

Supplemental Digital Content is available in the text
Keywords: cone-rod dystrophy, cosegregation analysis, CRX mutation, sanger sequencing, whole-exome sequencing
Abstract
Background:
Cone-rod dystrophy (CORD) is an inherited, progressive retinal disorder with genetic and phenotypic heterogeneity. Here, we aimed to identify the pathogenic mutation in affected individuals in a Chinese family with autosomal dominant cone-rod dystrophy (adCORD).
Methods:
Genomic DNA and clinical examination results were collected from a Chinese family presenting with adCORD. The candidate disease-causing mutations were screened with whole-exome sequencing (WES) and bioinformatics analyses. Sanger sequencing was used for validation and cosegregation analysis.
Results:
A novel frameshift mutation (NM_000554.4; c.538dupG:p.Val180fs) in exon 4 of the CRX gene was identified in all affected individuals in the Chinese family with adCORD. Cosegregation analysis confirmed that this mutation was cosegregated with the disease. This variant, which results in premature termination of the protein, was absent from all public variant databases or internal exome databases.
Conclusions:
We used whole-exome sequencing to identify a novel CRX mutation causing adCORD in a Chinese family. This study broadens the known pathogenic mutation spectrum of the CRX gene and shows the potential of WES in identifying the pathogenic mutations of CORD disease.
1. Introduction
Cone-rod dystrophy (CORD) is an inherited, progressive retinal disorder with a prevalence of approximately 1 in 40,000.[1] The predominant symptoms include progressively declining visual acuity, photophobia, color vision disturbances and night blindness, and nystagmus is often present. Electroretinogram (ERG) shows cone photoreceptor dysfunction or loss at an early stage, later followed by rod photoreceptor disorder. Fundus examination reveals retinal pigmentation and various degrees of retinal atrophy.[2,3] CORD is genetically and phenotypically heterogeneous and is transmitted through 3 forms of inheritance: autosomal dominant (AD), autosomal recessive (AR), and X-linked (XL) inheritance.[4–7] At present, >30 genes are reported to be associated with cone and cone-rod dystrophy in the RetNet database (http://www.sph.uth.tmc.edu/retnet/). No therapies are yet available to treat these diseases.
Here, we report a 4-generation Chinese family of 8 patients with adCORD. We used whole-exome sequencing (WES) to identify the pathogenic mutations in these affected individuals and discovered a novel mutation in the CRX gene.
2. Methods
2.1. Subjects and examinations
This research project was approved by the ethics committee of the National Research Institute for Family Planning. Written informed consent adhered to the tenets of the Declaration of Helsinki and the ARVO statement on the use of human subjects.
A 4-generation Chinese family including 8 affected individuals was enrolled in this study (Fig. 1). All ophthalmic examinations, including visual acuity, fundus examination, fluorescein fundus angiography (FFA), optical coherence tomography (OCT), visual field examination, and full field electroretinography (F-ERG), were performed at the Maternal and Child Health Hospital, Haidian, Beijing, where the diagnosis was made.
Figure 1.
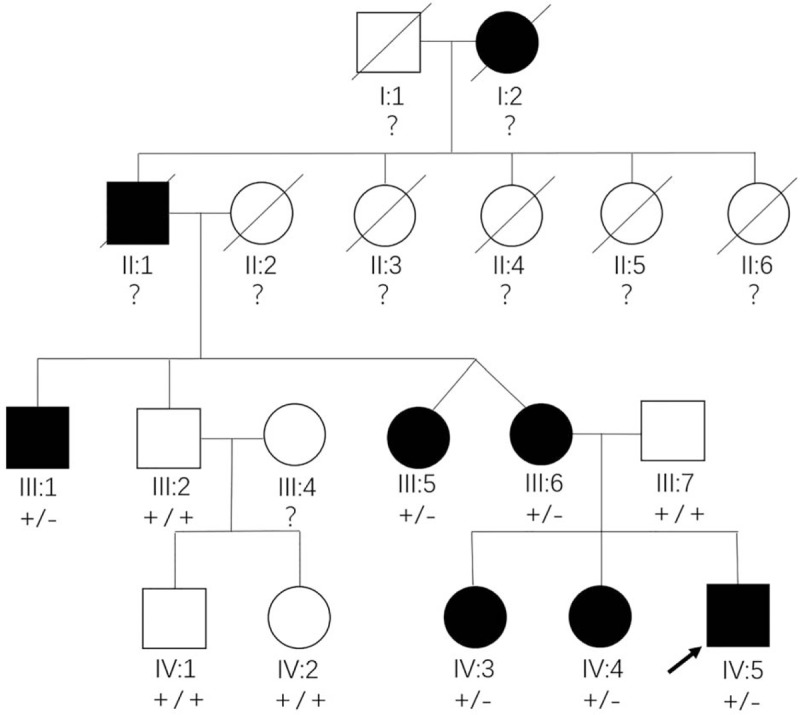
Pedigree of a 4-generation Chinese family with autosomal dominant cone-rod dystrophy. Square: male, circle: female, filled symbol: patient, unfilled symbol: unaffected member, slash: deceased, black arrow: the proband, plus: normal allele, minus: mutant allele, question marks: members whose genotype could not be examined.
2.2. Whole-exome sequencing and variant analyses
Peripheral venous blood was collected from 10 participants and genomic DNA was extracted with the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany). The DNA concentrations were measured with a NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA). WES was performed by a commercial company, Macrogen (Shenzhen, China). Briefly, all exons were captured with the SureSelect Human All Exon V5 Kit (Agilent, Santa Clara, CA) and the enriched DNA fragments were sequenced with a HiSeq 4000 sequencer (Illumina, San Diego, CA). The Burrows-Wheeler Alignment (BWA, http://bio-bwa.sourceforge.net/bwa.shtml) algorithm was used to align the sequencing reads against the human reference genome. The Genome Analysis Toolkit (GATK, https://www.broadinstitute.org/gatk/) was used to call variants such as single nucleotide variants (SNVs) and indels. SnpEff (http://snpeff.sourceforge.net/SnpEff.html) was then used to annotate the variants and predict their functional effects.
The candidate pathogenic variants were filtered as follows: the exonic non-synonymous mutations in known retinal disease-causing genes in the RetNet database were extracted from the WES results, including the missense, nonsense, frameshift, and splice-site mutations; and mutations with a minor allele frequency (MAF) ≥0.01 in the 1000 Genomes (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/), ESP6500 (http://evs.gs.washington.edu/EVS/), and ExAC (http://exac.broadinstitute.org/) databases were excluded. The impact of the filtered candidate mutations on the proteins was predicted with 4 online programs: PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), MutationTaster (http://www.mutationtaster.org/), and SNPs&GO (http://snps.biofold.org/snps-and-go/).
2.3. Sanger sequencing
Sanger sequencing was used to validate the presence of the filtered mutations for the proband. The forward and reverse primers for the BBS4, CRX, and RAX2 genes were as follows: 5′-ATGCTCTTATTCCCAGTG-3′, 5′-CTTAGGGTACAAGGTGTCT-3′ (BBS4); 5′-CCCCATCTCCGCTCTTAT-3′, 5′-TCTGAAACTTCCAGGCACTC-3′ (CRX); and 5′-TGGGCTTAGGGAACGCTTGG-3′, 5′-GGTCACCTCCTGCCTGACACT-3′ (RAX2). Cosegregation analysis of the mutations and disease phenotype was performed with Sanger sequencing for all available pedigree members.
3. Results
3.1. Clinical manifestations
The proband (Fig. 1, IV:5), a 19-year-old man, first presented 10 years ago with binocular vision loss, photophobia, and impaired vision in the dark without any obvious cause. The uncorrected visual acuity of each eye was 0.5 and no significant improvement was observed with spectacles. Since then, the proband's visual acuity has decreased progressively. Over the past 4 years, binocular vision has decreased significantly and the night blindness has become severe. The ocular examination results showed that uncorrected visual acuity was 0.15 (right eye) and 0.1 (left eye), with no improvement observed with correction. We found no abnormality in the anterior segment. Fundus examination showed a clear optic disc border, slightly light color, a thinning artery, mid-peripheral diffuse retinal pigment epithelium (RPE) atrophy, gold foil-like reflection, and geographic pigment epithelium atrophy foci in each macular area (Fig. 2A). FFA showed delayed retinal arterial filling, diffuse transmitted fluorescence, and irregularly geographic hypofluorescence in the macular area (Fig. 2B). OCT revealed a thinning retinal nerve epithelium layer in the binocular macular area, a significantly thinning outer nuclear layer of the fovea centralis, and an absent fovea centralis, with the structure of the inner segment/outer segment (IS/OS) layer being difficult to distinguish (Fig. 2C). Visual field examination revealed quadrant defects in the binocular nasal field (Fig. 3A). F-ERG showed the binocular rod cell response amplitude and maximum response amplitude were both decreased, and both the cone cell reaction and 30 Hz scintillation reaction indicated the absence of an effective waveform (Fig. 3B).
Figure 2.
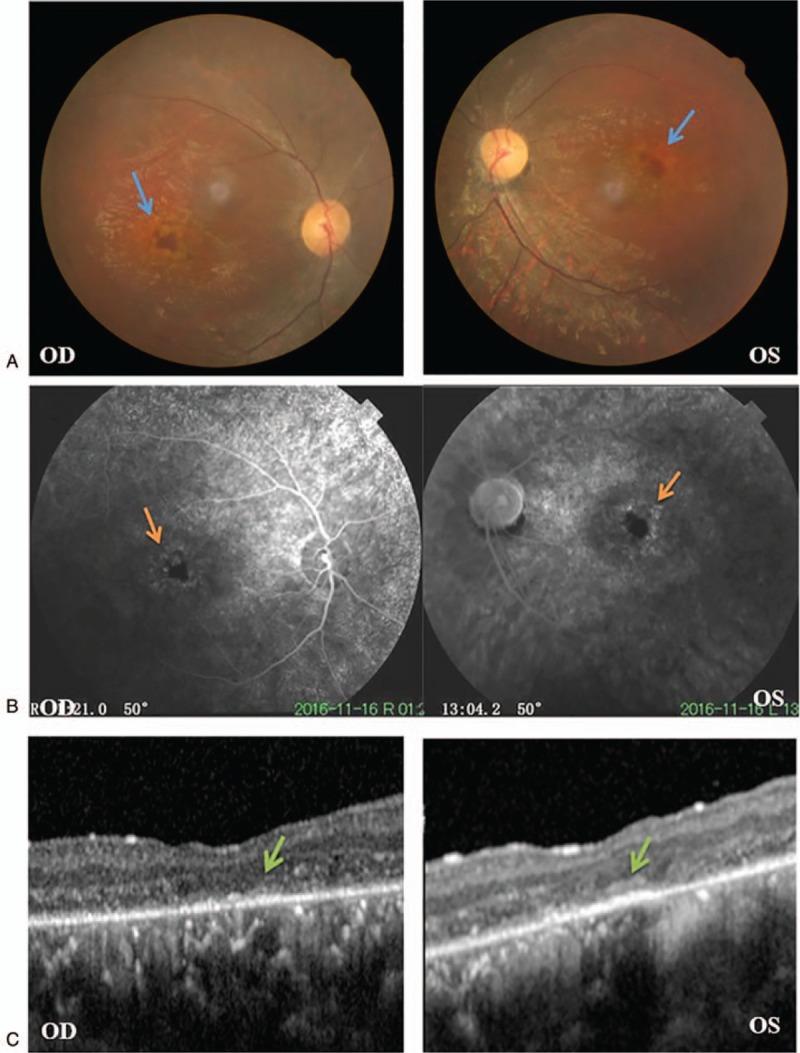
Ophthalmic examination results of the proband. A: Fundus images showed retinal pigment epithelium atrophy and gold foil-like reflection in the macular area. Blue arrow: geographic atrophy foci. B: Fluorescein fundus angiography showed diffuse transmitted fluorescence and geographic hypofluorescence in the macular area (orange arrow). C: Optical coherence tomography showed an absent fovea centralis and an undefined IS/OS layer (green arrow). OD: right eye, OS: left eye.
Figure 3.
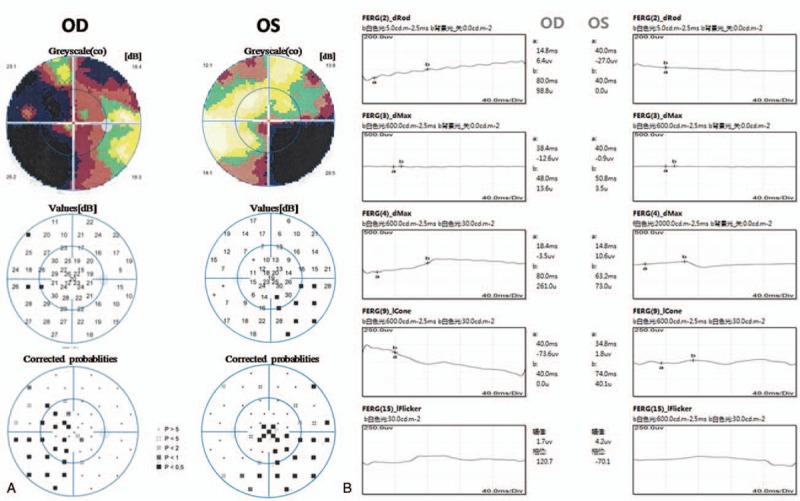
Further ophthalmic examination results of the proband. A: Visual field examination showed quadrant defects in the binocular nasal field. B: Full field electroretinography showed the binocular rod cell response amplitude and maximum response amplitude were both decreased, and both the cone cell reaction and 30 Hz scintillation reaction indicated the absence of an effective waveform. OD: right eye, OS: left eye.
The proband's sisters (IV:3, IV:4), mother (III:6), maternal aunt (III:5), maternal uncle (III:1), and maternal grandfather (II:1) all have a similar history of eye disease. His sister (IV:3) started to experience progressive binocular vision loss and photophobia at the age of 15, followed by night blindness symptoms. She was diagnosed with CORD at the Beijing Tongren Hospital in 2013. The results of her ocular examinations were an uncorrected visual acuity of 0.2 (right eye) and 0.05 (left eye). Fundus examination showed diffuse RPE atrophy and geographic atrophy foci in the macular area (Supplemental Figure 1A). FFA showed hypofluorescence in the fovea centralis of the macular area surrounded by punctate hyperfluorescence, with no fluorescence leakage (Supplemental Figure 1B). OCT revealed an undefined IS/OS layer, and a significantly thinning outer nuclear layer of the fovea centralis (Supplemental Figure 1C). Visual field examination revealed quadrant defects (Supplemental Figure 2A). And the sister's F-ERG results were similar to those of the proband (Supplemental Figure 2B).
3.2. Candidate pathogenic variants
A total of 78,471 SNPs and 9078 indels were identified in the proband (IV:5) by WES. After applying the first 2 filtering steps described in the Methods, 3 candidate mutations including 2 missense mutations and 1 frameshift mutation in 3 known retinal disease-causing genes were obtained. These mutations were c.233G > T in BBS4 (Gene ID: 585, OMIM: 600374), c.538dupG in CRX (Gene ID: 1406, OMIM: 602225), and c.235C > T in RAX2 (Gene ID: 84839, OMIM: 610362). These mutations were all absent from or were present at an extremely low frequency in the 1000 Genomes, ESP6500, and ExAC databases. The functional effects of these mutations, predicted with the online tools PolyPhen2, SIFT, MutationTaster, and SNPs&GO, indicated a deleterious impact for all 3 candidate mutations on their respective proteins (Table 1). Subsequent Sanger sequencing results validated the presence of these filtered mutations in the proband. Cosegregation analysis among all available individuals in the proband's family showed that only the CRX gene mutation (c.538dupG:p.Val180fs) was present in all affected members but not in any of the unaffected members (Fig. 4A, B); that is, only the CRX gene mutation was observed to cosegregate with the family disease phenotype. CRX is one of the pathogenic genes of cone-rod dystrophy.[8] Therefore, this mutation was identified as the pathogenic variant in this Chinese family with cone-rod dystrophy.
Table 1.
Candidate mutations in known retinal disease-causing genes identified by whole-exome sequencing.
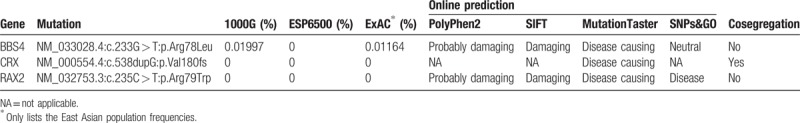
Figure 4.
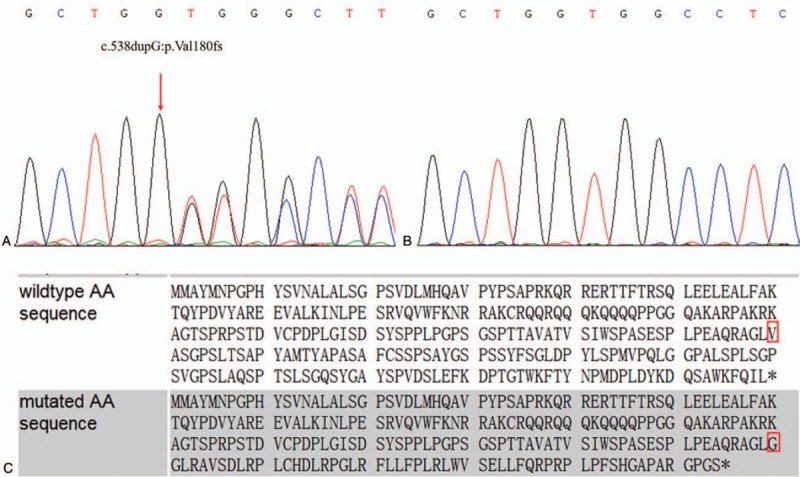
Sanger sequencing results and CRX mutation prediction. A: The sequencing result for affected members. B: The sequencing result for unaffected members. C: The amino acid sequence in the presence and absence of the CRX mutation. Red square: the mutation site.
The frameshift mutation we identified in the CRX gene in this family (c.538dupG:p.Val180fs) was absent in all public databases. It was predicted by MutationTaster to result in a frameshift and premature termination. The 1 bp guanine duplication at position 538 in the open reading frame alters the reading frame, resulting in the valine at position 180 changing to a glycine thereby causing premature protein termination at codon 235 (Fig. 4C). Based on the ACMG standards and guidelines for the interpretation of sequence variants, this mutation satisfies 5 different pathogenic evidence including PVS1, PM2, PM4, PP1, and PP3, and it is classified as pathogenic.[9]
4. Discussion
Retinal diseases exhibit extensive genetic and phenotypic diversity, with links to more than 250 genes (https://sph.uth.edu/retnet/). This poses a challenge for specific diagnosis based on clinical manifestations and for using Sanger sequencing to identify the pathogenic mutations. Thus, we narrowed our search by selecting all known retinal disease-causing genes only, and by using WES as the sequencing analysis tool in this study. Three candidate pathogenic mutations in 3 retinal disease-causing genes were identified as a result of the initial WES of the proband. Cosegregation analysis excluded 2 of these mutations, as they did not cosegregate with the disease in the proband's family. The remaining CRX variant was therefore identified as the disease-causing mutation in this Chinese adCORD family. This mutation was predicted to result in a frameshift and premature termination that would deleteriously impact normal CRX protein function.
The human cone-rod homeobox gene CRX is located on 19q13.33 and contains 4 exons encoding a 299-amino acid homeodomain transcription factor.[8] Animal experiments have shown that this gene is preferentially expressed in vertebrate photoreceptor cells of the retina and pinealocytes of the pineal gland.[10,11] The CRX protein plays an essential role in regulating the expression of specific genes in mammalian rod and cone photoreceptors to maintain the normal functioning of the cones and rods.[12] In Crx–/– mice, which lack photoreceptor outer segments, ERG shows the absence of normal rod and cone activity.[12]
Mutations of the human CRX gene have been reported to cause not only adCORD but also autosomal dominant inherited retinitis pigmentosa (RP) and Leber congenital amaurosis (LCA), which have different severities and ages of onset.[8,13,14] Many CRX frameshift mutations are known retinal disease-causing variants.[4,15,16] Huang et al[17] reported that the 49 reported CRX mutations are composed of 38.78% missense, 4.08% nonsense, 36.73% deletion, 16.33% insertion, and 4.08% indel. Mammalian CRX protein includes 5 conserved functional domains, comprising (from the N- to the C-terminus) the homeodomain (HD) and glutamine-rich (Gln) domain, and the basic, WSP, and OTX-tail motifs. The HD is associated with DNA binding while the C-terminal region of the protein, with the basic to OTX-tail domains, is associated with transcriptional regulation.[18] The reported missense mutations that cosegregate with disease phenotype lie within the HD domain, while frameshift and nonsense mutations are restricted to the C-terminal region of the protein.[19] Thus, the frameshift mutation observed in this Chinese family (c.538dupG:p.Val180fs) would impact normal CRX protein function by effecting the transcriptional regulation of photoreceptor-specific genes.
In conclusion, with the use of WES and Sanger sequencing, we revealed a novel frameshift mutation (NM_000554.4:c.538dupG:p.Val180fs) in exon 4 of the CRX gene in a Chinese family with adCORD. This study broadens the known pathogenic mutation spectrum of the CRX gene and contributes to improved genetic counseling for adCORD patients. In addition, this work showed that whole-exome sequencing is an efficient tool for identifying the pathogenic variants of a hereditary disease associated with multiple pathogenic genes.
Acknowledgments
The authors are grateful to all the study participants.
Author contributions
Data curation: Lihua Wang, Jingjing Feng.
Investigation: Lihua Wang.
Writing – original draft: Lihua Wang, Anhui Qi.
Formal analysis: Anhui Qi.
Methodology: Anhui Qi, Hong Pan.
Supervision: Hong Pan, Beihong Liu, Jingjing Feng.
Visualization: Beihong Liu.
Project administration: Wei Chen, Binbin Wang.
Validation: Wei Chen, Binbin Wang.
Resources: Binbin Wang.
Supplementary Material
Footnotes
Abbreviations: AD = autosomal dominant, adCORD = autosomal dominant cone-rod dystrophy, AR = autosomal recessive, CORD = cone-rod dystrophy, ERG = Electroretinogram, F-ERG = full field electroretinography, FFA = fluorescein fundus angiography, HD = homeodomain, IS/OS = inner segment/outer segment, LCA = Leber congenital amaurosis, MAF = minor allele frequency, OCT = optical coherence tomography, RP = retinitis pigmentosa, RPE = retinal pigment epithelium, SNVs = single nucleotide variants, WES = whole-exome sequencing, XL = X-linked.
LW and AQ have contributed equally to this work.
This study was supported by National Science and Technology Basic Work (2014FY130100), National Infrastructure of Chinese Genetic Resources (YCZYPT [2017]01-6), and the National Key Research and Development Program of China (2016YFC1000307-6).
The authors have no competing interests to declare.
Supplemental Digital Content is available for this article.
References
- [1].Thiadens AA, Phan TM, Zekveld-Vroon RC, et al. Clinical course, genetic etiology, and visual outcome in cone and cone-rod dystrophy. Ophthalmology 2012;119:819–26. [DOI] [PubMed] [Google Scholar]
- [2].Oishi M, Oishi A, Gotoh N, et al. Next-generation sequencing-based comprehensive molecular analysis of 43 Japanese patients with cone and cone-rod dystrophies. Mol Vis 2016;22:150–60. [PMC free article] [PubMed] [Google Scholar]
- [3].Hamel C. Cone rod dystrophies. Orphanet J Rare Dis 2007;2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kitiratschky V, Nagy D, Zabel T, et al. Cone and cone-rod dystrophy segregating in the same pedigree due to the same novel CRX gene mutation. Br J Ophthalmol 2008;92:1086–91. [DOI] [PubMed] [Google Scholar]
- [5].Huang L, Zhang Q, Li S, et al. Exome sequencing of 47 chinese families with cone-rod dystrophy: mutations in 25 known causative genes. PLoS ONE 2013;8:e65546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cremers F, van de Pol D, van Driel M, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum Mol Genet 1998;7:355–62. [DOI] [PubMed] [Google Scholar]
- [7].Demirci F, Rigatti B, Wen G, et al. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet 2002;70:1049–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Freund C, Gregory-Evans C, Furukawa T, et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997;91:543–53. [DOI] [PubMed] [Google Scholar]
- [9].Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen S, Wang Q, Nie Z, et al. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron 1997;19:1017–30. [DOI] [PubMed] [Google Scholar]
- [11].Furukawa T, Morrow E, Cepko C. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 1997;91:531–41. [DOI] [PubMed] [Google Scholar]
- [12].Furukawa T, Morrow E, Li T, et al. Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat Genet 1999;23:466–70. [DOI] [PubMed] [Google Scholar]
- [13].Sohocki M, Daiger S, Bowne S, et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat 2001;17:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Freund C, Wang Q, Chen S, et al. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Genet 1998;18:311–2. [DOI] [PubMed] [Google Scholar]
- [15].Rivolta C, Peck N, Fulton A, et al. Novel frameshift mutations in CRX associated with Leber congenital amaurosis. Hum Mutat 2001;18:550–1. [DOI] [PubMed] [Google Scholar]
- [16].Lines M, Hébert M, McTaggart K, et al. Electrophysiologic and phenotypic features of an autosomal cone-rod dystrophy caused by a novel CRX mutation. Ophthalmology 2002;109:1862–70. [DOI] [PubMed] [Google Scholar]
- [17].Huang L, Xiao X, Li S, et al. CRX variants in cone-rod dystrophy and mutation overview. Biochem Biophys Res Commun 2012;426:498–503. [DOI] [PubMed] [Google Scholar]
- [18].Chau K, Chen S, Zack D, et al. Functional domains of the cone-rod homeobox (CRX) transcription factor. J Biol Chem 2000;275:37264–70. [DOI] [PubMed] [Google Scholar]
- [19].Tran N, Chen S. Mechanisms of blindness: animal models provide insight into distinct CRX-associated retinopathies. Dev Dyn 2014;243:1153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.