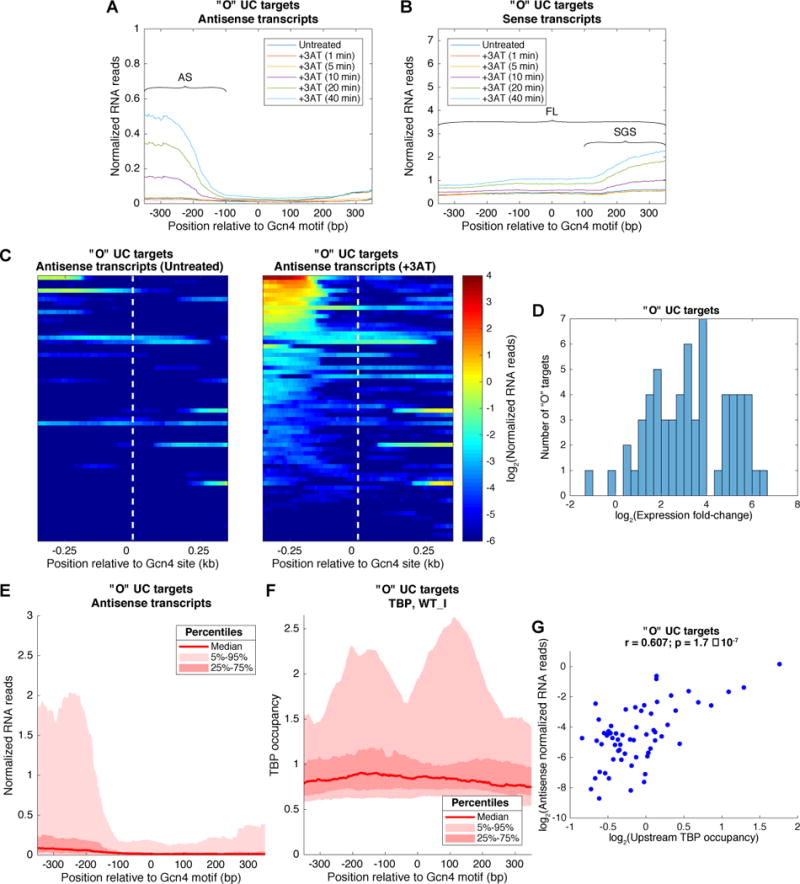
(A-B) Average RNA reads for AS (A) or sense (B) transcripts surrounding Gcn4 motifs at induced “O” UC target genes during a time course of 3AT treatment, indicating inferred positions of AS, SGS, and FL transcripts. (C) Heat maps showing AS transcript abundance and position relative to Gcn4 motifs at the “O” UC targets, before (left panel) and after (right panel) 40min of 3AT treatment, sorting genes by transcript abundance. (D) Histogram of fold-change in AS expression for “O” UC targets. (E) Distribution of AS transcript abundance upstream and downstream of the Gcn4 motifs after 40min of 3AT treatment for the “O” UC targets, showing the median number of reads (dark red line), the 25-75 percentiles (dark pink) encompassing the spread in read numbers for the 50% of genes closest to the median, and the 5-95 percentiles (light pink) for the 90% of genes closest to the median. While the median level is relatively low, genes in the top 25 percentiles (above upper boundary between light and dark pink areas, produce AS transcripts at levels comparable to the mean FL sense transcript level for this group of genes depicted in (B). RNA reads were normalized such that the genomic average RNA abundance is one. (F) TBP occupancy distribution near “O” UC targets. The median and indicated percentiles of TBP occupancy are shown as in panel (E). (G) Scatter plot representing the log2 of the average density of AS RNA reads in the 350bp regions upstream of the Gcn4 motifs versus the log2 of the average occupancy of TBP in the same loci. Pearson r = 0.607; F-test for a linear fit gives a p-value = 1.7 × 10−7; null hypothesis: coefficient of proportionality is equal to zero. See also Fig. S2 and S3.
As expected from the bidirectionality of conventional UASs, we also observed 3-AT induction of AS transcripts mapping upstream of 5′ Gcn4 peaks at T genes, again ~10-fold less abundant than the corresponding FL sense transcripts (Fig. S2A–E), consistent with previous studies (Jin et al., 2017) (Neil et al., 2009; Xu et al., 2009). Similar results were obtained from RNA-seq analysis of WT cells treated with SM under the same conditions used for Gcn4 ChIP-seq analysis (Fig. S3A–E); although the abundance of internal AS transcripts from induced “O” genes is reduced compared to that observed with 40min 3-AT treatment (Fig. S3A & C vs. Fig. 5A&C).
Supporting the RNA-seq data, ChIP-seq analysis of myc-tagged TBP on an isogenic WT strain revealed SM-induced TBP-myc occupancy peaks, mapping ~150-200 bp upstream and downstream of the Gcn4 binding motifs, at ~50% of the same group of induced “O” genes (Fig. 5F). Presumably, the twin peaks represent TBP recruitment to distinct promoters flanking the internal Gcn4 peaks driving bidirectional transcription (Rhee and Pugh, 2012). Again, a minority fraction of the genes was responsible for most of the induced TBP-myc occupancy (Fig. 5F); and the TBP occupancies upstream of the Gcn4 motifs positively correlated with the amounts of 3AT-induced AS transcription (Fig. 5G). The TBP-myc occupancies at these induced “O” genes are lower than observed in the promoters of T genes, but appear at similar locations relative to the Gcn4 binding sites for both groups of genes (cf. Figs. 5F and S2F).
At the UC genes POS5, SNX41, and COG1, the Gcn4 and associated induced TBP-myc occupancy peaks map between the 5′ ends of the divergent 3AT-induced sub-genic transcripts produced from the CDSs of these genes (Figs. 4A–B & D). At other UC genes, e.g. LEU3 and BIO3, the Gcn4 peak is close to the 5′ or 3′ end of the CDS, and the induced TBP peak resides in the abutting non-coding sequences, where it appears to mark a promoter driving induction of the adjacent genes (Fig. 4E-F).
Internal Gcn4 occupancy peaks do not reside in pre-existing NDRs but evoke moderate histone eviction
We asked next whether internal Gcn4 peaks reside in pre-existing NDRs inside CDS by interrogating histone H3 ChIP-seq data obtained using the same WT strain and SM induction conditions as above, employing micrococcal nuclease digestion to fragment the chromatin. As expected, the Gcn4 motifs at T genes generally map in the middle of the NDRs upstream of these genes, and SM evokes a marked symmetrical decline in H3 occupancy centered on the motifs (Fig. 6A, Ind. 5′ _I vs. Ind.5′_U); whereas a much smaller reduction in H3 occupancy is evoked by SM for a group of uninduced genes with 5′ Gcn4 peaks (Fig. 6B, Unind-5′_I vs Unind-5′_U).
Figure 6. Gcn4 binding in CDS does not occur in preexisting NDRs and evokes moderate nucleosome eviction.
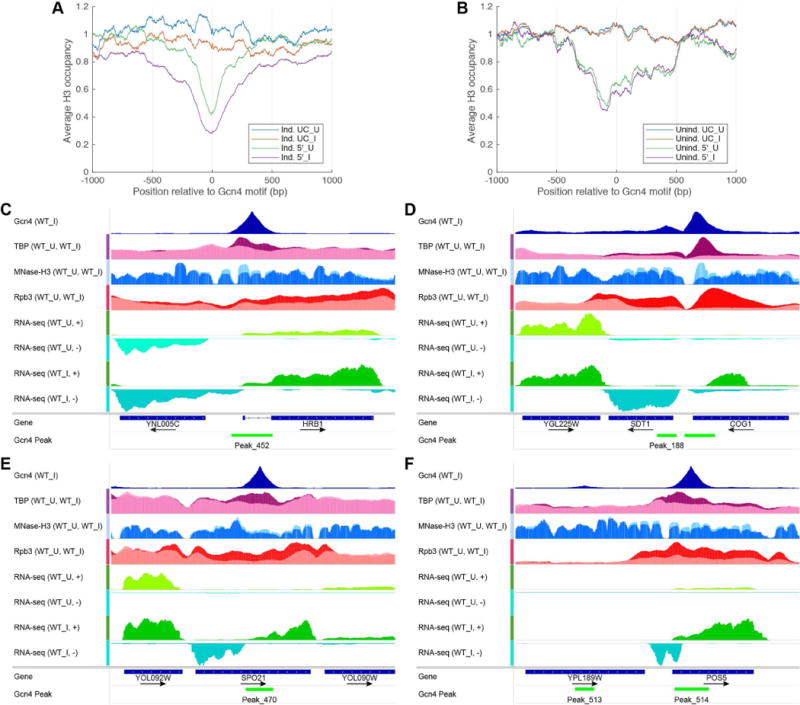
(A-B) MNase-H3-ChIP occupancies averaged and plotted relative to the Gcn4 motifs for (A) UC and 5′ induced targets and (B) UC and 5′ uninduced gene, in untreated (_U) and SM treated (_I) conditions, all listed in Data S4. All profiles were normalized so that the average occupancy for each chromosome was equal to one. (C-F) Exemplar genes showing eviction of nucleosomes from CDS surrounding Gcn4 internal peaks, depicted as in Fig. 4 except that normalized H3 occupancies are shown from WT_I cells (dark blue) overlayed on WT_U cells (light blue); and TBP-myc occupancies from WT_I cells (magenta) are overlayed on WT_U cells (pink).
Considering next SM-induced UC genes, the Gcn4 motifs mapping within CDSs are not located in pre-existing NDRs; however, a moderate reduction in H3 occupancy accompanies SM-induction of Gcn4 binding to these sites (Fig. 6A, Ind. UC_I vs Ind. UC_U); as seen for HRB1, COG1, POS5 and SPO21 in Figs. 6C-F. As expected, H3 occupancy was not reduced for a group of uninduced genes containing internal Gcn4 peaks (Fig. 6B, Unind. UC_I vs Unind. UC_U). These findings suggest that Gcn4 binding within CDS generally occurs in the absence of a pre-existing NDR but evokes moderate nucleosome eviction in the surrounding region.
Mutation of internal Gcn4 binding sites impairs transcription of the corresponding genes
To provide additional evidence that Gcn4 occupancy peaks within CDS activate transcription, we made precise chromosomal mutations that eliminate the consensus motifs at 12 induced UC genes, and conducted Rpb3 ChIP-seq on the resulting Gcn4 binding site (GBS) mutants. Rpb3 occupancy over the CDS of the mutated gene in 2 biological replicates was compared to that measured for the same gene in 2 replicates of the other 11 strains harboring different GBS mutations, and also 3 WT replicates. The availability of 25 replicates from 12 strains containing the unmutated allele facilitated identification of significant changes conferred by each GBS mutation (Data S5).
In 6 of 12 GBS mutants, Rpb3 occupancy over the mutated gene was reduced relative to that of the corresponding WT alleles in control strains (Table S1, rows 1-12), with the most dramatic results observed for the pos5-GBS mutation (Fig. 7A, summarized in Fig. 7D and Table S1, row 1). The GBS mutation did not decrease Rpb3 occupancies at genes immediately surrounding pos5-GBS (Fig. S4A), nor (as expected) at ARG1 or any of the other 11 UC genes mutated in different GBS mutants (Data S5, col. D–E). The reduction in Rpb3 occupancy extends throughout the 5′-noncoding region of POS5, where a strong 3AT-induced AS transcript maps (Fig. 7A), consistent with the mutation inactivating AS transcription extending upstream from the GBS. Conventional ChIP assays confirmed that the pos5-GBS mutation reduced Gcn4 binding to the CDS of this gene (Fig. S4A, bar plot on right).
Figure 7. Elimination of internal Gcn4 binding sites reduces Rpb3_I occupancies and 3AT-induced transcripts at UC target genes.

(A-C) Gcn4 occupancies from SM-induced WT; Rpb3 occupancies from uninduced or SM-induced WT, SM-induced GBS mutant, or gcn4Δ cells; and RNA read densities from uninduced or 3AT-induced WT cells; all depicted as in Fig. 4. Below IGV tracks are locations of amplicons produced from total mRNA and quantified by qRT-PCR from uninduced (WT_U) or SM-induced (WT_I) WT cells, or SM-induced GBS mutant cells. Mean (±SD) relative mRNA levels, normalized to actin mRNA, were determined from 2-3 biological replicates. (D-E) Summary of Rpb3 ChIP-seq measurements of -GBS mutant strains under SM-induction. For each gene, mean (±SD) Rpb3 occupancies are plotted for (i) 3 replicates of WT and 22 replicates of 11 mutants with GBS mutations in other genes (blue); (ii) 2 replicates of the GBS mutant for that gene (orange); (iii) 3 replicates of the gcn4Δ strain (green). Asterisks mark GBS mutant values that are ≤0.76 of control WT results and also significant at P<0.05(*) or P<0.01(**) in a 2-tailed, unpaired t-test. See also Fig. S4–S5 and Table S1–S2.
Elimination of the internal GBSs at VPS41, HIS2, YFR045W, ROT2 and TYR1 reduced Rpb3 occupancies in the CDSs of these genes too, to levels similar to those in WT_U and gcn4Δ_I cells (Fig. 7B–C & S4B–E; summarized in Fig. 7E and Table S1, rows 2-5 & 7), without affecting Rpb3 levels at surrounding genes (Fig. S4B–E) or other genomic loci (Data S5, cols. F-M, P-Q). The GBS mutations essentially eliminated Gcn4 binding to the relevant CDSs at all of these genes (Fig. S4B–E, right). Interestingly, the largest reduction in Rpb3 occupancies at HIS2 occurred at the 5′ end of the gene, including the region encoding a strong AS transcript (Fig. 7C), consistent with impaired Gcn4 induction of both the internal promoter and canonical 5′ promoter driving the FL HIS2 transcript. Remarkably, mutation of the GBS within BIO4 reduced Rpb3 levels in the adjacent gene, BIO3, even though induction of BIO4 itself was not significantly reduced (Fig. S4F; Fig. 7D–E, Table S1, rows 8, 19–20; Data S5, cols. R–S). The tyr1-GBS mutation also reduced Rpb3 occupancies in the adjacent gene (UBS1) in addition to TYR1 (Fig. 7D–E; Table S1, rows 17–18; Data S5, cols P-Q).
To support these conclusions, we conducted real-time qRT-PCR analysis of SM-induced RNA for the GBS mutants. For each mutated gene, we used primer pairs complementary to sequences located upstream or downstream of the GBS to amplify cDNAs corresponding to AS and SGS transcripts, respectively; whereas a pair of primers flanking the GBS was used to amplify the FL sense transcript. Considering pos5-GBS, the AS transcript amplicon POS5_1 was eliminated by the GBS mutation, and the FL and SGS amplicons POS5_GBS and POS5_2, respectively, were each reduced by ~3-fold (Fig. 7A), indicating that the GBS drives SM-induction of all three transcripts. Similarly, the GBS mutation at VPS41 reduced two amplicons for AS transcription, the FL transcript amplicon, and both SGS transcript amplicons (Fig. 7B); and the GBS mutation at HIS2 reduced the abundance of the AS and FL amplicons, and both SGS amplicons (Fig. 7C) (summarized in Table S2).
qRT-PCR analysis of the yfr045w-gbs allele revealed reductions in the three AS amplicons YFR045W_1, _2, and _3, and the FL amplicon YRF045W_GBS (Fig. S5A). An induced AS transcript initiates in the adjacent gene YFR046C and extends into YFR045W; and the reduction in amplicon _4 coupled with no effect on amplicon _5 by the GBS mutation suggests a specific decrease in this unusual AS transcript (Fig. S5A, Table S2). qRT-PCR analysis of bio4-GBS confirmed our previous conclusion (Fig. 7D-E) that induction of adjacent BIO3, but not BIO4 itself, was impaired by this mutation (Fig. S5B, Table S2). Similarly, cog1-GBS showed impaired induction of the adjacent gene, SDT1, and the AS transcript mapping in COG1, but not the FL COG1 transcript (Fig. S5C, Table S2). Interestingly, the gyp8-GBS mutation eliminates a Gcn4 binding site in the 3′ non-coding sequences of this gene, which reduces induction of the FL GYP8 transcript in addition to a GYP8 AS transcript, without affecting induction of downstream CAF16 (Fig. S5D, Table S2). qRT-PCR analyses of additional -GBS alleles (Table S2) provides further evidence that eliminating internal Gcn4 binding sites impairs induction of FL and sub-genic transcripts (rot2-GBS and sol1-GBS); of AS and SGS mRNAs, but not the FL transcript, of the gene harboring the GBS (spo21-GBS); or reduces transcription of the adjacent gene but not the gene containing the GBS (hmg2-GBS, LEU3). In summary, the GBSs in the CDS of 11 of 16 genes analyzed make significant contributions to induction of full-length or sub-genic transcripts from these or adjacent genes.
Little evidence for direct transcriptional repression by Gcn4
Expression of many genes encoding ribosomal proteins (RPGs) is reduced by amino acid starvation in a manner dependent on GCN4 function (Natarajan et al., 2001) and there is evidence that Gcn4 acts directly as a repressor at RPG promoters (Joo et al., 2010). However, we found that RPS13, RPL26A, RPL36B, and RPL37B are the only genes among 69 RPGs associated with a Gcn4 peak that also exhibited >1.5-fold repression of mRNA by SM (Saint et al., 2014); moreover, none of these RPGs contains the nearby Gcn4 peak in the promoter, and only RPS13 and RPL37B showed slight (~12%) repression of Rpb3 occupancy in SM-treated cells (‘Repressed genes’, Data S1, light or dark green). RPS14B contains a strong UC Gcn4 binding site in the intron with no apparent impact on transcription (Fig. 3F). While this work was in progress, ChIP-seq analysis of overexpressed GST-tagged Gcn4 was reported (Mittal et al., 2017), which idenfied extensive Gcn4 binding in CDS in addition to promoters, as observed here. It was proposed that Gcn4 acts directly to repress transcription from some of its target genes, including two RPGs (RPL14B and RPS24A). While we observed an “O,5” Gcn4 peak upstream of RPL14B (Peak-244), the increased Gcn4 binding was not accompanied by reduced Rpb3 occupancy on SM treatment (‘Rpb3 ChIP-seq’, Data S2). Thus, our results lend little support to the model that Gcn4 acts directly to repress RPG transcription, despite the widespread repression of these genes on ILV starvation (Saint et al., 2014).
DISCUSSION
ChIP-seq analysis of native Gcn4 has revealed 546 occupancy peaks in the yeast genome induced by SM treatment, but strikingly, only ~42% reside in 5′ non-coding sequences in the canonical locations of UAS elements. Our ChIP-seq analysis of Rpb3 occupancies and previous microarray measurements of mRNA expression (Natarajan et al., 2001) in WT, gcn4Δ or GCN4c cells indicated that only ~50% of genes with 5′ peaks exhibit Gcn4-dependent transcriptional induction on amino acid starvation, comprising the 149 T genes defined here. Thus, only ~25% of all identified Gcn4 peaks appear to function as conventional UASGCN4 elements.
While some of the ~150 uninduced genes with 5′ Gcn4 peaks might be explained by excessive distance between the Gcn4 binding site and promoter, or their presence in transcriptionally silenced chromatin environments, 35 belong to pairs of divergently transcribed genes where only one of the two exhibits induced Rpb3 occupancies, suggesting that the promoter sequence or chromatin structure of the other gene makes it unresponsive to Gcn4. In fact, many yeast Pol II promoters are bidirectional and produce divergent non-coding (nc) transcripts that are generally less abundant than the coding transcripts, which partly reflects weaker promoter activity in the nc direction (Churchman and Weissman, 2011; Rhee and Pugh, 2012). Early transcription termination by the Nrd1/Nab1/Sen1 (NNS) system can also reduce transcription in the non-coding direction (Mischo et al., 2011; Vasiljeva and Buratowski, 2006; Wei et al., 2011); and binding of an asymmetric repressor protein near the unresponsive promoter is also plausible.
Remarkably, ~60% of the Gcn4 peaks do not occur in promoter regions, with ~90% of these unconventional (UC) peaks mapping within CDS. While most of UC peaks appear to be nonfunctional, we identified 74 Gcn4-induced genes with only a UC peak detected in proximity to the TSS, and marshalled several lines of evidence that many of these UC peaks activate transcription. RNA-seq analysis revealed starvation-induced sub-genic AS and SGS transcripts that initiate within the CDS upstream and downstream of the internal Gcn4 peaks, suggesting that Gcn4 activates cryptic promoters flanking its UC binding sites. These internal AS/SGS transcripts occurred at some level for most of the induced “O” genes and were abundant at ~20% of them. Because cryptic transcripts are generally unstable, the low abundance of many such AS and SGS transcripts could underestimate the amount of transcription initiating from the internal promoters. Consistent with activation of cryptic promoters, we observed induction of TBP peaks in proximity to the internal Gcn4 peaks at ~50% of the induced “O” genes. Importantly, eliminating the GBSs in the CDS of 9 genes diminished the FL sense transcripts, sub-genic AS/SGS transcripts, or both; and in three instances, impaired induction of the FL transcript of an adjacent gene. Thus, internal GBSs at certain target genes contribute to induction of the FL transcripts encoding the proteins instrumental in the Gcn4-mediated starvation response. While there are a few reported instances of functional activator binding sites within yeast transcription units (Mellor et al., 1987) (Fantino et al., 1992), notably in Ty1 retrotransposons (Curcio et al., 2015), the extent of the phenomenon observed here for Gcn4 is unprecedented. Note however that mutating the internal GBSs at five Gcn4-induced genes had no effect on Rpb3 occupancies, suggesting that promoter binding by Gcn4 below the detection limit, or binding of other transcriptional activators induced by Gcn4, mediates activation of these, and possibly other, induced genes where we detected only UC Gcn4 peaks.
Examining nucleosome occupancies in SM-induced cells revealed that Gcn4 generally binds to internal UC binding sites not present in pre-existing NDRs, presumably reflecting the absence of GRF binding sites within CDS. Having observed a preference for Gcn4 binding to consensus motifs located distal to nucleosome dyads, we surmise that Gcn4 binds most efficiently in CDS when its binding motif resides within a linker separating adjacent nucleosomes. On the whole, the UC Gcn4 peaks are less efficient than conventional 5′ Gcn4 peaks in activating transcription. This was evident in the relatively weak recruitment of TBP to internal promoters compared to induced 5′ promoters, and might be rationalized by noting that Gcn4 binding to UC sites does not evoke substantial nucleosome eviction in the surrounding region compared to what occurs on Gcn4 binding in NDRs. As such, the internal promoters will be relatively more occluded by nucleosomes and less accessible to the transcription machinery. The relatively inefficient activation of canonical 5′ promoters by UC binding sites might also reflect interference by AS transcription originating from the internal promoter (Kim et al., 2016; Wei et al., 2011).
In closing, we have shown that Gcn4 activation of a target gene can be mediated, at least in part, by Gcn4 binding within the CDS or 3′ non-coding region of that gene, or an adjacent gene; and that internal Gcn4 binding sites also frequently activate cryptic bidirectional transcription within the CDS. Given that cryptic transcripts can be translated (Cheung et al., 2008), the protein products of certain sub-genic transcripts induced by Gcn4 could have biological functions. Given recent findings of carbon source regulation of cryptic transcription (Kim et al., 2016), it seems likely that the occurrence of functional activator binding sites within CDS will not be confined to Gcn4.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Alan G. Hinnebusch (ahinnebusch@nih.gov)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All Saccharomyces cerevisiae strains used in this study are listed in Table S4. They were maintained on either YPD plates or SC agar plates with appropriate selection for transformants.
METHOD DETAILS
Yeast strains
Yeast strains employed are listed in Table S4 and were either purchased from Research Genetics, described previously (Kim et al., 2006; Qiu et al., 2004), or constructed as described below.
ChIP-seq identification of Gcn4 binding sites
ChIP-seq analysis of Gcn4 occupancy
WT strain BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) and isogenic gcn4Δ::kanMX4 strain F731, both purchased from Research Genetics, were cultured in synthetic complete medium lacking isoleucine and valine (SC-Ilv) to log-phase (OD600=0.6-0.8) and SM was added at 1 μg/ml for 25 min to induce Gcn4 synthesis. ChIP-seq was conducted and DNA libraries for Illumina paired-end sequencing were prepared as described previously (Cole et al., 2014) with the modifications in (Qiu et al., 2015) except that chromatin samples containing 5 μg DNA were immunoprecipiteted overnight using Gcn4 antibodies (Zhang et al., 2008). Paired-end sequencing (50 nt from each end) was conducted by the DNA Sequencing and Genomics core facility of the NHLBI, NIH. Sequence data were aligned to the sacCer3 version of the genome sequence using alignment software Bowtie 2 (Langmead and Salzberg, 2012), and PCR duplicates were removed using the samtools rmdup package. Numbers of unique paired-end reads from Gcn4 ChIP-seq are summarized in Table S3. Gcn4 occupancy profiles were obtained from the alignment (.bam) files using the bioinformatics toolbox from MATLAB. To allow the comparison between different samples, each profile was normalized such that the average occupancy for each chromosome was equal to one.
MACS2 (http://liulab.dfci.harvard.edu/MACS/) was employed to identify Gcn4 binding sites from the Gcn4 ChIP-seq data using two replicate WT_I cultures (samples AGH32-86 and -87) and three replicate WT_U cultures (samples AGH32-45, -46, -47), using a threshold for the q-value of 10−4. The Gcn4 occupancy peaks called by MACS2 were assessed manually in the Integrative Genomics Viewer (IGV, Broad Institute) by comparing Gcn4 ChIP-seq results for replicate WT_I, WT_U, and gcn4_I samples with our published Rpb3 ChIP-seq results for replicate WT_I and WT_U cultures of the same WT strain analyzed here (Qiu et al., 2015). We identified 64 hyper-ChIP-able loci (Teytelman et al., 2013), which exhibit a high Gcn4 background over the entire gene regardless of SM induction or even in gcn4Δ cells. All but 4 peaks found in the hyper-ChIP-able loci were eliminated from consideration, as were 36 other called peaks, based on findings that their signals were essentially equivalent in WT_U, WT_I, and gcn4Δ_I cells (Fig. S1A). Manual inspection further suggested that 46 peaks identified by MACS2 using a higher threshold for the q-value of 10−2 were authentic peaks (elevated in all WT_I versus WT_U replicates and absent in all gcn4Δ_I samples) and were added to a final curated list of 546 induced, GCN4-dependent occupancy peaks, listed in Data S1, sheet “All Gcn4 sites”.
Gcn4 occupancies for each peak were calculated in MATLAB and summarized as the average “rmdup” reads from five biological replicates of WT_I cells (AGH32-86, -87, AGH24-04, -05, -06), three replicates of WT_U cells (AGH32-45, -46, -47), and gcn4 _I cells (AGH24-01, -02, -03). The fastq files of replicates were merged and processed together, and the results are listed in Data S1 (‘Gcn4 occups’). Pearson correlation coefficients for genome-wide occupancy profiles of the different replicates are listed in Table S3.
Analysis of nucleosome occupancies of Gcn4 peaks
To understand how the binding of Gcn4 is affected by the position of the Gcn4 motif relative to the nucleosome dyad, we aligned all Gcn4 motif matches reported by FIMO, and sorted them according to their distance from the nearest nucleosome occupancy peak from MNase-seq data in WT_U cells (Qiu et al., 2015). We used the findpeaks function from MATLAB to identify the nearest nucleosomes to the Gcn4 motifs, and separated the Gcn4 motifs into two classes: bound and unbound by Gcn4, according to whether the motif overlapped with a Gcn4 peak called by MACS2 or not (Fig. 3A). Using the same sorting order in the Gcn4 motif alignments, we also plotted the distribution of Gcn4 and Rpb3 occupancies before (WT_U) and after (WT_I) induction (Fig. 1E).
Support vector machine (SVM) classification of Gcn4 motifs
In supervised machine learning, support vector machines (SVMs) are frequently used for non-linear classification (Press, 2007). We used a type 1 SVM algorithm (also known as C-SVM) to classify the Gcn4 binding status of all Gcn4 motifs predicted by FIMO. We used the binary labels of the motifs (Gcn4-bound and Gcn4-unbound) indicating whether the motifs overlapped with the Gcn4 peaks identified by MACS2 or not, and as features the nucleosome occupancy at the corresponding motifs and the motif scores reported by FIMO (log-likelihood ratio score, assuming a null model in which sequences are generated at random with per-letter background frequencies characteristic of the yeast genome). The R implementation of SVM was employed: the ksvm function from kernlab library, using a Bessel kernel and 5-fold cross validation. The Gcn4 motifs were split into two sets: a training set containing the Gcn4 motifs from chromosomes I and II, and a testing set containing the motifs from chromosomes III – XVI.
To identify other features that might improve the classification accuracy of the Gcn4 motifs, we used the following published data sets: Abf1 ChIP-seq (Paul et al., 2015); ATAC-seq (Schep et al., 2015); DNase-seq (Hesselberth et al., 2009); H2A.Z ChIP-seq (Woo et al., 2013); Reb1 ChEC-seq (Zentner et al., 2015); Rsc8, Snf2, Ioc3 ChIP-seq (Parnell et al., 2015); and TBP ChIP-seq (Zentner and Henikoff, 2013). We computed the average signals (occupancy for ChIP-seq and ChEC-seq data, and cleavage density for ATAC-seq and DNase-seq data) at all Gcn4 motifs, and analyzed the differences between Gcn4-bound and Gcn4-unbound motifs. The following signals showed a significant difference between the two classes of Gcn4 motifs: ATAC-seq, DNase-seq, Reb1, Snf2 and TBP. Next, we added these five new features to the motif score and nucleosome occupancy, which we used in the previous SVM classification, and tested a large variety of supervised machine learning classification algorithms to see whether the extra features and the new algorithms can improve the classification accuracy that we obtained using SVM. Testing more than 120 different classification algorithms, all available in the caret library in R, the accuracy of the classification was improved by only ~4% (Table S6), the best performing algorithm being one from the SVM family (svmRadialWeights method of the caret library; see the caret documentation at http://topepo.github.io/caret/).
Identification of T and UC Gcn4 target genes
We used R to identify the gene whose TSS is nearest to each Gcn4 peak (nearest method from GenomicRanges package), and each Gcn4 site was examined individually in IGV to assess whether it falls within non-coding (NC) sequences present upstream (code 5′) or downstream (code 3′) of that gene, within the CDS (O, for ORF) of that gene (code O,O) or within the adjacent upstream or downstream gene (codes O,5′ and O,3′, respectively) (see Fig. 2A). Thus, 5′ sites occur in the canonical positions within non-coding sequences upstream of the nearest TSS, whereas O,5 sites occur in the CDS of the adjacent gene located 5′ of the nearest TSS. These designations for the 546 Gcn4 peaks are tabulated in columns 7-8 of ‘All Gcn4 sites’ of Data S1.
Based on inspection of Rpb3 induction levels of genes surrounding the Gcn4 peaks, and allowing for the possibility of activation by Gcn4 peaks located in unconventional locations, it was realized that a different gene than the one harboring the nearest annotated TSS could be the target of Gcn4 activation. Additionally, one of the two genes in pairs of divergently transcribed genes was not captured by the “nearest TSS” identification script. Hence, to generate the most likely list of potential Gcn4 target genes, the list of genes closest to the 546 Gcn4 peaks was expanded to include the most likely induced target gene when it differed from that belonging to the nearest TSS; and the second gene in pairs of divergently arranged genes were included when transcriptional induction of both genes was suggested by the Rpb3 ChIP-seq data. The result was a list of 652 potential Gcn4 target genes tabulated in ‘Potential Target Scoring’ of Data S1. Note that some genes were tabulated there more than once owing to the presence of more than one Gcn4 peak in their vicinity, e.g. one located 5′ and another located 3′ of the same gene. Accordingly, a number of the 546 Gcn4 binding sites are associated with more than one potential target gene, e.g. being located 5′ to one gene and 3′ to another.
To determine which of the potential Gcn4 target genes most likely exhibit Gcn4-dependent transcriptional activation, we conducted ChIP-seq analysis of Rpb3 on three replicate cultures of gcn4Δ strain F731 (Table S3) using the same SM treatment employed previously to analyze Rpb3 occupancies in the isogenic WT strain employed here. Rbp3 occupancies were normalized to the average occupancy per chromosome and the average occupancy per nucleotide was determined for each annotated ORF, as described previously (Qiu et al., 2015). Pearson correlation coefficients for genome-wide occupancy profiles of different replicates are listed in Table S3. A student’s t-test was conducted to identify genes exhibiting a significant difference in Rpb3 occupancies between gcn4Δ and WT strains (p<0.05) (‘Rpb3 ChIP-seq’, Data S2). We also interrogated the previously published compilation of mRNA expression microarray analysis, which had identified 512 genes induced by starvation for histidine (by treatment with 3AT) or Ile/Val (via excess leucine addition), or in response to a dominant constitutively activated GCN4c allele (Natarajan et al., 2001) (‘Natarajan 2001 targets, Data S1). We identified 223 genes associated with Gcn4 peaks, in either 5′, ORF, 3′ locations, which, showed significant Gcn4-dependent induction of mRNA as judged by either (i) significantly greater mRNA expression measured in a WT versus gcn4Δ strain both starved for histidine by 3-AT treatment (p<0.05) or (ii) significantly greater mRNA expression measured in a GCN4c versus WT strain under non-starvation conditions (p<0.05); or (iii) ≥1.5-fold greater Rpb3 occupancies in a WT versus gcn4Δ strain both treated with SM (p<0.05) and also ≥1.7-fold SM-induction of mRNA in WT cells in the study of (Saint et al., 2014) (‘Induced 5′ genes’, Data S1). Two genes that met criteria (i) but not (ii) were excluded because there was no evidence of Rpb3 or mRNA induction (YNL179C), or evidence of Rpb3 repression rather than induction (YKL055C). Of these 223 induced genes, 149 contain peaks 5′ of the CDS and were designated direct “T” target genes; the remaining 74 genes with proximal Gcn4 peaks in unconventional locations were designated induced UC target genes (‘T and UC targets’, Data S1). Among the 223 T and induced UC target genes (in red or dark orange sectors of Fig. 2C), 97% exhibited induction of mRNA or Rpb3, or reduced expression in gcn4Δ versus WT cells, under conditions of SM treatment. The remaining 7 genes were shown to be induced by 3AT treatment in WT but not gcn4Δ cells, or by the GCN4c allele (Natarajan et al., 2001), making it likely that their activation by Gcn4 is dampened in response to ILV starvation.
Gene ontology analysis was conducted using the on-line tool Funspec found at http://funspec.med.utoronto.ca/.
RNA-seq analysis
WT strain YDC111 (MATa ade2–1 can1-100 leu2-3,112 trp1-1 ura3-1) (Kim et al., 2006) and BY4741 were grown to log-phase (A600 ~0.7) at 30°C in synthetic complete medium without histidine and lacking isoleucine and valine (SC-Ilv) respectively. To induce Gcn4, 10 mM 3-aminotriazole (3-AT) (Sigma 61-82-5) was added to YDC111 cultures and aliquots (6 ml) of cells were removed after 0, 1, 5, 20 or 40 min, whereas from BY4141 cultures cells were removed before or after 1 μg/ml SM treatment for 25 min. Cells were rapidly cooled by immediate transfer to tubes containing ice and collected by centrifugation at 4°C and stored at 80°C until RNA was extracted, using the YeaStart RNA kit (Zymo Research R1002) or RNeasy Mini kit (Qiagen 74104). Libraries were prepared and sequenced using an Illumina NextSeq500 machine by ACGT Inc. RNA reads are normalized by combining all reads on both DNA strands for genes on the same chromosome and setting the average read density per nucleotide to one. Read densities are reported relative to the average read density per nucleotide on that chromosome. For analysis of 3AT-induced divergent transcription directed by class “O” Gcn4 peaks within CDS, we selected 62 genes containing class “O” occupancy peaks, belonging to the group of 74 UC target genes, which contain a significant match to the Gcn4 consensus binding site (listed in ‘O UC targets’, Data S3). For peaks containing more than one motif, the motif closest to the peak occupancy observed in the Gcn4 ChIP-seq data, or (when occupancy differences were not discriminatory) the motif with the greatest similarity to the consensus sequence in Fig. S1B, was selected as the Gcn4 binding site in that peak for further analysis. The number of RNA-seq reads covering each nucleotide in the sense or AS direction (relative to the coding sequence of the gene) were tabulated and shown in Figs. 5A-E. The same approach was used to analyze a group of 117 genes from the group of 149 T target genes, excluding one of the two genes in pairs of divergently arranged genes that exhibits lesser SM-induction of Rpb3 occupancy or mRNA expression compared to the partner gene (listed in ‘T targets w-o diverg prom’, Data S1), yielding the results in Figs. S2A–E. For this latter group of T genes, Gcn4 motifs mapping in the CDS were eliminated from consideration.
ChIP-seq analysis of TBP-myc13
SPT15-myc13::HIS3 strain HQY366 (Qiu et al., 2004), isogenic to BY4741 was cultured in the presence or absence of SM treated and subjected to ChIP-seq analysis as described previously (Qiu et al., 2015) except that chromatin samples containing 5.0μg DNA were immunoprecipitated with anti-myc antibodies (Roche). Paired-end sequencing libraries were prepared from immunoprecipitated DNA using Illumina paired-end kits from New England Biolabs (cat. #E7370 and #E7335). Numbers of aligned paired reads from TBP-myc13 ChIP-seq and reproducibility of replicates are summarized in Table S3. The distribution of TBP (median occupancy, and the ranges corresponding to 5-95 and 25-75 percentiles) was depicted in Fig. 5F for the 62 class “O” UC target genes in ‘O UC targets’, Data S3.
MNase-ChIP-seq of histone H3
WT strain BY4741 was cultured in the presence or absence of SM as described above. Chromatin preparation and titrations of MNase digestion were performed as described previously (Wal and Pugh, 2012), except that digestions were performed at 30°C for 10 min. ChIP and paired-end sequencing library preparation were performed as described previously (Qiu et al., 2015) for sonicated chromatin except that chromatin samples containing 5.0 μg DNA were immunoprecipited overnight with anti-H3 antibodies. A comprehensive analysis of these data will be described in a future publication.
Construction and verification of GBS mutants
The delitto perfetto technique was employed to conduct in vivo site-specific mutagenesis (Stuckey et al., 2011) of WT strain BY4741 to replace 11 bp sequences encompassing the Gcn4 binding motif for each of 16 different UC target genes with the sequence 5′ AGGATCCA 3′. This 8-bp sequence introduces a novel BamHI site, for screening purposes, without altering the CDS reading frame or introducing a binding site for any known transcription factor (based on the the Yeast Transcription Factor Specificity Compendium at http://yetfasco.ccbr.utoronto.ca/. For the YFR045W, two Gcn4 motifs and the intervening 25bp were replaced with the same 8-bp sequence, reducing the CDS length by 13 codons. The sequences of 11 or 25 bp replaced by 5′ AGGATCCA 3′ for the 17 genes are listed in column 4 of Supplementary Table S4. Strains YR201-VV005 in Table S4 contain the core cassette PGAL1SCE1-hyg-KlURA3 inserted in place of the 11 or 25 bp sequences containing the Gcn4 binding sites at the specified genes; whereas strains YR216-VV010 contain the 8 bp sequence containing the BamHI site inserted in place of the Gcn4 binding site comprising the indicated -GBS alleles. Primers for conducting the two stages of delitto perfetto to generate the 17 -GBS alleles are listed in the two three sections of Table S5. The mutagenized alleles were confirmed by sequencing DNA fragments amplified from the relevant chromosomal loci that encompassed ~200-400 bps surrounding the GBS mutation employing the primers listed in the third section of Table S5 (Primers to confirm -GBS mutant alleles). Quantitative ChIP analysis of Gcn4 was conducted to confirm loss of Gcn4 binding to the mutagenized Gcn4 consensus motif for each -GBS mutant. To this end, mutant and parental WT strains were cultured in SM medium, as above, and Gcn4 ChIP was performed as previously described using antibodies against Gcn4 (Qiu et al., 2015). DNA samples from immunoprecipitated and input chromatin samples (diluted to DNA concentrations comparable to the immunoprecipitated samples) were quantified by SYBR green-based real-time qPCR using primer pairs flanking the Gcn4 BS in each gene (listed in see Table S5, Primers for qRT-PCR analysis of RNA expression and Gcn4 binding for -GBS alleles, primers for amplifying GBS amplicons), normalizing the results to an amplicon of POL1 shown previously not to exhibit Gcn4 binding (Swanson et al., 2003).
Changes in mRNA expression conferred by GBS mutations were measured by real-time qRT-PCR of total RNA isolated from WT and GBS mutant strains cultured and treated with SM as described above for Gcn4 ChIP-seq analysis. Total RNA isolation, cDNA synthesis and qPCRs were performed as previously described (Rawal et al., 2014). To examine diverse sets of amplicons, we used SYBR green based Brilliant III Ultra-Fast SYBR Green qPCR master mix (Agilient Technology, cat. #600882), employing an amplicon of ACT1 for normalization. Primer pairs used to detect full length (FL), sub-genic sense (SGS) and anti-sense (AS) transcripts are listed in (Table S5, Primers for qRT-PCR analysis of RNA expression and Gcn4 binding for -GBS alleles).
DATA AND SOFTWARE AVAILABILITY
Raw and analyzed data have been deposited in the NCBI GEO database under the accession numbers GSE107532 (ChIP-seq) and GSE110413 (RNA-seq).
Supplementary Material
Table S5. Primers used in this study, related to STAR methods.
Table S6. Results of testing different supervised machine learning classification algorithms for ability to improve classification accuracy of Gcn4 motifs as bound or unbound using SVM, related to STAR methods.
Data S1. Lists of Gcn4 binding sites, Gcn4 motifs and their chromosomal coordinates; analyses of transcriptional activation of genes proximal to Gcn4 peaks, and lists of canonical (T) and unconventional (UC) target genes; lists and properties of different groups of genes with 5′ or UC binding sites, or repressed genes; and summary of GO enrichment of T genes, related to Fig. 1, 2, 3 and S1.
Data S2. Compilation of datasets of Gcn4-dependent transcriptional activation, assessed by Rpb3 ChIP-seq or expression microarray analysis of mRNA, related to Fig. 2.
Data S3. Lists of class “O” UC targets and T targets analyzed for starvation-induced mRNAs and TBP-myc occupancies, related to Fig. 5, S2 and S3.
Data S4. Lists of 5′ and UC Gcn4 binding site coordinates examined for histone H3 occupancies, related to Fig. 6.
Data S5. Statistical analysis of the CDS Rpb3 occupancies at relevant genes in WT and GBS mutants, related to Fig. 7, S4, S5 and Table S1.
Highlights.
Most occupied binding sites for yeast activator Gcn4 reside within coding sequences
Gcn4 binding to internal sites occurs without a nucleosome-depleted region (NDR)
Gcn4 binding within coding sequences frequently activates cryptic internal promoters
Internal Gcn4 binding can also activate nearby canonical 5′-positioned promoters
Acknowledgments
This study utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov), and was supported by the Intramural Research Program of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization, Y.R., R.V.C., V.V., H.Q., J.O., D.J.C.; and A.G.H.; Methodology, Y.R., R.V.C., V.V., H.Q., J.O., D.J.C.; and A.G.H.; Software, R.V.C.; Validation, Y.R., R.V.C., V.V., H.Q., and J.O.; Formal Analysis, Y.R., R.V.C., V.V. and A.G.H.; Investigation, Y.R., V.V., H.Q., J.O.; Writing – Original Draft, A.G.H.; Writing – Review & Editing, Y.R., R.V.C., D.J.C. and A.G.H.; Visualization, Y.R. and R.V.C.; Supervision, D.J.C. and A.G.H.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Bai L, Ondracka A, Cross FR. Multiple Sequence-Specific Factors Generate the Nucleosome-Depleted Region on CLN2 Promoter. Mol Cell. 2011;42:465–476. doi: 10.1016/j.molcel.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, J J, Grant CE, Noble WS. The MEME Suite. Nucleic Acids Res. 2015;43:W39–49. doi: 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung V, Chua G, Batada NN, Landry CR, Michnick SW, Hughes TR, Winston F. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol. 2008;6:e277. doi: 10.1371/journal.pbio.0060277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole HA, Howard BH, Clark DJ. Activation-induced disruption of nucleosome position clusters on the coding regions of Gcn4-dependent genes extends into neighbouring genes. Nucleic Acids Res. 2011;39:9521–9535. doi: 10.1093/nar/gkr643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole HA, Ocampo J, Iben JR, Chereji RV, Clark DJ. Heavy transcription of yeast genes correlates with differential loss of histone H2B relative to H4 and queued RNA polymerases. Nucleic Acids Res. 2014;42:12512–12522. doi: 10.1093/nar/gku1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio MJ, Lutz S, Lesage P. The Ty1 LTR-Retrotransposon of Budding Yeast, Saccharomyces cerevisiae. Microbiol Spectr. 2015;3 doi: 10.1128/microbiolspec.MDNA3-0053-2014. MDNA3-0053-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin C, Tice-Baldwin K, Shore D, Arndt KT. RAP1 is required for BAS1/BAS2- and GCN4-dependent transcription of the yeast }U}HIS4}u} gene. Mol Cell Biol. 1991;11:3642–3651. doi: 10.1128/mcb.11.7.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantino E, Marguet D, Lauquin GJ. Downstream activating sequence within the coding region of a yeast gene: specific binding in vitro of RAP1 protein. Mol Gen Genet. 1992;236:65–75. doi: 10.1007/BF00279644. [DOI] [PubMed] [Google Scholar]
- Good PD, Kendall A, Ignatz-Hoover J, Miller EL, Pai DA, Rivera SR, Carrick B, Engelke DR. Silencing near tRNA genes is nucleosome-mediated and distinct from boundary element function. Gene. 2013;526:7–15. doi: 10.1016/j.gene.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L, Hoar E. Upstream activation sites of the CYC1 gene of Saccharomyces cerevisiae are active when inverted but not when placed downstream of the “TATA box”. Proc Natl Acad Sci U S A. 1984;81:7860–7864. doi: 10.1073/pnas.81.24.7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol. 2005;59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- Jia MH, Larossa RA, Lee JM, Rafalski A, Derose E, Gonye G, Xue Z. Global expression profiling of yeast treated with an inhibitor of amino acid biosynthesis, sulfometuron methyl. Physiol Genomics. 2000;3:83–92. doi: 10.1152/physiolgenomics.2000.3.2.83. [DOI] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Eser U, Struhl K, Churchman LS. The Ground State and Evolution of Promoter Region Directionality. Cell. 2017;170:889–898 e810. doi: 10.1016/j.cell.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo YJ, Kim JH, Kang UB, Yu MH, Kim J. Gcn4p-mediated transcriptional repression of ribosomal protein genes under amino-acid starvation. Embo J. 2010;30:859–872. doi: 10.1038/emboj.2010.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, P G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lee BB, Oh YM, Zhu C, Steinmetz LM, Lee Y, Kim WK, Lee SB, Buratowski S, Kim T. Modulation of mRNA and lncRNA expression dynamics by the Set2-Rpd3S pathway. Nat Commun. 2016;7:13534. doi: 10.1038/ncomms13534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, McLaughlin N, Lindstrom K, Tsukiyama T, Clark DJ. Activation of Saccharomyces cerevisiae HIS3 results in Gcn4p-dependent, SWI/SNF-dependent mobilization of nucleosomes over the entire gene. Mol Cell Biol. 2006;26:8607–8622. doi: 10.1128/MCB.00678-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levo M, Avnit-Sagi T, Lotan-Pompan M, Kalma Y, Weinberger A, Yakhini Z, Segal E. Systematic Investigation of Transcription Factor Activity in the Context of Chromatin Using Massively Parallel Binding and Expression Assays. Mol Cell. 2017;65:604–617 e606. doi: 10.1016/j.molcel.2017.01.007. [DOI] [PubMed] [Google Scholar]
- Li H, H B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Lee CK, Granek JA, Clarke ND, Lieb JD. Whole-genome comparison of Leu3 binding in vitro and in vivo reveals the importance of nucleosome occupancy in target site selection. Genome Res. 2006;16:1517–1528. doi: 10.1101/gr.5655606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor J, Dobson MJ, Kingsman AJ, Kingsman SM. A transcriptional activator is located in the coding region of the yeast PGK gene. Nucleic Acids Res. 1987;15:6243–6259. doi: 10.1093/nar/15.15.6243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischo HE, Gomez-Gonzalez B, Grzechnik P, Rondon AG, Wei W, Steinmetz L, Aguilera A, Proudfoot NJ. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol Cell. 2011;41:21–32. doi: 10.1016/j.molcel.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal N, Guimaraes JC, Gross T, Schmidt A, Vina-Vilaseca A, Nedialkova DD, Aeschimann F, Leidel SA, Spang A, Zavolan M. The Gcn4 transcription factor reduces protein synthesis capacity and extends yeast lifespan. Nat Commun. 2017;8:457. doi: 10.1038/s41467-017-00539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan K, Meyer MR, Jackson BM, Slade D, Roberts C, Hinnebusch AG, Marton MJ. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol Cell Biol. 2001;21:4347–4368. doi: 10.1128/MCB.21.13.4347-4368.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil H, Malabat C, d’Aubenton-Carafa Y, Xu Z, Steinmetz LM, Jacquier A. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature. 2009;457:1038–1042. doi: 10.1038/nature07747. [DOI] [PubMed] [Google Scholar]
- Parnell TJ, Schlichter A, Wilson BG, Cairns BR. The chromatin remodelers RSC and ISW1 display functional and chromatin-based promoter antagonism. Elife. 2015;4:e06073. doi: 10.7554/eLife.06073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul E, Tirosh I, Lai W, Buck MJ, Palumbo MJ, Morse RH. Chromatin mediation of a transcriptional memory effect in yeast. G3 (Bethesda) 2015;5:829–838. doi: 10.1534/g3.115.017418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press WHT, Saul A, Vetterling William T, Flannery BP. Section 16.5 Support Vector Machines. 3rd. New York: Cambridge University Press; 2007. [Google Scholar]
- Qiu H, Chereji RV, Hu C, Cole HA, Rawal Y, Clark DJ, Hinnebusch AG. Genome-wide cooperation by HAT Gcn5, remodeler SWI/SNF, and chaperone Ydj1 in promoter nucleosome eviction and transcriptional activation. Genome Res. 2015 doi: 10.1101/gr.196337.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Hu C, Yoon S, Natarajan K, Swanson MJ, Hinnebusch AG. An array of coactivators is required for optimal recruitment of TATA binding protein and RNA polymerase II by promoter-bound Gcn4p. Mol Cell Biol. 2004;24:4104–4117. doi: 10.1128/MCB.24.10.4104-4117.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ, Winston F. Chromatin and transcription in yeast. Genetics. 2012;190:351–387. doi: 10.1534/genetics.111.132266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawal Y, Qiu H, Hinnebusch AG. Accumulation of a threonine biosynthetic intermediate attenuates general amino acid control by accelerating degradation of Gcn4 via Pho85 and Cdk8. PLoS Genet. 2014;10:e1004534. doi: 10.1371/journal.pgen.1004534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature. 2012;483:295–301. doi: 10.1038/nature10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint M, Sawhney S, Sinha I, Singh RP, Dahiya R, Thakur A, Siddharthan R, Natarajan K. The TAF9 C-terminal conserved region domain is required for SAGA and TFIID promoter occupancy to promote transcriptional activation. Mol Cell Biol. 2014;34:1547–1563. doi: 10.1128/MCB.01060-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, Greenleaf WJ. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 2015;25:1757–1770. doi: 10.1101/gr.192294.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K. Genetic properties and chromatin structure of the yeast gal regulatory element: an enhancer-like sequence. Proc Natl Acad Sci U S A. 1984;81:7865–7869. doi: 10.1073/pnas.81.24.7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuckey S, Mukherjee K, Storici F. In vivo site-specific mutagenesis and gene collage using the delitto perfetto system in yeast Saccharomyces cerevisiae. Methods Mol Biol. 2011;745:173–191. doi: 10.1007/978-1-61779-129-1_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MJ, Qiu H, Sumibcay L, Krueger A, Kim SJ, Natarajan K, Yoon S, Hinnebusch AG. A Multiplicity of coactivators is required by Gcn4p at individual promoters in vivo. MolCellBiol. 2003;23:2800–2820. doi: 10.1128/MCB.23.8.2800-2820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teytelman L, Thurtle DM, Rine J, van Oudenaarden A. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc Natl Acad Sci U S A. 2013;110:18602–18607. doi: 10.1073/pnas.1316064110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiljeva L, Buratowski S. Nrd1 interacts with the nuclear exosome for 3′ processing of RNA polymerase II transcripts. Mol Cell. 2006;21:239–248. doi: 10.1016/j.molcel.2005.11.028. [DOI] [PubMed] [Google Scholar]
- Wal M, Pugh BF. Genome-wide mapping of nucleosome positions in yeast using high-resolution MNase ChIP-Seq. Methods Enzymol. 2012;513:233–250. doi: 10.1016/B978-0-12-391938-0.00010-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Pelechano V, Jarvelin AI, Steinmetz LM. Functional consequences of bidirectional promoters. Trends Genet. 2011;27:267–276. doi: 10.1016/j.tig.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo S, Zhang X, Sauteraud R, Robert F, Gottardo R. PING 2.0: an R/Bioconductor package for nucleosome positioning using next-generation sequencing data. Bioinformatics. 2013;29:2049–2050. doi: 10.1093/bioinformatics/btt348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Munster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Morse RH. Chromatin opening and transactivator potentiation by RAP1 in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:5279–5288. doi: 10.1128/mcb.19.8.5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Henikoff S. Mot1 redistributes TBP from TATA-containing to TATA-less promoters. Mol Cell Biol. 2013;33:4996–5004. doi: 10.1128/MCB.01218-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Kasinathan S, Xin B, Rohs R, Henikoff S. ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat Commun. 2015;6:8733. doi: 10.1038/ncomms9733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Gaur NA, Hasek J, Kim SJ, Qiu H, Swanson MJ, Hinnebusch AG. Disrupting vesicular trafficking at the endosome attenuates transcriptional activation by Gcn4. Mol Cell Biol. 2008;28:6796–6818. doi: 10.1128/MCB.00800-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, L T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Byers KJ, McCord RP, Shi Z, Berger MF, Newburger DE, Saulrieta K, Smith Z, Shah MV, Radhakrishnan M, et al. High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res. 2009;19:556–566. doi: 10.1101/gr.090233.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S5. Primers used in this study, related to STAR methods.
Table S6. Results of testing different supervised machine learning classification algorithms for ability to improve classification accuracy of Gcn4 motifs as bound or unbound using SVM, related to STAR methods.
Data S1. Lists of Gcn4 binding sites, Gcn4 motifs and their chromosomal coordinates; analyses of transcriptional activation of genes proximal to Gcn4 peaks, and lists of canonical (T) and unconventional (UC) target genes; lists and properties of different groups of genes with 5′ or UC binding sites, or repressed genes; and summary of GO enrichment of T genes, related to Fig. 1, 2, 3 and S1.
Data S2. Compilation of datasets of Gcn4-dependent transcriptional activation, assessed by Rpb3 ChIP-seq or expression microarray analysis of mRNA, related to Fig. 2.
Data S3. Lists of class “O” UC targets and T targets analyzed for starvation-induced mRNAs and TBP-myc occupancies, related to Fig. 5, S2 and S3.
Data S4. Lists of 5′ and UC Gcn4 binding site coordinates examined for histone H3 occupancies, related to Fig. 6.
Data S5. Statistical analysis of the CDS Rpb3 occupancies at relevant genes in WT and GBS mutants, related to Fig. 7, S4, S5 and Table S1.