

ABSTRACT
Vesicle trafficking regulates epithelial cell migration by remodeling matrix adhesions and delivering signaling molecules to the migrating leading edge. Membrane fusion, which is driven by soluble N-ethylmaleimide-sensitive factor associated receptor (SNARE) proteins, is an essential step of vesicle trafficking. Mammalian SNAREs represent a large group of proteins, but few have been implicated in the regulation of cell migration. Ykt6 is a unique SNARE existing in equilibrium between active membrane-bound and inactive cytoplasmic pools, and mediating vesicle trafficking between different intracellular compartments. The biological functions of this protein remain poorly understood. In the present study, we found that Ykt6 acts as a negative regulator of migration and invasion of human prostate epithelial cells. Furthermore, Ykt6 regulates the integrity of epithelial adherens and tight junctions. The observed anti-migratory activity of Ykt6 is mediated by a unique mechanism involving the expressional upregulation of microRNA 145, which selectively decreases the cellular level of Junctional Adhesion Molecule (JAM) A. This decreased JAM-A expression limits the activity of Rap1 and Rac1 small GTPases, thereby attenuating cell spreading and motility. The described novel functions of Ykt6 could be essential for the regulation of epithelial barriers, epithelial repair, and metastatic dissemination of cancer cells.
Keywords: Junctions, SNARE; motility
Introduction
Intracellular vesicle trafficking plays a vital role in the regulation of epithelial homeostasis. In differentiated epithelial layers, it mediates the establishment of apico-basal cell polarity and the assembly of adherens junctions (AJ) and tight junctions (TJ), thereby enabling the transport and barrier functions of various epithelia [1]. Furthermore, vesicle trafficking is known to regulate epithelial morphogenesis and tissue repair by controlling epithelial cell migration [2,3]. In migrating cells, vesicle trafficking regulates several events. The most studied event is the remodeling of integrin-based focal adhesions (FA), which mediates cell attachment to the extracellular matrix (ECM). In motile cells, FA assemble at the migrating leading edge and disassemble at the trailing end of the cell [4]. This process involves the endocytosis of molecular components of disassembling FA and their subsequent exocytosis to the leading edge, to be reused in nascent ECM adhesions. Inhibition of the exocytosis and recycling of FA components could profoundly disrupt ECM adhesion and interfere with cell motility [5–8]. Vesicle trafficking is also important for the delivery of signaling molecules, such as active Src kinase and Rac1 GTPase, to the migrating leading edge, where these molecules regulate actin filament remodeling and the formation of membrane protrusions [9,10]. That abnormal vesicle trafficking can contribute to the pathogenesis of different diseases has been well documented. For example, defects of epithelial vesicle trafficking machinery are associated with impaired embryonic morphogenesis and tissue repair [2,3]. On the other hand, overactivation of this process can be responsible for the accelerated motility of cancer cells, driving tumor metastasis [11]. Therefore, elucidating vesicle trafficking mechanisms that regulate cell motility is fundamentally important for understanding the basic biology of epithelial barriers and the pathophysiology of tissue injury and tumorigenesis.
Intracellular vesicle trafficking occurs via a multistep process involving vesicle formation, tethering, docking, and fusion with targeted membranes [12]. Every step of this process is controlled by specific protein machinery that determines correct membrane recognition and fusion. Membrane fusion is the final and rate-limiting step of vesicle trafficking, mediated by the SNARE (Soluble N-ethylmalemide-sensitive factor Associated Receptor) protein complex [13,14]. Mammalian cells express 38 different SNAREs that regulate vesicle fusion with various membrane compartments. Direct interactions of distinct SNARE proteins located on both the vesicle and the target membrane bring the two lipid bilayers into proximity and force their fusion. These interactions are mediated by special SNARE motifs that are capable of self-association, creating a stable helical core complex [13–15]. Such complex is usually formed by three or four SNARE proteins, contributing four structurally different SNARE motifs. Several previous studies addressed the involvement of SNARE proteins in controlling the migration and invasion of epithelial, endothelial, and cancer cells. Interestingly, the ECM adhesion and motility of these cells appears to be orchestrated by SNAREs located in different membrane compartments including plasma membrane resident syntaxins 3, 4, 13, and SNAP23 [9,16], endosomal vesicle-associated membrane proteins (VAMP) 3 and 7 [8,17], and Golgi/endosomal syntaxin 6 [18,19]. The described studies focused on just a subset of all mammalian SNAREs that primarily mediate the post-Golgi steps of vesicle trafficking. Much less is known about the biosynthetic trafficking of adhesion proteins through the endoplasmic reticulum (ER) and Golgi, or how ER-Golgi resident SNAREs could control the assembly of ECM adhesions and epithelial cell motility.
Ykt6 was originally described as a component of the ER-Golgi SNARE machinery with unusual biochemical properties and poorly-defined cellular functions [20,21]. Unlike the majority of SNAREs, it lacks the transmembrane domain, and interacts with membrane compartments via farnesyl and palmitoyl anchors [20,21]. A significant pool of Ykt6 has been shown to accumulate in the cytoplasm, in a self-folded inhibited conformation [21–24]. This suggests the existence of regulatory mechanisms that switch the Ykt6 molecule into an open conformation, leading to its activation and translocation to endomembranes. The precise intracellular localization and functions of Ykt6 remain controversial. In mammalian epithelial cells and keratinocytes, it is enriched in the Golgi and the ER-Golgi intermediate compartment, and participates in ER-Golgi and intra-Golgi trafficking [20,25–28]. In contrast, in neuronal cells, the majority of Ykt6 was found in peripheral vesicles, presumably associated with the endocytic, not secretory, pathway [22,29]. Consistent with this role, a siRNA screen in Drosophila cells identified Ykt6 as an essential regulator of parasite phagocytosis [30]. Yet in other studies, Ykt6 was shown to mediate the secretion of lysosome-derived exosomes [31] and regulate fusion of constitutive secretory carriers with the plasma membrane [32]. Since Ykt6 is likely to participate in various stages of intracellular vesicle trafficking, this SNARE protein may play essential roles in controlling membrane dynamics during cell adhesion and migration. However, the involvement of Ykt6 in the regulation of cell motility has not been previously addressed. The present study was designed to fill this knowledge gap and to elucidate the roles of Ykt6 in mediating the collective migration and invasion of epithelial cells. Our data elevates Ykt6 as an important negative regulator of cell motility that acts via controlling the expression of Junctional Adhesion Molecule (JAM)-A and activity of Rap1 and Rac1 small GTPases.
Methods
Antibodies and other reagents
The following primary polyclonal (pAb) and monoclonal (mAb) antibodies were used to detect trafficking, signaling, junctional, and cell-matrix adhesion proteins: anti-Ykt6 rat mAb [22,29]; anti-JAM-A mAb (gift from Dr. C.A. Parkos, University of Michigan); anti β1-integrin mAb and anti Ykt6 pAb (Novus Biologicals, Littleton, CO); E-cadherin, β-catenin, p120 catenin, afadin and total paxillin, mAbs (BD Biosciences, San Jose, CA); talin and vinculin mAbs (Sigma-Aldrich, St. Louis, MO); anti-phospho-paxilin, total FAK, phospho-FAK, c-Src, phospho-c-Src, GAPDH and β4-integrin pAbs (Cell Signaling, Danvers, MA); anti-ZO-1, cadherin-11 and EEA1 pAbs, and anti-Claudin-4 mAb (Life Technologies); anti α-catenin mAb, anti-JAM-A and Rab7 pAbs (Abcam, Boston, MA). Anti-Rap1 and Rac mAbs were from Cell Biolabs (San Diego, CA) and anti-cadherin-6 and P-cadherin mAbs were from Merck-Millipore (Billerica, MA). Anti-TGN46 pAb was from Bio-Rad Laboratories (Hercules, CA) and anti Giantin pAb was from BioLegend (San Diego, CA). Alexa Fluor-488-conjugated donkey-anti-rabbit and donkey-anti-goat secondary antibodies, Alexa Fluor-555-conjugated donkey-anti-mouse, and donkey-anti-sheep secondary antibodies, and Alexa Fluor-488 and Fluor-555-labeled phalloidin were obtained from Life Technologies. Horseradish peroxidase-conjugated goat-anti-rabbit and anti-mouse secondary antibodies were acquired from Bio-Rad Laboratories. EHT 1864 was purchased from Bio-Techne (Minneapolis, MI). CE3F4 and 8-pCTP-2-O-Me-cAMP-AM were acquired from Tocris Bioscience (Bristol, UK). All other chemicals were obtained from Sigma-Aldrich.
Cell culture
DU145 prostate epithelial cells (American Type Culture Collection) were grown in RPMI media (Invitrogen) supplemented with 10% FBS, 5 mM pyruvate, and antibiotics. M12 and p69 prostate epithelial cells (gifts from Dr. Zendra Zehner, Virginia Commonwealth University) were grown in RPMI supplemented with 5% FBS 5 mM pyruvate, 1X ITS supplement (Invitrogen), and antibiotics. Phoenix 293 cells were grown in high-glucose DMEM. Cells were grown in T75 flasks, and for immunolabeling, the cells were seeded on either collagen-coated permeable polycarbonate filters (0.4 µm pore size, Costar Cambridge, MA) or on collagen-coated coverslips. For biochemical studies, cells were cultured on 6-well plates.
RNA interference and micro-RNA targeting
Ykt6 expression was transiently downregulated using gene-specific Dharmacon On-Target−Plus small interfering (si) RNAs (Thermo-Fisher). Either siRNA SmartPool or individual duplexes with the following sequences were used: duplex (D) 1-CUAAAGUGCAGGCCGAACU, D2-AUACCAGAACCCACGAGAA, D3-CUAUAAAACUGCCCGGAAA, D4-GCUCAAAGCCGCAUACGAU. Noncoding siRNA duplex 2 was used as a control. Dharmacon siRNA SmartPools were used to downregulate the expression of JAM-A (M-005053–01) and Rap 1 (M-003623–02). E-cadherin expression was downregulated by using Dharmacon siRNAs: D1-GGAGAGCGGUGGUCAAAGA, D2-ACCAGAACCUCGAACUAUA, D3-GAGAACGCAUUGCCACAUA, D4-GCAGUACAUUCUACACGUA. Paxillin specific siRNAs with DNA target sequences: D1-CCTGTGATTTATGCCAATAAA (SI00044625) and D2-CTGCTGGAACTGAACGCTGTA (SI04713562), as well as β1-integrin siRNAs with target sequences: D1-TACGTATTCAGTGAATGGGAA (SI00034377) and D2-TACGGAGGAAGTAGAGGTTAT (SI00034384) were obtained from Qiagen (Hilden, Germany). microRNA (miR)-145 (IH300,613–06) hairpin inhibitor, as well as miRIDIAN miR-145 mimetic (C-300613–05) were purchased from Dharmacon. miR hairpin inhibitor negative control #1 (IN-001005–1) and miR mimetic negative control #1 (CN-0010000–01), both from Dharmacon, were used as appropriate controls.
Cells were seeded in 6-well plates at approximately 60% confluence and transfected with siRNA or micro-RNAs using DharmaFect 1 transfection reagent as previously described [7,33]. The final siRNA concentration for any single siRNA transfection was either 50 or 100 nM. Cotransfections, involving either two different siRNAs, or siRNA/microRNA pairs, were performed with the final concentration of each oligonucleotide at 50 nm. Cells were utilized for experiments on days 3 and 4 post-transfection.
Quantitative real-time RT-PCR
Total RNA was isolated using an RNeasy mini kit (QIAGEN, Valencia CA), followed by DNase treatment to remove genomic DNA. Total RNA (1 µg) was reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad Laboratories). MicroRNAs (miR) were isolated using a PureLink miRNA isolation kit (Life technologies) and treated with DNAase. miR (0.4 µg) was reverse transcribed using a qScript MicroRNA cDNA Synthesis Kit (Quanta BioSciences, Beverly, MA). Quantitative real-time RT-PCR was performed using iTaq Universal SYBR Green Supermix (Bio-Rad Laboratories) and a 7900HT Fast Real-time PCR System (Applied Biosystems; Foster City, CA). The following primers were used to amplify the microRNAs: hsa-miR-145–5p: 5'-CAGTTTTCCCAGGAATCCCTAA-3'; hsa-miR-146a-5p: 5'-TGAGAACTGAATTCCATGGGTTA-3'; hsa-miR-495–5p: 5'-CAAACAAACATGGTGCACTTC-3'; and universal reverse primer: 5'-GCATAGACCTGAATGGCGGGTA-3'. The results were normalized to levels of U6 snRNA (forward primer: 5'-CTCGCTTCGGCAGCACA-3'; reverse primer: 5'-AACGCTTCACGAATTTGCGT-3'). The following primers were used for JAM-A: forward: 5'-TCGAGAGGAAACTGTTGTCC-3'; and reverse: 5'-ACCAGTTGGGAAGAAGGTA-3'. JAM-A expression levels were normalized to the house-keeping gene GAPDH (forward: 5'-CATGTTTGTGATGGGTGTGAACCA-3'; reverse: 5'-AGTGATGGCATGGACTGTGGTCAT-3'. The threshold cycle number (Ct) for specific genes of interest and the housekeeping gene was determined based on the amplification curve representing a plot of the fluorescent signal intensity versus the cycle number. The relative expression of each gene was calculated with a comparative Ct method that is based on the inverse proportionality between Ct and the initial template concentration (2-ΔΔCt), as previously described [34]. This method is based on the two-step calculation of ΔCt = Ct target gene – Ct GAPDH and ΔΔCt = ΔCte-ΔCtc where index e refers to the sample from any control or Ykt6 siRNA or corresponding miR treated cells, and index c refers to a sample from a control siRNA or control miR-treated cells assigned as an internal control.
Generation of stable cell lines overexpressing human ykt6
DU145 cell lines with stable overexpression of human Ykt6 were generated using a Gateway compatible N-TAP retroviral expression system. The initial full-length clone of human Ykt6 was obtained from the PlasmID Repository service of the DNA Resource Core (Harvard Medical School, Boston, MA) and was verified with sequencing. Afterwards, the Ykt6 sequence was transferred into an pMSCV-NTAP retroviral expression vector using a Gateway cloning system. For retroviral production, Phoenix cells were transfected with blend of 2 µg of N-TAP-Ykt6 or control retroviral expression plasmid, 0.1 µg of pENV, and 0.1 µg of pGagPol in a 0.25 ml transfection mix containing 25 μl of Trans-IT 2020 (MirusBio), according to the manufacturer’s instructions. Viral supernatants were collected 36 h and 48 h after transfection. The retroviral supernatant was used to infect 1 × 106 DU145 cells in the presence of 4 μg/ml polybrene. Infected cells were selected with 5 μg/ml puromycin.
Rap1 and rac1 activation assays
The activity of Rap1 and Rac1 small GTPases was determined by using activation assay kits (Cell Biolabs, San Diego, CA) according to manufacturer instructions.
Immunoblotting analysis
Total cell lysates were obtained by homogenizing the cells with RIPA cell lysis buffer (Tris 20 mM, NaCl 150 mM, EDTA 2 mM, EGTA 2 mM, Na deoxycholate 1%, SDS 0.1%, Triton 1%), supplemented with phosphatase inhibitor cocktails 2 & 3 (1:200) and protease inhibitor cocktail (1:100) (Sigma-Aldrich). The homogenized samples were cleared by centrifugation, mixed with an equal volume of 2X SDS sample buffer and boiled. The total cell lysates were separated by SDS-Polyacrylamide gel electrophoresis with 10–20 µg of protein loaded into each well. The separated proteins were then transferred by standard electroblotting technique onto either nitrocellulose or polyvinylidene difluoride membranes. After transfer, the membranes were incubated with primary and HRP-conjugated secondary antibodies, and the proteins were visualized using standard enhanced chemiluminescence reagents and x-ray film. Protein expression was quantified by densitometry (Epson Perfection V500 photo scanner and ImageJ 1.47v software [National Institute of Health, Bethesda, MD]) of three immunoblot images, each representing an independent experiment. Data is presented as normalized values assuming the expression levels in control siRNA-treated groups as 1. Statistical analysis was performed with row densitometric data using Microsoft Excel.
Immunofluorescence labeling and confocal microscopy
Control and Ykt6-depleted epithelial cells cultured on collagen-coated coverslips were fixed in 100% methanol for 20 minutes at −20°C. Fixed cells were immunolabeled with antibodies specifically recognizing junctional proteins and Golgi markers as described previously [33,35]. Immunolabeled cells were examined using a Zeiss LSM 700 Laser Scanning Microscope (Zeiss Microimaging Inc.; Thornwood; NY). The Alexa Fluor-488 and Alexa Fluor-555 signals were imaged sequentially in frame-interlace mode to eliminate cross talk between channels. Image analysis was conducted using imaging ZEN 2011 (Carl Zeiss Microscopy Inc.; Thornwood; NY) and Adobe Photoshop software. Colocalization analysis was performed using the Coloc-2 module of Fuji Image J software. Images shown are representative of three independent experiments with multiple images taken per slide.
Scratch wound assay
Epithelial cells were plated in 6-well plates and grown for 3 days until confluency. A pipette tip was used to make a thin scratch wound in the cell monolayer. The plates were then washed with a complete medium and marked on the bottom by drawing lines across the newly-formed wounds. The images of wound monolayers were acquired at 0 and 8 hours after wounding, using an inverted bright field microscope equipped with a camera. The width of the wounded area along the established marks was measured using ImageJ software [36].
Matrigel invasion assay
Epithelial cells were detached using TrypLETM Express reagent (Thermo-Fisher), re-suspended in 1% FBS containing RPMI medium, and counted. Approximately 3 × 107 cells were loaded into the upper chamber of BioCoat Matrigel coated membrane-inserts (BD Biosciences). The upper chamber was filled with serum-free RPMI medium, while the lower chamber was filled with RPMI containing 10% FBS as a chemoattractant. Cells were allowed for overnight invasion through Matrigel at 37°C. Thereafter, plates were washed with Hank’s Balanced Salt Solution (HBSS) and fixed with methanol for 10 minutes. Fixed cells were stained using Eosin Y (10 minutes) followed by Azure A/Azure B (10 minutes). Non-invaded cells were removed from the upper surface of Matrigel by wiping with a cotton bud. The invaded labeled cells were counted under a bright field microscope.
Cell spreading assay
On day 4 post-transfection, adherent cells were released from plates using TrypLETM Express reagent and counted. Next, 105 cells were seeded in collagen-coated 24-well plates. Cells were allowed to attach and spread over a collagen I-coated surface for 1 h and attached cells were examined under an inverted bright field microscope equipped with a camera. The surface area of spread cells was measured using the ImageJ program. In each experiment, 50 randomly selected cells were measured for every experimental group. The results shown are representative of two independent experiments.
Cell proliferation assay
Control and Ykt6 transfected cells in exponential growth were harvested and plated at a density of 3 × 104 cells per well in 24-well plates. On day 2 and 3 post-plating, both floating and attached cells were harvested by trypsinization and collected by centrifugation. Cell pellets were resuspended in fresh media, and the total cells number for each well were counted using a hemocytometer. The results shown are representative of two independent experiments.
Statistics
All data are expressed as means ± standard error (se) from three independent experiments. Statistical analysis was performed by using a one-way ANOVA to compare obtained numerical values in the control and two experimental groups (knockdown with two different Ykt6 siRNAs). If the ANOVA test showed significant differences, a post-hoc t-test was used to compare the difference between the control and each Ykt6-depleted group. P values < 0.05 were considered statistically significant.
Data availability
Materials, data and associated protocols will be available upon request from the corresponding author.
Results
Downregulation of ykt6 expression promoted cell migration and impaired the architecture of epithelial junctions
The roles of Ykt6 in the regulation of epithelial cell motility were investigated using a DU145 prostate epithelial cell line. DU145 are well-differentiated epithelial cells capable of individual and collective migration and it is a commonly used model to study different modes of epithelial cell motility. Expression of Ykt6 was transiently downregulated in DU145 cells by using either siRNA SmartPool or four individual siRNA duplexes. All siRNA duplexes and the SmartPool resulted in a very efficient (more than 80%) Ykt6 knockdown on day 4 post-transfection (Figure 1a). Two individual Ykt6 siRNA duplexes (D1 and D3) were selected for the subsequent experiments, though pilot experiments demonstrated similar effects for all four duplexes and the siRNA SmartPool on DU145 cell migration (data not shown). Different approaches were used to investigate the roles of Ykt6 in epithelial cell motility. One approach is a scratch wound assay that examines collective migration of epithelial sheets, and the other is a Matrigel invasion assay that involves the migration of individual epithelial cells or their small clusters through a 3-D matrix. Downregulation of Ykt6 expression significantly accelerated both the collective migration and Matrigel invasion of DU145 cells (Figure 1(b-e)). This effect was especially robust in cell invasion, where the loss of Ykt6 caused more than two-fold increase in cell motility (Figure 1(e)). Additionally, Ykt6 knockdown significantly promoted wound closure in p69 and M17 prostate epithelial cell monolayers (Suppl. Fig. 1), thereby indicating that the observed effects are not peculiar responses of DU145 cells. Together, these results suggest a novel anti-migratory role of Ykt6 in prostate epithelial cells.
Figure 1.
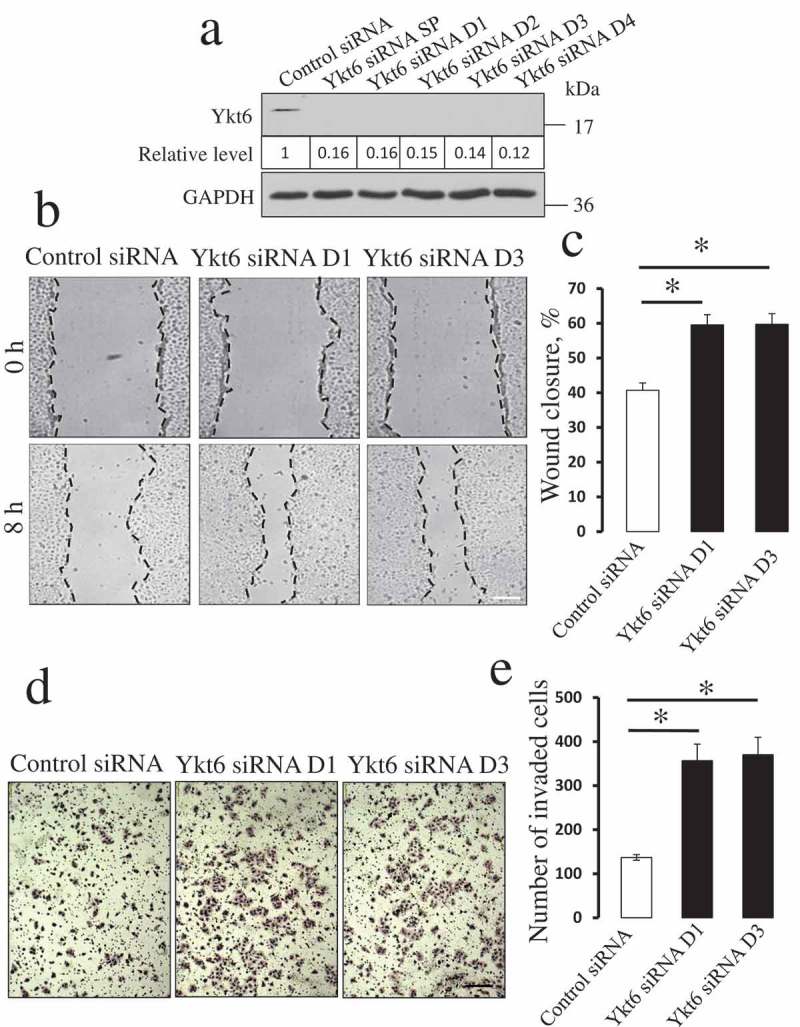
Depletion of Ykt6 enhances planar migration and matrix invasion of prostate epithelial cells.
DU145 prostate epithelial cells were transfected with either control siRNA, Ykt6-specific siRNA SmartPool (SP), or individual Ykt6 siRNA duplexes (D1-D4). (a) Immunoblotting analysis displays the efficiency of Ykt6 knockdown on day 4 post-transfection. Numbers indicate the average values for the Ykt6 relative expression calculated from 3 independent experiments. (b) Representative images of wound healing in DU145 cell monolayers transfected with either control, or two Ykt6-specific, siRNA duplexes (D1 and D3). (c) Quantitation of wound closure during 8 h of cell migration. (d,e) Representative images of Matrigel-invaded control and Ykt6-depleted epithelial cells and a quantitative analysis showing the numbers of cells invaded through Matrigel. Data are presented as mean ± SE (n = 3); *P < 0.01 as compared to the control siRNA-treated group. Scale bars, 200 µm.
Since the migration of well-differentiated epithelial cells is known to be regulated by the assembly of intercellular junctions [37], we next investigated the effect of Ykt6 depletion on the integrity of two major junctional complexes, namely adherens junctions (AJ) and tight junctions (TJ). AJ integrity was evaluated with the immunolabeling of E-cadherin and P-cadherin, whereas TJ organization was determined by immunolabeling of junctional adhesion molecule (JAM)-A, and ‘zonula occludens’ (ZO)-1. Confocal microscopy demonstrated a characteristic ‘chicken wire’ labeling pattern of the selected AJ and TJ proteins in control siRNA-transfected DU145 cells (Figure 2, arrows). Interestingly, Ykt6 knockdown induced multiple changes in the localization of junctional proteins including a dramatic loss of E-cadherin labeling intensity, the accumulation of P-cadherin into cytosolic vesicles, and the disruption of continuous JAM-A and ZO-1 labeling at areas of cell-cell contact (Figure 2, arrowheads and asterisks). We also sought to understand whether the observed intracellular accumulation of P-cadherin was caused by the defective exocytosis, or the accelerated endocytosis, of this AJ protein in Ykt6-depleted cells. Dual immunofluorescence labeling of P-cadherin with exocytosis/Golgi marker, TGN46, or endosomal markers early endosomal antigen (EEA)-1 and Rab7, with subsequent confocal microscopy, were performed to distinguish between these two mechanisms. Intracellular P-cadherin markedly colocalized with TGN46 (Suppl. Fig. 2, arrows), based on the calculated Pearson’s coefficient of 0.7 ± 0.1 (n = 10) for the correlation between red and green signals. By contrast, little or no colocalization of P-cadherin with EEA1 and Rab7 was detected (Suppl. Fig 2), which demonstrates selective retention of P-cadherin within the exocytic pathway. Together, our data indicate that Ykt6 depletion impairs the assembly of epithelial junctions by either causing mislocalization or by decreasing the expression of major AJ and TJ proteins.
Figure 2.
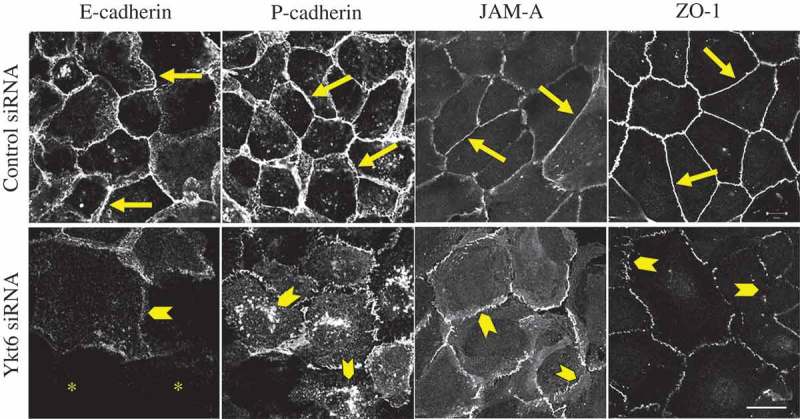
Loss of Ykt6 disrupts the integrity of epithelial apical junctions. DU145 cells were transfected with either control siRNA or Ykt6-specific siRNA D1 and, on day 4 post-transfection, were fixed and immunolabeled for different AJ (E-cadherin, P-cadherin) and TJ (JAM-A, ZO-1) proteins. Arrows indicate normal AJ and TJ structure in control cells. Arrowheads show AJ and TJ disassembly, as well as intracellular accumulation of P-cadherin in Ykt6-depleted cells. Asterisks mark cells with very low levels of E-cadherin labeling. Scale bar, 20 µm.
Increased motility of ykt6-depleted epithelial cells depended on the selective upregulation of JAM-A expression
To gain insight into the mechanisms mediating the accelerated motility and apical junction disassembly caused by Ykt6 knockdown, we examined the expression of different ECM adhesion and junctional proteins. Immunoblotting analysis revealed altered expression of a number of adhesion molecules in Ykt6-depleted DU145 cells. These changes included an up to 49% reduction of E-cadherin expression, a 24–52% decrease in claudin-4 protein level, 56–61% decrease in paxillin phosphorylation, and the increased intensity of the lower band of the β1-integrin doublet (Figure 3(a,b)), which indicates accumulation of the non-glycosylated precursor of this protein [7]. However, one of the most remarkable effects of Ykt6 knockdown was a dramatic upregulation of JAM-A expression. Indeed, JAM-A protein level was 3–4 fold higher in DU145 cells transfected with anti-Ykt6 siRNAs as compared to control siRNA-transfected cells (Figure 3(b)). Interestingly such JAM-A upregulation was evident at the protein, but not at the mRNA level (Figure 3(c)).
Figure 3.
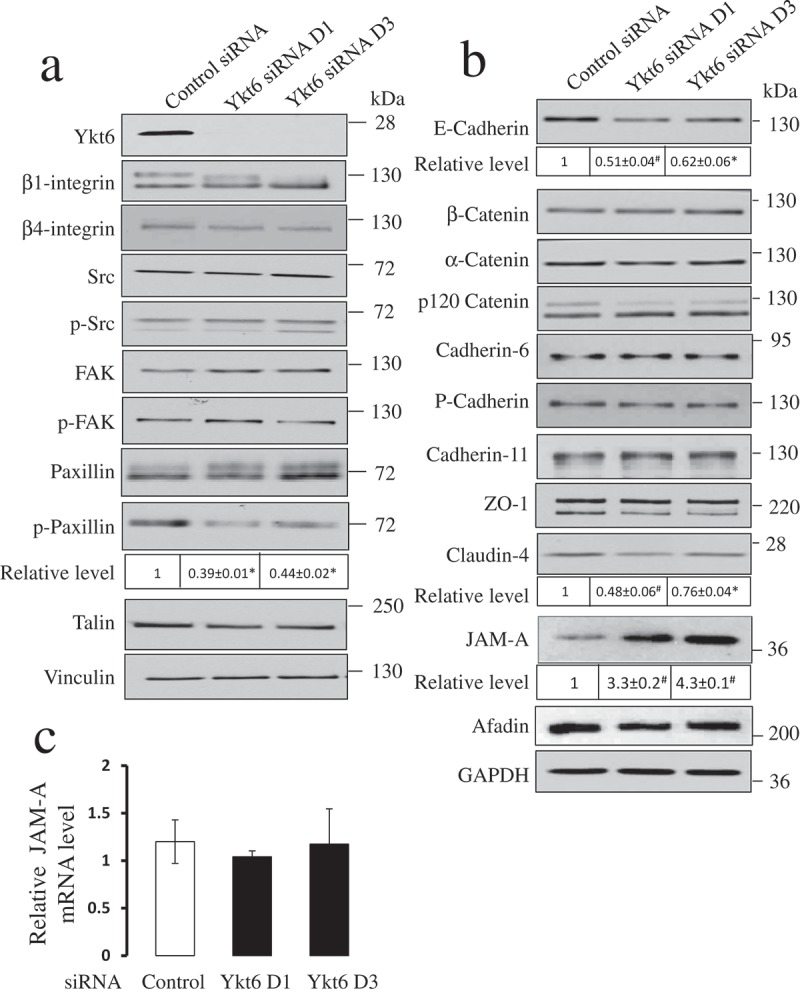
Depletion of Ykt6 has differential effects on the expression of junctional and focal adhesion proteins.
Immunoblotting analysis shows the expression and phosphorylation of major focal adhesion proteins (a), as well as the expression of different AJ and TJ proteins (b) in control and Ykt6-depleted DU145 cells, on day 4 post-transfection. Numbers indicate the average values for the relative expression of select proteins calculated from 3 independent experiments. (c) Quantitative RT-PCR analysis of JAM-A mRNA level in control and Ykt6-depleted DU145 cells on day 4 post-transfection. Data are presented as mean ± SE (n = 3); *P < 0.01; # P < 0.001 as compared to the control siRNA-treated group.
To ensure that the observed effects of Ykt6 knockdown on epithelial cell motility and adhesion protein expression are not related to possible off-target effects of siRNA transfection, we performed rescue experiments. These experiments involved the creation of a DU145 cell line with stable overexpression of a HA-tagged human Ykt6 (Suppl. Fig. 3a). Ykt6 overexpression itself did not affect epithelial wound closure (Suppl. Fig. 3b,c) or Matrigel invasion (Suppl. Fig. 3d,e), but it increased a steady-state level of E-cadherin protein (Suppl. Fig. 3a). Furthermore, Ykt6-overexpression prevented the increased motility of DU145 cells and JAM-A upregulation caused by Ykt6-specific siRNAs (Suppl. Fig. 3). The results of these rescue experiments strongly suggest that loss of Ykt6 specifically stimulates expression of JAM-A protein and enhances different modes of epithelial cell migration.
JAM-A is a known accelerator of epithelial cell motility [38–41]. Since upregulation of this protein appears to be the most pronounced molecular event that positively correlates with increased cell motility following Ykt6 knockdown, we sought to determine if such JAM-A upregulation plays a causal role in driving the migration of Ykt6-depleted cells. Knockdown of JAM-A alone caused a modest but significant inhibition of wound closure, but did not attenuate Matrigel invasion of control DU145 cells (Figure 4). In contrast, co-knockdown of JAM-A and Ykt6 completely reversed the increased motility of Ykt6-depleted cells. Indeed JAM-A/Ykt6-co-depleted cells closed wounds at significantly slower rate as compared to control DU145 cells, whereas their invasion was not significantly different from the control group (Figure 4). To ensure that the observed JAM-A dependence is a unique mechanism driving the migration of Ykt6-deficient cells, we performed several control experiments. First, we evaluated the possible contribution of altered cell proliferation, but observed no effects of Ykt6-depletion on this process (Suppl. Fig. 4). Next, we examined whether decreased E-cadherin expression could be responsible for the accelerated motility of Ykt6-depleted cells by weakening their intercellular adhesions. E-cadherin level was downregulated in DU145 cells by four different siRNA duplexes. Interestingly, such downregulation did not affect epithelial wound healing (Suppl. Fig. 5). Finally, we investigated if the observed misprocessing of β1-integrin or the dephosphorylation of paxillin could promote cell motility by altering ECM adhesions. However, knockdown of either β1-integrin, or paxillin, with two different siRNA duplexes for each protein target, had no effect on the wound healing ability of DU145 cells (Suppl. Fig. 6). Together, our results highlight the upregulation of JAM-A expression as a unique mechanism that accelerates the collective migration and Matrigel invasion of Ykt6-depleted epithelial cells.
Figure 4.
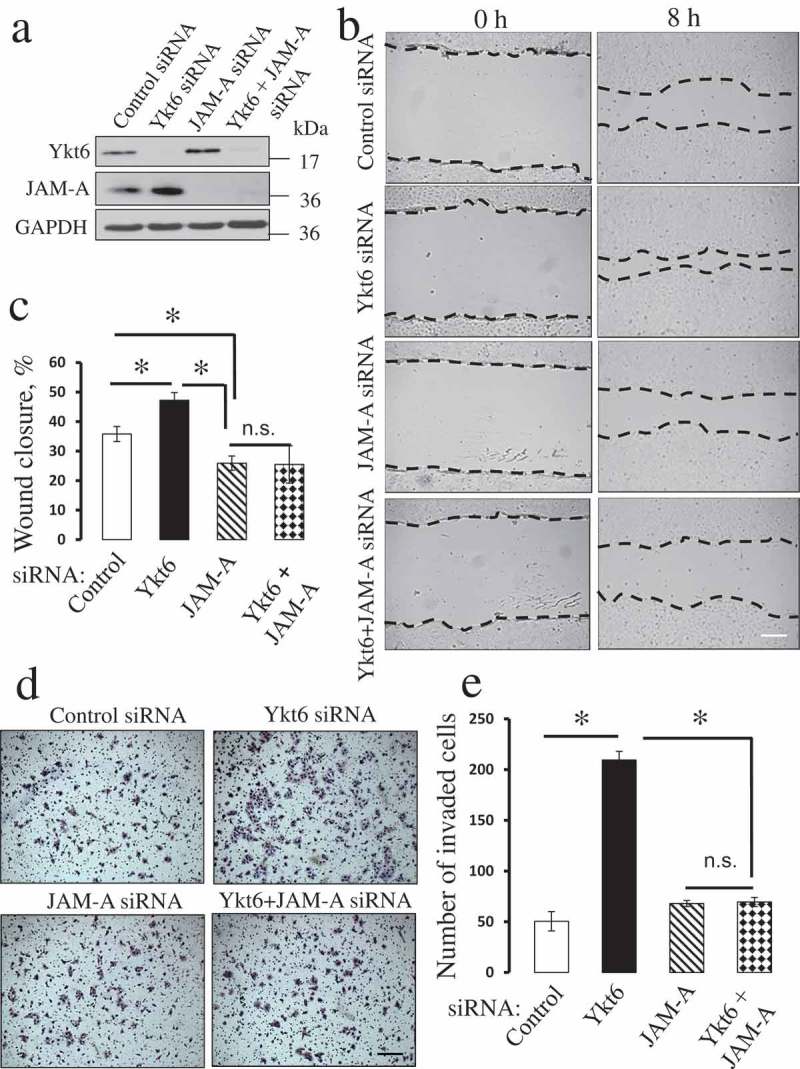
Downregulation of JAM-A expression reverses the accelerated migration of Ykt6-depleted epithelial cells.
DU145 cells were co-transfected with one of the following siRNA pairs: control-control, control-Ykt6 D1, control-JAM-A, and Ykt6 D1-JAM-A. (a) Expression of Ykt6 and JAM-A in total cell lysates was determined on day 4 post-transfection. Cell motility was examined by using wound healing (b,c) and Matrigel invasion (d,e) assays. Data are presented as mean ± SE (n = 3); *P < 0.01. Scale bars, 200 µm.
Figure 5.
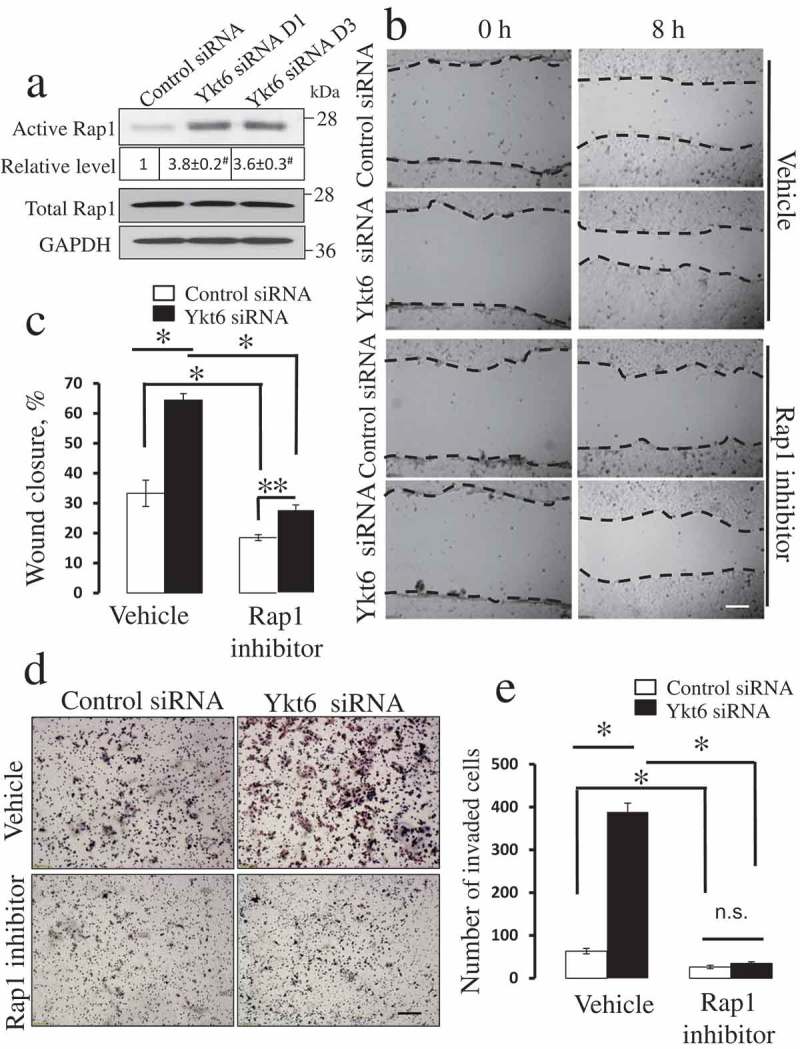
Pharmacological inhibition of Rap1 GTPase attenuates the accelerated motility of Ykt6-depleted epithelial cells.
DU145 cells were transfected with either control or Ykt6 siRNAs (D1 and D3 for the Rap1 activation assay and D1 for the functional studies). (a) The amount of active and total Rap1 was determined in total cell lysates on day 4 post-transfection. Numbers indicate the average values for the active Rap1 relative expression calculated from 3 independent experiments. The effects of pharmacological Rap1 inhibitor (CE3F4, 50 µM) on cell motility were determined using wound closure (b,c) and Matrigel invasion (d,e) assays. Data are presented as mean ± SE (n = 3); **P ≤ 0.05; *P ≤ 0.01; # P ≤ 0.001. Scale bars, 200 µm.
Figure 6.
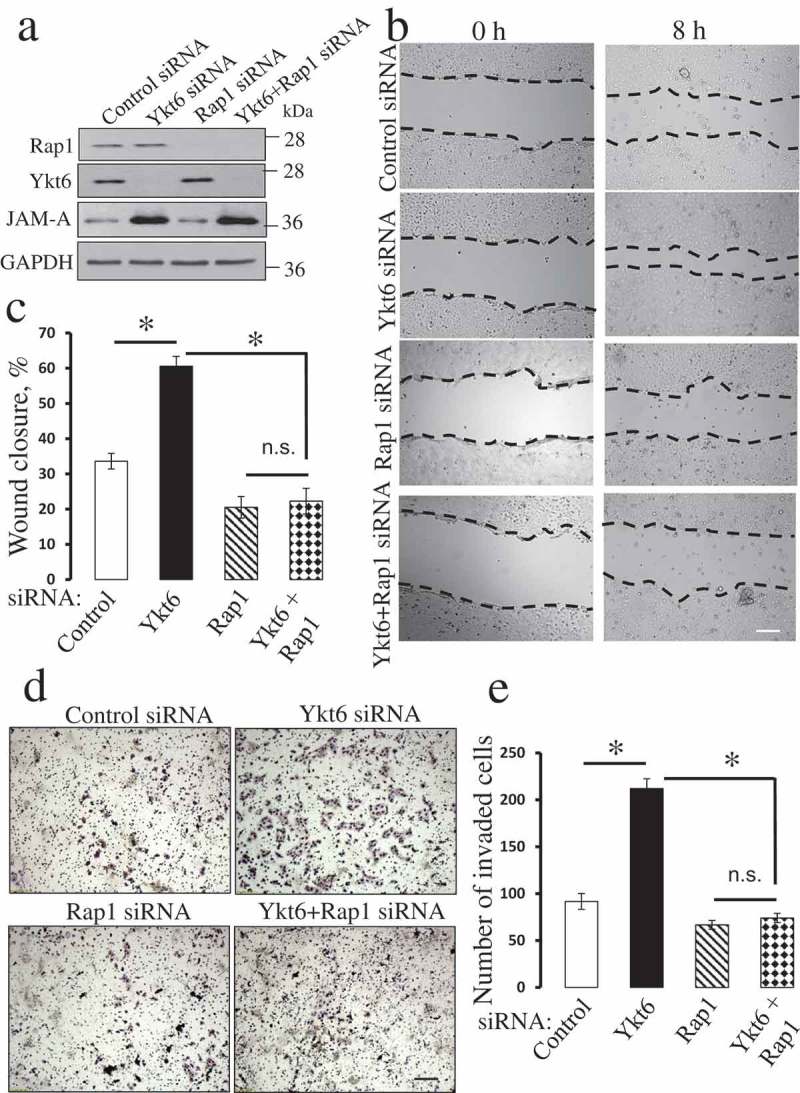
Downregulation of Rap1 expression reverses the accelerated migration of Ykt6-depleted epithelial cells.
DU145 cells were co-transfected with one of the following siRNA pairs: control-control, control-Ykt6 D1, control-Rap1, and Ykt6 D1-Rap1. (a) The expression of Ykt6, Rap1 and JAM-A in total cell lysates was determined on day 4 post-transfection. Cell motility was examined using wound healing (b,c) and Matrigel invasion (d,e) assays. Data are presented as mean ± SE (n = 3); *P < 0.01. Scale bars, 200 µm.
Ykt6 knockdown accelerated epithelial cell motility by activating rap1 and rac1 small GTPases
The next series of experiments was designed to elucidate the down-stream signaling events by which upregulated JAM-A could drive the motility of Ykt6-depleted epithelial cells. Since activation of Rap1 small GTPase was found to be a crucial mediator of the promigratory activity of JAM-A [38,40,41], we sought to investigate if this mechanism also plays a role in our experimental system. A biochemical pull-down assay demonstrated more than 3 fold increase in the amount of active Rap1 in DU145 cells transfected with Ykt6 siRNA D1 and D3, respectively, as compared to control cells (Figure 5(a)). Consistently, pharmacological activation of Rap1 with 8-pCTP-2-O-Me-cAMP-AM promoted wound healing in control DU145 cell monolayers (data not shown). Exposure of either control or Ykt-6 depleted DU145 cells to a specific pharmacological Rap1 inhibitor, CE3F4 (50 µM) [42], significantly attenuated their wound healing (Figure 5(b,c)) and Matrigel invasion (Figure 5(d,e)). Importantly, the Rap1 inhibitor significantly decreased the magnitude of accelerated wound healing in Ykt6-depleted cells as compared to the vehicle-treated cells (from ~ 94% to 50%) and completely blocked the increased cell invasion caused by Ykt6 knockdown (Figure 5). Consistently with these pharmacological inhibition data, a simultaneous siRNA-mediated knockdown of Ykt6 and Rap1 in DU154 cells also reversed the accelerated wound closure (Figure 6(a-c)) and Matrigel invasion (Figure 6(d,e)) of Ykt6-deficient cells. siRNA-mediated knockdown of Rap1 did not prevent the upregulation of JAM-A expression following Ykt6 depletion (Figure 6(a)), which places Rap1 activation downstream of JAM-A upregulation under these experimental conditions.
How does Rap1 activation promote the motility of Ykt6-depleted DU145 cells? Previous studies suggested that Rap1 controls epithelial cell migration by regulating either β1-integrin expression [38,40,41] or Rac1-depended cell spreading [43,44]. Since β1-integrin expression was not increased in Ykt6-depleted DU145 cells (Figure 3(a)), we disregarded this mechanism and focused on the involvement of Rac1 GTPase. Figure 7(a) shows that cell transfection with anti-Ykt6 siRNA in a 2–4 fold increase in Rac1 activity as compared to the control group. This Rac1 activation was accompanied by the increased spreading of Ykt6-depleted cells on the collagen I substrate (Suppl. Fig. 7). To demonstrate a causal link between Rac1 activation and the increased motility of Ykt6 depleted cells, we used a selective Rac inhibitor, EHT 1864, which displaces bound nucleotides and locks Rac GTPases in an inactive state [45]. Treatment with EHT 1864 (50 µM) reversed the accelerated wound healing (Figure 7(b,c)) and Matrigel invasion (Figure 7(d,e)) caused by Ykt6 depletion. Together, these data suggest that increased JAM-A expression in Ykt6-depleted epithelial cells promotes cell motility by activating Rap1 and Rac1.
Figure 7.
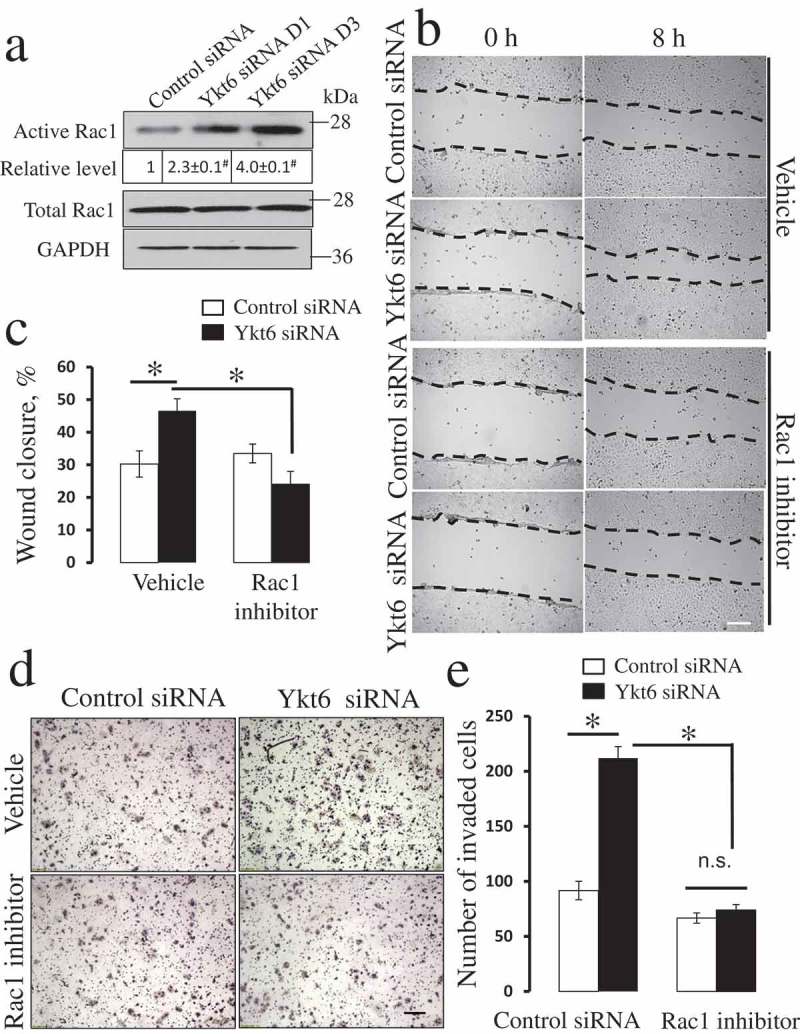
Pharmacological inhibition of Rac1 GTPase attenuates the accelerated motility of Ykt6-depleted epithelial cells.
DU145 cells were transfected with either control or Ykt6 siRNAs (D1 and D3 for the Rac1 activation assay and D1 for the functional studies). (a) The amount of active and total Rac1 was determined in total cell lysates on day 4 post-transfection. Numbers indicate the average values for the active Rac1 relative expression calculated from 3 independent experiments. The effects of a pharmacological Rac1 inhibitor (EHT1864, 50 µM) on cell motility were examined using wound closure (b,c) and Matrigel invasion (d,e) assays. Data are presented as mean ± SE (n = 3); *P < 0.01; # P < 0.001. Scale bars, 200 µm.
Ykt6 knockdown increased the level of JAM-A protein by inhibiting mir-145 expression
A key question that needed to be addressed relates to the mechanism by which Ykt6 knockdown increases JAM-A levels. Since the expression of JAM-A mRNA was not affected by Ykt6 depletion, such upregulation must occur post-transcriptionally. Several lines of evidence suggest the involvement of microRNAs in this process. First, microRNAs are known to regulate the expression of various proteins by controlling the translation of their mRNAs [46,47]. Second, previous studies documented the important role of several microRNAs in the regulation of JAM-A expression [48–51]. Finally, the processing and activity of different microRNAs depends on their association with the ER, Golgi, and endosomal membranes, and it could be impaired if such membrane association is perturbed [52,53]. We reasoned that Ykt6 might disrupt ER and Golgi-dependent vesicle trafficking, thereby impairing microRNA homeostasis. To test this hypothesis, we first examined the effect of Ykt6 knockdown on ER-Golgi vesicle trafficking by analyzing the Golgi integrity. Control DU145 cells do not form a compact Golgi complex, instead appearing as a cluster of perinuclear vesicles containing corresponding protein markers, such as Giantin and TGN46 (Suppl. Fig. 8, arrows). However, Ykt6 depletion markedly disrupted the localization of Golgi markers in DU145 cells, resulting in diffuse Giantin labeling and the scattering/aggregation of TGN46-containing vesicles (Suppl. Fig. 8, arrowheads). In HeLa cells, normally possessing a more compact Golgi complex, its integrity was also disrupted by Ykt6 depletion, as manifested by the fragmentation and vesiculation of perinuclear Giantin and TGN46-containing tubules (Suppl. Fig. 9). It is noteworthy that the observed defect of Golgi-dependent glycosylation of β1-integrin (Figure 3(a)) also suggests abnormal ER-Golgi or intra-Golgi vesicular transport in Ykt6-depleted cells.
Figure 8.
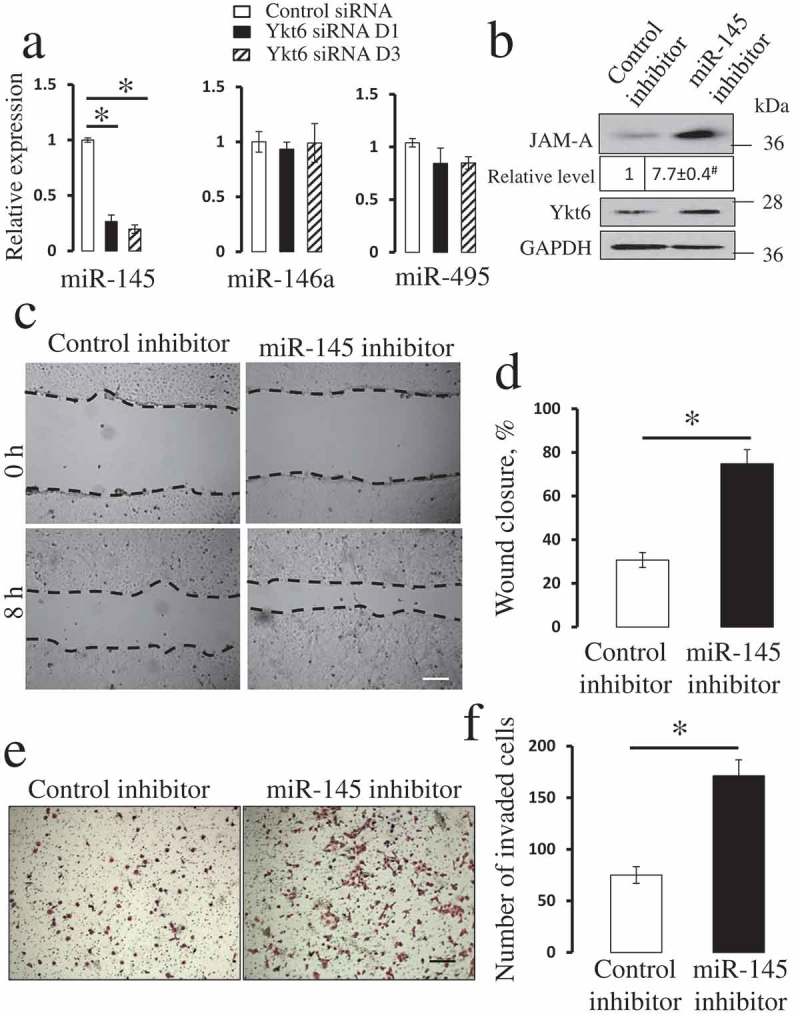
Inhibition of miR-145 phenocopies the effects of Ykt6 depletion on JAM-A expression and epithelial cell motility.
(a) The expression of miR-145, miR-146a, and miR-495 was determined in control and Ykt6-depleted DU145 cells by quantitative RT-PCR. (b-e) DU145 cells were transfected with either control miR inhibitor or with a specific miR-145 inhibitor. The effects of miR-145 inhibitor on the expression of JAM-A and Ykt6, as well as on epithelial cell motility, were determined by immunoblotting analysis (b), wound healing (c,d), and Matrigel invasion (e,f) assays. Data are presented as mean ± SE (n = 3); *P < 0.01; # P < 0.001 as compared to the control groups. Scale bars, 200 µm.
Figure 9.
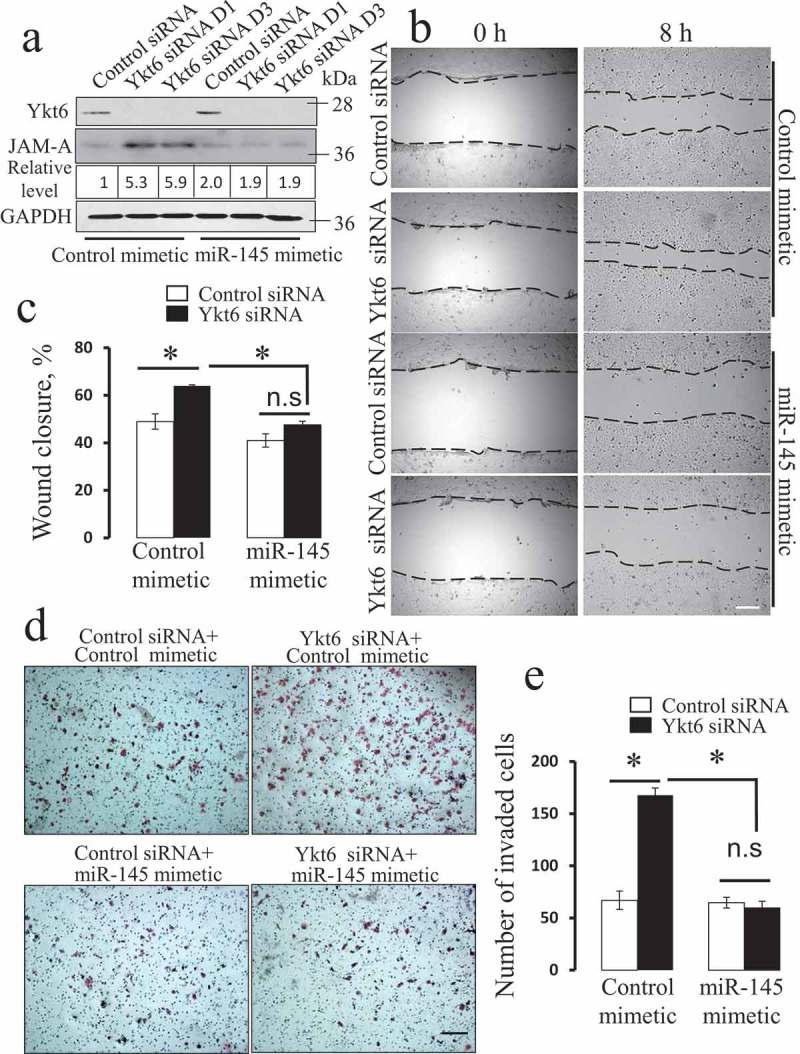
MiR-145 mimetic reverses the effect of Ykt6 depletion on JAM-A expression and the motility of prostate epithelial cells.
DU145 cells were transfected with either control siRNA, or Ykt6 siRNA D1, and co-transfected with either miR-145 mimetic or its control. (a) The expression of Ykt6 and JAM-A was determined by immunoblotting on day 4 post-transfection. Numbers indicate the average values for the JAM-A relative expression calculated from 3 independent experiments. Cell motility was examined using wound healing (b,c) and Matrigel invasion (d,e) assays. Data are presented as mean ± SE (n = 3); *P < 0.01. Scale bars, 200 µm.
Next, we used quantitative RT-PCR analysis to examine the cellular levels of three microRNAs previously implicated in the regulation of JAM-A expression [48–51]. Figure 8(a) demonstrates that loss of Ykt6 markedly decreased the expression of miR-145, without having significant effect on miR-146a and miR-495 levels. Since miR-145 is known to inhibit JAM-A expression [48,50,51], loss of this microRNA could be responsible for the increased JAM-A level in Ykt6-depleted cells. To test this hypothesis, we examined the effects of miR-145 inhibition on control DU145 cells. Cells transfected with a miR-145 inhibitor showed more than 7 fold higher level of JAM-A protein as compared to control miR-transfected cells (Figure 8(b)). Furthermore, miR-145 inhibition promoted both wound healing and Matrigel invasiveness of DU145 cells, thereby phenocopying the effects of Ykt6 knockdown (Figure 8(c-f)). Consistently, transfection of DU145 cells with miR-145 mimetic prevented the upregulation of JAM-A expression (Figure 9(a)) and reversed the increase in epithelial wound healing and Matrigel invasion caused by Ykt6 depletion (Figure 9(b-e)). Collectively, these results strongly suggest that loss of Ykt6 stimulates JAM-A expression and promotes prostate epithelial cell motility by decreasing the expression of miR-145.
Discussion
Ykt6 is a unique SNARE protein implicated in the regulation of vesicle trafficking between the ER, the Golgi, endosomes, and the plasma membrane [20,21,26,28,31,54]. While recent biochemical and biophysical studies revealed the molecular details of the structure and the molecular plasticity of Ykt6 [24], little is known about this protein’s roles in regulating cellular homeostasis. Our study reveals two novel functions of Ykt6 in human prostate epithelial cells: maintenance of the integrity of apical junctions and inhibition of cell motility (Suppl. Fig. 10). The first function can be directly linked to the membrane fusion activity of Ykt6, which mediates the trafficking of different AJ and TJ proteins to the plasma membrane. This notion is supported by the observed phenotype of Ykt6-depleted cells characterized either by the mislocalization of junctional components into trans-Golgi network-derived vesicles or by the decreased expression of some AJ and TJ proteins, which may also result from their abnormal trafficking and degradation (Figures 2 & 3; Suppl. Fig. 2). These effects of Ykt6 knockdown on apical junctions recapitulate the epithelial AJ/TJ disassembly caused by the loss of other SNARE proteins and regulators [33,55–57].
Unexpectedly, Ykt6 knockdown had a gain-of-function effect on epithelial cell motility, as demonstrated both by increased collective migration during wound healing and accelerated individual cell invasion into Matrigel (Figure 1). These results indicate that Ykt6 suppresses epithelial cell migration: an unusual activity among vesicle trafficking proteins, the majority of which are positive regulators of cell motility [2,11]. Intriguingly, our data suggest that Ykt6 inhibits epithelial cell migration via a specific mechanism involving downregulation of JAM-A expression (Figures 3 & 4). JAM-A is an immunoglobulin-like transmembrane protein that engages in multiple homotypic and heterotypical interactions on the cell surface and in the cellular cortex [58,59]. In confluent epithelial cell monolayers, JAM-A is a component of TJs, playing an essential role in the control of barrier permeability [58,59]. However, in migrating cells, JAM-A acts a potent modulator of cell motility. Thus, a number of previous studies have demonstrated that JAM-A accelerates the migration of intestinal epithelial [40,41], breast cancer [38,39,50], and endothelial cells [60,61]. Consistent with this data, we found that the increased migration and invasion of Ykt-6-depleted DU145 cells was accompanied by a dramatic upregulation of JAM-A expression (Figure 3). More importantly, the accelerated motility of epithelial cells caused by Ykt6 depletion was completely suppressed by JAM-A knockdown (Figure 4).
Our data suggest that the increase in JAM-A expression following Ykt6 knockdown drives epithelial cell motility by stimulating Rap1 small GTPase. Indeed, Rap1 was markedly activated in Ykt6-depleted cells, and either pharmacological inhibition or siRNA-mediated knockdown of this GTPase reversed the pro-motile effects of Ykt6 knockdown (Figures 5 & 6). These findings are consistent with previous studies conducted with intestinal epithelial and breast cancer cells, which implicated Rap1 activation in JAM-A-dependent acceleration of cell migration [38,59]. While we did not examine mechanisms of JAM-A-dependent stimulation of Rap1 activity in Ykt6-depleted DU145 cells, they could be similar to the mechanisms unraveled in the described studies. For example, in intestinal and mammary epithelial cells, JAM-A molecules were shown to homodimerize at the plasma membrane, giving rise to a multiprotein complex consisting of a scaffolding protein, afadin, Rap1, and a guanine nucleotide exchange factor (GEF) for Rap1, PDZ-GEF2 [38,59]. Assembly of this complex brings Rap1 and PDZ-GEF2 into proximity, resulting in Rap1 activation.
How could Rap1 activation promote the migration of Ykt6-depleted epithelial cells? Previous studies indicated that after being activated by JAM-A, Rap1 stimulates cell motility via integrin-dependent mechanisms, either by upregulating β1-integrin expression in epithelial cells [38,40,41] or by activating αV/β3 integrin in the vascular endothelium [60,61]. However, such integrin dependent mechanisms are unlikely to be involved in the accelerated motility of Ykt6-depleted cells. Indeed, in our experimental system, increased expression of JAM-A was not accompanied by the upregulation of β1-integrin level. Moreover, Ykt6 depletion impaired the maturation of β1-integrin by decreasing its Golgi-dependent glycosylation (Figure 3(a)), which likely results in impaired β1-integrin trafficking and function [7]. The observed decrease in paxillin phosphorylation in Ykt6-depleted epithelial cells (Figure 3(a)) is also inconsistent with the activation of integrin-dependent signaling. Finally, the expressional downregulation of either β1-integrin or paxillin failed to recapitulate the effects of Ykt6 knockdown on the motility of DU145 cells (Suppl. Fig. 6).
Our data suggest alternative molecular events that link JAM-A-dependent Rap1 activation and the accelerated motility of Ykt6-depleted epithelial cells. These events involve the activation of Rac1 GTPase and increased cell spreading (Figure 7 and Suppl. Fig. 7). Previous studies in epithelial cells and fibroblasts demonstrated that Rap1 could activate Rac1 to promote cell spreading and migration [43,44]. Rap1 was shown to interact with Rac1 GEFs, such as TIAM1 and VAV2, and to localize them to the leading edge of migrating cells [43]. Since Rac1 is a major trigger of actin polymerization at the cell cortex, its activation accelerates the formation of actin-rich protrusions (lammelipodia), thereby stimulating cell motility.
One of the most interesting findings of this study is the selective upregulation of JAM-A protein level in Ykt6-depleted epithelial cells. This phenomenon is caused by the derepression of JAM-A expression due to the downregulation of miR-145 (Figures 8 & 9). MiR-145 is a known repressor of JAM-A in breast cancer [50], glioblastoma [48], and endothelial cells [62]. Interestingly, downregulation of JAM-A expression was previously implicated in the attenuated migration of miR-145-overexpressing breast cancer cells [50]. This is consistent with our observation that the decrease in miR-145 level is responsible for the enhanced JAM-A expression and the accelerated motility of Ykt6-depleted DU145 cells (Figure 8). Loss of miR-145 in Ykt6-depleted DU145 cells was associated with increased level of JAM-A protein, but not mRNA (Figure 3). This suggests that miR-145 does not affect the stability of JAM-A mRNA, but rather regulates its translation. Such translational regulation has also been reported for other protein targets of miR-145 [63–65].
Ykt6-dependent regulation of microRNA expression represents a novel function for this membrane fusion protein. While the precise mechanisms underlying such Ykt6 activity remain to be elucidated, we propose that they could be linked to Ykt6 roles in regulating ER-Golgi vesicle trafficking and integrity of these organelles. Indeed the final steps in microRNA biogenesis, and their resultant functional activity, are regulated by a number of cytoplasmic proteins, most notably by RNAse III enzyme, dicer, and the RNA-induced silencing complex (RISC) [66]. The Argonaute protein family is the major constituent of the RISC complex directly interacting with micro-RNAs [66]. Several studies have demonstrated the association of dicer and Argonauts with different membrane compartments such as the ER, the Golgi and endosomes [67–71]. The loss of such associations between dicer, RISC, and endomembranes impairs microRNA dependent processing and activity [69,70]. Importantly, our present results (Suppl. Figs. 8 & 9), along with a previous study [28], revealed perturbations in ER-Golgi trafficking and the Golgi integrity in Ykt6-depleted cells. This may disrupt the association of the micro-RNA processing machinery with the ER/Golgi membranes, thereby inhibiting the processing of some micro-RNAs, including miR-145. It should be noted that our data provides only indirect support for this particular hypothetical mechanism, which would be tested with future studies. While no previous studies implicated SNAREs in the regulation of mRNA processing, a recent report has demonstrated that a lysosomal SNARE protein, Sec-22, modulates RNA interference in Caenorhabdis elegans [72]. It is likely therefore that eukaryotic SNARE proteins may play previously unanticipated roles in regulating intracellular biogenesis and activity of small RNAs.
The results of our study implicating Ykt6 in the regulation of cell motility and epithelial junctions allows for the prediction of possible pathophysiological consequences of altered Ykt6 expression and activity. For example, downregulation or otherwise inactivation of this membrane fusion protein may lead to the disruption of epithelial barriers during tissue inflammation and to the increased metastatic dissemination of cancer cells. Although we did not observe the effects of Ykt6 depletion on prostate epithelial cell proliferation (Suppl. Fig. 4), a previous study reported increased mitotic activity of Ykt6-overexpressing renal epithelial cells [73]. This raises the possibility that Ykt6, if overexpressed, could promote tumor growth. Little is known about Ykt6 expression and activity in cancer or other human diseases. A recent study reports Ykt6 overexpression in non-small lung cancer, which poorly correlated with patient survival [74]. An Oncomine database demonstrates a variegated pattern of Ykt6 expression in different cancers: it is downregulated in prostate and breast cancer and upregulated in head and neck cancer, lymphoma, and bladder cancer. However, the cellular localization and activity of Ykt6 could be strongly modulated by its post-translational modifications [20]. Therefore, the expression of Ykt6 mRNA may not serve as a good indicator of its functional activity in cancer cells. Interestingly, a recent study identified Ykt6 as a susceptibility locus for predisposition to diabetes and microalbuminuria [75]. This may link genetic polymorphism of Ykt6 with tissue barrier defects in diabetes, although further study is needed to prove such an association and to explore its causality and mechanisms.
Funding Statement
This work was supported by the National Institute of Neurological Disorders and Stroke [5P30NS047463]and National Cancer Institute [P30CA016059].
Acknowledgments
We thank Drs. Jesse C. Hay, Charles A. Parkos and Zendra Zehner for providing reagents for this study and Dr. Kevin Hogan for editing the manuscript. Microscopy was performed at the VCU Department of Anatomy and Neurobiology Microscopy Facility, supported, in part, with funding from the National Institute of Health (NIH)-NINDS Center core grant 5P30NS047463 and NIH-NCI Cancer Center Support Grant P30CA016059.
Disclosure statement
No potential conflict of interest was reported by the authors.
Competing Financial interests
The authors declare no competing financial interests
SUPPLEMENTAL DATA
Supplemental materials for this article can be accessed here.
References
- [1].Bryant DM,Mostov KE. From cells to organs: building polarized tissue. Nat Rev Mol Cell Biol. 2008;9:887–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fletcher SJ, Rappoport JZ. The role of vesicle trafficking in epithelial cell motility. Biochem Soc Trans. 2009;37:1072–1076. [DOI] [PubMed] [Google Scholar]
- [3].Wirtz-Peitz F, Zallen JA. Junctional trafficking and epithelial morphogenesis. Curr Opin Gen Develop. 2009;19:350–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. [DOI] [PubMed] [Google Scholar]
- [5].Luftman K, Hasan N, Day P, et al. Silencing of VAMP3 inhibits cell migration and integrin-mediated adhesion. Biochem Biophys Res Commun. 2009;380:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mendoza P, Diaz J, Torres VA. On the role of Rab5 in cell migration. Curr Mol Med. 2014;14:235–245. [DOI] [PubMed] [Google Scholar]
- [7].Naydenov NG, Feygin A, Wang L, et al. N-ethylmaleimide-sensitive factor attachment protein alpha (αSNAP) regulates matrix adhesion and integrin processing in human epithelial cells. J Biol Chem. 2014;289:2424–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Williams KC, Cappolino MG. Phosphorylation of membrane type 1-matrix metalloproteinase (MT1-MMP) and its vesicle-associated membrane protein 7 (VAMP7)-dependent trafficking facilitate cell invasion and migration. J Biol Chem. 2011;286:43405–43416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Williams KC, Cappolino MG. SNARE-dependent interaction of Src, EGFR and beta1 integrin regulates invadopodia formation and tumor cell invasion. J Cell Sci. 2014;127:1712–1725. [DOI] [PubMed] [Google Scholar]
- [10].Williamson RC, Cowell CA, Hammond CL, et al. Coronin-1C and RCC2 guide mesenchymal migration by trafficking Rac1 and controlling GEF exposure. J Cell Sci. 2014;127:4292–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lanzetti L, Di Fiore PP.. Behind the Scenes: endo/Exocytosis in the Acquisition of Metastatic Traits. Cancer Res. 2017;77:1813–1817. [DOI] [PubMed] [Google Scholar]
- [12].Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. [DOI] [PubMed] [Google Scholar]
- [13].Hong W. SNAREs and traffic. Biochim Biophys Acta. 2005;1744:493–517. [PubMed] [Google Scholar]
- [14].Malsam J, Kreye S, Sollner TH. Membrane fusion: sNAREs and regulation. Cell Mol Life Sci. 2008;65:2814–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Malsam J, Sollner TH. Organization of SNAREs within the Golgi stack. Cold Spring Harb Perspect Biol. 2011;3:a005249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Day P, Riggs KA, Hasan N, et al. Syntaxins 3 and 4 mediate vesicular trafficking of α5β1 and α3β1 integrins and cancer cell migration. Int J Oncol. 2011;39:863–871. [DOI] [PubMed] [Google Scholar]
- [17].Kean MJ, Williams KC, Skalski M, et al. VAMP3, syntaxin-13 and SNAP23 are involved in secretion of matrix metalloproteinases, degradation of the extracellular matrix and cell invasion. J Cell Sci. 2009;122:4089–4098. [DOI] [PubMed] [Google Scholar]
- [18].Garcia-Melero A, Reverter M, Hoque M, et al. Annexin A6 and Late Endosomal Cholesterol Modulate Integrin Recycling and Cell Migration. J Biol Chem. 2016;291:1320–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tiwari A, Jung JJ, Inamdar SM, et al. Endothelial cell migration on fibronectin is regulated by syntaxin 6-mediated α5β1 integrin recycling. J Biol Chem. 2011;286:36749–36761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fukasawa M, Varlamov O, Eng WS, et al. Localization and activity of the SNARE Ykt6 determined by its regulatory domain and palmitoylation. Proc Natl Acad Sci U S A. 2004;101:4815–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].McNew JA, Sogaard M, Lampen NM, et al. Ykt6p, a prenylated SNARE essential for endoplasmic reticulum-Golgi transport. J Biol Chem. 1997;272:17776–17783. [DOI] [PubMed] [Google Scholar]
- [22].Hasegawa H, Zinsser S, Rhee Y, et al. Mammalian ykt6 is a neuronal SNARE targeted to a specialized compartment by its profilin-like amino terminal domain. Mol Biol Cell. 2003;14:698–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tochio H, Tsui MM, Banfield DK, et al. An autoinhibitory mechanism for nonsyntaxin SNARE proteins revealed by the structure of Ykt6p. Science. 2001;293:698–702. [DOI] [PubMed] [Google Scholar]
- [24].Wen W, Yu J, Pan L, et al. Lipid-Induced conformational switch controls fusion activity of longin domain SNARE Ykt6. Mol Cell. 2010;37:383–395. [DOI] [PubMed] [Google Scholar]
- [25].Parlati F, Varlamov O, Paz K, et al. Distinct SNARE complexes mediating membrane fusion in Golgi transport based on combinatorial specificity. Proc Natl Acad Sci U S A. 2002;99:5424–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Santos AJ, Raote I, Scarpa M, et al. TANGO1 recruits ERGIC membranes to the endoplasmic reticulum for procollagen export. Elife. 2015;4:e10982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Volchuk A, Ravazzola M, Perrelet A, et al. Countercurrent distribution of two distinct SNARE complexes mediating transport within the Golgi stack. Mol Biol Cell. 2004;15:1506–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang T, Hong W. Ykt6 forms a SNARE complex with syntaxin 5, GS28, and Bet1 and participates in a late stage in endoplasmic reticulum-Golgi transport. J Biol Chem. 2001;276:27480–27487. [DOI] [PubMed] [Google Scholar]
- [29].Hasegawa H, Yang Z, Oltedal L, et al. Intramolecular protein-protein and protein-lipid interactions control the conformation and subcellular targeting of neuronal Ykt6. J Cell Sci. 2004;117:4495–4508. [DOI] [PubMed] [Google Scholar]
- [30].Peltan A, Briggs L, Matthews G, et al. Identification of Drosophila gene products required for phagocytosis of Leishmania donovani. PLoS ONE. 2012;7:e51831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gross JC, Chaudhary V, Bartscherer K, et al. Active Wnt proteins are secreted on exosomes. Nat Cell Biol. 2012;14:1036–1045. [DOI] [PubMed] [Google Scholar]
- [32].Gordon DE, Chia J, Jayawardena K, et al. VAMP3/Syb and YKT6 are required for the fusion of constitutive secretory carriers with the plasma membrane. PLoS Genetics. 2017;13:e1006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Naydenov NG, Brown B, Harris G, et al. A membrane fusion protein αSNAP is a novel regulator of epithelial apical junctions. PLoS ONE. 2012;7:e34320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ivanov AI, Pero RS, Scheck AC, et al. Prostaglandin E(2)-synthesizing enzymes in fever: differential transcriptional regulation. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1104–17. [DOI] [PubMed] [Google Scholar]
- [35].Naydenov NG, Harris G, Morales V, et al. Loss of a membrane trafficking protein αSNAP induces non-canonical autophagy in human epithelia. Cell Cycle. 2012;11:4613–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Friedl P, Mayor R. Tuning collective cell migration by cell-cell junction regulation. Cold Spring Harb Perspect Biol. 2017;9:a029199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].McSherry EA, Brennan K, Hudson L, et al. Breast cancer cell migration is regulated through junctional adhesion molecule-A-mediated activation of Rap1 GTPase. Breast Cancer Res. 2011;13:R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McSherry EA, McGee SF, Jirstrom K, et al. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int J Cancer. 2009;125:1343–1351. [DOI] [PubMed] [Google Scholar]
- [40].Severson EA, Jiang L, Ivanov AI, et al. Cis-dimerization mediates function of junctional adhesion molecule A. Mol Biol Cell. 2008;19:1862–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Severson EA, Lee WY, Capaldo CT, et al. Junctional adhesion molecule A interacts with Afadin and PDZ-GEF2 to activate Rap1A, regulate β1 integrin levels, and enhance cell migration. Mol Biol Cell. 2009;20:1916–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Courilleau D, Bisserier M, Jullian JC, et al. Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J Biol Chem. 2012;287:44192–44202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Arthur WT, Quilliam LA, Cooper JA. Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. J Cell Biol. 2004;167:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Takahashi M, Rikitake Y, Nagamatsu Y, et al. Sequential activation of Rap1 and Rac1 small G proteins by PDGF locally at leading edges of NIH3T3 cells. Genes Cells. 2008;13:549–569. [DOI] [PubMed] [Google Scholar]
- [45].Shutes A, Onesto C, Picard V, et al. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007;282:35666–35678. [DOI] [PubMed] [Google Scholar]
- [46].Mathonnet G, Fabian MR, Svitkin YV, et al. MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex eIF4F. Science. 2007;317:1764–1767. [DOI] [PubMed] [Google Scholar]
- [47].Pillai RS, Bhattacharyya SN, Artus CG, et al. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–1576. [DOI] [PubMed] [Google Scholar]
- [48].Alvarado AG, Turaga SM, Sathyan P, et al. Coordination of self-renewal in glioblastoma by integration of adhesion and microRNA signaling. Neuro Oncol. 2016;18:656–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cao M, Nie W, Li J, et al. MicroRNA-495 induces breast cancer cell migration by targeting JAM-A. Protein Cell. 2014;5:862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gotte M, Mohr C, Koo CY, et al. miR-145-dependent targeting of junctional adhesion molecule A and modulation of fascin expression are associated with reduced breast cancer cell motility and invasiveness. Oncogene. 2010;29:6569–6580. [DOI] [PubMed] [Google Scholar]
- [51].Miyata R, Kakuki T, Nomura K, et al. Poly(I:C) induced microRNA-146a regulates epithelial barrier and secretion of proinflammatory cytokines in human nasal epithelial cells. Eur J Pharmacol. 2015;761:375–382. [DOI] [PubMed] [Google Scholar]
- [52].Kim YJ, Maizel A, Chen X. Traffic into silence: endomembranes and post-transcriptional RNA silencing. EMBO J. 2014;33:968–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Smalheiser NR. Regulation of mammalian microRNA processing and function by cellular signaling and subcellular localization. Biochim Biophys Acta. 2008;1779:678–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gordon DE, Bond LM, Sahlender DA, et al. A targeted siRNA screen to identify SNAREs required for constitutive secretion in mammalian cells. Traffic. 2010;11:1191–1204. [DOI] [PubMed] [Google Scholar]
- [55].Ikari A, Tonegawa C, Sanada A, et al. Tight junctional localization of claudin-16 is regulated by syntaxin 8 in renal tubular epithelial cells. J Biol Chem. 2014;289:13112–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jung JJ, Inamdar SM, Tiwari A, et al. Syntaxin 16 regulates lumen formation during epithelial morphogenesis. PLoS ONE. 2013;8:e61857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sharma N, Low SH, Misra S, et al. Apical targeting of syntaxin 3 is essential for epithelial cell polarity. J Cell Biol. 2006;173:937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Luissint AC, Nusrat A, Parkos CA. JAM-related proteins in mucosal homeostasis and inflammation. Semin Immunopathol. 2014;36:211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Severson EA, Parkos CA. Structural determinants of Junctional Adhesion Molecule A (JAM-A) function and mechanisms of intracellular signaling. Curr Opin Cell Biol. 2009;21:701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Naik MU, Naik UP. Junctional adhesion molecule-A-induced endothelial cell migration on vitronectin is integrin αv β3 specific. J Cell Sci. 2006;119:490–499. [DOI] [PubMed] [Google Scholar]
- [61].Peddibhotla SS, Brinkmann BF, Kummer D, et al. Tetraspanin CD9 links junctional adhesion molecule-A to αvβ3 integrin to mediate basic fibroblast growth factor-specific angiogenic signaling. Mol Biol Cell. 2013;24:933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Schmitt MM, Megens RT, Zernecke A, et al. Endothelial junctional adhesion molecule-a guides monocytes into flow-dependent predilection sites of atherosclerosis. Circulation. 2014;129:66–76. [DOI] [PubMed] [Google Scholar]
- [63].Ikemura K, Yamamoto M, Miyazaki S, et al. MicroRNA-145 post-transcriptionally regulates the expression and function of P-glycoprotein in intestinal epithelial cells. Mol Pharmacol. 2013;83:399–405. [DOI] [PubMed] [Google Scholar]
- [64].Wu H, Xiao Z, Wang K, et al. MiR-145 is downregulated in human ovarian cancer and modulates cell growth and invasion by targeting p70S6K1 and MUC1. Biochem Biophys Res Commun. 2013;441:693–700. [DOI] [PubMed] [Google Scholar]
- [65].Zhang J, Guo H, Zhang H, et al. Putative tumor suppressor miR-145 inhibits colon cancer cell growth by targeting oncogene Friend leukemia virus integration 1 gene. Cancer. 2011;117:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Barbato C, Ciotti MT, Serafino A, et al. Dicer expression and localization in post-mitotic neurons. Brain Res. 2007;1175:17–27. [DOI] [PubMed] [Google Scholar]
- [68].Barman B, Bhattacharyya SN. MRNA targeting to endoplasmic reticulum precedes ago protein interaction and MicroRNA (miRNA)-mediated translation repression in mammalian cells. J Biol Chem. 2015;290:24650–24656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lee YS, Pressman S, Andress AP, et al. Silencing by small RNAs is linked to endosomal trafficking. Nat Cell Biol. 2009;11:1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Pepin G, Perron MP, Provost P. Regulation of human Dicer by the resident ER membrane protein CLIMP-63. Nucl Acids Res. 2012;40:11603–11617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Stalder L, Heusermann W, Sokol L, et al. The rough endoplasmatic reticulum is a central nucleation site of siRNA-mediated RNA silencing. EMBO J. 2013;32:1115–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhao Y, Holmgren BT, Hinas A. The conserved SNARE SEC-22 localizes to late endosomes and negatively regulates RNA interference in Caenorhabditis elegans. RNA. 2017;23:297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Thayanidhi N, Liang Y, Hasegawa H, et al. R-SNARE ykt6 resides in membrane-associated protease-resistant protein particles and modulates cell cycle progression when over-expressed. Biol Cell. 2012;104:397–417. [DOI] [PubMed] [Google Scholar]
- [74].Ruiz-Martinez M, Navarro A, Marrades RM, et al. YKT6 expression, exosome release, and survival in non-small cell lung cancer. Oncotarget. 2016;7:51515–51524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Choi JW, Moon S, Jang EJ, et al. Association of prediabetes-associated single nucleotide polymorphisms with microalbuminuria. PLoS One. 2017;12:e0171367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Materials, data and associated protocols will be available upon request from the corresponding author.