

ABSTRACT
Protein aggregates, and in particular amyloids, are generally considered to be inherently irreversible aberrant clumps, and are often associated with pathologies, such as Alzheimer’s disease, Parkinson’s disease, or systemic amyloidosis. However, recent evidence demonstrates that some aggregates are not only fully reversible, but also perform essential physiological functions. Despite these new findings, very little is known about how these functional protein aggregates are regulated in a physiological context. Here, we take the yeast pyruvate kinase Cdc19 as an example of a protein forming functional, reversible, solid, amyloid-like aggregates in response to stress conditions. Cdc19 aggregation is regulated via an aggregation-prone low complexity region (LCR). In favorable growth conditions, this LCR is prevented from aggregating by phosphorylation or oligomerization, while upon glucose starvation it becomes exposed and allows aggregation. We suggest that LCR phosphorylation, oligomerization or partner-binding may be general and widespread mechanisms regulating LCR-mediated reversible protein aggregation. Moreover, we show that, as predicted by computational tools, Cdc19 forms amyloid-like aggregates in vitro. Interestingly, we also observe striking similarities between Cdc19 and its mammalian counterpart, PKM2. Indeed, also PKM2 harbors a LCR and contains several peptides with high amyloidogenic propensity, which coincide with known phosphorylation sites. Thus, we speculate that the formation of reversible, amyloid-like aggregates may be a general physiological mechanism for cells to adapt to stress conditions, and that the underlying regulatory mechanisms may be conserved from yeast to humans.
KEYWORDS: Protein aggregation, amyloid, functional amyloids, reversible protein aggregates, pyruvate kinase, low complexity region, stress granules
Protein aggregation is a biochemical process in which proteins alter their conformation and condense to form membrane-less inclusions that can be very diverse in localization, structure, composition and stoichiometry. Until recently, this process was generally assumed to be restricted to a set of proteins that due to mutations or environmental stresses form inherently irreversible and toxic clumps, which are commonly associated to a wide range of pathologies, from neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD) to systemic or localized amyloidosis [1–4]. This strong pathological association gave rise to the pervasive view that protein aggregation is an intrinsically irreversible and pathological process.
However, it has recently become clear that in several cases protein aggregation is not an aberrant and irreversible process, but rather a highly regulated physiological mechanism that contributes to a variety of complex cellular processes [5–11], ranging from fertilization [10], storage of hormones [9] and adaptation to stress conditions [5,8,11]. In fact, cells have been shown to harness protein aggregation to tune their proteomes by specifically regulating the compartmentalization [12], activity [13], storage [8,9] and degradation [5] of several proteins. Moreover, besides performing physiological functions, some aggregates have also been shown to be fully resolubilized in a regulated way. Altogether, these new findings forward the exciting notion that protein aggregates may be central actors in a previously-unrecognized and potentially widespread dynamic mechanism for cellular organization and regulation [14].
These findings thus challenge the way we consider and study protein aggregates, and raise new questions and problems. First, we need to re-think and re-define the features characterizing protein aggregates. Indeed, the “classical” protein aggregate definition encompassing only irreversible, inactive, non-functional, and in some cases infective aggregates does not hold for all cases. Thus, in order to precisely characterize the nature of a protein aggregate, one has to carefully evaluate several properties like function, reversibility, infectivity, localization, composition, and structure (Figure 1). Second, these new findings also prompt us to reflect on our terminology. Several new terms have been coined to indicate the different types of protein clumps formed in a physiological context, such as phase separations, biomolecular condensates, quinary assemblies, functional aggregates, etc [15]. However, this variegated nomenclature is unfortunately often leading to confusion and disagreements. Because all of these newly described inclusions, as well as the well-known disease-linked aggregates ultimately consist of condensed proteins (and often other components such as RNA), it would be appropriate to find an all-spanning term that encompasses all these structures. We suggest that the term “aggregates” may be the most appropriate when referring to both pathological and physiological inclusions. In fact, while the term “aggregate” has a negative connotation for historical reasons, its literal meaning does not necessarily indicate a pathological structure and can therefore be used also when referring to functional inclusions. Moreover, physiological and pathological aggregates share striking similarities, and both inclusion types fall within our initial definition of protein aggregation. In addition, it has also been hypothesized that physiological aggregation might be a precursor of pathological events. In this article we thus refer to protein aggregates as any membrane-less structure containing condensed proteins (and often also RNA) in variable stoichiometry that is stable enough to be isolated biochemically, and whose formation often involves polyvalent interactions and significant conformational changes in the proteins. This definition thus comprises a large range of structures which can have very different properties, functions and material states, from solid-like assemblies to less stable and more dynamic structures, like stress granules, Cajal bodies and the nucleolus [16]. Our definition however excludes a number of protein assemblies, including multisubunit complexes such as the recently identified TOROIDs [17], proteasomal bodies [18] and higher order structures of metabolic enzymes [19,20], whose formation does not involve major conformation changes. These assemblies, despite being of high interest, present very different dynamics and properties and will not be discussed further. In this article, we focus mainly on the more stable and solid class of protein aggregates, with particular attention to the amyloid fold.
Figure 1.
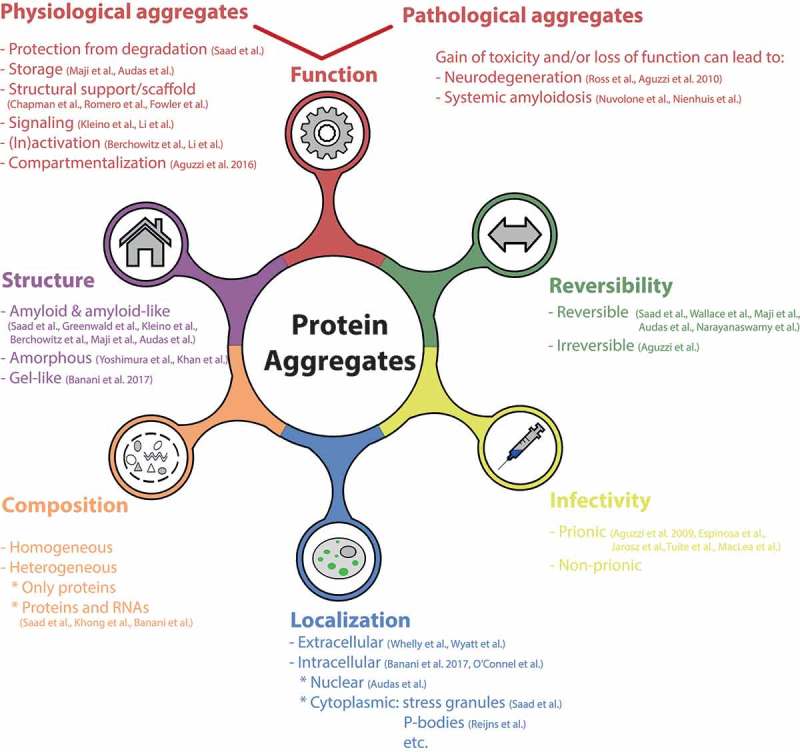
Features characterizing protein aggregates. Physiological and pathological protein aggregates are very diverse and multifaceted, and may be categorized based on features like structure [5,8,9,13,15,21,22,38,44], function [1,2,5,8,9,12,13,23–26,44–46], reversibility [5–9], infectivity [27–31], localization [5,8,15,32–34,47] and composition [5,35,36].
Amyloids: aggregates at the verge between pathology and physiology
One of the best-studied types of protein aggregates is amyloid fibrils. Amyloids consist of highly ordered, biochemically stable intermolecular β-sheets aligned to form elongated and un-branched fibrils, typically considered irreversible [4,37]. These aggregates gained the spotlight as they are the principal hallmark of several devastating neurodegenerative pathologies like Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s diseases (HD) and amyotrophic lateral sclerosis (ALS), but have also been associated to amyloidosis, both localized as with type II diabetes or systemic as with senile systemic amyloidosis [4,38–41]. In all these cases, amyloid formation is linked to degeneration of the affected tissue, but the exact mechanism of toxicity is often poorly understood.
Due to the well-established connection between amyloids and diseases, one of the perhaps most surprising discoveries in recent years was that some amyloid-like assemblies are required to carry out physiological functions. Indeed, in filamentous fungi amyloid fibrils are involved in heterokaryon formation [42], in Drosophila amyloid-like oligomers are implicated in memory persistence [43] and protection from pathogens [44], and in mammals amyloids have been shown to play an important role in a broad range of physiological processes such as melanin production [45], peptide hormones storage [9], programmed cell necrosis [46], fertilization of oocytes by sperm [10,47], antimicrobial responses [48] and cellular responses to stress [8]. Moreover, some of these functional amyloids were shown to be particularly remarkable because they can be fully resolubilized under specific physiological conditions, instead of being irreversible as previously thought [16].
This dual nature of amyloids as irreversible pathological clumps and as reversible physiological assemblies thus raised several crucial questions: how is functional amyloid formation and maintenance regulated in vivo? How are physiological amyloids re-solubilized and re-folded into the native state? How are the physiological and pathological aggregates related? Are physiological amyloids the precursors of pathological inclusions? And what goes wrong in the context of pathology? Answering these questions will undoubtedly provide a better understanding of the mechanisms governing physiological protein aggregation, and could also shed new light on the processes underlying the onset and progression of protein aggregation diseases. In particular, identification of the molecular machinery necessary for the dissolution of physiological amyloids may allow devising novel strategies to treat aggregation diseases.
Functional amyloids: towards an understanding of their regulation in yeast
In recent years, the yeast S. cerevisiae has been one of the favorite model organisms to study reversible protein aggregation. This simple organism not only offers powerful genetic and genomic tools, but has also been shown to extensively use protein aggregation to regulate cell fate decisions [49,50], meiosis progression [13], adaptation to stress [5,11,51] and aging [52]. Indeed, several studies reported the formation of reversible aggregates on a genome-wide scale, and showed that a surprisingly large number of proteins can undergo reversible aggregation in yeast [6,7,11]. These observations led to the hypothesis that reversible aggregates could constitute a previously-unrecognized widely used level of cellular organization.
Studies in yeast also allowed us to get a first glimpse on how physiological aggregation is regulated. We now know that functional aggregation generally depends on the presence of a prion-like, glutamine/asparagine-rich domain, or a region of low compositional complexity (LCR), which interestingly are often found also in pathological aggregates [53,54]. Indeed, several proteins able to undergo reversible aggregation in yeast were shown to contain a predicted LCR, which likely triggers aggregate formation. As an example, the RNA-binding protein Rim4 has been demonstrated to undergo developmentally regulated, LCR-dependent aggregation. During meiosis I, Rim4 mediates translational repression of several mRNAs involved in gametogenesis, including the B-type cyclin CLB3, by regulated aggregation of the protein in complex with its target mRNAs. At the onset of meiosis II, aggregates are cleared and mRNA translation is resumed to allow progression through gametogenesis. Thus, this LCR-dependent regulation of Rim4 plays an important role in gametogenesis by ensuring proper cell cycle progression through meiosis. Interestingly, careful biochemical analysis has revealed that Rim4 forms amyloid fibrils in vitro, in an LCR-dependent manner. In summary, this example not only shows that amyloids can be functional and reversible, but also gives a first indication on how these structures are regulated in a physiological context. Even though the initial starvation signal triggering Rim4 aggregation still needs to be identified, we now know that the formation of Rim4 amyloids is developmentally regulated and is dependent on an LCR.
Functional, reversible amyloid-like aggregates: the example of the yeast pyruvate kinase Cdc19
The amyloid-like structure of Cdc19 aggregates in vitro
Among the long list of proteins undergoing reversible aggregation in yeast cells, metabolic enzymes stand out as one of the largest clusters [55]. Thus, to better understand the regulatory mechanisms underlying reversible protein aggregation, we have recently studied the formation of yeast pyruvate kinase Cdc19 aggregates. Pyruvate kinase is a pivotal metabolic enzyme that catalyzes the final rate-limiting and irreversible step of glycolysis, transferring a phosphate group from phosphoenolpyruvate (PEP) to adenosine diphosphate (ADP), thus producing pyruvate and adenosine triphosphate (ATP). Upon glucose starvation or heat shock, this enzyme forms reversible aggregates, which are essential for adaptation and survival to stress and are readily resolubilized after stress release [5]. As in the case of Rim4, also Cdc19 aggregation depends on a short aggregation-prone LCR, which is necessary and sufficient to trigger aggregation [5]. In our previous studies, to characterize Cdc19 aggregation dynamics, we used live-cell microscopy to follow formation and re-solubilization of Cdc19 aggregates upon acute glucose starvation and glucose re-addition or upon heat shock. These experiments revealed rapid induction of cytoplasmic foci that increased in number and size upon prolonged starvation and that were re-solubilized within minutes once glucose was re-added to the medium [5]. Interestingly, here we expand this observation by reporting that these foci are initially highly mobile and can fuse with each other, while after a prolonged stress they tend to be more static and do not fuse anymore. Indeed, already within 4 hours of starvation a significant amount of Cdc19 condensed in a few, more static, bright dots (Figure 2(a)). We observed similarly growing foci forming not only upon acute and prolonged glucose starvation but also when cells slowly enter stationary phase, suggesting that also in this case cells initially form small motile foci, which then fuse together to form fewer, bigger and more static assemblies. Heat shock could also induce Cdc19 aggregation, but several other stresses such as nitrogen starvation, oxidative stress or osmotic stress do not, which suggests that this response is specific and highly regulated [5]. Interestingly, we also showed that Cdc19 not only co-localizes to stress granules, but is also able to modulate the dynamics of their formation and dissolution [5,55]. This suggested that Cdc19 is an integral component of these membrane-less organelles and may be a part of the regulatory core machinery necessary for their formation. Unexpectedly, we observed that Cdc19-containing stress granules were completely solid after 2 days in stationary phase as judged by fluorescence recovery after photobleaching (FRAP). Formation of solid aggregates was reminiscent of irreversible, pathological aggregates, yet these aggregates retained full reversibility upon stress relief, which was independent of aggregate degradation or de novo protein synthesis. Initial biochemical characterization was compatible with the formation of amyloid-like structures by Cdc19. Indeed, purified Cdc19 formed Thioflavin T-positive aggregates in vitro in an LCR-dependent manner. However, unraveling the exact biophysical and structural nature of these aggregates in vitro and in particular the corresponding Cdc19 aggregate structures in vivo prompted further studies.
Figure 2.
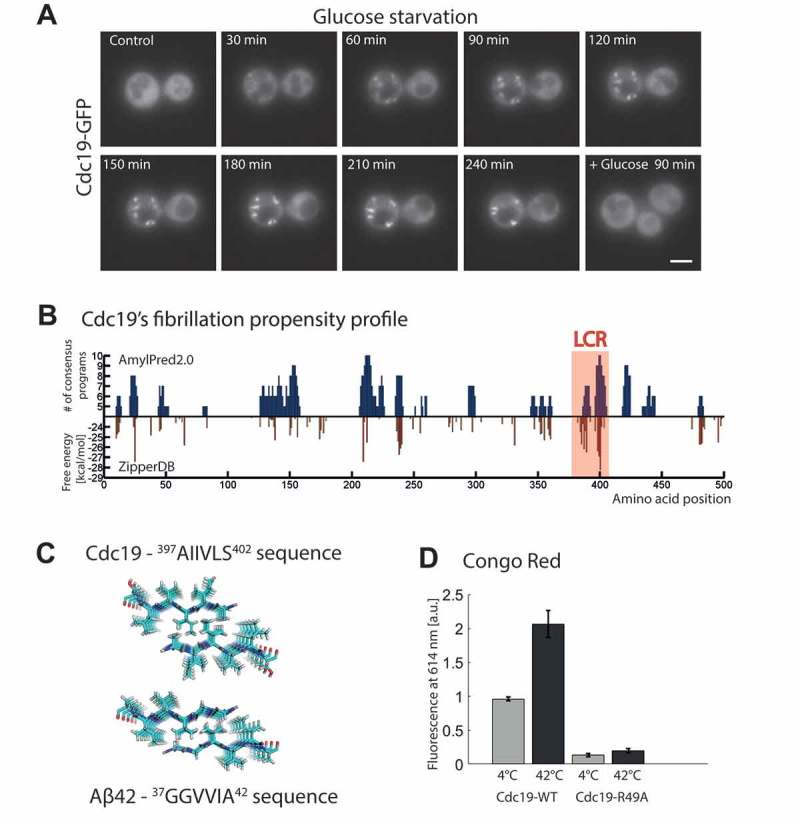
The yeast pyruvate kinase Cdc19 forms reversible, amyloid-like aggregates upon stress. (a) Cdc19 aggregates reversibly upon stress. Cells expressing Cdc19-GFP were grown in SD-full (synthetic complete medium containing glucose) and followed during shifts to glucose starvation media and subsequent nutrient repletion in microfluidic chips, as described previously [5,60]. Time-lapse fluorescence microscopy was performed as described previously [60] and images were taken every 10 min. To monitor successful switch of media, glucose starvation media was supplemented with a fluorescent dye (Alexa Fluor 680-Dextran, 3,000 MW, Invitrogen). Representative images of three independent experiments showing cells during exponential phase in glucose-rich medium (control), glucose starvation (every 30 min for 4 h) and 90 min after re-addition of glucose are shown. Scale bar, 3 μm. (b) Cdc19 contains several stretches of amino acids that are computationally predicted to form amyloid-like fibrils. The region with highest fibrillation propensity correlates with the experimentally validated Cdc19 LCR (highlighted in the red box) [5]. The amino acid sequence of Cdc19 is plotted along the x-axis. On the y-axis, the fibrillation propensities of Cdc19 segments are shown, as computed using the consensus tool AmylPred2.0 (upper part, (http://aias.biol.uoa.gr/AMYLPRED2/) [57]), and the structure-based ZipperDB prediction method (lower part, (http://services.mbi.ucla.edu/zipperdb/) [56]). AmylPred2.0 combines 11 different prediction tools. A peptide is considered amyloidogenic if the consensus of at least 5 out of 11 methods is reached. With ZipperDB, fibrillation propensities for every possible six-residue peptide not containing a proline from the protein sequence of interest are computed using a structure-based algorithm. Proline-containing hexapeptides are not included in the profile, as prolines are β-strand breakers and thus incompatible with the structure required for amyloid fibrils formation. Based on experimental data, a predicted energy of −23 kcal/mol was chosen as threshold and thus segments with energies below this value are considered to have high fibrillation propensity. (c) Predicted fibrillar structure of the hexamer with highest fibrillation propensity in Cdc19 compared to the fibrillar structure of a classical amyloidogenic peptide of Aβ42. (d) Cdc19 aggregates are stained by the amyloid-binding dye Congo Red. Cdc19 was purified as described previously [5]. A 1 mM stock solution of Congo Red (Sigma-Aldrich) was prepared in protein lysis buffer and filtered (0.2 μm, Millipore). Before each assay aliquots of purified wild-type Cdc19 (Cdc19-WT) or of the non-aggregating Cdc19-R49A mutant (Cdc19-R49A) were thawed on ice and cleared by centrifugation (at 4°C for 10 min at 21,000 g). The proteins (0.3 mg/ml) were then incubated 20 min at 42°C to induce aggregation or kept on ice. Heat shocked and control samples were incubated with Congo Red solution (final concentration of 100 μM) and fluorescence intensity was measured in 96-well half-area non-binding polystyrene plates (Corning). The Congo Red fluorescence signal was recorded at 30°C in a plate reader (Clariostar, BMG Labtech) by monitoring the emission signal at 614 ± 15 nm after excitation at 560 ± 30 nm. Within an individual experiment the fluorescence measurements were carried out in triplicate. Three independent repetitions were performed using three different protein aliquots for Cdc19-WT and two different aliquots for the non-aggregating control Cdc19-R49A. Data are represented as mean ± s.e.m. of three independent experiments.
Here we now show a first step towards a better characterization of the potential amyloid nature of Cdc19 aggregates. In order to identify probable amyloidogenic regions within Cdc19 we applied different aggregation-predicting computational tools. Interestingly, we found that several peptides within the protein were predicted to have amyloidogenic properties. For example, Figure 2(b) depicts the fibrillation propensity profile of Cdc19, as predicted by two independent tools: ZipperDB [56] and AmylPred2.0 [57]. The ZipperDB database contains calculations of the likelihood that a particular amino acid sequence can form a “steric zipper” (two self-complementary β-sheets), and thus give rise to the spine of amyloid fibrils. This tool is thus structure-based and uses a threshold chosen based on experimental data, which can be readily verified experimentally [56,58]. AmylPred2.0 instead combines 11 different algorithms that have been specifically developed to predict the tendency of a specific amino acid sequence to form amyloid fibrils and provides a consensus estimation combining their outputs. These methods are based on multiple principles and take into account different features of the given sequence like structure, sequence, average packing density, folding energy, etc [57]. Thus, the combination of ZipperDB and AmylPred2.0 offers a diversified evaluation of fibrillation propensities, and should predict probable “amyloidogenic determinant” peptides for a given protein with high confidence. Indeed, here we show that both methods identified several regions spread over the Cdc19 sequence as amyloid-prone (Figure 2(b)), and the consensus regions identified by the two methods are largely overlapping. These amyloidogenic segments are not clustered in a specific region but distributed on different areas of the 3D structure. Importantly however, the hexamers with the highest fibrillation propensity identified by ZipperDB (i.e. lowest predicted energy) and with the highest confidence by AmyPred2.0 are identical and correspond to the experimentally-validated LCR of Cdc19. The highest scoring hexamer is predicted to form a highly ordered β-sheet, which closely resembles the one formed by the classical amyloid-forming protein Aβ42 (Figure 2(c)). This might imply that short, highly amyloidogenic stretches, like Cdc19 LCR, are nucleators of the aggregation reaction, and are thus necessary and sufficient for aggregates formation, while other parts of the protein might be involved in facilitating and stabilizing the amyloid fibrils.
To experimentally support these predictions, amyloid-like aggregates can be assessed by SDS-resistance assays and stained with amyloid-specific dyes like Congo Red or Thioflavin T. Moreover, their secondary structure can be analyzed with circular dichroism (CD), Fourier transform infrared spectroscopy (FT-IR) and high-resolution techniques such as X-ray crystallography and solid-state nuclear magnetic resonance (ssNMR). Together these methods emerged as gold standards for detecting amyloid structures in vitro [59]. Here we now show Congo Red-dependent fluorescence of heat-shock induced Cdc19 aggregates (Figure 2(d)), supporting our published observations using Th-T and CD [5]. As expected, heat shock strongly induced Congo Red-dependent fluorescence of wild-type Cdc19 (Cdc19-WT), while our non-aggregating Cdc19-R49A mutant was not stained by this dye. Since in the Cdc19-R49A mutant the LCR is constitutively shielded [5], the fact that this mutant does not aggregate and is not stained by Congo Red further supports the notion that although Cdc19 contains several amyloidogenic regions, monomerization and solvent exposure of a single LCR is necessary for amyloid-like aggregation in vitro and in vivo. In summary, we show that Cdc19 aggregates induced by heat shock in vitro score positive in all biochemical and biophysical assays generally applied to characterize amyloids, and suggest that reversible Cdc19 aggregates in vivo may also have an amyloid-like structure.
The challenges of characterizing the structure of Cdc19 aggregates in vivo
Characterizing the structure of protein aggregates in vivo is experimentally very challenging due to the complex nature and heterogeneity of these inclusions within cells. For example, Cdc19 aggregates accumulate in stress granules, which contain hundreds of proteins and RNAs. While for in vitro studies with single recombinant proteins several very accurate techniques are available to predict, detect and characterize amyloids and amyloid-like aggregates, most of these approaches cannot be applied in vivo [59]. Below, we thus discuss different methods that can be used towards achieving this aim.
As a first test to examine the potential of proteins to form amyloids in vivo, a b-ISOX precipitation assay from cell lysates is commonly used. The b-ISOX compound forms ordered crystals resembling amyloid structures that can recruit and precipitate amyloidogenic proteins [61]. Indeed, we previously found that Cdc19 precipitated with b-ISOX in an LCR-dependent manner from cell extracts. Even though these results provide a first indication that Cdc19 forms amyloid-like aggregates also in vivo, additional evidence is required to extend and firmly establish these findings.
An additional possibility to detect amyloid-like aggregates in vivo is their direct visualization by fluorescence microscopy using genetically encoded fusion tags and conformational-sensitive fluorescent dyes, like Thioflavin S (Th-S) or Congo Red [62]. Similar to Th-T, Th-S displays increased intensity and shifts the fluorescence maximum of the spectra upon binding to amyloids fibrils. Whereas Th-T is usually preferred for in vitro studies, Th-S and Congo Red are the gold standard used for in vivo staining of amyloids as they have a better ability to penetrate biological membranes and to accumulate inside cells [59]. Indeed, Th-S has successfully been used to detect amyloids within intact bacteria and in combination with fluorescence microscopy and/or flow cytometry could thus also be explored to study amyloid formation in eukaryotes [59,63].
Another way to detect amyloid fibrils in vivo is to directly visualize them using different types of electron microscopy. For instance, correlative light-electron microscopy (CLEM), a technique that allows high-resolution imaging by EM of the same structure observed by light microscopy using fluorescence detection, has already been effectively used to prove the existence of fibrillar structures of Sup35-GFP prions in yeast [PSI+] cells [64]. One limitation of this technique is that EM analysis only assesses the morphology of the aggregates, but cannot directly connect fibrillar structures with amyloids containing cross β-sheets due to a lack of spatial resolution. Moreover, like in the case of Cdc19, many aggregates co-localize with other stress granule components, which could make it difficult to determine which proteins form amyloid stripes in such granules. Besides direct visualization, also assessing the resistance to SDS of protein aggregates formed in vivo using SDS-AGE assays is commonly used as readout for amyloid formation. Although SDS resistance is one of the established conditions for (pathological) amyloid structures, it cannot be excluded that some false positive results may arise due to the potential trapping of other proteins in amyloid-containing large assemblies. Finally, the recently developed method of limited proteolysis coupled to mass spectrometry (LiP-SRM) allows directly analyzing protein structural changes in complex biological samples [65,66]. In this protocol, cell extracts are first digested in non-denaturing conditions by a broad-specificity protease, so that the cuts are dictated by the structural features of the proteins. Then, the sample is denatured and completely digested by trypsin. As a control, the same proteome is subjected to trypsinization only. Cell extracts from different conditions (e.g. exponentially growing vs starved cells) can then be compared and protein structural changes detected. This method has already been successfully used to probe conformational changes of the Parkinson’s disease-linked protein α-synuclein [65]. LiP-SRM may thus be promising to study the different conformational changes that drive the conversion of reversible aggregates and possibly amyloids in vivo.
Taken together, at present there is no simple method to directly detect whether a specific protein forms amyloid-like structures in vivo. Therefore, it will be necessary to combine several distinct and unrelated techniques such as CLEM, staining with amyloid-specific dyes and LiP-SRM, and to compare conformations of aggregates formed in vivo and in vitro with recombinant proteins to generate several lines of evidence supporting the hypothesis of amyloids in vivo. In the future it will be indispensable to develop methods to directly and unequivocally detect the presence of amyloid assemblies in vivo.
Regulation of Cdc19 aggregation
Another crucial challenge in future years will be understanding the mechanisms regulating the assembly and disassembly of functional aggregates. Since reversible aggregation is often triggered by exposure of an aggregation-prone LCR, aggregate formation can conceptually be regulated by LCR accessibility. This accessibility can be reduced for example by phosphorylation, oligomerization, binding to other structures and/or other post-translational modifications, which would thus prevent aggregation. Upon specific clues, removal of these protection mechanisms may expose the LCR and allow aggregation (Figure 3(a)). For example, Cdc19 exists in vivo in an equilibrium between monomers and homotetramers, and tetramer formation is allosterically regulated by binding to the metabolite fructose-1,6-bisphosphate (FBP). In the tetrameric form, the LCR of Cdc19 is hidden between the subunits (Figure 3(b)), but in the monomer it is exposed to the surface. This region is necessary and sufficient to allow aggregation upon stress, and consistently, mutations preventing the formation of tetramers drastically enhance aggregation, while mutations stabilizing the tetrameric form of Cdc19 (Cdc19-R49A) largely suppress aggregation [5]. Interestingly, phosphoproteomic analysis identified a cluster of phosphorylation sites in the LCR (Figure 3(c)) that are rapidly dephosphorylated upon stress. Together, phosphorylation and oligomerization prevent unscheduled aggregation of Cdc19 under glucose-rich conditions. Upon glucose starvation, the LCR gets dephosphorylated and this allows reversible aggregation. Here we now show that Cdc19’s phosphorylation sites are not clustered only in the LCR but are also present within other predicted amyloidogenic regions. It is thus tempting to speculate that phosphorylation within amyloidogenic regions may be a more general mechanism to regulate the dynamics of aggregation in response to stress and/or to control re-solubilization of protein aggregates upon stress relief. A combination of post-translational modifications might be used to adapt and fine-tune the aggregation response to the specific needs of the cell.
Figure 3.

LCR-mediated reversible protein aggregation can be regulated by several mechanisms, including LCR phosphorylation, oligomerization and/or interaction with a binding partner. (a) Model showing possible mechanisms to prevent LCR-mediated aggregation. Unscheduled aggregation can be avoided by protecting aggregation-prone LCRs by means of post-translational modifications (e.g. phosphorylation), oligomerization and/or interaction with a binding partner. Upon stress, these protections are removed and the LCR becomes exposed, thus allowing functional, reversible aggregation. (b) In vivo, Cdc19 is in an equilibrium between a monomeric form (left) and a tetrameric form (right). The red region corresponds to the predicted LCR identified by the SEG program (http://mendel.imp.ac.at/METHODS/seg.server.html) [67], using a trigger window length [W] = 25, trigger complexity [K(1)] = 3.0 and extension complexity [K(2)]) = 3.3. Blue regions correspond to amyloidogenic peptides as predicted by AmylPred2.0 (http://aias.biol.uoa.gr/AMYLPRED2/) [57]. (c) Schematic representation of Cdc19 showing that empirically measured phosphosites taken from our previously published data [5]) often map within Cdc19 LCR (in red) or in predicted amyloidogenic regions (in blue) .
Regulation of functional amyloids: from yeast to mammals
Functional amyloid-like aggregates are not unique to yeast, but are instead widespread in nature, and can be found in several different organisms, ranging from bacteria to mammals [68,69]. Some functional amyloid-like aggregates (like Balbiani bodies or nuclear amyloid bodies) are particularly remarkable as they have been shown to be fully reversible [16]. Also other transiently forming proteinaceous organelles like stress granules are not a prerogative of yeast but can be found also in several higher eukaryotes, including humans. However, the components, functions and underlying regulatory mechanisms of these structures in mammalian cells have yet to be fully understood. Our recent advances in understanding how such structures are regulated in yeast might provide useful information to better understand their regulation in mammalian cells. Indeed, it is tempting to speculate that also in mammalian cells some proteins might form reversible, functional, amyloid-like aggregates, and that these structures might be regulated similarly to Cdc19. As a support for this hypothesis, yeast and mammalian stress granules share several characteristics. They are both formed by RNAs and proteins, and analysis of their proteomes revealed a significant overlap in composition. Abundant components are for instance translation factors and RNA-binding proteins, several of which contain LCRs [5,70,71]. Structurally, mammalian stress granules are comprised of a more stable core and a rather dynamic shell, while yeast stress granules are mostly formed by a core part and possibly a proportionally smaller shell. In both organisms, the cores are established early during stress granule assembly, suggesting that they may be necessary for seeding the liquid-like shell [70]. Although the core stress granule machinery is conserved from yeast to mammals and may contribute to the regulation of reversible aggregation, systematic screens for reversibly aggregating proteins are still missing. Moreover, stress granule formation in mammals is usually studied upon arsenite treatment or inhibition of translation, and the physiological triggers that lead to stress granule formation in mammalian cells remain poorly understood. In particular, it is unclear whether acute glucose starvation and/or heat shock, two conditions that very efficiently initiate stress granule formation in yeast, also trigger reversible protein aggregation in mammalian cells. Thus, the exact physiological function of these membrane-less organelles is still under debate. As mentioned earlier, in yeast, reversible aggregation of Cdc19 into stress granules upon heat shock or carbon starvation emerged as a protective mechanism to prevent its degradation during stress. Avoiding Cdc19 degradation allows rapid restoration of cellular functions upon stress release without the need for de novo protein synthesis, and is essential for survival. Given the similarities between yeast and mammalian pyruvate kinase and stress granules components, it is tempting to speculate that reversible aggregation in mammals may similarly contribute to stress adaptation. Moreover, developmental transitions are often accompanied by global metabolic changes, including B-cell activation and fertilization. It will thus be interesting to examine whether reversible aggregation of metabolic enzymes may similarly contribute to the regulation of these processes.
Mammalian pyruvate kinase shares a number of critical features with Cdc19 that may suggest its ability to reversibly aggregate in mammalian cells. In mammals, pyruvate kinase is encoded by two genes that produce a total of four pyruvate kinase isoforms (M1, M2, L and R) [72]. While the L and R isoforms are only found in specific tissues, the M isoforms are rather ubiquitously expressed. PKM1 and PKM2 are generated by alternative splicing and differ only by a short stretch of 23 amino acids, yet they present quite different characteristics. Indeed, while PKM1 functions in differentiated tissues and is insensitive to FBP, PKM2 is enriched in rapidly proliferating cells and, as Cdc19, can be allosterically activated by FBP [73–75]. PKM2 expression has been shown to be particularly upregulated in many human cancers, but the functional significance of this regulation remains subject of active debate [72,76]. Interestingly, here we now show (Figure 4) that the alternative exon usage generating PKM2 not only alters sensitivity to FBP, but also introduces a short LCR that is missing in PKM1, suggesting that PKM2 represents the functional homologue of Cdc19. Moreover, we also show that predictions of amyloidogenic regions identify several stretches in the sequence of both PKM1 and PKM2 with high amyloidogenic propensities (Figure 4). Interestingly, the LCR in PKM2 scores the highest, suggesting that, as in the case of Cdc19, this region may be the trigger for the formation of PKM2 amyloid-like aggregates. This is supported by literature data, where PKM2 has been precipitated with b-ISOX from HEK293 cells lysates [77] and purified rabbit PKM2 has been shown to be Th-T positive [78]. Similarly, here we also report a surprising overlap between known PKM2 phosphorylation sites and amyloidogenic regions, suggesting that analogous to Cdc19 amyloid formation may be regulated by phosphorylation. Taken together, these structural and functional similarities, as well as the predictions shown in Figure 4, strongly suggest that PKM2 might have the ability to form reversible aggregates similar to Cdc19. We hypothesize that the short stretch of amino acids identified as an LCR might be essential and sufficient to cause aggregation, and that phosphorylation sites might be able to regulate aggregation. Since PKM2 critically contributes to the regulation of cell growth it is also tempting to speculate that regulated aggregation of key metabolic enzymes may control critical growth transitions in mammalian cells similar to Cdc19. Identifying physiological conditions that trigger aggregation of PKM2 may allow genetic dissection of the contribution of phosphorylation sites for the formation of amyloid-like aggregates.
Figure 4.
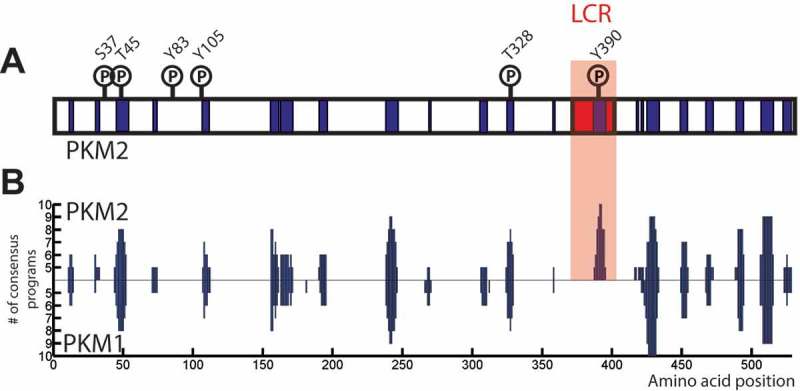
The mammalian pyruvate kinase PKM2 might also form amyloid-like aggregates, which could be regulated similar to Cdc19. A-B) Both PKM1 and PKM2 contain several stretches of amino acids that are predicted to form amyloids. However, only PKM2 presents a predicted LCR, which overlaps with the region with highest fibrillation propensity (highlighted in the red box). In addition, known PKM2 phosphorylation sites show a remarkable overlap with predicted amyloidogenic regions, suggesting that PKM2 might aggregate in a regulated way similar to Cdc19. The fibrillation profiles of PKM2 (upper part) and PKM1 (lower part) were computed using AmylPred2.0 (http://aias.biol.uoa.gr/AMYLPRED2/) [57]. Predicted amyloidogenic regions are shown in in blue. PKM2 phosphorylation sites were found in the Phospho.ELM database (http://phospho.elm.eu.org) [79]. PKM2’s predicted low complexity region was identified by the SEG program (http://mendel.imp.ac.at/METHODS/seg.server.html) [67], using a trigger window length [W] = 25, trigger complexity [K(1)] = 3.0 and extension complexity [K(2)] = 3.3., PKM2 LCR is highlighted in red.
Conclusions and outlook
In conclusion, recent results demonstrate that functional protein aggregation constitutes an additional regulatory layer used by cells to control multiple processes. New data reveal that this physiological organizational process is more complex, widespread and multifaceted than we thought [16]. Indeed, we now know that regulated protein aggregation can be triggered by several different stimuli (carbon starvation [5], heat shock [5], cytoplasm acidification [11], etc.), it involves a large number of proteins [6,7], it is controlled by complex and diverse mechanisms [5], it leads to different outputs that can range from minimal [11,19] to outstanding [5] structural rearrangements, and it serves very different purposes (protein inactivation [13], protein protection [5], signaling [44], etc.). Moreover, the recently discovered ability of cells to reverse aggregation and resolubilize protein aggregates showed that these processes are highly dynamic and constitute a simple yet very efficient way to transiently and specifically rearrange and reorganize proteomes in a flexible and tightly regulated manner.
Despite these recent findings, our understanding of reversible functional protein aggregates is still in its infancy. Identified physiological aggregates are diverse in terms of structure, function and regulation, and to-date only a small fraction has been systematically investigated. Several different structures, such as cytoplasmic stress granules, Cajal bodies and nuclear speckles have been described (and extensively reviewed in [16,80,81]). However, how exactly they are regulated is still largely unclear. Some of these structures are particularly remarkable, as they take an amyloid fold, which was previously thought to be intrinsically irreversible and linked to pathology. Examples of reversible, functional amyloids are Balbiani bodies found in oocytes, nuclear amyloid bodies (A-bodies) and cytoplasmic Cdc19 aggregates in yeast cells [5,14,16]. These bodies contain several proteins and serve several functions, from maintaining cell dormancy, to survive transient stress conditions. The discovery of widespread reversible physiological amyloidogenic programs opened a series of new questions. As shown by the case of the yeast pyruvate kinase Cdc19, recent studies in simple model organisms allowed us to take the first steps towards a mechanistic understanding of the regulation of reversible functional amyloids, and highlighted the importance of LCRs and post-translational modifications in these processes. However, more detailed mechanistic characterizations and in-depth studies also in mammalian cells are still lacking.
Moving forward, we envisage two main pressing questions. First, it will be crucial to understand how reversible functional aggregates are regulated in vivo. The discovery of widespread physiological amyloidogenic programs shows that amyloids are a fundamental component of cellular organization. Understanding how cells cope, control and harness protein aggregates is thus crucial to understand the biology of the cell. Recently, thanks also to the study of these processes in simple organisms such as budding yeast, we finally start understanding the molecular determinants, regulatory mechanisms and physiological roles of such reversible aggregates. The second fundamental challenge for the future will be to investigate what is the relationship between pathological and physiological amyloids. Even though some structurally reversible aggregates resemble amyloids, firm evidence for their assembly state in vivo is lacking. So, how does the structure and assembly properties of reversible aggregates really compare to pathological assemblies? Moreover, although tempting to speculate, it remains to be examined if pathological amyloids emerge from reversible, physiological precursors and if deregulation of these processes might contribute to disease. Interestingly however, phosphorylation and LCR protection may also play important roles in the formation of pathologic aggregates, as in the case of the RNA-binding protein FUS, which is often mutated in patients suffering of amyotrophic lateral sclerosis (ALS). Indeed, recent in vitro experiments suggest that phosphorylation of FUS-LCRs interferes with droplet formation, inhibits their incorporation into hydrogels and even dissolves already formed droplets [82,83]. Moreover, other ALS proteins are also found in stress granules, and it will be interesting to explore their link to disease formation.
In summary, understanding how cells regulate and harness protein aggregation will not only open new insights into exciting cell biology, but, quite unexpectedly, might also help to better understand disease mechanisms and allow devising novel treatments. Pathologic protein assemblies are associated with more than 30 diseases, and amyloid-related pathologies are currently estimated to affect 45 million people worldwide, a number predicted to triple by 2050 mainly due to ageing of the population [84]. Thus, as there is an urgent need to understand and ultimately cure or prevent these irreversible processes, the study of protein aggregation has become a central area of research. While many questions remain, research on reversible functional aggregation in simple organisms such as budding yeast moved the first steps towards a mechanistic understanding of how cells regulate protein aggregation and might pave the way for a better understanding, treatment and prevention of aggregation-based diseases.
Funding Statement
This work was supported by the Eidgenössische Technische Hochschule Zürich [ETH-46 16-1];European Research Council[268930];Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung [310030B_160312];Schweizerischer Nationalfonds zur Förderungder Wissenschaftlichen Forschung [31003A_166513];Stiftung Synapsis - Alzheimer Forschung Schweiz AFS
Acknowledgments
We thank A. Smith, S. Kroschwald, P. Kimmig, F. van Drogen, P. Arosio, A. Sengör and L. Garbani Marcantini for critical manuscript reading and helpful discussions. Work in the Dechant and Peter laboratories is funded by independent SNSF research grants (31003A_166513 and 310030B_160312), and MP is supported by the ETHZ (ETH-46 16-1), an ERC senior award (268930) and the Synapsis foundation.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Aguzzi A, O’Connor T.. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9(3):237–248. [DOI] [PubMed] [Google Scholar]
- [2].Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10 Suppl:S10–S17. [DOI] [PubMed] [Google Scholar]
- [3].Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296(5575):1991–1995. [DOI] [PubMed] [Google Scholar]
- [4].Chiti F, Dobson CM. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu Rev Biochem. 2017;86:27–68. [DOI] [PubMed] [Google Scholar]
- [5].Saad S, Cereghetti G, Feng Y, et al. Reversible protein aggregation is a protective mechanism to ensure cell cycle restart after stress. Nat Cell Biol. 2017;19(10):1202–1213. [DOI] [PubMed] [Google Scholar]
- [6].Wallace EW, Kear-Scott JL, Pilipenko EV, et al. Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell. 2015;162(6):1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Narayanaswamy R, Levy M, Tsechansky M, et al. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc Natl Acad Sci U S A. 2009;106(25):10147–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Audas TE, Audas DE, Jacob MD, et al. Adaptation to stressors by systemic protein amyloidogenesis. Dev Cell. 2016;39(2):155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Maji SK, Perrin MH, Sawaya MR, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325(5938):328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guyonnet B, Egge N, Cornwall GA. Functional amyloids in the mouse sperm acrosome. Mol Cell Biol. 2014;34(14):2624–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Munder MC, Midtvedt D, Franzmann T, et al. A pH-driven transition of the cytoplasm from a fluid- to a solid-like state promotes entry into dormancy. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Aguzzi A, Altmeyer M. Phase separation: Linking cellular compartmentalization to disease. Trends Cell Biol. 2016;26(7):547–558. [DOI] [PubMed] [Google Scholar]
- [13].Berchowitz LE, Kabachinski G, Walker MR, et al. Regulated formation of an amyloid-like translational repressor governs gametogenesis. Cell. 2015;163(2):406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang M, Audas TE, Lee S. Disentangling a bad reputation: Changing perceptions of amyloids. Trends Cell Biol. 2017;27(7):465–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Banani SF, Lee HO, Hyman AA, et al. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18(5):285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Woodruff JB, Hyman AA, Boke E. Organization and function of non-dynamic biomolecular condensates. Trends Biochem Sci. 2018;43(2):81–94. [DOI] [PubMed] [Google Scholar]
- [17].Prouteau M, Desfosses A, Sieben C, et al. TORC1 organized in inhibited domains (TOROIDs) regulate TORC1 activity. Nature. 2017;550(7675):265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Peters LZ, Hazan R, Breker M, et al. Formation and dissociation of proteasome storage granules are regulated by cytosolic pH. J Cell Biol. 2013;201(5):663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Petrovska I, Nüske E, Munder MC, et al. Filament formation by metabolic enzymes is a specific adaptation to an advanced state of cellular starvation. Elife. 2014;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jin M, Fuller GG, Han T, et al. Glycolytic enzymes coalesce in G bodies under hypoxic stress. Cell Rep. 2017;20(4):895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yoshimura Y, Lin Y, Yagi H, et al. Distinguishing crystal-like amyloid fibrils and glass-like amorphous aggregates from their kinetics of formation. Proc Natl Acad Sci U S A. 2012;109(36):14446–14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Khan MV, Zakariya SM, Khan RH. Protein folding, misfolding and aggregation: A tale of constructive to destructive assembly. Int J Biol Macromol. 2018;112:217–229. [DOI] [PubMed] [Google Scholar]
- [23].Chapman MR, Robinson LS, Pinkner JS, et al. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. 2002;295(5556):851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Romero D, Aguilar C, Losick R, et al. Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc Natl Acad Sci U S A. 2010;107(5):2230–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nuvolone M, Merlini G. Systemic amyloidosis: novel therapies and role of biomarkers. Nephrol Dial Transplant. 2017;32(5):770–780. [DOI] [PubMed] [Google Scholar]
- [26].Nienhuis HL, Bijzet J, Hazenberg BP. The prevalence and management of systemic amyloidosis in western countries. Kidney Dis (Basel). 2016;2(1):10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev. 2009;89(4):1105–1152. [DOI] [PubMed] [Google Scholar]
- [28].Espinosa Angarica V, Ventura S, Sancho J. Discovering putative prion sequences in complete proteomes using probabilistic representations of Q/N-rich domains. BMC Genomics. 2013;14:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jarosz DF, Lancaster AK, Brown JCS, et al. An evolutionarily conserved prion-like element converts wild fungi from metabolic specialists to generalists. Cell. 2014;158(5):1072–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tuite MF, Serio TR. The prion hypothesis: from biological anomaly to basic regulatory mechanism. Nat Rev Mol Cell Biol. 2010;11(12):823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].MacLea KS. What makes a prion: infectious proteins from animals to yeast. Int Rev Cell Mol Biol. 2017;329:227–276. [DOI] [PubMed] [Google Scholar]
- [32].Wyatt AR, Yerbury JJ, Poon S, et al. Therapeutic targets in extracellular protein deposition diseases. Curr Med Chem. 2009;16(22):2855–2866. [DOI] [PubMed] [Google Scholar]
- [33].O’Connell JD, Zhao A, Ellington AD, et al. Dynamic reorganization of metabolic enzymes into intracellular bodies. Annu Rev Cell Dev Biol. 2012;28:89–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Reijns MA, Alexander RD, Spiller MP, et al. A role for Q/N-rich aggregation-prone regions in P-body localization. J Cell Sci. 2008;121(Pt 15):2463–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Khong A, Matheny T, Jain S, et al. The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol Cell. 2017;68(4):808–820.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Banani SF, Rice AM, Peeples WB, et al. Compositional control of phase-separated cellular bodies. Cell. 2016;166(3):651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sawaya MR, Sambashivan S, Nelson R, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447(7143):453–457. [DOI] [PubMed] [Google Scholar]
- [38].Greenwald J, Riek R. Biology of amyloid: structure, function, and regulation. Structure. 2010;18(10):1244–1260. [DOI] [PubMed] [Google Scholar]
- [39].Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. [DOI] [PubMed] [Google Scholar]
- [40].Hoppener JW, Lips CJ. Role of islet amyloid in type 2 diabetes mellitus. Int J Biochem Cell Biol. 2006;38(5–6):726–736. [DOI] [PubMed] [Google Scholar]
- [41].Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15(6):384–396. [DOI] [PubMed] [Google Scholar]
- [42].Dos Reis S, Coulary-Salin B, Forge V, et al. The HET-s prion protein of the filamentous fungus Podospora anserina aggregates in vitro into amyloid-like fibrils. J Biol Chem. 2002;277(8):5703–5706. [DOI] [PubMed] [Google Scholar]
- [43].Majumdar A, Cesario WC, White-Grindley E, et al. Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell. 2012;148(3):515–529. [DOI] [PubMed] [Google Scholar]
- [44].Kleino A, Ramia NF, Bozkurt G, et al. Peptidoglycan-sensing receptors trigger the formation of functional amyloids of the adaptor protein imd to initiate drosophila NF-κB signaling. Immunity. 2017;47(4):635–647.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fowler DM, Koulov AV, Alory-Jost C, et al. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006;4(1):e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li J, McQuade T, Siemer AB, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150(2):339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Whelly S, Johnson S, Powell J, et al. Nonpathological extracellular amyloid is present during normal epididymal sperm maturation. PLoS One. 2012;7(5):e36394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jang H, Arce FT, Mustata M, et al. Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link. Biophys J. 2011;100(7):1775–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Caudron F, Barral Y. A super-assembly of Whi3 encodes memory of deceptive encounters by single cells during yeast courtship. Cell. 2013;155(6):1244–1257. [DOI] [PubMed] [Google Scholar]
- [50].Schlissel G, Krzyzanowski MK, Caudron F, et al. Aggregation of the Whi3 protein, not loss of heterochromatin, causes sterility in old yeast cells. Science. 2017;355(6330):1184–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Franzmann TM, Jahnel M, Pozniakovsky A, et al. Phase separation of a yeast prion protein promotes cellular fitness. Science. 2018;359(6371):eaao5654. [DOI] [PubMed] [Google Scholar]
- [52].Saarikangas J, Barral Y. Protein aggregates are associated with replicative aging without compromising protein quality control. Elife. 2015;4: pii: e06197. doi: 10.7554/eLife.06197 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sant’Anna R, Fernández MR, Batlle C, et al. Characterization of amyloid cores in prion domains. Sci Rep. 2016;6:34274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sabate R, Rousseau F, Schymkowitz J, et al. What makes a protein sequence a prion? PLoS Comput Biol. 2015;11(1):e1004013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kramer K, Sachsenberg T, Beckmann BM, et al. Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nat Methods. 2014;11(10):1064–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Goldschmidt L, Teng PK, Riek R, et al. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci U S A. 2010;107(8):3487–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tsolis AC, Papandreou NC, Iconomidou VA, et al. A consensus method for the prediction of ‘aggregation-prone’ peptides in globular proteins. PLoS One. 2013;8(1):e54175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Saelices L, Johnson LM, Liang WY, et al. Uncovering the mechanism of aggregation of human transthyretin. J Biol Chem. 2015;290(48):28932–28943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Villar-Piqué A, Espargaró A, Ventura S, et al. Screening for amyloid aggregation: in-silico, in-vitro and in-vivo detection. Curr Protein Pept Sci. 2014;15(5):477–489. [DOI] [PubMed] [Google Scholar]
- [60].Dechant R, Binda M, Lee SS, et al. Cytosolic pH is a second messenger for glucose and regulates the PKA pathway through V-ATPase. EMBO J. 2010;29(15):2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kato M, Han TW, Xie S, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149(4):753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kimura Y, Koitabashi S, Fujita T. Analysis of yeast prion aggregates with amyloid-staining compound in vivo. Cell Struct Funct. 2003;28(3):187–193. [DOI] [PubMed] [Google Scholar]
- [63].Espargaro A, Sabate R, Ventura S. Thioflavin-S staining coupled to flow cytometry. A screening tool to detect in vivo protein aggregation. Mol Biosyst. 2012;8(11):2839–2844. [DOI] [PubMed] [Google Scholar]
- [64].Kawai-Noma S, Pack C-G, Kojidani T, et al. In vivo evidence for the fibrillar structures of Sup35 prions in yeast cells. J Cell Biol. 2010;190(2):223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Feng Y, De Franceschi G, Kahraman A, et al. Global analysis of protein structural changes in complex proteomes. Nat Biotechnol. 2014;32(10):1036–1044. [DOI] [PubMed] [Google Scholar]
- [66].Polverino de Laureto P, Taddei N, Frare E, et al. Protein aggregation and amyloid fibril formation by an SH3 domain probed by limited proteolysis. J Mol Biol. 2003;334(1):129–141. [DOI] [PubMed] [Google Scholar]
- [67].Wootton JC. Non-globular domains in protein sequences: automated segmentation using complexity measures. Comput Chem. 1994;18(3):269–285. [DOI] [PubMed] [Google Scholar]
- [68].Fowler DM, Koulov AV, Balch WE, et al. Functional amyloid–from bacteria to humans. Trends Biochem Sci. 2007;32(5):217–224. [DOI] [PubMed] [Google Scholar]
- [69].Pallares I, Iglesias V, Ventura S. The rho termination factor of clostridium botulinum contains a prion-like domain with a highly amyloidogenic core. Front Microbiol. 2015;6:1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wheeler JR, Jain S, Khong A, et al. Isolation of yeast and mammalian stress granule cores. Methods. 2017;126:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Jain S, Wheeler JR, Walters RW, et al. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell. 2016;164(3):487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Gupta V, Bamezai RN. Human pyruvate kinase M2: a multifunctional protein. Protein Sci. 2010;19(11):2031–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005;44(27):9417–9429. [DOI] [PubMed] [Google Scholar]
- [74].Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–233. [DOI] [PubMed] [Google Scholar]
- [75].Mazurek S, Boschek CB, Hugo F, et al. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15(4):300–308. [DOI] [PubMed] [Google Scholar]
- [76].Israelsen WJ, Dayton TL, Davidson SM, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155(2):397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kwon I, Kato M, Xiang S, et al. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell. 2013;155(5):1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Guerrero-Mendiola C, Oria-Hernandez J, Ramirez-Silva L. Kinetics of the thermal inactivation and aggregate formation of rabbit muscle pyruvate kinase in the presence of trehalose. Arch Biochem Biophys. 2009;490(2):129–136. [DOI] [PubMed] [Google Scholar]
- [79].Dinkel H, Chica C, Via A, et al. Phospho.ELM: a database of phosphorylation sites–update 2011. Nucl Acids Res. 2011;39(Database issue):D261–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Weber SC. Sequence-encoded material properties dictate the structure and function of nuclear bodies. Curr Opin Cell Biol. 2017;46:62–71. [DOI] [PubMed] [Google Scholar]
- [81].Rabouille C, Alberti S. Cell adaptation upon stress: the emerging role of membrane-less compartments. Curr Opin Cell Biol. 2017;47:34–42. [DOI] [PubMed] [Google Scholar]
- [82].Murray DT, Kato M, Lin Y, et al. Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell. 2017;171(3):615–627.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Han TW, Kato M, Xie S, et al. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 2012;149(4):768–779. [DOI] [PubMed] [Google Scholar]
- [84].Arosio P, Meisl G, Andreasen M, et al. Preventing peptide and protein misbehavior. Proc Natl Acad Sci U S A. 2015;112(17):5267–5268. [DOI] [PMC free article] [PubMed] [Google Scholar]