

Abstract
Background
Tofacitinib is an oral Janus kinase inhibitor. This open-label, long-term extension (LTE) study (NCT00658359) evaluated long-term tofacitinib treatment in stable kidney transplant recipients (n = 178) posttransplant.
Methods
Patients who completed 12 months of cyclosporine (CsA) or tofacitinib treatment in the phase IIb parent study (NCT00483756) were enrolled into this LTE study, evaluating long-term tofacitinib treatment over months 12 to 72 posttransplant. Patients were analyzed by tofacitinib less-intensive (LI) or more-intensive (MI) regimens received in the parent study. For both groups, tofacitinib dose was reduced from 10 to 5 mg twice daily by 6 months into the LTE. Patients were followed up through month 72 posttransplant, with a focus on month 36 results.
Results
Tofacitinib demonstrated similar 36-month patient and graft survival rates to CsA. Biopsy-proven acute rejection rates at month 36 were 11.2% for CsA, versus 10.0% and 7.4% (both P > 0.05) for tofacitinib LI and MI, respectively. Least squares mean estimated glomerular filtration rates were 9 to 15 mL/min per 1.73 m2 higher for tofacitinib versus CsA at month 36. The proportions of patients with grade 2/3 interstitial fibrosis and tubular atrophy in month 36 protocol biopsies were 20.0% for LI and 18.2% for MI (both P > 0.05) versus 33.3% for CsA. Kaplan-Meier cumulative serious infection rates at month 36 were numerically higher for tofacitinib LI (43.9%; P = 0.45) and significantly higher for MI (55.9%; P < 0.05) versus CsA (37.1%).
Conclusions
Long-term tofacitinib continued to be effective in preventing renal allograft acute rejection and preserving renal function. However, long-term tofacitinib and mycophenolic acid product combination was associated with persistent serious infection risk.
Chronic allograft nephropathy, histologically described as kidney allograft interstitial fibrosis and tubular atrophy (IFTA), is understood to be driven by multiple factors, including immune injury, ischemia reperfusion, donor disease, and immunosuppressive drug toxicity.1,2 The extent to which calcineurin inhibitors (CNI) contribute to allograft IFTA over the long term has been disputed.3,4 Nevertheless, concern over potential nephrotoxic and deleterious metabolic effects of CNI has prompted interest in CNI minimization and avoidance of immunosuppressive regimens.5-7 Although some CNI-sparing regimens have achieved improvement in renal function,8-12 there was suboptimal prevention of acute allograft rejection.8-11,13-17
Tofacitinib is an oral Janus kinase (JAK) inhibitor for the treatment of rheumatoid arthritis, psoriatic arthritis, and ulcerative colitis. Phase II studies in kidney transplant recipients showed that a CNI-free regimen using tofacitinib was effective in preventing acute allograft rejection, improving renal function, and reducing chronic allograft histologic injury, and was associated with a lower risk of developing diabetes in the 6- to 12-month period posttransplant.18-20 However, these studies demonstrated an increased risk of serious infections and posttransplant lymphoproliferative disease (PTLD), although it has subsequently been demonstrated that these risks were associated with higher tofacitinib exposure.20 Whether the clinical benefits and risks persist during long-term treatment with tofacitinib in kidney transplant patients is unknown.
De novo kidney transplant recipients who completed 12 months of treatment in a phase IIb study comparing tofacitinib with cyclosporine (CsA) (NCT00483756)19,20 were eligible to participate in a long-term extension (LTE) study and were followed up for an additional 5 years.
Here, we report data from this LTE study describing the long-term efficacy and safety of tofacitinib in stable kidney transplant recipients.
MATERIALS AND METHODS
Patients
Patients were 18 to 70 years of age and were recipients of primary renal allografts from deceased donors or human leukocyte antigen-mismatched living donors. Patients must have completed 12 months of treatment with tofacitinib or CsA in A3921030 (NCT00483756).20
Study Design and Treatment
This was a phase IIb, randomized, multicenter, open-label LTE study (A3921050; NCT00658359) evaluating the efficacy and safety of tofacitinib versus CsA in kidney transplant recipients completing 12 months of tofacitinib or CsA treatment in A3921030 (NCT00483756).20 The open-label LTE study was initiated in August 2008, through to the last patient visit and completion in June 2015. This was an international, multicenter study, with a total of 43 investigational centers randomizing patients for participation in the study. Patients continued prior treatment assigned in the parent study through month 72 posttransplant (Figure 1). All tofacitinib-treated patients received tofacitinib 10 mg twice daily (BID) at study entry (month 12), regardless of dose in the parent study, reducing to 5 mg BID by month 18 posttransplant. However, patients retained their categorization from the parent study—tofacitinib less intensive (LI) and more intensive (MI)—for analysis. Mycophenolic acid (MPA) products were continued through month 72 posttransplant. Corticosteroids could be discontinued after month 12 at the investigator’s discretion.
FIGURE 1.
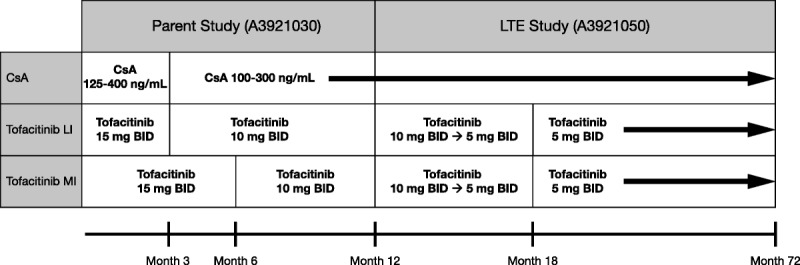
Treatment groups and dosing regimens are shown from the full analysis set, including the parent study (A3921030; NCT00483756) and the LTE study (A3921050; NCT00658359). CsA 125 to 400 ng/mL and 100 to 300 ng/mL represent the target 12-hour trough whole blood levels. All patients received concomitant mycophenolic acid product and corticosteroid taper through month 72.
Reported clinical outcomes in tofacitinib-treated patients primarily reflected experience in patients maintained on tofacitinib 5 mg BID after months 12 to 18 posttransplant, with background MPA products and corticosteroids.
A protocol amendment was implemented to discontinue patients with above-median exposure (AME) of tofacitinib, due to potential association with PTLD. Patients with AME exposure within the first 6 months posttransplant were discontinued. After discontinuation, patients were followed up for 12 months, including a follow-up evaluation 2 months (±14 days) after the last tofacitinib dose. Additional follow-up visits (or a minimum of a telephone call) were arranged every 3 months for 12 months after the last dose of tofacitinib, to determine new-onset serious infections, malignancy, graft loss, or death.
This study was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practice guidelines. The final protocol, amendments, and informed consent documentation were reviewed and approved by the institutional review boards and independent ethics committees of the investigational centers. All patients provided written, informed consent.
Efficacy and Safety: Objectives and Endpoints
The objective of this LTE study was to evaluate the long-term efficacy and safety of tofacitinib in stable kidney transplant recipients. Efficacy outcomes included: patient and allograft survival rates, incidence of first biopsy-proven acute rejection (BPAR) and IFTA (as determined by a blinded central pathologist), incidence of treated clinical acute rejection (episodes were diagnosed by the study site, and patients received antirejection treatment), and glomerular filtration rate (GFR). Allograft biopsy and GFR measured using iohexol were required at month 36.
Safety endpoints included: adverse events (AEs), serious AEs (SAEs), serious infections, malignancies, PTLD, polyomavirus-associated nephropathy (PVAN), new-onset diabetes mellitus (NODM), and herpes zoster virus (HZV) infections. Laboratory evaluations included: hemoglobin (Hgb) levels, white blood cell (WBC) count, absolute lymphocyte count (ALC), absolute neutrophil count (ANC), and the proportions of patients with Epstein-Barr virus (EBV) and BK virus (BKV).
Statistical Analysis
Efficacy and safety analyses were based on the full analysis set, which included all patients who received 1 dose or more of study treatment in the LTE study.
Kaplan-Meier (KM) curves were fitted for time-to-event data for: patient and allograft survival, serious infections, HZV infection, BPAR, treated clinical acute rejection, PVAN, and NODM. Kaplan-Meier rate differences were compared between tofacitinib doses and active comparators, based on the Wald test, at each time point. Although KM analysis was applied to all data available through month 72, interpretation of the data is limited after month 36, owing to the decreasing number of patients in the tofacitinib group (partly as a result of discontinuations required by the protocol amendment); in-text discussion of results therefore focuses on month 36 data.
Estimated GFR (eGFR) was calculated using the modification of diet in renal disease (MDRD) formula, with last observation carried forward (LOCF) and an imputation of death and graft loss as zero eGFR. For continuous data collected over time, a linear mixed-effects model with repeated measures was used. The model included treatment, visit, and treatment-by-visit interaction as fixed effects, and baseline (as appropriate) as covariates.
Binary variables were analyzed using large sample approximation or exact methods for endpoints with sparse cells.
Exploratory Exposure Analysis
As a post hoc exploratory analysis, patients receiving tofacitinib LI or MI regimens were recategorized, according to their pharmacokinetic exposure, into below-median exposure (BME) or AME, within 6 months posttransplant. This enabled investigation of the relationship between tofacitinib concentrations and both efficacy and safety endpoints. In each evaluable tofacitinib-treated patient, available 2-hour postdose concentrations (C2) over the first 6 months posttransplant were normalized by dose and a median C2 was calculated. Following this, the median C2 (for each individual patient) was adjusted for the dose and weighted for the duration of treatment with the particular dose.19 For exposure analysis, the total number of patients available for evaluation over time for groups CsA, BME, and AME, respectively, were as follows: baseline, n = 64, 62, and 50; month 12, n = 64, 59, and 49; month 36, n = 52, 45, and 0; and month 72, n = 31, 27, and 0.
RESULTS
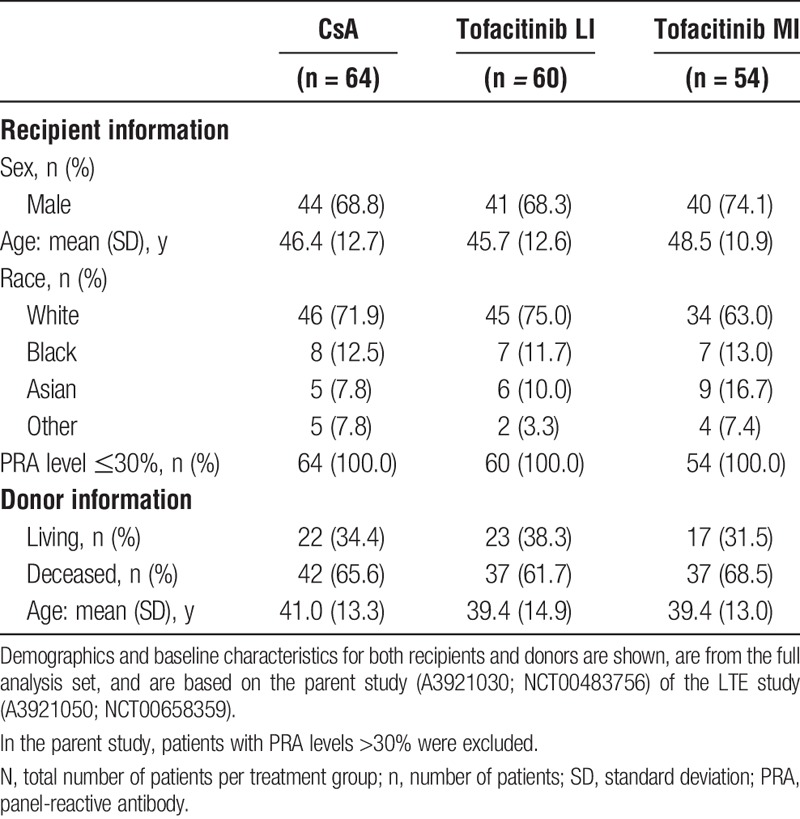
Patient Demographics and Baseline Characteristics
A total of 178 patients were enrolled and treated, of whom 119 patients discontinued; patient disposition is shown in Figure 2. A summary of patient demographics and baseline characteristics at the time of transplantation is shown in Table 1. Baseline characteristics were generally similar among the treatment groups. The median treatment duration for patients was 66.1 months (range, 12.4-72.9) for the CsA group, 53.7 months (range, 12.1-74.9) for the tofacitinib LI group, and 28.4 months (range, 12.3-73.8) for the tofacitinib MI group.
FIGURE 2.
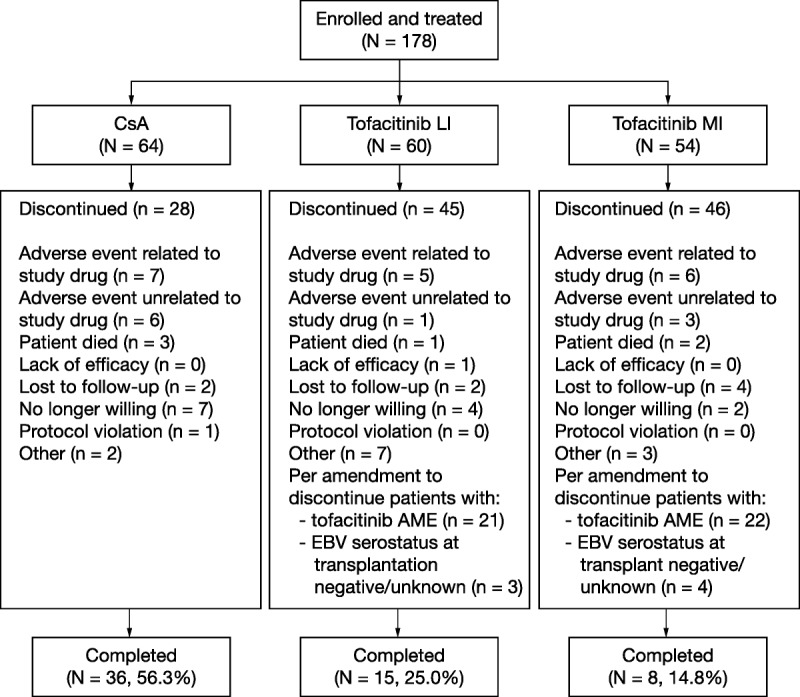
Patient disposition data, presented from the LTE study only (A3921050; NCT00658359). N, total number of patients per treatment group; n, number of patients.
TABLE 1.
Demographics and baseline characteristics at the time of transplantation
Efficacy Outcomes
Patient and Allograft Survival
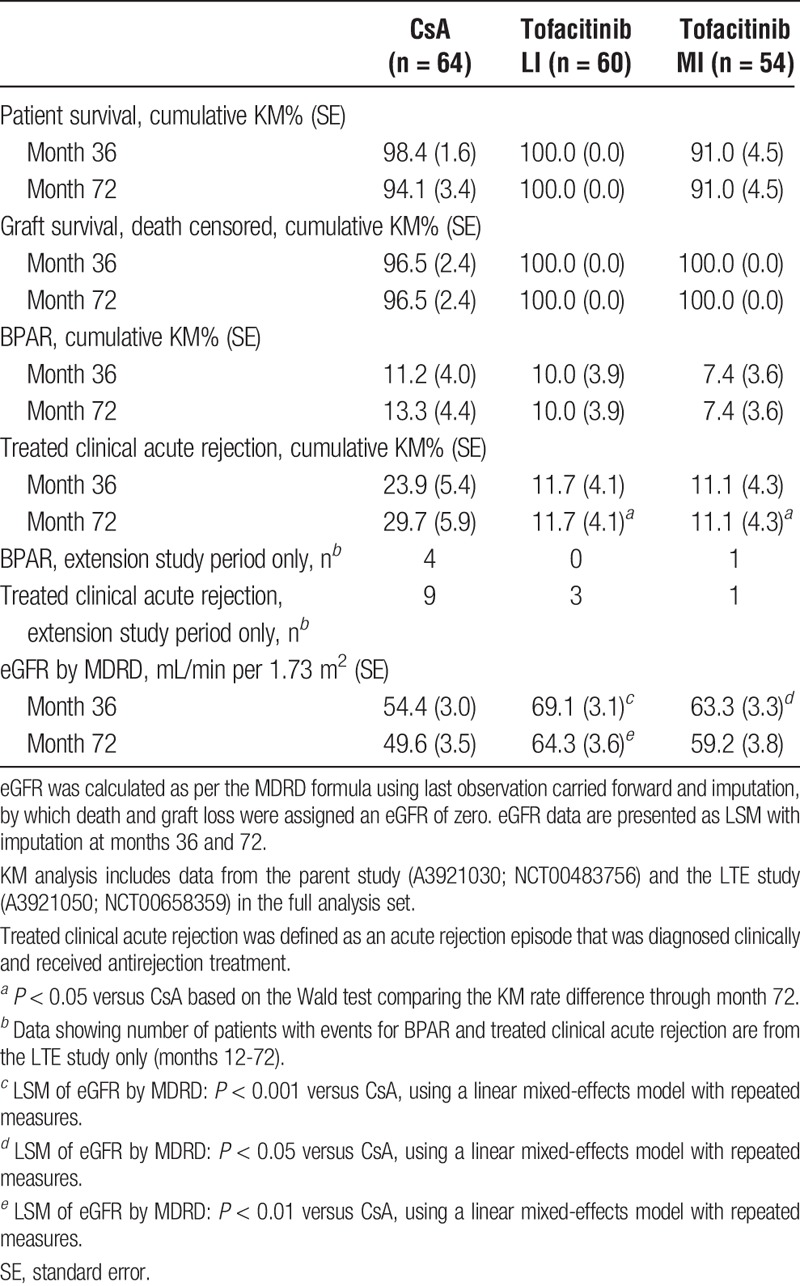
Among patients who entered this LTE study, KM estimates showed no significant differences in patient survival and death-censored allograft survival (to month 36) for either of the tofacitinib groups versus CsA (Table 2).
TABLE 2.
Efficacy outcomes through month 72
First BPAR
Before study entry at month 12, first BPAR was reported in 4, 6, and 3 patients in the CsA, tofacitinib LI, and tofacitinib MI groups, respectively. From month 12 posttransplant through month 72, first BPAR was reported in 4 patients in the CsA group and in 1 patient in the tofacitinib MI group; no patients in the tofacitinib LI group experienced BPAR. At month 36, KM estimates were 11.2% for CsA versus 10.0% (P = 0.83) and 7.4% (P = 0.48) for tofacitinib LI and MI, respectively (Table 2). Although BPAR events continued to accumulate in the CsA group after month 12, the difference versus tofacitinib did not reach statistical significance (Figure 3). For tofacitinib exposure-based analysis, through months 12 to 72 first BPAR was reported in 1 patient in the BME group and in no patients in the AME group. For patients with BME at month 36, KM estimates for first BPAR were 11.3% (P = 0.98) versus 11.2% for CsA.
FIGURE 3.
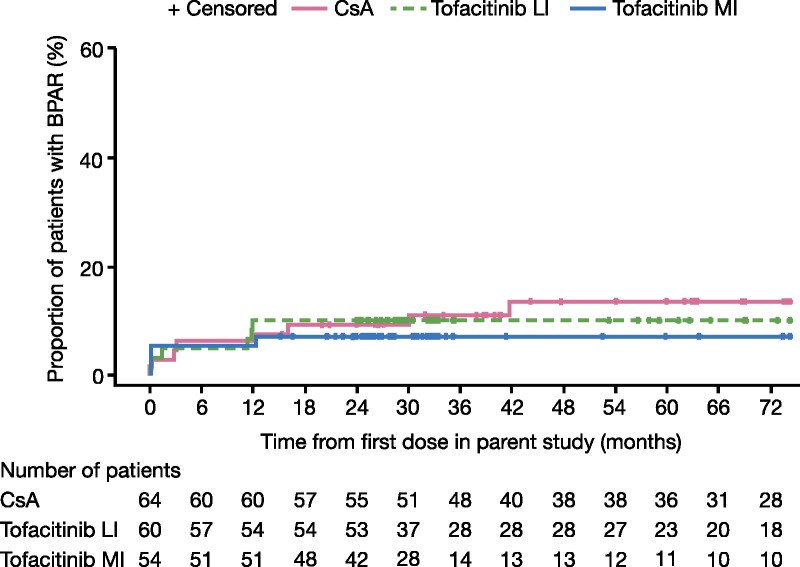
KM estimates of BPAR by dose groups. BPAR was defined as acute/active cellular rejection as interpreted by the central blinded pathologist, according to the Banff 97 working classification.21 Data are based on all biopsies (including for-cause and protocol biopsies). Data in graphs show the first occurrence of BPAR. Data presented are from the full analysis set for the parent study (A3921030; NCT00483756), months 0 to 12, and the LTE study (A3921050; NCT00658359), months 12 to 72.
Treated Clinical Acute Rejection
Treated clinical acute rejection was reported in 9, 4, and 5 patients in the CsA, tofacitinib LI, and tofacitinib MI groups, respectively, in the parent study. From month 12 through month 72, additional treated clinical acute rejection was reported in 9, 3, and 1 patients in the CsA, tofacitinib LI, and tofacitinib MI groups, respectively. At month 36, the KM rates of clinical acute rejection were 11.7% (P = 0.07) and 11.1% (P = 0.06) for tofacitinib LI and MI, respectively, versus 23.9% for CsA (Table 2).
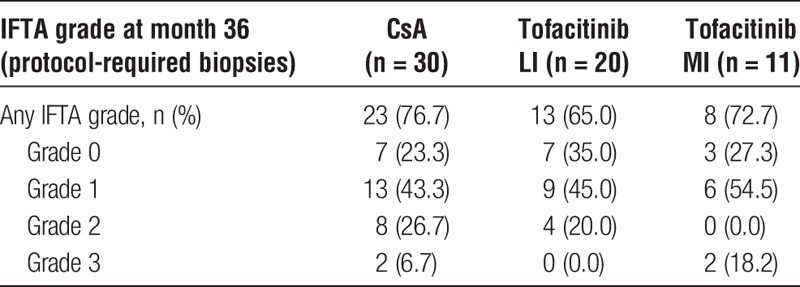
Rates of IFTA
Only 61 of 178 enrolled patients completed the protocol-required allograft biopsy at month 36, and 44 patients showed findings consistent with IFTA. Most IFTA cases were classified as mild (grade 1). The proportions of patients with grade 2/3 IFTA in month 36 protocol biopsies were 20.0% for tofacitinib LI and 18.2% for tofacitinib MI versus 33.3% for CsA (Table 3). For patients with BME at month 36, based on protocol-required biopsies, the proportions of grade 2/3 IFTA were 19.4% versus 33.3% for CsA.
TABLE 3.
Proportion of patients with IFTA by severity grades in the protocol-required allograft biopsy at month 36
Glomerular Filtration Rate
Only 13 patients in the tofacitinib MI group completed measured GFR at month 36 versus 30 patients in the tofacitinib LI group and 39 patients in the CsA group. Although least squares means (LSM) of measured GFR were numerically higher at month 36 for the tofacitinib LI and MI groups (76.9 mL/min [P = 0.07] and 75.9 mL/min [P = 0.2], respectively) versus CsA (67.6 mL/min), the differences were not statistically significant. However, LSM of MDRD-estimated eGFR (LOCF plus imputation) were numerically higher in the tofacitinib groups versus CsA at all visits, reaching statistical significance at month 15 through month 36 for tofacitinib MI and at month 15 through month 72 for tofacitinib LI (P < 0.0001 to P < 0.05; Figure 4). At month 36, LSM eGFRs were approximately 9 to 15 mL/min per 1.73 m2 higher in the tofacitinib groups versus CsA. Least squares means eGFRs were generally maintained through 72 months, with values approximately 10 to 15 mL/min per 1.73 m2 higher in the tofacitinib groups versus CsA at month 72. For patients with BME at month 36, LSMs of measured GFR were 78.6 mL/min (P = 0.02) versus 67.7 mL/min for CsA. Similarly, MDRD-estimated eGFR (LOCF plus imputation) was significantly higher in the BME group versus CsA from month 15 through month 72 and was approximately 13 mL/min per 1.73 m2 higher than CsA at month 72.
FIGURE 4.
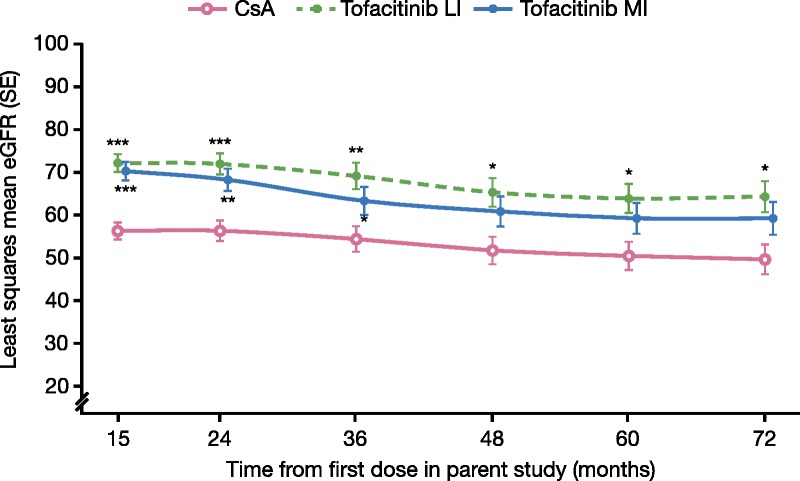
LSM of eGFR (mL/min per 1.73 m2) by MDRD over time, with LOCF plus imputation of death and graft loss as zero eGFR. ***P < 0.0001; **P < 0.001; *P < 0.05 for tofacitinib versus CsA. P value from linear mixed model with treatment, visit, and treatment by visit interaction as fixed effects. An unstructured variance-covariance was used. These data represent LSM eGFR by MDRD equation with LOCF plus imputation (patients with death or graft loss were imputed as eGFR = 0) over time and corresponding SE. Data presented are from the LTE study (A3921050; NCT00658359), months 12 to 72.
Safety Outcomes
Adverse Events
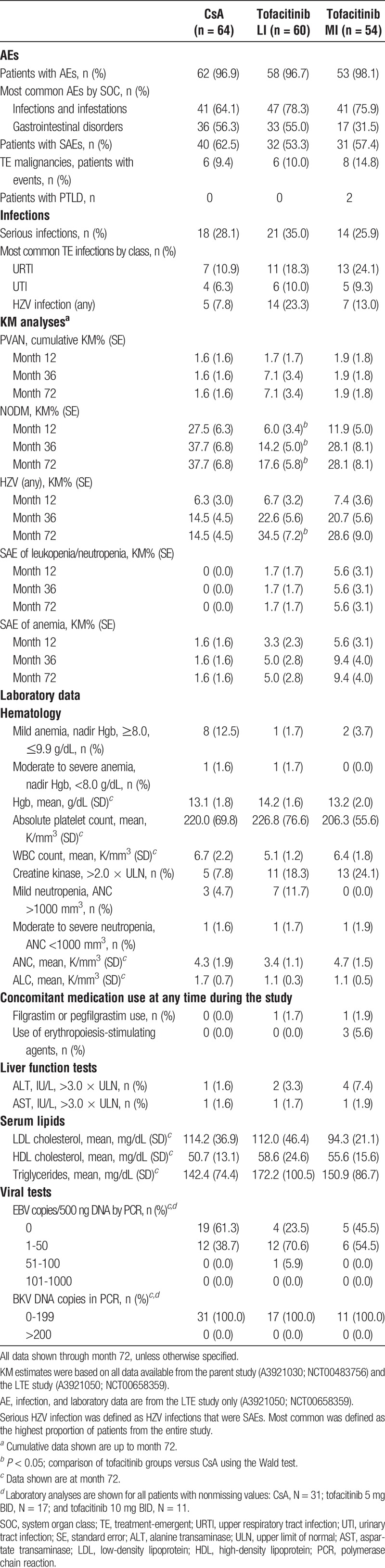
Adverse events were reported in 96.9%, 96.7%, and 98.1% of the CsA, tofacitinib LI, and tofacitinib MI groups, respectively (Table 4). The most common types of AEs were infections. The most common AE terms for tofacitinib LI and MI were HZV infection (23.3% and 13.0% vs CsA 7.8%) and upper respiratory tract infection (18.3% and 24.1% vs CsA 10.9%) (Table 4). Broadly similar proportions of patients among treatment groups discontinued due to AEs for CsA (18.8%), tofacitinib LI (10.0%), and tofacitinib MI (18.5%). The most common types of SAEs for patients receiving tofacitinib were infections (35.0% for LI and 25.9% for MI versus 28.1% for CsA). The most common individual SAE terms for the tofacitinib LI group were kidney transplant rejection, pneumonia, BK viral nephropathy, and urinary tract infection (all 5%), whereas sepsis was the most common (5.6%) for the tofacitinib MI group.
TABLE 4.
Safety outcomes through month 72
At study entry (month 12), serious infection rates were similar in the CsA (26.6%) and tofacitinib LI groups (25.0%), but were numerically higher for tofacitinib MI (38.9%, P = 0.15 vs CsA). Kaplan-Meier estimates of serious infection rates increased over time in each group and were significantly higher for tofacitinib MI versus CsA from month 24 through month 36 (range, P = 0.02-0.05). At month 36, KM estimates were numerically higher for tofacitinib LI (43.9%; P = 0.45) and significantly higher for tofacitinib MI (55.9%; P < 0.05) versus CsA (37.1%; Figure 5).
FIGURE 5.
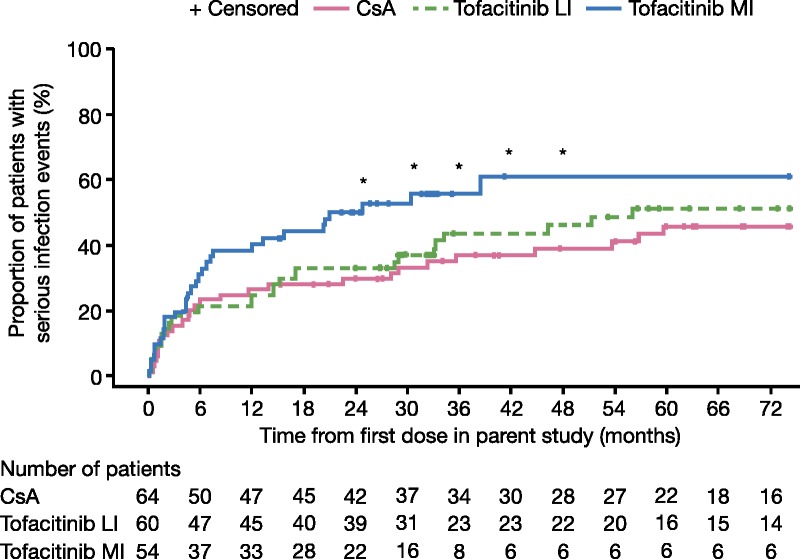
KM estimates of serious infection events in the study period. *P < 0.05 for comparison of CsA and tofacitinib MI; Wald test comparing rate differences. Data represent KM estimates of serious infection events over the full analysis set for the parent study (A3921030; NCT00483756), months 0 to 12, and the LTE study (A3921050; NCT00658359), months 12 to 72.
Exposure-based analysis comparing rates of serious infections in the AME and BME tofacitinib groups showed numerically higher rates for AME (44.0%) versus CsA (26.6%) and BME (22.6%) at month 12. Similar to the dose-based analysis, the KM serious infection rate increased in all groups over time after month 12. At the last evaluable time point for the AME group (month 30), the cumulative serious infection rate (53.1%, P = 0.04) was significantly higher versus CsA (33.4%), and the cumulative serious infection rate in the BME group was numerically higher versus CsA (38.8%, P = 0.53). At month 36, the cumulative serious infection rate was 45.8% (P = 0.33) for the BME group versus 37.1% for CsA.
Kaplan-Meier estimates of HZV infection rate increased over time. At month 36, rates were numerically higher in both tofacitinib groups (LI, 22.6%, P = 0.25; MI, 20.7%, P = 0.38) versus CsA (14.5%), although they did not reach significance. For serious HZV infections, although rates also increased over time, no significant differences were observed among the treatment groups at month 36, with KM rates of 1.7% for tofacitinib LI and 5.6% for tofacitinib MI versus 4.8% for CsA. For the BME group, rates of serious HZV infections were 1.6% versus 4.8% for CsA.
From month 12 through month 72, 6 (9.4%), 6 (10.0%), and 8 (14.8%) patients reported malignancy in the CsA, tofacitinib LI, and tofacitinib MI groups, respectively (Table 4). Of the 31 malignancy events recorded, most (22/31) were nonmelanoma skin cancer (basal cell or squamous cell skin cancer).
Two patients in the tofacitinib MI group developed PTLD after month 12. Both patients, and the 3 patients in parent study A3921030 who experienced PTLD, belonged to the AME group. No additional cases of PTLD were observed after introduction of the protocol amendment that required discontinuation of 43 AME patients. Among the patients who were discontinued from tofacitinib, no PTLD cases were reported during 12 months of postdose follow-up. At month 36, KM analysis showed significantly lower rates of NODM for tofacitinib LI (14.2%; P = 0.006) and numerically lower rates for tofacitinib MI (28.1%; P = 0.37) versus CsA (37.7%) (Table 4). Three cases of BK viral nephropathy occurred in patients receiving tofacitinib LI.
Laboratory Data
A summary of laboratory data is included in Table 4. There was a higher proportion of patients receiving CsA who at any time in the study experienced mild anemia (nadir Hgb ≥8.0 and ≤9.9 g/dL; 12.5%) compared with the tofacitinib groups (1.7-3.7%). Moderate to severe anemia (nadir Hgb levels <8 g/dL) was reported in 1 patient (1.6%) receiving CsA and in 1 patient (1.7%) receiving tofacitinib LI. Concomitant use of erythropoiesis-stimulating agents was reported in 3 patients (5.6%) in the tofacitinib MI group. Mean platelet counts, WBC, and ANC were generally similar across the treatment groups. The proportions of patients with ANC less than 1000/mm3 were as follows: CsA, 1.6%; tofacitinib LI, 1.7%; and tofacitinib MI, 1.9%. Concomitant use of filgrastim or pegfilgrastim was reported in 1 (1.7%) patient receiving tofacitinib LI and in 1 (1.9%) patient receiving tofacitinib MI. At month 72, mean ALC was higher for CsA (1.7 K/mm3) than in each of the tofacitinib groups (both 1.1 K/mm3).
Mean serum high-density lipoprotein-cholesterol values were modestly higher at month 72 in the tofacitinib groups compared with CsA, and mean triglyceride values were modestly higher in the tofacitinib LI group than in the CsA group. In contrast, mean serum low-density lipoprotein-cholesterol values in the tofacitinib groups were generally comparable to those in the CsA group. The use of lipid-lowering agents was common with 42.4% or greater of patients in each of the treatment groups receiving these medications; there was no statistically significant difference between the tofacitinib groups and the CsA group at any time point.
The proportion of patients with no BKV or low-grade viremia was similar among the tofacitinib and CsA groups. A higher proportion of patients in the tofacitinib groups had low-grade EBV. At month 72, more patients in the tofacitinib groups versus CsA had EBV counts of 1 to 50 copies/500 ng DNA (DNA) or 51 to 100 copies/500 ng DNA.
DISCUSSION
Tofacitinib is an oral JAK inhibitor that targets inflammation by reducing proinflammatory cytokine signaling and production and has been approved for the treatment of rheumatoid arthritis, psoriatic arthritis, and ulcerative colitis.22 Tofacitinib has been evaluated as a substitute for CNIs for rejection prophylaxis in de novo kidney transplantation.18-20 The objectives of the phase II LTE study described here were to evaluate the long-term efficacy and safety of tofacitinib.
The results of this LTE study suggest that tofacitinib treatment beyond the first 12 months posttransplant continued to be effective in preventing acute allograft rejection, with few cases of late-onset BPARs, and cumulative rates of BPAR and treated clinical acute rejection no higher than CsA at month 36. A low risk of mortality and graft loss was also observed in all treatment groups. In the exploratory analysis by tofacitinib exposure, similar efficacy to CsA at month 36 was also demonstrated in the patients with lower tofacitinib drug exposure (BME).
Tofacitinib LI continued to demonstrate a significantly higher MDRD-calculated eGFR than CsA at every time point after month 12, with tofacitinib groups having eGFR 9 to 15 mL/min per 1.73 m2 higher than CsA at month 36, and values approximately 10 to 15 mL/min/1.73 m2 higher at month 72. The BME tofacitinib group also showed significantly higher MDRD-calculated eGFR than CsA for months 15 to 72 in the exploratory analysis. These allograft function findings were similar to those of the recent phase III BENEFIT study investigating long-term outcomes for kidney transplant recipients receiving a CNI-free regimen including belatacept versus CsA.11 Similarly, the ZEUS study comparing long-term efficacy of everolimus with CsA also showed sustained, although more modest, improvements in eGFR up to 5 years posttransplantation.23 However, both the BENEFIT and ZEUS studies reported significantly higher initial rates of acute rejection versus the CsA arm.14,23
Unfortunately, the small number of patients who underwent protocol biopsy at month 36 precluded adequate assessment of the rate of progression and severity of IFTA over time. The patients in this study were not evaluated for the development of donor-specific antibodies, thus preventing the assessment of an immunologic contribution to IFTA progression.
Similar rates of AEs were reported for the tofacitinib groups versus CsA. The most common types of AEs were infections and gastrointestinal disorders. Rates of serious infections increased in all treatment groups over time after month 12, such that the cumulative rates of serious infections during month 24 through month 36 were numerically higher for tofacitinib LI and significantly higher for tofacitinib MI versus CsA. In the parent study, 12-month serious infection rates were reported to be 25.3% for CsA, 37.0% for tofacitinib LI, and 44.5% for tofacitinib MI,20 whereas the cumulative rates of serious infections reported here at month 36 were 37.1% for CsA, 43.9% for tofacitinib LI, and 55.9% for tofacitinib MI, suggesting that the magnitude of the risk relative to CsA persisted. Although the proportion of patients with serious infections continued to increase over time in all treatment groups in this LTE study, among the tofacitinib-treated patients, the increase in the risk of serious infection appeared to slow over time with dose reduction. Specifically, in the first 12 months, when the tofacitinib-treated patients received 10 to 15 mg BID, serious infection rates were 25.0% and 38.9%, respectively, for the LI and MI patients who entered this LTE study. Between months 24 and 36, when all tofacitinib-treated patients received 5 mg BID, the cumulative rates of serious infection only increased by approximately 5% to 10% in the LI and MI groups.
Exposure-based analysis suggested that the risk of serious infection could be related to tofacitinib exposure, with the AME group having a statistically higher cumulative rate of serious infections versus CsA at the last evaluable time point (month 30) (53.1% vs 33.4%, P = 0.04). In contrast, the BME group maintained a generally similar cumulative serious infection rate versus CsA at month 36 (45.8% vs 37.1%), though serious infection risk increased with time in all treatment groups. These findings suggest that a risk of serious infection persists with long-term tofacitinib and CsA treatment.
Consistent with previous preliminary exposure-based analyses,19 there was an increased risk of developing PTLD in the tofacitinib AME group, which included all 5 patients with PTLD. As further confirmation, after implementation of a protocol amendment requiring discontinuation of all remaining AME patients, no additional cases of PTLD were reported.
There were higher cumulative rates of hematologic SAEs (eg, neutropenia/leukopenia or anemia) in the tofacitinib groups versus CsA at month 36. However, most of these hematologic SAEs occurred within the first 12 months posttransplant. In the parent study, lower MPA clearance was observed in patients receiving tofacitinib versus patients receiving CsA,20 which may have contributed to the higher rate of hematologic SAEs in the first 12 months. Mycophenolic acid area under the curve was not determined due to insufficient pharmacokinetic data. The extent to which concomitant MPA administration contributed to infection or hematologic risks of long-term tofacitinib treatment is unknown.
This is the first study to report long-term data on kidney transplant recipients treated with a JAK inhibitor. Nonetheless, our evaluation had several limitations. The implementation of the protocol amendment to discontinue patients with tofacitinib AME decreased patient numbers in the tofacitinib groups by approximately 50% across the tofacitinib groups. The lack of additional transplant studies with tofacitinib also likely prompted investigators and patients to discontinue from this study. The decrease in participating patients over time reduced the statistical power to assess the long-term safety profile of tofacitinib and introduced an imbalance in patient numbers between tofacitinib and CsA groups, confounding between-group comparisons. The use of CsA was also a limitation, preventing the comparison of tofacitinib with the current standard of care (tacrolimus). Also, only clinically stable patients who completed the parent study were eligible for enrollment in the LTE study, potentially resulting in a selection bias. The objective of this LTE study was to evaluate the clinical outcomes of long-term tofacitinib treatment, with an emphasis on events occurring after the first 12 months posttransplant. Although the use of KM analysis allowed assessment of the cumulative event rate through month 72, patient withdrawals that occurred at earlier time points and were censored could present a different risk profile to that presented for patients that remained in the study, which would violate the noninformative censoring assumption required by the KM analysis.
The findings from this LTE phase II study showed that long-term tofacitinib treatment continued to be effective in preventing acute rejection of renal allografts and preserving renal function. The current data confirmed an increased risk of PTLD associated with higher exposure of tofacitinib, as no other cases developed after discontinuation of the AME group. Long-term treatment with tofacitinib with MPA products was also associated with a persistent risk of serious infections. The long-term risk-benefit of a CNI-free regimen based on tofacitinib in kidney transplant patients has yet to be conclusively determined.
ACKNOWLEDGMENTS
The authors would like to thank the study investigators, research coordinators, patients, and study teams. Medical writing support, under the guidance of the authors, was provided by Rebecca Douglas, PhD, at CMC CONNECT, a division of Complete Medical Communications Ltd, Macclesfield, UK and was funded by Pfizer Inc, New York, NY in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med. 2015;163:461-464).
Footnotes
Published online 8 August, 2018.
S.B. has received grants from Novartis and has acted as a consultant for Genentech. F.G.V. has received grants from Amgen, Astellas, Bristol-Myers Squibb, Novartis, and Pfizer Inc. H.T.S. has received grants from Bristol-Myers Squibb, Novartis, and Pfizer Inc, and has acted as a consultant for, and received payment for lectures from, Bristol-Myers Squibb, Novartis, and Pfizer Inc. P.J.O. has acted as a consultant for, and has received travel support from, Pfizer Inc and has received payment for lectures and travel support from Astellas. A.Y., S.S., E.N.B., and Y.S.K. declare no conflicts of interest. J.J.F. has received grants from Pfizer Inc and consulting fees from Novartis, Sanofi, and Transplant Genomics Inc. KB has received research funds and/or honoraria from AbbVie, Alexion, Astellas, Bristol-Myers Squibb, Chiesi, Fresenius, Genentech, Hexal, Novartis, Otsuka, Pfizer, Roche, Siemens, Teva, and Veloxis Pharma. C.M.H., H.L., and G.C. are employees of Pfizer Inc and hold stock/stock options in Pfizer Inc.
The study described in this article was sponsored by Pfizer Inc.
All authors contributed to the concept, design, and development of the article, and approved the final article for submission.
ClinicalTrials.gov Identifier: NCT00658359.
REFERENCES
- 1.Li X, Zhuang S. Recent advances in renal interstitial fibrosis and tubular atrophy after kidney transplantation. Fibrogenesis Tissue Repair. 2014;7:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boor P, Floege J. Renal allograft fibrosis: biology and therapeutic targets. Am J Transplant. 2015;15:863–886. [DOI] [PubMed] [Google Scholar]
- 3.Nankivell BJ, PʼNg CH, OʼConnell PJ, et al. Calcineurin inhibitor nephrotoxicity through the lens of longitudinal histology: comparison of cyclosporine and tacrolimus eras. Transplantation. 2016;100:1723–1731. [DOI] [PubMed] [Google Scholar]
- 4.Chapman JR. Chronic calcineurin inhibitor use is nephrotoxic. Clin Pharmacol Ther. 2011;90:207–209. [DOI] [PubMed] [Google Scholar]
- 5.Bamoulid J, Staeck O, Halleck F, et al. The need for minimization strategies: current problems of immunosuppression. Transpl Int. 2015;28:891–900. [DOI] [PubMed] [Google Scholar]
- 6.Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol. 2009;4:481–508. [DOI] [PubMed] [Google Scholar]
- 7.Almeida CC, Silveira MR, de Araújo VE, et al. Safety of immunosuppressive drugs used as maintenance therapy in kidney transplantation: a systematic review and meta-analysis. Pharmaceuticals (Basel). 2013;6:1170–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ekberg H, Bernasconi C, Tedesco-Silva H, et al. Calcineurin inhibitor minimization in the Symphony study: observational results 3 years after transplantation. Am J Transplant. 2009;9:1876–1885. [DOI] [PubMed] [Google Scholar]
- 9.Ekberg H, Grinyó J, Nashan B, et al. Cyclosporine sparing with mycophenolate mofetil, daclizumab and corticosteroids in renal allograft recipients: the CAESAR Study. Am J Transplant. 2007;7:560–570. [DOI] [PubMed] [Google Scholar]
- 10.Rostaing L, Vincenti F, Grinyó J, et al. Long-term belatacept exposure maintains efficacy and safety at 5 years: results from the long-term extension of the BENEFIT study. Am J Transplant. 2013;13:2875–2883. [DOI] [PubMed] [Google Scholar]
- 11.Vincenti F, Rostaing L, Grinyo J, et al. Belatacept and long-term outcomes in kidney transplantation. N Engl J Med. 2016;374:333–343. [DOI] [PubMed] [Google Scholar]
- 12.Ekberg H, Tedesco-Silva H, Demirbas A, et al. Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med. 2007;357:2562–2575. [DOI] [PubMed] [Google Scholar]
- 13.Vincenti F, Charpentier B, Vanrenterghem Y, et al. A phase III study of belatacept-based immunosuppression regimens versus cyclosporine in renal transplant recipients (BENEFIT study). Am J Transplant. 2010;10:535–546. [DOI] [PubMed] [Google Scholar]
- 14.Vincenti F, Larsen CP, Alberu J, et al. Three-year outcomes from BENEFIT, a randomized, active-controlled, parallel-group study in adult kidney transplant recipients. Am J Transplant. 2012;12:210–217. [DOI] [PubMed] [Google Scholar]
- 15.Lee J, Lee JJ, Kim BS, et al. A 12-month single arm pilot study to evaluate the efficacy and safety of sirolimus in combination with tacrolimus in kidney transplant recipients at high immunologic risk. J Korean Med Sci. 2015;30:682–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flechner SM, Gurkan A, Hartmann A, et al. A randomized, open-label study of sirolimus versus cyclosporine in primary de novo renal allograft recipients. Transplantation. 2013;95:1233–1241. [DOI] [PubMed] [Google Scholar]
- 17.Flechner SM, Glyda M, Cockfield S, et al. The ORION study: comparison of two sirolimus-based regimens versus tacrolimus and mycophenolate mofetil in renal allograft recipients. Am J Transplant. 2011;11:1633–1644. [DOI] [PubMed] [Google Scholar]
- 18.Busque S, Leventhal J, Brennan DC, et al. Calcineurin-inhibitor-free immunosuppression based on the JAK inhibitor CP-690,550: a pilot study in de novo kidney allograft recipients. Am J Transplant. 2009;9:1936–1945. [DOI] [PubMed] [Google Scholar]
- 19.Vincenti F, Silva HT, Busque S, et al. Evaluation of the effect of tofacitinib exposure on outcomes in kidney transplant patients. Am J Transplant. 2015;15:1644–1653. [DOI] [PubMed] [Google Scholar]
- 20.Vincenti F, Tedesco Silva H, Busque S, et al. Randomized phase 2b trial of tofacitinib (CP-690,550) in de novo kidney transplant patients: efficacy, renal function and safety at 1 year. Am J Transplant. 2012;12:2446–2456. [DOI] [PubMed] [Google Scholar]
- 21.Racusen LC, Solez K, Colvin RB, et al. The Banff 97 working classification of renal allograft pathology. Kidney Int. 1999;55:713–723. [DOI] [PubMed] [Google Scholar]
- 22.Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem. 2014;57:5023–5038. [DOI] [PubMed] [Google Scholar]
- 23.Budde K, Lehner F, Sommerer C, et al. Five-year outcomes in kidney transplant patients converted from cyclosporine to everolimus: the randomized ZEUS study. Am J Transplant. 2015;15:119–128. [DOI] [PubMed] [Google Scholar]