

Supplemental Digital Content is available in the text
Keywords: differential methylation sites, gene expression profile, protein–protein interaction network
Abstract
We attempted to investigate the mechanism and susceptibility genes for diabetes in Han and Kazak ethnic individuals.
The abdominal omental adipose tissues were obtained from diabetic cases and healthy controls. The gene expression and methylation data were produced for Kazak and Han individuals, respectively, and analyzed by bioinformatics analysis.
We obtained 921 differentially expressed genes (DEGs) in Han group and 1772 in Kazak group. DEGs in Han group were significantly related with type 2 diabetes mellitus, and biosynthesis of amino acids, while the DEGs specific to Kazak patients were significantly enriched in metabolism-related pathways such as carbon metabolism, propanoate metabolism, and 2-oxocarboxylic acid metabolism. Major facilitator superfamily domain containing 1 (MFSD1) was found to be a methylation associated gene at hypermethylation site of cg16289538 in Han group. Rho guanine nucleotide exchange factor 1 (ARHGEF1) was the susceptible gene corresponding to the methylation sites of cg18800192 and cg00759295 in Kazak group. ARHGEF1 was also a node in protein–protein interaction network and significantly enriched in hsa04270: vascular smooth muscle contraction pathways.
The molecular mechanism of diabetes may be different in Han and Kazak patients. MFSD1 and ARHGEF1 may be the diabetes susceptible genes.
1. Introduction
Diabetes mellitus (DM) is a metabolic disease, which is characterized by high sugar in blood for a long period. The high blood sugar in DM results from insulin deficiency or cell response disorder. For different pathogenesis, diabetes can be classified into 2 subtypes, type 1 and 2 DM.[1] The classic signs of the 2 types of DM are weight loss, polyuria, polydipsia, and polyphagia.[2] Long-term diabetes can result in damage and dysfunction in eyes, kidney, blood vessels, and other tissues. It is reported that 387 million people are expected to be diagnosed with diabetes in 2014 globally.[3] It is estimated that diabetes cause deaths of about 1.5 to 4.9 million people each year from 2012 to 2014. Moreover, there will be an increasing trend of diabetes incidence from 2000 to 2030.[4] DM has become a global burden in recent years.
Although type 2 diabetes (T2D) patients can be grouped to obese and nonobese types, 80% patients are obese with an abdominal distribution of fat. Regional distribution of body fat has been proposed to contribute to T2D development.[5] It is reported that abdominal obesity has strong association with T2D incidence among ethnicities.[6] Abdominal obesity has been applied as an index for predicting glucose tolerance abnormalities for Chinese adults.[7] Recently, it is found that adipose tissue-derived biomolecules, also served as adipokines, can modulate the chronic diseases such as T2D.[8] Thus, the investigations for adipose tissue may help understanding the mechanism of T2D development. However, the ethnic differences in T2D development have not been clarified yet.
DNA methylation by drugs or alkylation reagents may result in other methylation sites and cause mismatch pairing, which may play a role in the pathogenesis of various diseases like cancer and possibly diabetes.[9] Microarray technology has been widely applied to give a genetic landscape of complex diseases.[10] Microarray analysis of gene expression or methylation profiling has been performed to investigate the T2D-related molecular mechanism.[11,12] In this article, we analyzed the diabetes-related genes and DNA methylation sites in Han and Kazak ethnic individuals, respectively. In the present study, we attempted to compare the gene expression and methylation patterns in diabetics of 2 ethnicities.
2. Methods
2.1. Subjects
In our study, Han and Kazak male patients admitted in Xinjiang Uygur Autonomous Region People's Hospital Department were investigated. T2D patients were diagnosed based on the World Health Organization (WHO) 1999 diagnostic criteria and International Obesity Task Force Asian adult standard 2000. Cases with type 1 diabetes, tumor, acute inflammation, and liver and kidney disease were excluded in our study and those took drugs that affected the glucolipid metabolism were also excluded. Finally, patients with T2D (fasting plasma glucose ≥ 7.0 mmol/L, 2h plasma glucose ≥ 11.1) were included in this study. Healthy participants underwent standard physical examination in our hospital were recruited as controls.
In total, 36 abdominal omental adipose tissues from 12 ethnic Han males (6 T2D and 6 controls) and 12 ethnic Kazak males (6 T2D and 6 controls) were obtained under abdominal operation. All the samples were ranged into 3 groups: obesity group (n = 12, 6 Han and 6 Kazak), diabetes group (n = 12, 6 Han and 6 Kazak), and normal group (n = 12, 6 Han and 6 Kazak).
The study procedure was approved by Ethics Committee of Xinjiang Uygur Autonomous Region People's Hospital and performed in accordance with the ethical standards. All the subjects had given the written informed consent before study.
2.2. RNA microarray
Total RNA was extracted from fat tissues of diabetes and normal individuals using TRIZOL reagent (Cat. No. 15596-026, Life Technologies, Carlsbad, CA) and purified by using an RNeasy mini kit (Cat. No. 74106, QIAGEN, GmBH, Hilden, Germany). Then, RNA was amplified and labeled using a Low RNA Input Linear Amplification kit (Cat. No. 5184-3523, Agilent Technologies, Santa Clara, CA), 5-(3-aminoallyl)-UTP (Cat. No. AM8436, Ambion, Austin, TX), and Cy3 NHS ester (Cat. No. PA13105, GE healthcare Biosciences, Pittsburgh, PA), according to the manufacturer's instructions. The gene expression profiles were produced based on the Agilent array platform.
2.3. DNA methylation microarray
DNA methylation array data of fat tissues were developed from a replication set of 5 Han diabetic patients, 3 Kazak diabetic cases, 5 Han healthy controls, and 5 Kazak healthy controls. Genomic DNA was extracted by standard phenol–chloroform methodologies. The quality of DNA was evaluated by a Spectrophotometer (Nano-Drop Technologies, Wilmington, DE). Total of 1.8 g DNA from each sample was bisulfate converted by EZ-DNA methylation kit (Zymo Research Corporation, California) according to manufacturers’ protocols. The methylation data were measured based on the platform of Illumina HumanMethylation450 BeadChip.
2.4. Gene expression data analysis
After removing unreliable probe sets’ signals, the expression array data were preprocessed by Limma package,[13] including background correction, quantile normalization, gene symbol transformation, and summarization.[14]
2.5. DNA methylation array data preprocessing
The DNA methylation array data were processed by IMA package[15] (https://www.rforge.net/IMA/). Beta matrix represents that the degree of DNA methylation variability was obtained. Then, M-value matrix was calculated for quantifying methylation levels. Based on the annotated information of the platform of Illumina Human Methylation 450K, the methylation sites in X and Y chromosome and the probes containing single nucleotide polymorphism sites were deleted.[16]
2.6. Differentially expressed gene analysis
The differentially expressed genes (DEGs) in T2D patients were identified compared with controls by using Limma package in R. The cut-off value was set as |log2 fold change| > 0.585 and P value < .05. In order to evaluate the differential expression of DEGs in T2D and control cases, hierarchical clustering analysis was performed by pheatmap 1.0.8 package in R (https://cran.r-project.org/web/packages/pheatmap/index.html). The difference between DEGs lists originating from Han and Kazak diabetic cases were analyzed by Venn diagram.
2.7. Differential DNA methylation sites
Intergroup variation of DNA methylation was analyzed in Han ethnic and Kazak ethnic population, respectively. Differential methylation sites of T2D patients were identified by CpGassoc package 2.60[17] (https://cran.r-project.org/web/packages/CpGassoc/) with P value < .05. The difference of differential methylation sites was observed and the distribution of methylation sites in gene and chromosome was focused.
2.8. Integrated analysis of DEGs and differential methylation sites
To investigate the effect of DNA methylation variation on gene expression patterns, the gene expression profiles of genes at the site of aberrant DNA methylation were collected. The up-regulated genes that differentially expressed due to hypomethylated status and the down-regulated ones due to hypermethylated status were focused. The genes that closely related with methylation status were displayed by circos software[18] (http://circos.ca/).
2.9. TFBS analysis
The transcription factors (TFs) targeting the methylation-related genes were searched against ITFP (http://itfp.biosino.org/itfp) and TRANSFAC (http://www.gene-regulation.com/pub/databases.html) databases.
The TF binding motif and base sequences within upstream from 2k bp to downstream 100 bp were predicted by MotifDb package in R (Version 1.18.0, http://www.bioconductor.org/packages/release/bioc/html/MotifDb.html). Position weight matrix algorithm was used to predict the transcription factor binding sites (TFBS) in binding motif and gene promoter region with the score >85%. The methylation sites located in TFBS region (from 101 bp upstream to 10 bp downstream) of target genes were focused.
2.10. PPI and function analysis of the methylation-related DEGs
The protein–protein interactions (PPIs) of the methylation-related DEGs were analyzed based on STRING database 10.5 (https://string-db.org/).[19] The protein pairs with required confidence (combined score) > 0.4 were collected for PPI network construction with the application of Cytoscape software 3.4.0 (http://cytoscape.org/).[20] The connectivity degrees of all nodes in PPI network were calculated and hub nodes were selected.
The genes in PPI network were subjected to gene ontology (GO) function[21] and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis[22] by Database for Annotation, Visualization and Integrated Discovery online tool[23] (Version: 6.8, https://david.ncifcrf.gov/).
3. Results
3.1. Subjects
Based on the inclusion and exclusion criteria, a total of 24 subjects were included in this study. In Han group, there were 6 diabetes with mean age of 47.5 ± 5.6 years old (3 males and 3 females), and 6 paired controls (male:female = 1:1, mean age, 48.7 ± 4.4 years old). In Kazak group, 6 diabetes with mean age of 51.8 ± 5.7 years old and 6 controls (mean age: 49.0 ± 5.2 years old) were included. There were no difference in the age, height, and the level of total cholesterol, triglyceride, low-density lipoprotein, and high-density lipoprotein between diabetes and controls of the 2 ethnic group (P > .05). Besides, all the basic information of diabetes were similar between Han and Kazak groups (P > .05) (Table S1).
3.2. DEG analysis
After gene expression data preprocessing, we obtained 13,194 genes of 24 samples. In Han ethnic individuals, there were 921 DEGs (213 were up-regulated and 708 were down-regulated) in diabetic patients compared with controls. In Kazak diabetics, a total 1772 genes were obtained, including 631 up-regulated genes and 1141 down-regulated ones. Heat map of DEGs suggested that the diabetic and control samples were clearly separated by the expression profile of DEGs in Han ethnic people (Fig. 1A), while in Kazak individuals, the samples were relatively distinguished excepted for the TF301 sample (Fig. 1B).
Figure 1.
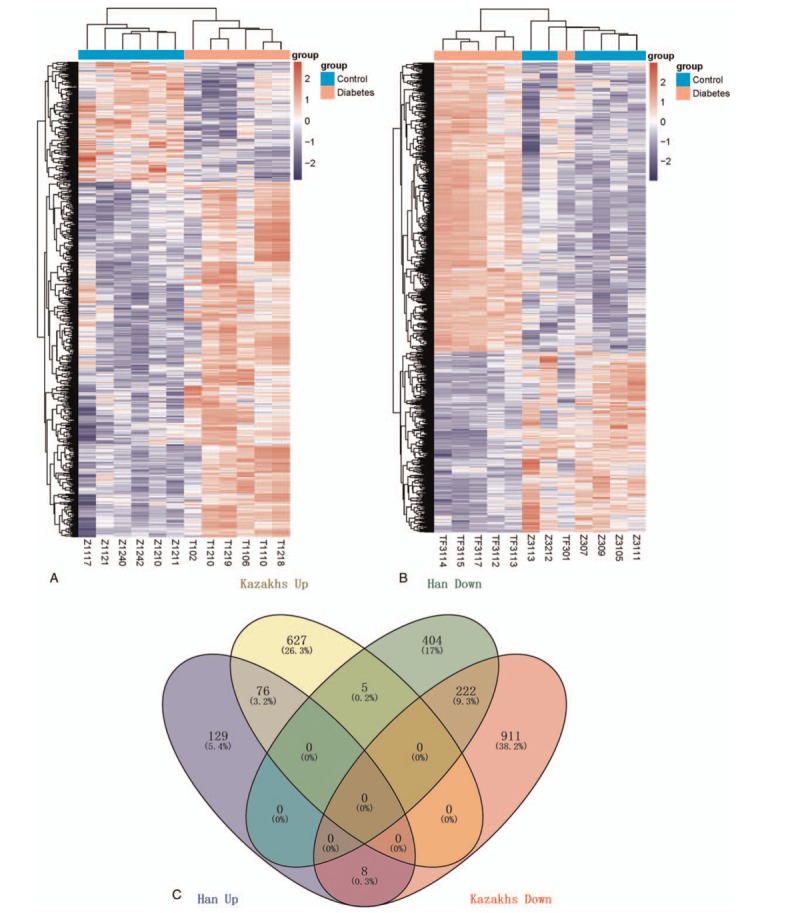
(A) Hierarchical clustering of differentially expressed genes in Han group; (B) hierarchical clustering of differentially expressed genes in Kazak group; (C) and Venn diagram of differentially expressed genes in Han and Kazak groups.
Venn diagram illustrates that 76 genes were commonly overexpressed in both Han and Kazak diabetic patients and 222 genes were commonly down-regulated in both ethnic peoples. In total, 129 genes were particularly up-regulated in Han diabetics and 404 genes were particularly down-regulated in Han diabetics; while in Kazak individuals, there were 627 up-regulated genes and 911 down-regulated genes in diabetics. Besides, 8 genes were up-regulated in Han patients, while reversely expressed in Kazak patients. Similarly, 5 genes were down-regulated in Han diabetics, while up-regulated in Kazak patients (Fig. 1C).
3.3. Functional analysis
Pathway and function annotation were performed for the common or ethnic-specific DEGs in the 2 ethnic groups. Figure 2A shows that many GO terms were overrepresented by both ethnic-specific DEGs and common DEGs. Up-regulated genes are closely related with positive regulation of secretion, regulation of blood circle and actin-mediated cell contraction. The down-regulated genes were mainly enriched in immune-related biological processes, such as neutrophil-mediated immunity, neutrophil activation, neutrophil activation involved in immune response, granulocyte activation, and T cell activation (Fig. 2A). The overrepresented pathways of the down-regulated genes included Osteoclast differentiation, antigen processing, and presentation. With regard to the up-regulated genes, Han ethnic-specific DEGs were mainly enriched in biosynthesis of amino acids, prostate cancer, and type 2 DM. Kazak ethnic-specific DEGs were closely related with metabolism pathways such as carbon metabolism, propanoate metabolism, and 2-oxocarboxylic acid metabolism, while the common up-regulated genes in both ethnic groups were significantly enriched in fatty acid metabolism (Fig. 2B).
Figure 2.

Function and pathway analysis of the differentially expressed genes. (A) Significant biological functions for Han, Kazak-specific genes and common genes and (B) significant pathways for Han, Kazak-specific genes and common genes.
3.4. Differential methylation sites identification
Based on methylation microarray data analysis, there were 46,871 differential methylation sites in Han ethnic diabetics compared with controls, which contained 7352 hypomethylation sites (corresponding to 4848 genes), and 39,519 hypomethylation sites (corresponding to 3825 genes). In Kazak diabetics, there were 22,046 differential methylation sites, including 6812 hypermethylation sites (corresponding to 3825 genes) and hypomethylation sites (corresponding to 15,234 genes). The hypermethylation site in Han ethnic group took account for 15.7% of all the differential methylation sites, while it was 30.9% in Kazak group (Fig. 3A). The rate of hypermethylation sites was higher in Kazak group than in Han ethnic group.
Figure 3.

The distribution of differential methylation sites. (A) PiePlot of rate of hypermethylation and hypomethylation sites in Han group; (B) distribution of differential methylation sites in genome of Han group; (C) PiePlot of the rate of hypermethylation and hypomethylation sites in Kazak group; and (D) distribution of differential methylation sites in genome of Kazak group.
The locations of methylated sites in genome were annotated. As shown in Fig. 3B, there was no significant difference in the distribution of differential methylation sites in 2 ethnic groups.
3.5. Integrated analysis of gene expression data and methylation data
In gene promoter region, the differential methylation sites with reversely expressed genes were collected. In Han ethnic group, there were 14 hypermethylation sites corresponding to 12 down-regulated genes and 5 hypomethylation sites corresponding to 5 down-regulated genes. In Kazak group, there were 150 hypermethylation sites (110 down-regulated genes) and 52 hypomethylation sites (43 up-regulated genes) (Fig. 4). No common methylation sites were observed in 2 ethnic groups.
Figure 4.
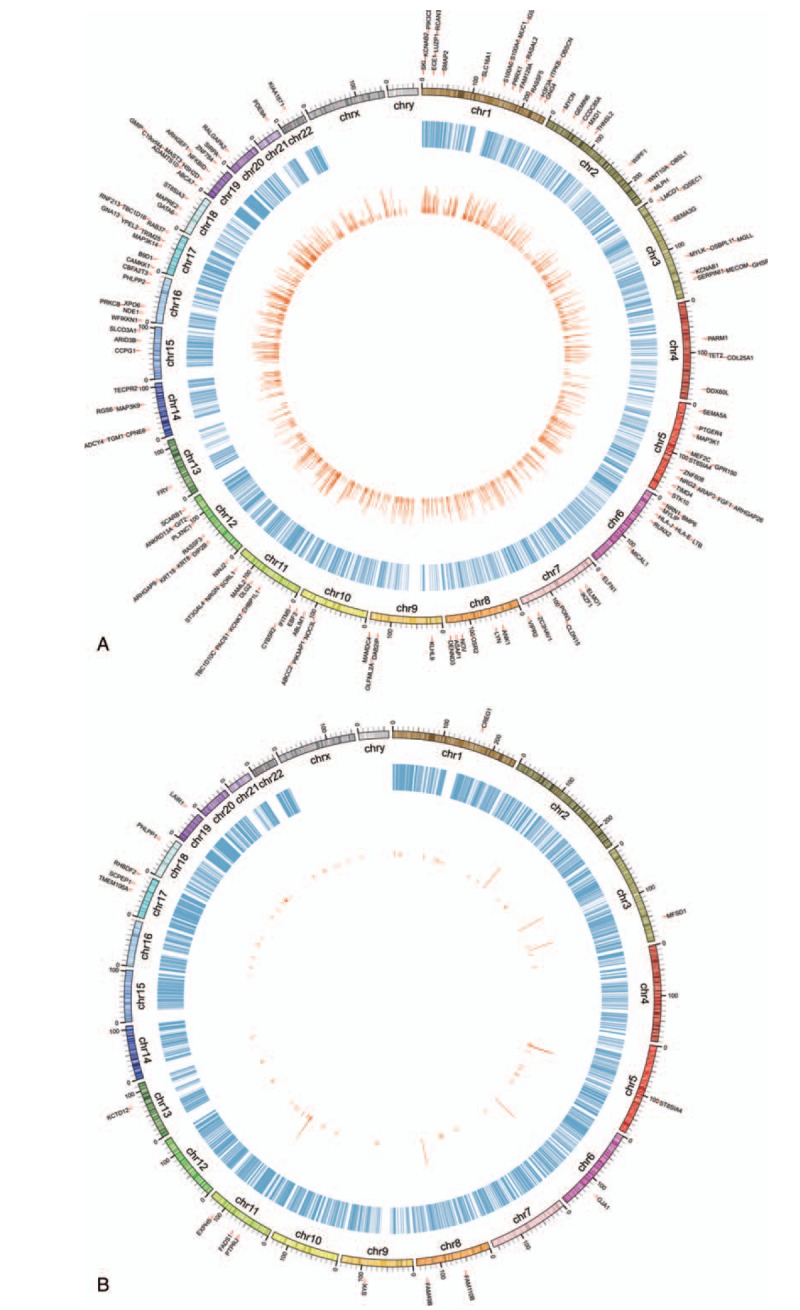
Circos diagram of the interactions between differential methylation sites and risk genes. (A) Han group and (B) Kazak group.
3.6. TFBS analysis
TFBS analysis was performed for genes that affected by aberrant methylation sites. The hypermethylation site cg16289538 in Han ethnic group, hypermethylation sites cg00759295, cg18800192, cg02142767 and hypomethylation site cg04251733 were located within from upstream 10 bp to downstream 10 bp of TFBS. The detailed information of the predicted TFBS was listed in Table 1. cg02142767 was located in scavenger receptor class B, member 1 (SCARB1, target gene) binding site of general transcription factor IIIC subunit 2 (GTF3C2, TF).
Table 1.
Transcriptional binding sites around the methylation sites.

3.7. PPI network
By searching the aberrant methylation-related genes against STRING database, 4 protein pairs were obtained in Han ethnic group, containing spleen tyrosine kinase (SYK)–protein tyrosine phosphatase, receptor type J (PTPRJ); SYK-PH domain and leucine-rich repeat protein phosphatase 1 (PHLPP1); PHLPP1-PTPRJ and PHLPP1-serine carboxypeptidase 1 (SCPEP1). In Kazak group, there were 87 protein pairs connecting 66 nodes. In the PPI network, PHLPP2, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta (PIK3CD), protein kinase C, beta (PRKCB), and G protein subunit gamma 4 (GNG4) were hub nodes (Fig. 5A). Genes in PPI network were closely related with inositol phosphate metabolic process, synapse assembly related biological process, inflammatory mediator regulation of TRP channels, inflammatory mediator regulation of TRP channels, and NF-kappa B signaling pathway (Fig. 5B and C).
Figure 5.
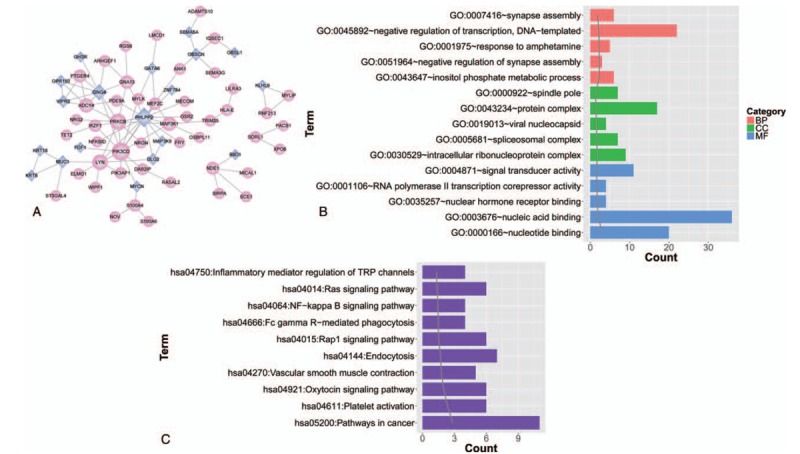
(A) Protein–protein interaction (PPI) network of risk genes in Kazak group; (B) significant gene ontology (GO) function terms enriched by genes in PPI network; and (C) significant pathways enriched by genes in PPI network.
4. Discussion
DM is a metabolic disease and has posed a serious health burden on a global scale. The prevalence of DM has been found vary among ethnic groups. In order to investigate the difference in genetic mechanism of T2D development between Han and Kazak ethnic groups, the microarray analyses of the gene expression and methylation pattern of abdominal omental adipose tissues in diabetics were performed. The feature genes in diabetics of different ethnic groups were analyzed.
Our data showed that total 921 genes were differentially expressed in Han ethnic diabetics and 1772 in Kazak ethnic group. The number of DEGs in Kazak group was larger than in Han ethnic group, which suggested that the gene expression profile was affected by different ethnic backgrounds.
The similar findings were observed in GO and pathway analysis. The up-regulated genes specific to Han patients were significantly related with type 2 DM and biosynthesis of amino acids. T2D-related pathways were dysregulated in Han ethnic patients, which suggested that our findings were significant. Different from Han ethnic group, the DEGs specific to Kazak patients were significantly enriched in metabolism-related pathways such as carbon metabolism, propanoate metabolism, and 2-oxocarboxylic acid metabolism. These mentioned above indicated that there were some differences in the mechanism of diabetes development between Han and Kazak ethnic patients.
Methylation plays a regulatory role in gene expression and the changes in methylation are involved in the dysfunction of biological processes. Genetic predisposition is the main risk factor for T2D development and is closely related with disease susceptibility. It is reported that differential DNA methylation has been found in the promoter of genes involved in glucose metabolism.[11] The genes that affected by differential DNA methylation were proposed to be the candidate genes for T2D development. DNA methylation pattern analysis facilitated the identification of new targets for T2D. In order to explore the candidate genes in T2D development, we performed integrated analysis of methylation analysis and gene expression profiling. The differential methylation sites-related genes were focused and the TFBS were predicted.
In Han ethnic group, the hypermethylation site (cg16289538) was located in the target gene major facilitator superfamily domain containing 1 (MFSD1) of TF (E2F transcription factor 4 [E2F4]), which indicated that the changes of methylation may affect the E2F4 binding to gene promoter that regulated in MFSD1 gene expression. A previous genome wide analysis showed that MFSD1 was a susceptible gene in chronic periodontitis.[24] In addition, MFSD1 was reported to interact with interleukin 8 (IL-8) that may share the common function with IL-8.[25]IL-8 is a proinflammatory cytokine, which was closely related with diabetes development. The IL-8 gene promoter polymorphisms increased the risk of T2D development and the genes that interacted with IL-8 could be the susceptible genes for T2D.[26] Thus, we speculated that MFSD1 gene at the hypermethylation sites could be the susceptible to T2D.
Similarly, rho guanine nucleotide exchange factor 1 (ARHGEF1) gene was the targets for TF (zinc finger protein 160, ZNF160) at 2 hypermethylation sites in Kazak ethnic patients. ARHGEF1 is a member of rho guanine nucleotide exchange factors and has effects on pulmonary leukocyte function, vascular tone, and blood pressure.[27] Evidence has suggested that ARHGEF11 R1467H polymorphism is closely associated with the risk of T2D in Chinese population.[28] Thus, we speculated that ARHGEF1 was a candidate risk gene for T2D development. The significant role of ARHGEF1 in T2D has been proved by PPI network analysis. PPI network was comprised of methylation susceptibility genes and ARHGEF1 was a node in PPI network, which indicated that ARHGEF1 had multiple interactions with others. The function annotation showed that ARHGEF1 was closely related with vascular smooth muscle contraction, which was consistent with the fact that ARHGEF1 regulated blood pressure by mediating angiotensin II.[29] Hypertension and diabetes shared the same characteristic of higher carotid/femoral pulse wave velocity.[30]ARHGEF1 has been suggested as the therapeutic target for hypertension. Thus, we speculate that ARHGEF1 may be a diabetes susceptibility gene.
In conclusion, the pathways involved in T2D progression may be different in different ethnic individuals. MFSD1 gene may be a T2D susceptible gene in Han ethnic group and ARHGEF1 may be a diabetes susceptibility gene for Kazak ethnic individuals. Our findings may provide genetic landscape of the mechanisms of diabetes in different ethnic patients. However, these results should be further confirmed by more samples in the future.
Author contributions
Conceptualization: Wei Li, Peng Xu, Zhiwei Zhang, Tingting Wang.
Formal analysis: Jun Li, Yan Wang, Siyuan Li.
Writing – original draft: Cuizhe Wang, Xiaodan Ha.
Writing – review & editing: Cuizhe Wang, Jianxin Xie, Jun Zhang.
Supplementary Material
Footnotes
Abbreviations: ARHGEF1 = rho guanine nucleotide exchange factor 1, DEG = differentially expressed gene, DM = diabetes mellitus, E2F4 = E2F transcription factor 4, GO = gene ontology, IL-8 = interleukin 8, MFSD1 = major facilitator superfamily domain containing 1, PHLPP1 = PH domain and leucine-rich repeat protein phosphatase 1, PPI = protein–protein interaction, PTPRJ = protein tyrosine phosphatase, receptor type J, SYK = spleen tyrosine kinase, T2D = type 2 diabetes, TFBS = transcription factor binding sites, TFs = transcription factors, ZNF160 = zinc finger protein 160.
CW and XH are co-first authors.
This study was supported by the Natural Science Foundation of China (No. 81360142); Applied Basic Research Project of Xinjiang Corps (No. 2015AG016); National Basic Research Program of China (973 Program) (No. 2009CB526505); and The Program for High-Level Personnel Start at Shihezi University (No. RCZX201230, gxjs2012-zdgg02).
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Mellitus D. Diagnosis and classification of diabetes mellitus. Diabetes Care 2005;28:S37–42. [DOI] [PubMed] [Google Scholar]
- [2].Cooke DW, Plotnick L. Type 1 diabetes mellitus in pediatrics. Pediatr Rev 2008;29:374–84. [DOI] [PubMed] [Google Scholar]
- [3].Beagley J, Guariguata L, Weil C, et al. Global estimates of undiagnosed diabetes in adults. Diabetes Res Clin Pract 2014;103:150–60. [DOI] [PubMed] [Google Scholar]
- [4].Wild S, Roglic G, Green A, et al. Global prevalence of diabetes estimates for the year 2000 and projections for 2030. Diabetes Care 2004;27:1047–53. [DOI] [PubMed] [Google Scholar]
- [5].Despres J. Abdominal obesity as important component of insulin-resistance syndrome. Nutrition 1992;9:452–9. [PubMed] [Google Scholar]
- [6].Freemantle N, Holmes J, Hockey A, et al. How strong is the association between abdominal obesity and the incidence of type 2 diabetes? Int J Clin Pract 2008;62:1391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].He Y, Zhai F, Ma G, et al. Abdominal obesity and the prevalence of diabetes and intermediate hyperglycaemia in Chinese adults. Public Health Nutr 2009;12:1078–84. [DOI] [PubMed] [Google Scholar]
- [8].Rega-Kaun G, Kaun C, Wojta J. More than a simple storage organ: adipose tissue as a source of adipokines involved in cardiovascular disease. Thromb Haemost 2013;110:641–50. [DOI] [PubMed] [Google Scholar]
- [9].Kou Y, Koag MC, Lee S. N7 methylation alters hydrogen bonding patterns of guanine in duplex DNA. J Am Chem Soc 2015;137:14067–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Heller MJ. DNA microarray technology: devices, systems, and applications. Annu Rev Biomed Eng 2002;4:129–53. [DOI] [PubMed] [Google Scholar]
- [11].Maier S, Olek A. Diabetes: a candidate disease for efficient DNA methylation profiling. J Nutr 2002;132(suppl):2440S–3S. [DOI] [PubMed] [Google Scholar]
- [12].Wilson KH, Eckenrode SE, Li QZ, et al. Microarray analysis of gene expression in the kidneys of new- and post-onset diabetic NOD mice. Diabetes 2003;52:2151–9. [DOI] [PubMed] [Google Scholar]
- [13].Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003;4:249–64. [DOI] [PubMed] [Google Scholar]
- [15].Wang D, Yan L, Hu Q, et al. IMA: an R package for high-throughput analysis of Illumina's 450K Infinium methylation data. Bioinformatics 2012;28:729–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sboner A, Demichelis F, Calza S, et al. Molecular sampling of prostate cancer: a dilemma for predicting disease progression. BMC Med Genomics 2010;3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Barfield RT, Kilaru V, Smith AK, et al. CpGassoc: an R function for analysis of DNA methylation microarray data. Bioinformatics 2012;28:1280–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Res 2009;19:1639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].von Mering C, Huynen M, Jaeggi D, et al. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res 2003;31:258–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Huang DW, Sherman BT, Tan Q, et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 2007;8:R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Teumer A, Holtfreter B, Völker U, et al. Genome-wide association study of chronic periodontitis in a general German population. J Clin Periodontol 2013;40:977–85. [DOI] [PubMed] [Google Scholar]
- [25].Rk G, Ms K, Kk G, et al. Evaluation of Hs-CRP levels and interleukin 18 ((137G/C) promoter polymorphism in risk prediction of coronary artery disease in first degree relatives. PLoS ONE 2015;10:e0120359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sun F, Kobayashi M, Fukushima T. Association between IL-18 gene promoter polymorphisms and CTLA-4 gene 49A/G polymorphism in Japanese patients with type 1 diabetes. J Autoimmun 2004;22:73–8. [DOI] [PubMed] [Google Scholar]
- [27].Peelman F, Tavernier J. ROCKing the JAKs. JAKSTAT 2013;2:e24074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu J, Chen X, Guo Q, et al. Association of ARHGEF11 R1467H polymorphism with risk for type 2 diabetes mellitus and insulin resistance in Chinese population. Mol Biol Rep 2011;38:2499–505. [DOI] [PubMed] [Google Scholar]
- [29].Bernstein KE, Fuchs S. Angiotensin II and JAK2 put on the pressure. Nat Med 2010;16:165–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Benetos A, Zervoudaki ASA. Effects of lean and fat mass on bone mineral density and arterial stiffness in elderly men. Osteoporos Int 2009;20:1385–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.