

Barrier tissue dysfunction is a fundamental component of chronic human inflammatory diseases1. Specialized epithelial subsets—including secretory and ciliated cells—differentiate from basal stem cells to collectively protect the upper airway2–4. There, allergic inflammation can develop from persistent activation5 of Type 2 immunity6 (T2I), resulting in chronic rhinosinusitis (CRS): ranging from rhinitis to severe nasal polyps7. Basal cell hyperplasia is a hallmark of severe disease7–9, yet how these progenitors2,10,11 contribute to clinical presentation and barrier tissue dysfunction in humans remains unexplored. Profiling primary human surgical CRS samples (18,036 cells, n=12) that span the disease spectrum with Seq-Well12 for massively-parallel single-cell RNA-sequencing (scRNA-seq), we report transcriptomes for human respiratory epithelial, immune, and stromal cell types/subsets from a T2I inflammatory disease, and map key mediators. Through comparison with nasal scrapings (18,704 cells, n=9), we define core, healthy, inflamed, and polyp secretory cell signatures. We find striking differences between the epithelial compartments of the non-polyp and polyp cellular ecosystems, identifying and validating a global reduction of cellular diversity in polyps characterized by basal cell hyperplasia, concomitant decreases in glandular cells, and phenotypic shifts in secretory cell antimicrobial expression. We detect an aberrant basal progenitor differentiation trajectory in polyps, and propose cell-intrinsic13, epigenetic14,15, and extrinsic factors11,16,17 that lock polyp basal cells into this uncommitted state. Finally, we functionally validate that basal cells ex vivo retain intrinsic memory of IL-4/IL-13 exposure, and test the potential for clinical administration of IL-4Rα blockade to modify basal and secretory cell states in vivo. Overall, we identify that a key element of T2I barrier tissue dysfunction is reduced epithelial diversity stemming from functional shifts in basal cells. Our data demonstrate that epithelial stem cells may contribute to the persistence of human disease by serving as repositories for allergic memories.
The T2I module6 regulates homeostatic processes18 (metabolism), host defense19 (parasites, venoms, allergens, toxins) and inflammatory tissue repair11. However, this module may become self-reinforcing in allergic inflammation leading to substantial alterations in gross tissue architecture20 as observed in polyps7. To investigate how the overall tissue cellular ecosystem shifts in composition and state during chronic respiratory T2I disease in humans, we used Seq-Well12 to profile the ethmoid sinus (EthSin) of patients spanning the CRS spectrum (Fig. 1a; Supplementary Table 1; Methods; Supplementary Information; n=12 samples: 6 non-polyp, 6 polyp). Deconstructing these tissues into their component cells provides a unique lens into the cellular ecosystem of human T2I, helping us to: 1. characterize each major cell type without biases typically introduced by marker pre-selection; 2. evaluate cell types/states with striking disease-associated transcriptional differences; and 3. reconstruct tissue-level dynamics.
Figure 1 |. Mapping the T2I inflamed human sinus cellular ecosystem by scRNA-seq.
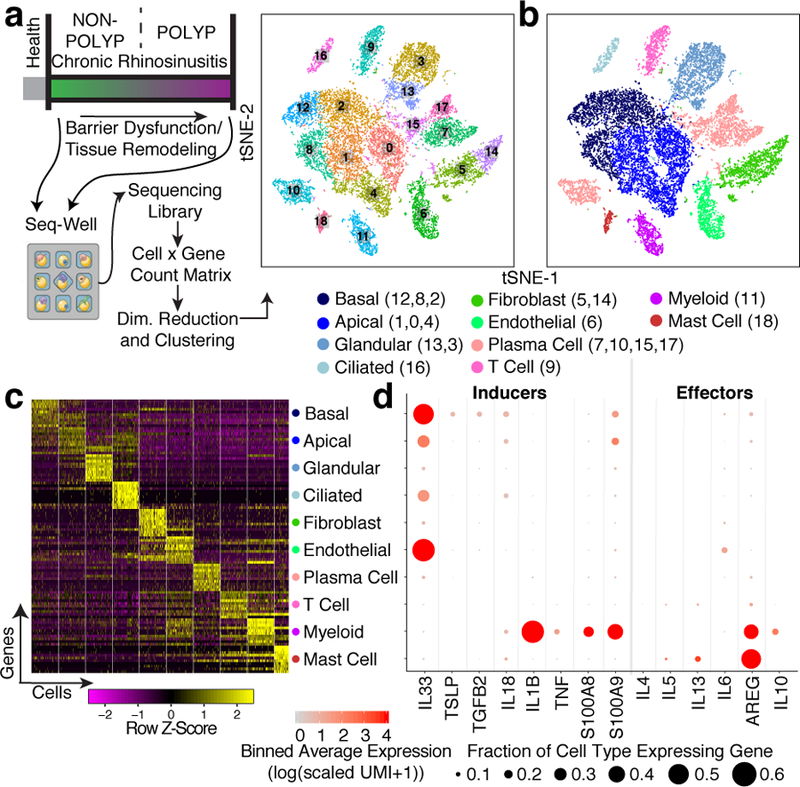
a, Clinical disease spectrum (n=12 samples) and experimental workflow leading to a tSNE plot displaying 18,036 single cells, colored by shared nearest neighbor (SNN) clusters and (b) cell types (ROC-test; Supplementary Table 3; Methods) from respiratory tissue. c, Heatmap of top-10 marker genes by ROC-test (AUC>0.73) for indicated cell types; maximum 500 cells/type (Extended Data Fig. 2a annotated; Supplementary Table 3 full gene list). d, Dot plot of T2I mediators mapped onto cell types across all samples (Extended Data Fig. 4 by disease state).
We derived a unified cells-by-genes expression matrix (18,036 cells) and performed dimensionality reduction and graph-based clustering (Fig. 1a; Extended Data Fig. 1a,b; Supplementary Table 2; Methods). We used complete lists of cluster-specific genes to identify epithelial2, stromal7,20, and immune cells4,6, recovering a reproducible distribution of cell types across patients (Fig. 1b,c; Extended Data Figs. 1c–e&2a–e; Supplementary Table 3; Methods; Supplementary Information). We highlight the major types recovered: basal (KRT5) and apical (KRT8)—which orient the pseudostratified epithelial division—further specialized ciliated (FOXJ1) and glandular3 (LTF) cells, and supportive endothelial (DARC), fibroblast (COL1A2), plasma (CD79A), myeloid (HLA-DRA), T (TRBC2), and mast cells (TPSAB1) (Extended Data Fig. 1e). For each cell type, sub-clustering revealed further, potentially meaningful heterogeneity, providing a useful reference atlas for studying human inflammatory diseases of barrier tissues (Extended Data Fig. 3a–c; Supplementary Information).
Next, we charted the cell-of-origin for chemokines and lipid mediators, which aid in the recruitment and positioning of lymphoid and myeloid cells in tissues during T2I21 (Extended Data Fig. 4a; Supplementary Information). For example, we found mast cells specifically enriched for HPGDS, PTGS2, and ALOX5, suggesting they may be a dominant source of prostaglandin D2, implicated in activation of T-helper 2 (Th2) cells4. Alongside these mediators, the production of instructive first-order cytokines primes recruitment and activation of effector mechanisms. In particular, IL-25, IL-33, and TSLP are broadly regarded as epithelial-derived cytokines4,5,16,20,22, yet little is known about their cell-of-origin in human disease. TSLP was uniquely restricted to basal cells, which may link increased basal cell numbers to activation of effector cells (Fig. 1d; Extended Data Figs. 3a&4b,c; Supplementary Information).
Second-order effector cytokines were identified in a subset of CD4+ T cells expressing IL4, IL5, IL13, and HPGDS, fitting the profile of allergen-specific Th2A cells23 (Fig. 1d; Extended Data Fig. 4c–e; Methods; Supplementary Information). Additionally, substantial numbers of mast cells produced IL5 and IL13, which along with myeloid cells, were the main expressers of the tissue-reparative cytokine AREG22. Notably, patients with or without polyps showed consistent cells-of-origin for T2I-related chemokines, lipids, and cytokines, except select mediators (Extended Data Fig. 4a,b; Supplementary Information). Several genome-wide association studies (GWAS)-implicated genes for allergic diseases24 were restricted to specific cell types. Thus, we mapped the expression of proposed risk genes including GATA2, IL1RL1 (IL-33R), CDHR3, KIF3A, TMEM232 and MYC (Extended Data Fig. 4f; Supplementary Information). Cellular maps of tissues frequently affected by inflammatory disease should aid in providing mechanistic insights into genotype-phenotype interactions.
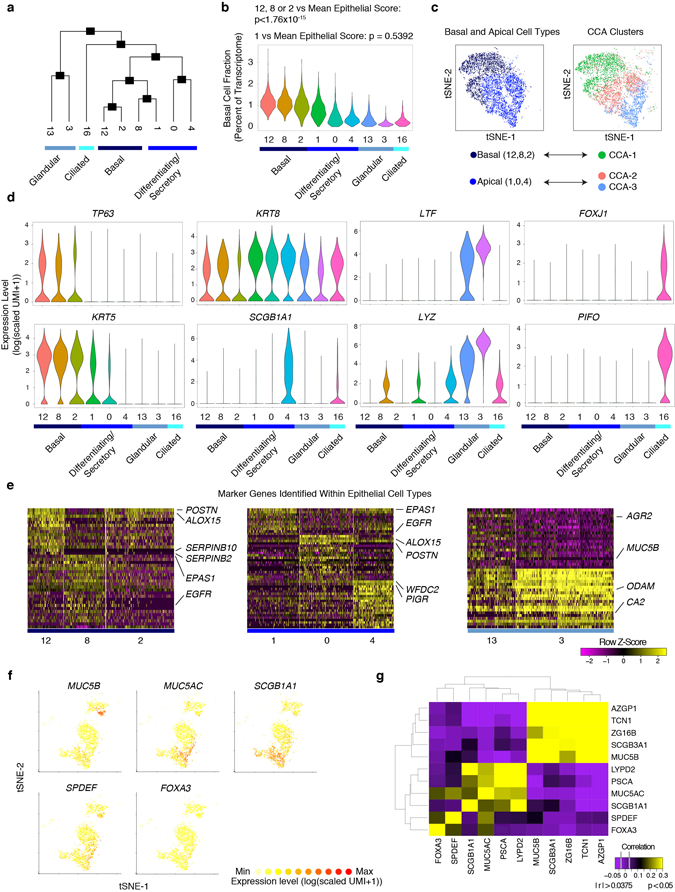
We further analyzed clusters within the broad epithelia (Fig. 2a; Extended Data Fig. 5a–c) providing single-cell human transcriptomes25 for basal, secretory, glandular, and ciliated cell types from a T2I ecosystem (Fig. 2a,b; Extended Data Fig. 5; Supplementary Table 3). Epithelial marker gene analysis identified conserved programs present in basal (clusters=3), differentiating/secretory (clusters=3), glandular (clusters=2) and ciliated (clusters=1) types (Fig. 2a,b; Extended Data Fig. 5a–d; Supplementary Table 3, Supplementary Information)2,3.
Figure 2 |. Single-cell transcriptomes of epithelial cells in T2I highlight shifts in secretory cell states across health and disease.
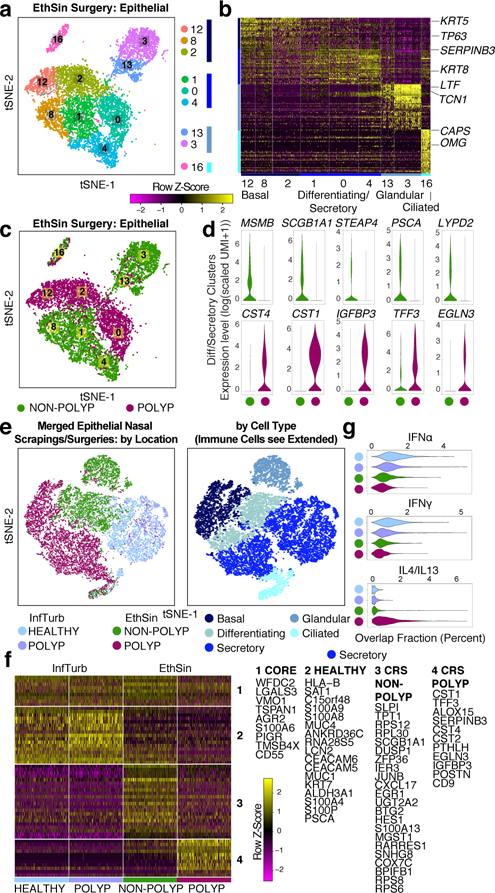
a, tSNE plot of 10,274 epithelial cells (n=12 samples), colored by SNN-clusters (Fig. 1; Extended Data Fig. 6 re-clustered) with blue color bars representing cell types determined per Extended Data Fig. 5, and (b) heatmap of marker genes by ROC (AUC>0.65; Supplementary Table 3, full list). c,d, tSNE plot (a) colored by disease (n=6 non-polyp, n=6 polyp samples) and (d) violin plots (Methods: all violins generated using standard Seurat implementation with default smoothing, density generated at >25% positive values, widest aspect centre of positive measures, minima/maxima within scale representing all points) for differentially expressed genes across disease state in differentiating/secretory cells; 2,566 cells, n=6 non-polyp; and 1,796 cells, n=6 polyp samples; *bimodal test, all p<2.03×10−55 or less with Bonferroni-correction (Supplementary Table 3, exact values). e, tSNE plot of 18,325 re-clustered single cells from merged nasal scrapings (n=9) and surgical samples (n=12) by (left) location (healthy InfTurb (3,681 cells, n=3 samples), polyp-bearing patient InfTurb (1,370 cells, n=4 samples), non-polyp EthSin surgical samples (5,928 cells, n=6 samples), and polyp surgical and scraping samples directly from polyp in EthSin (7,346 cells, n=8 samples)) and (right) cell type (3,152 basal, 3,089 differentiating, 8,840 secretory, 1,105 ciliated, and 2,139 glandular cells). f, Heatmap of secretory cells (1,000 cells/location) displaying select genes (AUC>0.65; Supplementary Table 3). g, Violin plots of IFNα, IFNγ, and IL-4/IL-13 gene signatures for secretory cells; healthy InfTurb (3,414 cells), polyp-bearing patient InfTurb (1,239 cells), non-polyp EthSin surgical samples (1,048 cells), polyp surgical and scraping samples directly from polyp in EthSin (3,139 cells); effect size −1.16, −1.05, 1.32, respectively polyp EthSin vs healthy; *Mann-Whitney U-test, p<2.2×10-16.
Observing striking polyposis-related differences across clusters within cell types (Fig. 2c; Extended Data Fig. 5e; Supplementary Information), we quantified the numerical over-representation of cells from non-polyp and polyp ecosystems within each cluster and type. The clusters comprising basal, differentiating/secretory, and glandular cells showed the most significant links to disease-state (p-values: Fisher’s with least-significant-difference; Fig. 2c; Supplementary Table 3). We compared transcriptomes of differentiating/secretory cells3 (containing KRT8 secretory and apical goblet cells), noting that secretory cells from polyps appear to supplant antimicrobial function with tissue-repair (Fig. 2d; Supplementary Table 3, Supplementary Information).
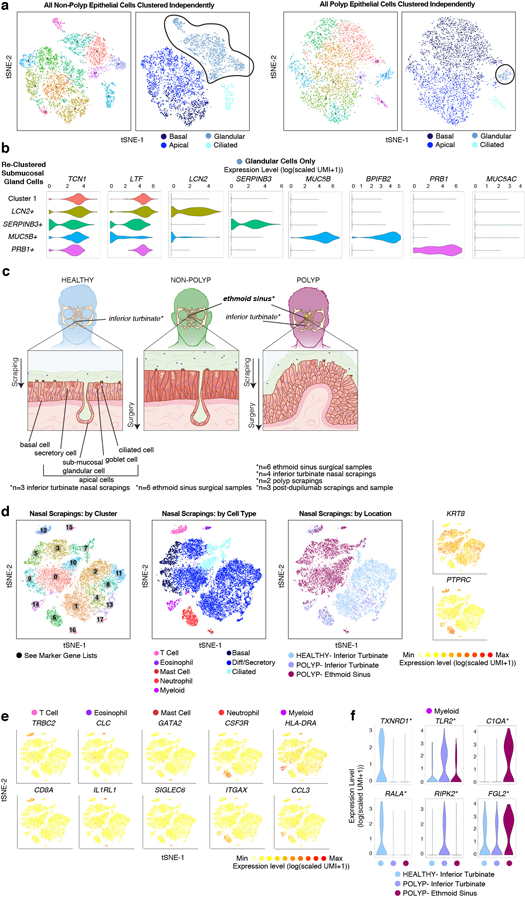
Intriguingly, we observe expression of MUC5B within glandular mucus cells (cluster 13), but MUC5AC in a distinct subset of secretory goblet cells co-expressing SCGB1A1 and FOXA3 (Fig. 2b; Extended Data Fig. 5f,g, Supplementary Information). This suggests the goblet cell program is layered atop a secretory cell base2. We also assessed glandular heterogeneity identifying five discrete subsets with variegated antimicrobial expression3 (Fig. 2a; Extended Data Fig. 6a,b; Supplementary Table 3; Supplementary Information). This compartmentalization may represent a mechanism for regulated secretion, with imbalances affecting innate host defense.
To contextualize disease-associated shifts, we turned to sino-nasal scrapings as a method of sampling healthy apical cells through Seq-Well (Extended Data Fig. 6c,d, Methods; Supplementary Tables 3&6; 18,704 additional cells: n=3 healthy inferior turbinate (InfTurb), n=4 polyp-patient InfTurb, n=2 polyp directly). We recovered immune cells, differentiating/secretory and ciliated epithelial cells from the InfTurb of patients with polyposis and healthy controls, but basal cells only in polyp scrapings (Extended Data Fig. 6d–f; Supplementary Table 3; Supplementary Information). By combining all epithelial cells from surgical resections with scrapings (Fig. 2a–c,e), we identified a conserved secretory core gene set present in all sites sampled, and healthy, CRS-InfTurb, CRS-EthSin-non-polyp and CRS-EthSin-polyp specific gene signatures. Overall, we note a shift from IFNα-/IFNγ-induced to IL-4/IL-13-induced genes with increasing disease severity (Fig. 2e–g; Supplementary Table 3; Supplementary Information). Secretory cells from involved CRS-EthSin tissue differ significantly from the InfTurb, and secretory cells in non-polyp and polyp EthSin reach distinct states whose altered functionality may be linked to disease severity.
As specialized epithelial cell types arise from basal progenitors2,10, we formally examined their distribution in each sample (Fig. 3a; Extended Data Fig. 7a). Our data indicate a significant loss of epithelial ecological diversity in nasal polyps by Simpson’s Index (Methods), largely driven by glandular and ciliated cell depletion, and an enrichment in basal cells (Fig. 3a,b; Extended Data Fig. 7a–d; Supplementary Information). This altered diversity tracked closely with rank-ordered pathology of patient tissue samples, which correlated positively with basal cell frequency (r=0.6252) and negatively with epithelial diversity (r=−0.6824; Extended Data Fig 7e). We speculate immune cells in polyps may represent an overcorrection in attempting to balance the epithelial compartment (Extended Data Fig. 7f; Supplementary Information).
Figure 3 |. Reduced epithelial ecological diversity and basal cell hyperplasia in nasal polyps.
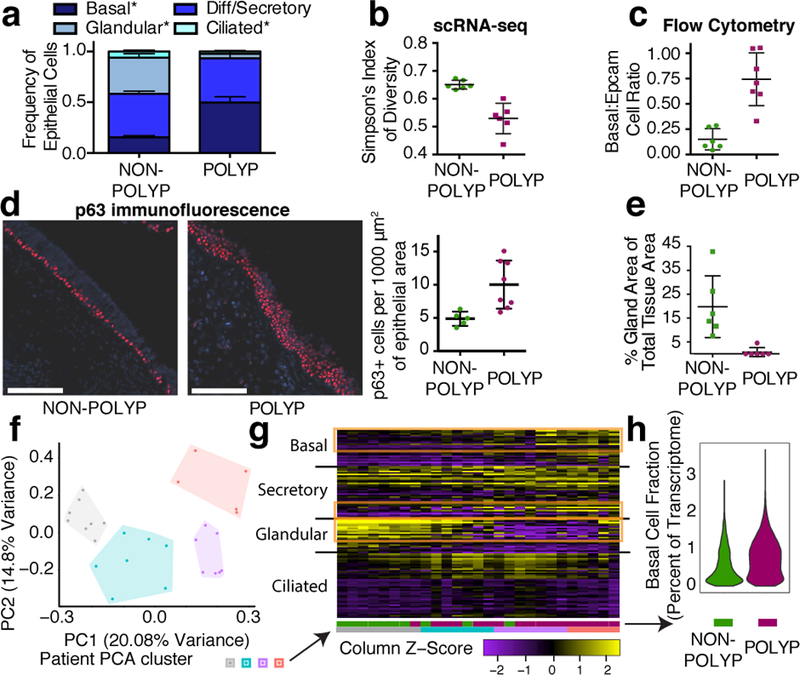
a, scRNA-seq cell frequency (Fig. 2a; Extended Data Fig. 7a individual points) calculated for each sample; basal cell p=0.00023, glandular p<0.0001, ciliated p=0.0387, non-polyp vs. polyp; and (b) Simpson’s index (Methods, p=0.0021); n=6 non-polyp, n=6 polyp samples; *t-test, two-sided, mean±s.e.m. c, Flow cytometry quantification (Extended Data Fig. 7, full gating), n=6 non-polyp, n=7 polyp samples; *t-test, two-sided, p=0.0005; mean±s.e.m. d, Immunofluorescence and quantification for basal cells normalized to 1,000μm2 of epithelium; n=5 non-polyp patients, 10 sections; n=8 polyp patients, 41 sections *Mann-Whitney U-test, p=0.0282, mean±s.d.; scale bar 100μm, and (e) quantification of glandular area, n=6 non-polyp, n=6 polyp patients; *t-test, two-sided, p=0.0022; (Extended Data Fig. 7, isotype and representative). f,g, Bulk-tissue RNA-seq deconvolution by (f) PCA and (g) heatmap over epithelial subset-specific genes (rows) with KNN-clusters (n=4; Methods), from n=10 non-polyp, n=17 polyp samples (columns). h, Violin plot of basal cell gene fraction in scRNA-seq epithelium (Methods, Supplementary Table 4); 5,928 cells, n=6 non-polyp; 4,346 cells, n=6 polyp; effect size 0.457, *Mann-Whitney U-test, p<2.2×10-16.
To confirm our epithelial findings, we utilized complementary approaches. With flow cytometry10, we demonstrated that the frequency of basal cells significantly increased in polyps at the expense of differentiated epithelial cells in 13 additional patients (Fig. 3c; Extended Data Fig. 7g,h). Using histology (not subject to dissociation-induced artifacts like scRNA-seq or flow cytometry), we confirmed8 a significant increase of p63+ cells per 1,000μm2 of epithelial area, and a striking loss of glands in polyps (Fig. 3d,e; Extended Data Fig. 7i,j; Methods). Finally, we utilized marker genes for specialized lineages to deconvolve bulk EthSin-tissue RNA-seq of another 27 individuals. We identified four patient clusters and confirmed glandular enrichment in non-polyps, and shifts in secretory cell states and the progressive acquisition of basal-associated transcripts in polyps (Fig. 2d,f; Fig. 3f,g; Supplementary Tables 1&3; Methods, Supplementary Information). We also validated these findings with publicly-available RNA-seq datasets containing normal human sinus tissue and polyps (Extended Data Fig. 7k,l; Supplementary Information).
To identify what mechanisms might account for decreased epithelial diversity in polyps, we compared the transcriptomes of non-polyp and polyp basal progenitors2,10, identifying elevated polyp expression of transcripts involved in extracellular matrix remodeling and chemo-attraction of effector cells, and a decrease in protease-inhibitor expression and metabolic genes (Fig. 4a). As some of these upregulated genes are IL-4/IL-13 responsive7, we assessed cytokine-induced gene sets. A combined IL-4/IL-13 signature is strongly induced not only in differentiated polyp epithelium, but also in basal cells, with a large effect size between disease states (Fig. 4b,c; Extended Data Fig. 8a; Methods; Supplementary Table 4). IFNα- and IFNγ-signatures—indicative of a Type-1 immune module6—have small effect sizes (Extended Data Fig. 8b; Supplementary Table 4). Furthermore, from specific hallmark genes, we observed altered balance between Wnt (CD44) and Notch (HEY1) signaling in polyp epithelium favoring Wnt26,27 (Fig. 4a,c; Supplementary Table 4). We further contextualized our basal cell findings by defining alterations in the fibroblast niche that correlate with basal hyperplasia, and identifying significant changes in myeloid and endothelial cell gene expression (Extended Data Figs. 7b&8c–f; Supplementary Information).
Figure 4 |. T2I cytokines and developmental pathways converge at the epigenetic level in basal cells to intrinsically impair differentiation in vivo.
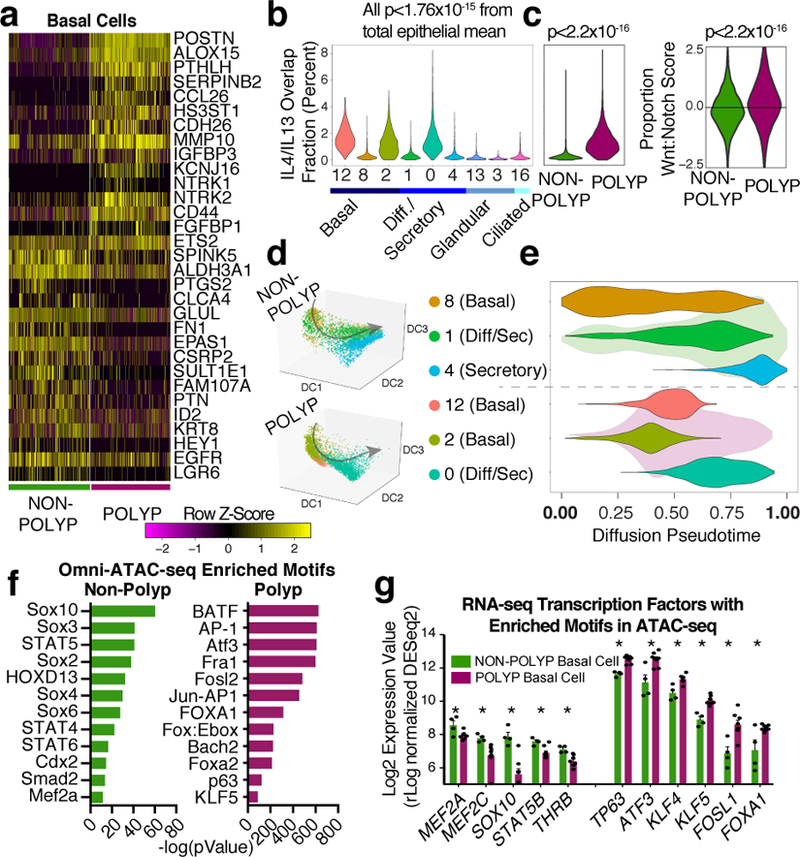
a, Heatmap of select genes over basal cell clusters 8 and 12; 860 cells, n=6 non-polyp, 858 cells, n=6 polyp samples; *bimodal test, all displayed genes p<1.97×10−39 or less with Bonferroni correction; (Supplementary Table 3). b, Violin plot (Methods; Extended Data Fig. 8; Supplementary Table 3 cell numbers, Supplementary Table 4 gene lists) for IL-4/IL-13 commonly-induced gene signature; *Mann-Whitney U-test, p<1.76×10−15, relative to mean score, with Bonferroni correction. c, Violins of shared IL-4/IL-13 signature (*Mann-Whitney U-test, p<2.2×10−16, effect size 1.305) and Wnt:Notch target gene proportion (*t-test, two-sided, p<2.2×10−16, effect size 0.334, NB: axis truncated, zero indicates equal scores) over 5,928 cells, n=6 non-polyp vs. 4,346 cells, n=6 polyp. d,e, Diffusion pseudotime (Methods) over epithelial cells with unified gene list (Supplementary Table 3 cell numbers, gene list) and (e) violin plot of pseudotime component with green (n=6 non-polyp) and purple (n=6 polyp) underlying distribution. f, Omni-ATAC-seq and HOMER motif enrichment over background peaks (Methods); all q-value<0.0002 Benjamini corrected, and (g) transcription factors from low-input RNA-seq on sorted basal cell populations; *t-test, two-sided, p<0.05 or less Holm-Sidak correction; mean±s.e.m; n=3 non-polyp (4 RNA-seq), n=7 polyp.
Next, we used diffusion pseudotime mapping (Methods), aligning and reconstructing how basal cells differentiate to mature secretory cells to identify where basal cells become “stuck” in polyps. In the non-polyp ecosystem, we observed that basal cells traverse a wider swath of common pseudotime, with the majority of secretory cells distributed towards the trajectory’s terminus (Fig. 4d,e; Extended Data Fig. 9a; Methods). Conversely, in polyps, basal cells accumulate shy of the trajectory’s midpoint, losing the true progenitor position occupied by cluster 8, yet failing to contribute towards later differentiation states (Fig 4d,e; Extended Data Fig. 9a). Ordering cells along this common axis, we identified several genes dysregulated in polyps during epithelial cell differentiation (Extended Data Fig. 9b; Supplementary Table 3; Supplementary Information).
We sorted basal cells (Extended Data Fig. 7h) from 3 non-polyp and 7 polyp tissues and performed Omni-assay for transposase accessible chromatin-(ATAC)-Seq to identify intrinsic epigenetic changes from the integration of extrinsic cellular signaling events28, and subsequent bulk RNA-seq to confirm/extend our findings (Methods). Polyp basal cells were enriched in peaks for bZIP transcription factor motifs, including various AP-1 family members11, such as JUN, FOXA1, ATF3, KLF5 and p63 itself associated with the maintenance of an undifferentiated state, chromatin opening, and oncogenesis (Fig. 4f; Extended Data Fig. 9b–f; Supplementary Table 5, Supplementary Information). Clustering of enriched motifs revealed changes in correlation by disease state (Extended Data Fig. 9c–f; Supplementary Information). We identified expressed candidate transcription factors that may bind to these accessible sites (Fig. 4f,g; Extended Data Fig. 9e,f; Supplementary Information). Collectively, our transcriptomic, pseudotemporal, and epigenetic studies led us to hypothesize that during chronic T2I, basal cell differentiation is intrinsically impaired through the influence of extrinsic cues (e.g., IL-4/IL-13 and Wnt pathway).
To functionally test for intrinsically altered differentiation potential in vitro, we first seeded basal cells from non-polyp or polyp tissue into air-liquid interface (ALI) cultures (Fig. 5a; Extended Data Fig. 9g; Supplementary Table 3; Methods; Supplementary Information). Our data suggest that basal cells from polyps can be released from their “stuck” state and differentiate towards a mixed-tissue secretory cell phenotype if provided with strong and sustained extrinsic cues, even in the presence of IL-13 (Fig. 5b; Extended Data Figs. 7h&9f,h,i; Supplementary Information).
Figure 5 |. Transcriptional memory of IL-4/IL-13 exposure retained by basal cells ex vivo and in vivo IL-4Rα blockade partially resets state in polyps.
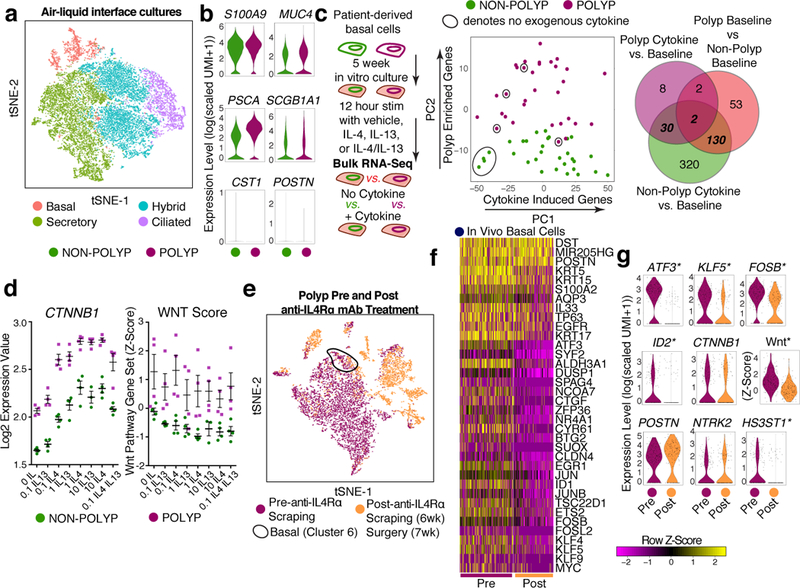
a, tSNE plot of cell types from ALI-cultures (Extended Data Fig. 9g) over 16,173 single cells (8,483 non-polyp; 7,690 polyp) and (b) violin plots for secretory genes (from Fig. 2f) on ALI secretory cells (3,277 non-polyp; 3,143 polyp); not significant (n.s.) except MUC4, PSCA and SCGB1A1 greater in polyp secretory cells; bimodal p<4.04×10−16, Bonferroni correction. c, Basal cell inflammatory memory to IL-4/IL-13 performed (Methods) and displayed as PCA over variable genes and Venn diagram overlaps of differential expression; *t-test, two-tailed, Bonferroni corrected p<0.05, Supplementary Table 3 full lists. d, CTNNB1 expression and Wnt pathway z-score (Methods) in basal cells from (c); n=4 samples/dose; 2-way ANOVA; p<0.0001 for CTNNB1; p<0.0282 for Wnt pathway, (a-d) n=2 basal cell donors each non-polyp and polyp. e-g, scRNA-seq on anti-IL-4Rα-treated individual and (e) tSNE plot of 8,764 single cells from nasal polyps colored by pre- (5,731 cells) and post- (3,033 cells) treatment samples (Extended Data Fig. 10 for more), (f) heatmap of select genes (AUC>0.68 core, p<2.46×10−5 or less with Bonferroni correction) over basal cells (200 pre, 151 post), and (g) violin plots for select genes; n.s. save *bimodal p<0.00087 with Bonferroni correction Supplementary Table 3 for full list; Wnt score Pre vs. Post: *t-test, two-tailed, p<2.2×10−16, effect size 0.942.
Second, as ALI cultures enforced strong terminal differentiation, we directly tested how IL-4/IL-13 act to induce rapid expression of genes in basal cells cultured 5 weeks ex vivo hypothesizing that polyp basal cells would respond more vigorously to exogenous cytokines than non-polyp ones14. Surprisingly, we identified 482 genes induced in non-polyp basal cells, but only 42 in polyps (Fig. 5c; Supplementary Table 3). PCA highlighted that while unstimulated non-polyp basal cells grouped together, those from polyp basal cells were distributed along PC1, which captured cytokine stimulation (Fig. 5c). Identifying overlaps in genes significantly induced by cytokine treatment in non-polyp basal cells with genes upregulated at baseline in polyp vs. non-polyp basal cells resolved 132 genes (Fig. 5c; Supplementary Table 3).
We focused on the central overlap of these three differential expression tests, which included CTNNB1 (β-catenin), the key effector of Wnt pathway activation26,27 (Fig. 5c). We highlight the fundamental finding that CTNNB1 was robustly induced in non-polyp and polyp basal cells in a dose-sensitive fashion to IL-4 and IL-13. Moreover, baseline CTNNB1 expression in polyp basal cells was equivalent to the levels induced by cytokine treatment of non-polyp cells (Fig. 5d). Wnt-pathway target genes were significantly upregulated across doses tested, confirming activation of the pathway overall, and of specific factors (CTGF) (Fig. 5d; Extended Data 9j; Supplementary Table 4; Supplementary Information). Based on polyp epithelial gene signatures (Fig. 4c), and our functional testing for IL-4/IL-13 induced genes “remembered” by polyp basal cells (Fig. 5c,d), we propose that chronic IL-4/IL-13 exposure in vivo can lead to persistent expression of Wnt/CTNNB1 target genes in a cell-intrinsic fashion, even in the absence of exogenous cytokine.
One polyp patient sampled through scraping commenced treatment with a monoclonal antibody (mAb) targeting the shared IL-4Rα subunit of the IL-4 and IL-13 receptors to treat atopic dermatitis, allowing us the chance to examine the in vivo relevance of our observational, mechanistic and functional data on how T2I cytokines influence basal cell states (Fig. 5e; Extended Data Fig. 10a,b; Methods). We compared cells recovered from pre- and 6-week-post-mAb scrapings, and through surgical intervention at 7 weeks post-mAb (Fig. 5e; Extended Data Fig. 10a,b; Supplementary Table 7; Supplementary Information). We identified basal cells and generated a heatmap containing their top marker genes, agnostic to treatment, followed by genes differentially expressed pre- and post-treatment, leveraging myeloid cells to identify basal-specific changes (Fig. 5f; Extended Data Fig. 10a–c; Supplementary Table 3; Supplementary Information).
Contextualizing these findings within our previous data, we identify several key gene sets, including a conserved core set of basal cell genes (Extended Data Fig. 10d). Intriguingly, transcription factors upregulated in polyp basal cells identified through Omni-ATAC-seq/RNA-seq (ATF3, KLF5, FOSB) were significantly decreased by treatment (Fig. 5g). While Wnt pathway target gene expression was globally reduced, CTNNB1 expression was notably retained, as were genes upregulated both in vitro and in vivo in polyp basal cells, suggesting that some genes in this patient, and at this timepoint, persist (Fig. 5g; Extended Data Fig. 10d; Supplementary Information).
Lastly, we sought to understand how changes in the basal epithelium propagated through to secretory cells. Within secretory cells recovered from scrapings of both InfTurb and accessible polyp tissue pre- and post-treatment, our data suggest that even though EthSin-CRS samples have unique secretory cell signatures (Fig. 2f), cytokine blockade leads to gene expression associated with healthy InfTurb secretory cells, even in polyp tissue (Extended Data Fig. 10e–h; Supplementary Table 8; Supplementary Information).
One goal of understanding the cellular and molecular pathways activated in T2I is to provide mechanisms which explain persistent chronic allergic inflammatory disease29. Utilizing scRNA-seq applied to patients across the CRS spectrum, our study provides descriptive, mechanistic and functional insights into an enigmatic basal cell state and productive differentiation of a barrier tissue. We show striking differences in antimicrobial expression by secretory cells relative to healthy tissue, a loss of glandular cell heterogeneity, and that IL-4/IL-13 strongly induce a transcriptional program already at the level of basal progenitor cells15. Our data may help to explain why nasal polyposis is associated with infections by specific microorganisms7, and how a mAb targeting the shared IL-4/IL-13 receptor can reduce nasal polyp burden (Methods).
Taken together with recent work in the murine intestinal tract and skin11,13,16,17,30, we provide human evidence for the emerging paradigm of stem cell dysfunction altering the set-point of barrier tissues, highlighting substantial overlap amongst putative driving transcription factors (ATF3, AP-1, TP63, and KLF5)13. This demonstrates that the principle of inflammatory memory28 underlying barrier tissue adaptation is a generalizable phenomenon observed in distinct anatomical locations, inflammatory modules, and species. We build on these findings by culturing basal cells ex vivo and identifying the indelible mark of IL-4/IL-13 as a baseline induction of the Wnt pathway. We propose that basal cells form “memories” of chronic exposure to an inflammatory T2I environment, shifting the entire cellular ecosystem away from productive differentiation, and propagating disease. Future work will seek to determine the relative contributions of memory stored in distinct cellular compartments to develop the most effective mechanisms by which to erase them.
METHODS
Study Participants and Design for Single-Cell Study from Ethmoid Sinus Tissue
Subjects between the ages of 18 and 75 years were recruited from the Brigham and Women’s Hospital (Boston, Massachusetts) Allergy and Immunology clinic and Otolaryngology clinic between May 2014 and March 2018 (Supplementary Table 1). The Institutional Review Board approved the study, and all subjects provided written informed consent. Ethmoid sinus (EthSin) tissue was collected at the time of elective endoscopic sinus surgery from patients with physician-diagnosed CRS with and without nasal polyps based on established guidelines31. Patients with polyps include both aspirin-tolerant chronic rhinosinusitis with nasal polyps (CRS polyp) and individuals with aspirin-exacerbated respiratory disease (AERD), both referred to as CRS-EthSin-polyp for the purposes of this study. Patients were suspected of having AERD if they had asthma, nasal polyposis, and a history of respiratory reaction on ingestion of a COX 1 inhibitor, with confirmation via a graded oral challenge to aspirin. Subjects with cystic fibrosis and unilateral polyps were excluded from the study. No distinctions were made between these two disease endotypes in our study as both present with polyposis, but we present the information of clinical diagnosis in Supplementary Table 1.
A tissue segment (one per patient) for bulk tissue RNA-seq was immediately placed in RNAlater (Qiagen) for RNA extraction, For patient samples loaded on Seq-Well and for flow-sorting to Omni-ATAC-seq/RNA-seq, tissue was received in-hand, placed in RPMI (Corning) with 10% FBS (ThermoFisher 10082–147) and immediately put on ice for transport. Details of the subjects’ characteristics included in scRNA-seq cohort, tissue RNA-seq cohort, and basal cell flow cytometry/ATAC-seq/RNA-seq cohort (including age, gender, medication use, and disease severity) are included in Supplementary Table 1.
NB: Originally, we enrolled a healthy control subject with no known history of CRS or nasal polyposis who was undergoing sinus surgery for concha bullosa. However, this subject upon pathology evaluation was noted to have mild eosinophilia. A chart review revealed a history of allergic rhinitis and asthma, and their diagnosis was updated to CRS non-polyp clinically by the surgeon upon follow-up visits so we updated their status accordingly in our study. Additionally, non-polyp patient 6 was sampled twice (denoted as 6A and 6B), representing distinct cells that were captured on two different Seq-Well arrays. As such, they should not be viewed as a technical replicate and are referred to as distinct samples.
Collection of Inferior Turbinate and Nasal Polyp Samples through Nasal Scraping
Nasal samples were collected from the inferior turbinate (InfTurb) of healthy control subjects and from the inferior turbinate and accessible polyp tissue in subjects with CRS-EthSin-polyps using the Rhino-Pro® Curette, a sterile, disposable, mucosal collection device, as described32,33. One sample was taken from the right and left mid-inferior portion of the inferior turbinate using a gentle scraping motion. In two subjects with CRS polyp, with accessible nasal polyp tissue, the polyp tissue was sampled using the Rhino-Pro® Curette under direct visualization. The nasal scrapings were placed directly in RPMI with 10% FBS and immediately put on ice for transport before loading on Seq-Well arrays. Details of the subjects’ characteristics (including age, gender, medication use, and disease severity) are included in Supplementary Table 1.
Nasal scraping allows for access to the superficial epithelial cell layer of the inferior turbinate34; in contrast, the surgical resections from ethmoid sinus we utilize as the central data set of this paper contain both epithelial cells and underlying tissue, including sub-mucosal glands34 (Extended Data Fig. 6c). Since scraping samples a proximal but distinct anatomical location with a distinct technique, in addition to collecting inferior turbinate scrapings from healthy controls (n=3), we also collected inferior turbinate scrapings from individuals with polyps (n=4), and, from two of these individuals, from accessible polyps protruding beyond the middle meatus (n=2).
One subject with CRS polyps and co-morbid severe atopic dermatitis was started on dupilumab35, a human monoclonal antibody that binds to the IL-4Rα subunit approved for severe atopic dermatitis36, and in a randomized, double-blind, placebo-controlled parallel-group study was shown to significantly reduce endoscopic nasal polyp burden after 16 weeks37. The inferior turbinate and nasal polyp tissue was sampled with the Rhino-Pro® Curette pre- and post-treatment with 3 doses of dupilumab, and through endoscopic sinus surgery as noted above.
Tissue Digestion
Single-cell suspensions from collected surgical specimens were obtained using a modified version of a previously published protocol38, described below in detail. Each specimen was received directly in hand and processed directly with an average time from patient to loading onto the Seq-Well platform of 3 total hours, and never exceeding 4 hours. Surgical specimens were collected into 30 mL of ice cold RPMI (Corning). Specimens were finely minced between two scalpel blades and incubated for 15 minutes at 37°C in a rotisserie rack with end-over-end rotation in 25 mL digestion buffer supplemented with 600 U/mL collagenase IV (Worthington) and 20 μg/mL DNAse 1 (Roche) in RPMI with 10% fetal bovine serum. After 15 minutes, samples were triturated five times using a syringe with a 16G needle and returned to the rotisserie rack for another 15 minutes. At the conclusion of the second digest period, samples were triturated an additional five times using a syringe with a 16G needle, at which point the digest process was stopped via the addition of EDTA to 20mM. Nasal scrapings were only dissociated with one 15 minute dissociation via collagenase and omission of the 16G needle trituration, instead replaced with P1000 pipette trituration, as typically cell yields were <20,000 total cells. Processing downstream remained identical. Samples were typically fully dissociated at this step and were filtered through a 70μM cell strainer and spun down at 500G for 10 minutes followed by a rinse with ice-cold PBS (ThermoFisher 10010023, Ca/Mg-free) to 30 mL total volume. Red blood cells (RBCs) were lysed using ACK buffer (ThermoFisher A1049201) for 3 minutes on ice to remove RBCs, even if no RBC contamination was visibly seen in order to maintain consistency across patient groups. Cells were then washed with sterile PBS and spun down at 500G for 5 minutes, resuspended in complete RPMI medium with 2% FCS (RPMI1640 [ThermoFisher 61870–127], 100 U/ml penicillin [ThermoFisher 15140–122], 100 μg/mL streptomycin [ThermoFisher 15140–122], 10 mM HEPES [ThermoFisher 15630–080], 2% FCS [ThermoFisher 10082–147], 50 μg/mL gentamicin [ThermoFisher 15750–060]), and counted to adjust concentration to 100,000 cells/mL for loading onto Seq-Well arrays.
Flow Cytometry, Cell Sorting, and Analysis
Single-cell suspensions in FACS Buffer (HBSS [ThermoFisher 14170161, Ca/Mg-free] supplemented with 2% FCS) were pre-incubated with Fc-Block (BD 564220) before staining for surface antigens. The following antibodies were used to identify basal cells via flow cytometry: FITC anti-human THY1 (Biolegend, clone 5E10), Brilliant Violet 421 anti-human CD45 (Biolegend, clone HI30), Brilliant Violet 650 anti-human EPCAM (Biolegend, clone 9C4), APC/Cy7 anti-human ITGA6 (Biolegend, clone GoH3), PE/Cy7 anti-human NGFR (Biolegend, clone ME20.4), APC anti-human PDPN (Biolegend, clone NC-08). Cells were stained for 30 minutes on ice in FACS buffer and then washed for immediate sorting. Cells were sorted on a BD FACSAria Fusion cell sorter using BD FACSDiva software. Up to 10,000 Basal cells were sorted into 100 μL BAM banker (Wako chemicals) for Omni-ATAC-seq and cooled to −80°C using a “Mr. Frosty” freezing container (Thermo scientific). Samples were stored at −80°C until thawed for Omni-ATAC-seq. 1,000 Basal cells for bulk RNAseq were sorted directly into 5 μL TCL buffer (Qiagen). FlowJo v10 by TreeStar was used to generate plots.
Histologic Analyses
Biopsies were fixed in 4% paraformaldehyde, embedded in paraffin, and 6 µm sections were prepared and stained with hematoxylin and eosin for quantification of glandular areas. Photomicrographs encompassing the entire area of each biopsy were taken. Total and glandular areas were measured with Image J software and expressed as glandular area as a percentage of total area. For p63 immunofluoresence, sections were quenched for 10 minutes in 1 mg/mL sodium borohydride in PBS. For antigen retrieval, slides were placed in a Coplin jar with preheated citrate target retrieval buffer (DAKO) at 95°C and transferred to a steamer for 60 minutes. Slides were cooled for 20 minutes at room temperature and then transferred to distilled water followed by PBS. Samples were blocked with serum free protein block (DAKO) containing 5% normal donkey serum for 60 minutes. Samples were incubated overnight at 4°C with purified anti-TP63 antibody (Biolegend, clone W15093A). After three washes in PBS-T, samples were incubated with 1:500 AlexaFluor 647-conjugated donkey anti-mouse IgG (Jackson immunoresearch, catalog# 715–605-150) and 1:10,000 Hoescht nuclear dye. Quantification of p63+ cells was performed in a blinded fashion and involved counting of p63+ nuclei relative to background staining with an isotype control primary antibody. As the epithelium can vary in length across sections, we normalized our quantification of total positive nuclei per 1,000μm2 area of epithelium as measured in ImageJ and report the final value as p63+ cells/1,000μm2 of epithelium.
Single-cell RNA-seq with Seq-Well
Once a single-cell suspension was obtained from freshly resected sinus tissue, or scrapings from inferior turbinate, we utilized the Seq-Well platform for massively parallel scRNA-seq to capture transcriptomes of single cells on barcoded mRNA capture beads. Full methods on implementation of this platform are available in Gierahn et al12. Briefly, 20,000 cells were loaded onto one array preloaded with barcoded mRNA capture beads (ChemGenes). The loaded arrays containing cells and beads were then sealed using a polycarbonate membrane with a pore size of 0.01 μm, which allows for exchange of buffers but retains biological molecules confined within each nanowell. Subsequent exchange of buffers allows for cell lysis, transcript hybridization, and bead recovery before performing reverse transcription en masse. Following reverse transcription using Maxima H Minus Reverse Transcriptase (ThermoFisher EP0753) and an Exonuclease I treatment (NewEngland Biolabs M0293L) to remove excess primers, PCR amplification was carried out using KAPA HiFi PCR Mastermix (Kapa Biosystems KK2602) with 2,000 beads per 50 μL reaction volume. Libraries were then pooled in sets of six (totaling 12,000 beads) and purified using Agencourt AMPure XP beads (Beckman Coulter, A63881) by a 0.6X SPRI followed by a 0.7X SPRI and quantified using Qubit hsDNA Assay (Thermo Fisher Q32854). Quality of WTA product was assessed using the Agilent hsD5000 Screen Tape System (Agilent Genomics) with an expected peak >1,000bp tailing off to beyond 5000bp, and a small/non-existent primer peak, indicating a successful preparation. Libraries were constructed using the Nextera XT DNA tagmentation method (Illumina FC-131-1096) on a total of 600 pg of pooled cDNA library from 12,000 recovered beads using index primers with format as in Gierahn et al12. Tagmented and amplified sequences were purified at a 0.6X SPRI ratio yielding library sizes with an average distribution of 650–750 base pairs in length as determined using the Agilent hsD1000 Screen Tape System (Agilent Genomics). Two arrays were sequenced per sequencing run with an Illumina 75 Cycle NextSeq500/550v2 kit (Illumina FC-404-2005) at a final concentration of 2.2–2.8pM. The read structure was paired end with Read 1 starting from a custom read 1 primer12 containing 20 bases with a 12bp cell barcode and 8bp unique molecular identifier (UMI) and Read 2 containing 50 bases of transcript information.
Single-cell RNA-seq Computational Pipelines and Analysis
Read alignment was performed as in Macosko et al39. Briefly, for each NextSeq sequencing run, raw sequencing data was converted to demultiplexed FASTQ files using bcl2fastq2 based on Nextera N700 indices corresponding to individual samples/arrays. Reads were then aligned to Hg19 genome using the Galaxy portal maintained by the Broad Institute for Drop-Seq alignment using standard settings. Individual reads were tagged according to the 12-bp barcode sequenced and the 8-bp UMI contained in Read 1 of each fragment. Following alignment, reads were binned onto 12-bp cell barcodes and collapsed by their 8-bp UMI. Digital gene expression matrices (e.g. cells-by-genes tables) for each sample were obtained from quality filtered and mapped reads, with an automatically determined threshold for cell count. UMI-collapsed data was utilized as input into Seurat40 (https://github.com/satijalab/seurat) for further analysis. Before incorporating a sample into our merged dataset, we individually inspected the cells-by-genes matrix of each as a Seurat object.
For analysis of all sequenced surgical ethmoid sinus resection samples, we merged UMI matrices across all genes detected in any condition and generated a matrix retaining all cells with at least 500 UMI detected (19,196 cells and 31,032 genes). This table was then utilized to setup the Seurat object in which any cell with at least 300 unique genes was retained and any gene expressed in at least 5 cells was retained (Supplementary Information: an R Script is included from this point to set up Seurat object and walk reader through dimensionality reduction and basic data visualization). The object was initiated with log-normalization, from a UMI+1 count matrix, scaling, and centering set to True. The total number of cells passing these filters captured across all patients was 18,624 cells with 22,575 genes, averaging 1,503 cells per sample with a range between 789 cells and 3,109 cells (Extended Data Fig. 1a,b, Supplementary Table 2). Before performing dimensionality reduction, data was subset to include cells with less than 12,000 UMI, and a list of 1,627 most variable genes was generated by including genes with an average normalized and scaled expression value greater than 0.13 and with a dispersion (variance/mean) greater than 0.28. We then performed principal component analysis (PCA) over the list of variable genes. For both clustering and t-stochastic neighbor embedding (tSNE), we utilized the first 12 principal components, as upon visual inspection of genes contained within, each contributed to a non-redundant cell type and this reflected the inflection point of the elbow plot. We used FindClusters within Seurat (which utilizes a shared nearest neighbor (SNN) modularity optimization based clustering algorithm) with a resolution of 1.2 and tSNE set to Fast with the Barnes-hut implementation to identify 21 clusters across the 12 input samples.
For analysis of all sequenced inferior turbinate scraping samples, the object was initiated with log-normalization, from a UMI+1 count matrix, scaling, and centering set to True. The total number of cells passing these filters captured across all patients was 18,704 cells with 24,842 genes, averaging 2,078 cells per sample with a range between 65 cells and 5,625 cells (NB: The 65 cell sample was a very mucus-laden polyp inferior turbinate sample, perhaps explaining the low cell yield, but clustered well within the three other samples each containing 253, 599, and 1,381 cells). Before performing dimensionality reduction, data was subset to include cells with less than 10,000 UMI, and a list of 1,499 most variable genes was generated by including genes with an average normalized and scaled expression value greater than 0.22 and with a dispersion (variance/mean) greater than 0.26. We then performed PCA over the list of variable genes. For both clustering and tSNE, we utilized the first 16 principal components, as upon visual inspection of genes contained within, each contributed to a non-redundant cell type and this reflected the inflection point of the elbow plot. We used FindClusters (which utilizes an SNN modularity optimization based clustering algorithm) with a resolution of 1 and tSNE set to Fast with the Barnes-hut implementation to identify 18 clusters across the 9 input samples.
For analysis of all sequenced ALI cultures, the object was initiated with log-normalization, from a UMI+1 count matrix, scaling, and centering set to True. The total number of cells passing these filters captured across all patients was 16,173 cells with 27,396 genes, averaging 2,448 cells per sample with a range between 1,980 cells and 3,009 cells. Before performing dimensionality reduction, data was subset to include cells with less than 25,000 UMI, and a list of 1,670 most variable genes was generated by including genes with an average normalized and scaled expression value greater than 0.35 and with a dispersion (variance/mean) greater than 0.35. We then performed PCA over the list of variable genes. For both clustering and tSNE, we utilized the first 16 principal components, as upon visual inspection of genes contained within, each contributed to a non-redundant cell state and this reflected the inflection point of the elbow plot. We used FindClusters (which utilizes an SNN modularity optimization based clustering algorithm) with a resolution of 0.6 and tSNE set to Fast with the Barnes-hut implementation to identify 11 clusters across the 4 input samples.
Cell Type Identification and within Cell Type Analysis
To identify genes which defined each cluster, we performed a ROC test implemented in Seurat with a threshold set to an area under the curve of 0.65. Top marker genes with high specificity were used to classify cell subsets into cell types (Fig. 1a–c; Extended Data Fig. 1e) based on existing biological knowledge. Three clusters were considered doublets (588 cells) based on co-expression of markers indicative of distinct cell types at ~1/2 the expression level detected in the parent cell cluster (e.g. T cell and myeloid cell) and removed from further analyses yielding a matrix with 18,036 cells used in all subsequent steps. Closely related clusters were merged to cell types based on biological curation and analysis of hierarchical cluster trees yielding ten total cell types (Fig. 1a–c; Extended Data Fig. 1e). We identified a much smaller number of eosinophils than expected in our single-cell data. Specifically, if we do not place bulk tissue immediately into RNA-later within 10 minutes, we cannot reliably detect eosinophil associated transcripts. However, flow cytometrically we recover from 0.5% to 5% of total cells fitting eosinophil profiles from polyps, and focused single-cell studies on granulocytes at the expense of the full ecosystem are possible and the topic of future work (data not shown). With the gentler tissue dissociation required for scrapings (Methods), we did recover a greater frequency of eosinophils from polyps in line with flow data (0.31% to 4.6% of cells; Extended Data Fig. 6d). We also did not find a distinct cluster of ILCs as they are around 0.01 to 0.1% of CD45 cells across the CRS spectrum per existing literature41 and extrapolating to the number of CD45 cells we captured, we would have detected between 0.8 and 8 ILCs. To investigate further granularity present within cell types, such as T cells, myeloid cells, fibroblasts, endothelial cells, and epithelial cells, we subset these cells from the Seurat object and re-ran dimensionality reduction and clustering (Extended Data Figs. 3, 4 and 6). The process used for clustering and subset identification was adapted for each cell type to optimize the parameters of variable genes, principal components, and resolution of clusters desired. Canonical correlation analysis42 (CCA) was also performed to validate epithelial cell type classification across disease states (Extended Data Fig. 5; Supplementary Information).
Differential Expression and Fractional Contribution of Gene Set to Transcriptome
To identify differentially expressed genes within cell types across non-polyp and polyp disease states, we utilized the ‘bimod’ setting in FindMarkers implemented in Seurat based on a likelihood ratio test designed for single-cell differential expression incorporating both a discrete and continuous component43. To determine the expression contribution to a cell’s transcriptome of a particular gene list, we summed the total log-normalized expression values for genes within a “list of interest” and divided by the total amount of log-normalized transcripts detected in that cell, giving the proportion of a cell’s transcriptome dedicated to producing those genes. For comparison of Wnt and Notch signaling, we z-scored the expression contribution metric and subtracted the value of Notch from Wnt yielding a metric centered on zero if both scores are equivalent, or weighted in the positive direction if enriched in Wnt. For reference gene lists used, including basal cell44, IFNα-, IFNγ-, IL-4-, IL-13-, IL-4/IL-13-induced genes45, Wnt and Notch please see Supplementary Table 4.
Simpson’s Index of Diversity, and Fibroblast Gene correlation with Basal Cell Frequency
To measure the “richness” of the epithelial ecosystem46, we employed Simpson’s Index of Diversity (D), which we present as (1-D), and ranges between 0 and 1, with greater values indicating larger sample diversity47. We used Simpson’s Index to characterize the composition of epithelial cells across basal, differentiating/secretory, glandular, and ciliated groupings in the non-polyp and polyp ethmoid sinus tissue ecosystems, as this metric accounts for both the number of distinct cell types present (e.g. species), and the evenness of the cellular composition across those cell types (e.g. relative abundance of species to each other). This measure takes into account the total number of members of a cell type, the number of cell types, and the total number of cells present. We calculate (1-D) for each sample. To determine genes correlated in specific cell types (e.g. fibroblasts) with the frequency of basal cells present in a cellular ecosystem, we correlated the average log-normalized single-cell count data for each gene to the rank of samples determined by increasing frequency of basal cells in each ecosystem (8.2% to 19.1% for non-polyp and 27.9% to 70.1% for polyp samples, Extended Data Fig. 7b).
Tissue and Sorted Basal Cell RNA-seq
Population RNA-seq was performed using a derivative of the Smart-Seq2 protocol for single cells48. In brief, tissue was collected directly into RNAlater (Qiagen) in the surgical suite and stored at −80°C until RNA isolation. RNA was isolated from 30 patients using phenol/chloroform extraction and normalized to 5ng as the input amount for a 2.2X SPRI ratio cleanup using Agencourt RNAClean XP beads (Beckman Coulter, A63987). RNA-seq from sorted basal cells was done as a bulk population using Smart-Seq2 chemistry starting with a 2.2X SPRI ratio cleanup. After oligo-dT priming, Maxima H Minus Reverse Transcriptase (ThermoFisher EP0753) was utilized to synthesize cDNA with an elongation step at 52°C before PCR amplification (15 cycles for tissue, 18 cycles for sorted basal cells) using KAPA HiFi PCR Mastermix (Kapa Biosystems KK2602). Sequencing libraries were prepared using the Nextera XT DNA tagmentation kit (Illumina FC-131-1096) with 250pg input for each sample. Libraries were pooled post-Nextera and cleaned using Agencourt AMPure SPRI beads with successive 0.7X and 0.8X ratio SPRIs and sequenced with an Illumina 75 Cycle NextSeq500/550v2 kit (Illumina FC-404-2005) with loading density at 2.2pM, with paired end 35 cycle read structure. Tissue samples were sequenced at an average read depth of 7.98 million reads per sample and 3 samples not meeting quality thresholds were excluded from further analyses yielding 27 total useable samples. Sorted basal cell samples were sequenced at an average read depth of 21.15 million reads per sample and all samples met quality thresholds regarding genomic and transcriptomic alignment.
Tissue and Sorted Basal Cell RNA-seq Data Analysis
Tissue and sorted basal cell samples were aligned to the Hg19 genome and transcriptome using STAR49 and RSEM50. 3 samples were excluded for low transcriptome alignment (<25%), so we retained 27 samples for further analyses. Differential expression analysis was conducted using DESeq2 package for R51. Genes regarded as significantly differentially expressed were determined based on an adjusted p-value using the Benjamini-Hochberg procedure to correct for multiple comparisons with a false discovery rate <0.05. We performed Ingenuity Pathway Analysis (IPA, Qiagen) through an instance available through the Broad Institute on the top 1,000 genes (all adjusted p<0.05) differentially expressed from our DESeq2 analysis, taking into account corresponding log-fold change for each gene. We also subset the tissue RNA-seq matrix based on genes found in Supplementary Table 3, which, from our single-cell marker discovery, were specific for basal, differentiating/secretory, glandular, or ciliated cells. We then ran PCA and KNN clustering implemented in R over these genes in order to identify the greatest vectors of variance across samples within the epithelial cell compartment (Fig. 3f,g).
For re-analysis of published data, we used two publicly-available RNA-seq data sets: one profiling normal human olfactory mucosa and the other assessing differences in gene expression between healthy, non-eosinophilic nasal polyps and eosinophilic nasal polyps7,52,53 NB: analysis is done per sample and as such no comparisons across the data sets or samples are made.
Diffusion Pseudotime Mapping for Differentiation Analysis
Using diffusion pseudotime54 mapping, which seeks to provide the most likely reconstruction for the developmental progression of a set of cells we built a trajectory for cells within basal and differentiating/secretory epithelial clusters (non-polyp clusters: 8-basal, 1-differentiating/secretory, 4-secretory; and polyp clusters: 12-basal, 2-basal, 0-differentiating/secretory; running several iterations starting from a random seed cell in cluster 8), over the combined basal and apical marker gene list (Fig. 4d; Extended Data Fig. 9a, Supplementary Table 3). By calculating a pseudotime trajectory for cells from both non-polyps and polyps together, we were then able to ask where cells from each disease state fall along a shared inferred temporal axis (Fig. 4d,e; Extended Data Fig. 9a). Diffusion pseudotime54 was calculated using the scanpy python package ‘dpt’ function on log-normalized data for clusters 8, 1 and 4 (predominantly non-polyp, Supplementary Table 3) and 12, 2, and 0 (predominantly polyp, Supplementary Table 3) together. A random root cell was chosen from cluster 8, as this was the basal cell cluster representative of the non-polyp (e.g. less aberrant) state, and we also ran iterations with random root cells chosen from the entire set of clusters and it assigned cluster 8 as the cluster most enriched at the beginning of the diffusion map, regardless. Plots were created with the seaborn, matplotlib, and pandas packages. Pearson correlations were then calculated for all genes in all cells tested, or for all genes in non-polyp cells and all genes in polyp cells, relative to pseudotime (Extended Data Fig. 9b). Differential correlation testing was performed using the cocor package to identify significance for the difference between correlation coefficients using Fisher’s 1925 z-statistic, account for number of cells.
Epigenetic Profiling of Basal Cells using Omni-ATAC-seq
Accessible chromatin profiling55 using the Omni-ATAC-seq protocol as described in Corces et al56 was performed on basal cells stored in 100 μL BAMBanker freezing media from 12 patients (n=4 non-polyp (3 retained after data quality filtering) and n=8 polyp). Cells (ranging from 1,000 to 10,000) were thawed quickly in a 37°C rock bath and 900 μL of ice-cold PBS supplemented with Roche complete-Mini Protease inhibitor was added immediately. Cells were split into two 1.5mL Eppendorf DNA lo-bind tubes to serve as technical replicates. Cells were centrifuged at 500g for 5 minutes at 4°C, washed once in PBS with protease inhibitor, centrifuged at 500g for 5 minutes at 4°C and supernatant was removed completely using two separate pipetting steps with extreme caution taken to avoid resuspension (e.g. smooth and consistent aspiration). The transposition reaction consisted of 20 μL total volume of the following mixture (10 μL 2X TD Buffer, 1 or 0.5 μL TDEnzyme, 0.1 μL of 2% digitonin, 0.2 μL of 10% Tween 20, 0.2 μL of 10% NP40, 6.6 μL of 1X PBS and 2.3 μL of nuclease free water). We performed replicates with two distinct concentrations of TDE since, when dealing with minute clinical samples, flow sorting can sometimes give variable cell numbers, and the ratio of TDE to cells is critical in determining the frequency with which cuts are made in the genome. We optimized in pilot experiments that for basal cell inputs in the range of 500 to 10,000 cells, the aforementioned two ratios gave expected patterns of nucleosome banding in gels (data not shown). We performed two reactions and then later, during in silico analysis, pooled peaks together for downstream analysis. The cells were resuspended into the transposition mixture and incubated at 37°C for 30 minutes in an Eppendorf Thermomixer with agitation at 300rpm. Transposed DNA was purified using a Qiagen MinElute Reaction Cleanup Kit with elution in 15 μL. Libraries were constructed from 10 μL of DNA using a 50 μL total reaction volume of NEB HF 2X PCR Master Mix with custom Nextera N700 and N500 index primers to barcode samples (also used in Smart-Seq2 protocol). We performed 14 cycles of PCR amplification and SPRI purified at 1.8X ratio. Based on the molarity of each library, we adjusted the number of subsequent PCR cycles to either 3, 4 or 5 more for each sample. We then performed a 0.25X reverse SPRI to remove larger fragments followed by a 1.7X SPRI to purify libraries for sequencing. Libraries were sequenced on an Illumina NextSeq with paired end 38 cycle read structure at a loading density of 1.95pM.
Omni-ATAC-seq Data Analysis
Reads were aligned using bowtie2 using the following flags: “-S -p 1 -X 2000 --chunkmbs 1000” then bams were created using samtools view with the following flags: “samtools view -bS -F 4 –”. Duplicates were removed with picard. Forward reads were shifted 4bp and negative reads were shifted 5bp using a custom python script and the pysam package as is recommended for ATAC-seq data. Samples for each patient were merged using samtools merge and all patients were downsampled to 3 million reads using custom python scripts and ‘samtools view’ with the ‘-b’ and ‘-s’ flags. MACS2 ‘callpeak’ command was used to call peaks on each sample with flags ‘“-f BAMPE’-q 0.001 --nomodel --shift −100 --extsize 200 -B –broad’. Peaks from all samples were merged into one peakfile with bedtools and counts of reads per peak for each sample was generated with bedtools multicov. DESeq2 was run with the design ~polyp, testing for significant differences between polyp and non-polyp samples on this peak matrix and differential peaks with Benjamini-Hochberg adjusted p-value less than 0.01 with ‘greater’ or ‘less’ null hypotheses were used in downstream analysis. Homer2 was run for known motif finding on differential peaks with the set of all peaks as background57. To determine a false discovery rate, Homer2 was run on sets of random peaks chosen with replacement from the set of all peaks.
Epithelial Cell Culture
Tissues were digested as described above from either non-polyp or polyp surgical resections from the ethmoid sinus. 1,000,000 digested cells were added to a 25 cm2 tissue culture flask (Corning) pre-coated with 0.03 mg/mL Type I bovine collagen solution (StemCell Technologies) and cultured in PneumaCult-Ex media (Stemcell Technologies, 05008). Media was changed every second day until cells reached confluence. Cells were subsequently frozen in 70% basal media with 20% FBS and 10% DMSO.
Air-liquid Interface Cultures
For air-liquid interface (ALI) cultures58, 100,000 cultured epithelial cells per well were added to 0.4μm pore 24-well polyester membrane inserts (Corning) pre-coated with 0.03 mg/mL Type I bovine collagen solution (StemCell Technologies) with Pneumacult-Ex media (Stemcell Technologies, 05008) on both sides of the membrane. After 24 hours, apical media was changed to remove dead cells. After 72 hours, apical media was removed completely and basal media was changed to Pneumacult-ALI (Stemcell Technologies, 05001) supplemented with 5 mL 100x penicillin-streptomycin (Fisher), 1 mL 500x gentamicin/amphotericin B (ThermoFisher), 1 mL 0.2% heparin sodium salt in PBS (Stemcell Technologies) and 2.5 mL 200x hydrocortisone stock solution (Stemcell Technologies) and 0, 0.1, 1 or 10 ng/mL IL-13 (Biolegend). Basal media was changed every 2–3 days for 21 days, after which membranes were removed and cells dissociated with Stempro Accutase Cell Dissociateion Reagent (Gibco) for Seq-Well or flow cytometry. After following scRNA-seq data analysis pipelines described above, cell states recovered in ALI cultures (Fig. 5a; Extended Data Fig. 9g) were related to in vivo cell types59.
Basal Cell Stimulation
Basal cells from non-polyp or polyp surgical resections from ethmoid sinus were placed into epithelial cell culture (e.g. “lateral expansion” in the absence of differentiation, see above, inspired by experiments in microglia60), passaged, and 10,000 cells seeded at passage 5 (e.g. 5 weeks ex vivo) and cultured at confluence in 96 well flat-bottom collagen-coated tissue culture plates (Corning, 3799) for 48 hours in Pneumacult-Ex serum-free media (Stemcell Technologies, 05008). Cytokines were added for 12 hours overnight at increasing doses (0, 0.1, 1, 10 ng/mL) of IL-4 (Biolegend 766205), IL-13 (Biolegend 571104), and (0.1, ng/mL) IL-4+IL-13 in combination (n=32 samples non-polyp and n=32 samples polyp basal cells over all conditions, each condition run as a biological duplicate, and a technical duplicate therein) before lysis using RLT + 1% BME (Qiagen and Sigma, respectively). Bulk RNA-seq was performed as described for sorted basal cells starting from lysates. Basal cell stimulation samples were sequenced at an average read depth of 3 million reads per sample and all samples met quality thresholds regarding genomic and transcriptomic alignment.
Statistical Analyses
Number of samples included in analyses are listed throughout figure legends and all represent distinct biological samples. The same surgeon performed surgeries on all individuals and was blinded to study design. The same allergist/immunologist performed nasal scrapings on all samples and was blinded to study design. Quantification of histological sections was performed in a blinded fashion. No samples or cells meeting quality thresholds were excluded from analyses. Where single-cell data was analyzed on a gene-level, the statistics were performed over the number of cells. Statistical analyses were performed using GraphPad Prism v7.0a, Seurat 1.4.0.1 implemented in RStudio, DESeq2 1.10.1 package implemented in RStudio, and Ingenuity Pathway Analysis run through the Broad Institute, and macs2, DESeq2 and Homer2 for Omni-ATAC-Seq. All violin plots were generated using standard Seurat code without modification to smoothing or density. Violin density only generated when >25% of cells in indicated sample have non-zero measurement for gene, widest aspect represents centre of positive measures, minima and maxima are represented within the scale with minima at 0 and maxima encompassing all points for the count-based expression level (log(scaled UMI+1)) of each gene. Exact values for all genes displayed and tested available in Supplementary Table 3 organized by panel. All violin plots contain at minimum 100 individual cells in any one cluster (Supplementary Table 3 for precise numbers of cells per cluster and type, most are included in figure legends where space allows), and have points suppressed for ease of legibility. Some violin plots with less than 100 cells have individual data points displayed and corresponding statistical metrics are available in accompanying figure legend and Supplementary Table 3. As some scores followed non-normal distributions as tested for using a Lilliefors normality test, we utilized a Mann-Whitney U-test where indicated for determining statistical significance. For scores in single-cell data, we report effect sizes in addition to statistical significance as an additional metric for the magnitude of the effect observed. The calculation was performed as Cohen’s d where: effect size d = (Mean1-Mean2)/(S.D. pooled). Unpaired two-tailed t-tests for direct comparisons and t-test with Holm-Sidak correction, Bonferroni correction, or Benjamini-Hochberg for multiple comparisons, depending on software package used, where appropriate. Mann-Whitney U-test for quantification of histological data due to non-normally distributed data. Pearson correlation thresholds were determined as significant through determination of asymptotic p-values through use of rcorr function in Hmisc, but exact corrected p-values by Holm-Sidak method for multiple comparisons are calculated for those highlighted in text using RcmdrMisc package. Comparison of Pearson correlation coefficients in pseudotime analyses was done using Fisher’s 1925 z-statistic accounting for the number of cells.
Data Availability Statement
The cells-by-genes matrix generated from ethmoid sinus surgical resections and analyzed during the current study is available along with the manuscript as Supplementary Table 2 along with R code for standard implementation of Seurat. A cells-by-genes matrix from inferior turbinate and polyp scraping data is also available as Supplementary Table 6. Dupilumab treatment cells-by-genes matrices as Supplementary Tables 7 and 8. A metadata table encompassing all scRNA-seq samples is provided as Supplementary Table 9. The count and TPM matrices and associated metadata from bulk tissue RNA-seq are available as Supplementary Tables 10, 11, and 12. FASTQ file format data will be available through dbGaP under accession number XXXX. Marker gene lists for cell types identified in Fig. 1a,b, and from resultant analyses in Fig. 2b, for frequencies of cell clusters and types in Fig. 2c, for cell types identified in Fig. 2e, Fig. 2f, Fig. 3g, Fig. 5a, Fig. 5e, Extended Data Fig. 3a,b,c, Extended Data Fig. 4c, Extended Data Fig. 5e, Extended Data Fig. 6b,d, Extended Data Fig. 10a, selected comparisons of differential expression in Fig. 2d, Fig. 4a, Fig. 5c, Fig. 5f, Extended Data Fig. 2c, Extended Data Fig. 10h, and pseudotime correlation Extended Data Fig. 9b, are available as tabs in Supplementary Table 3. Differential peak calling from epigenetic profiling available in Supplementary Table 5. Additional R code for analyses available on http://shaleklab.com/resources/.
Extended Data
Extended Data Figure 1 |. Consistency of cell capture and identification in surgical EthSin scRNA-seq patient cohort.
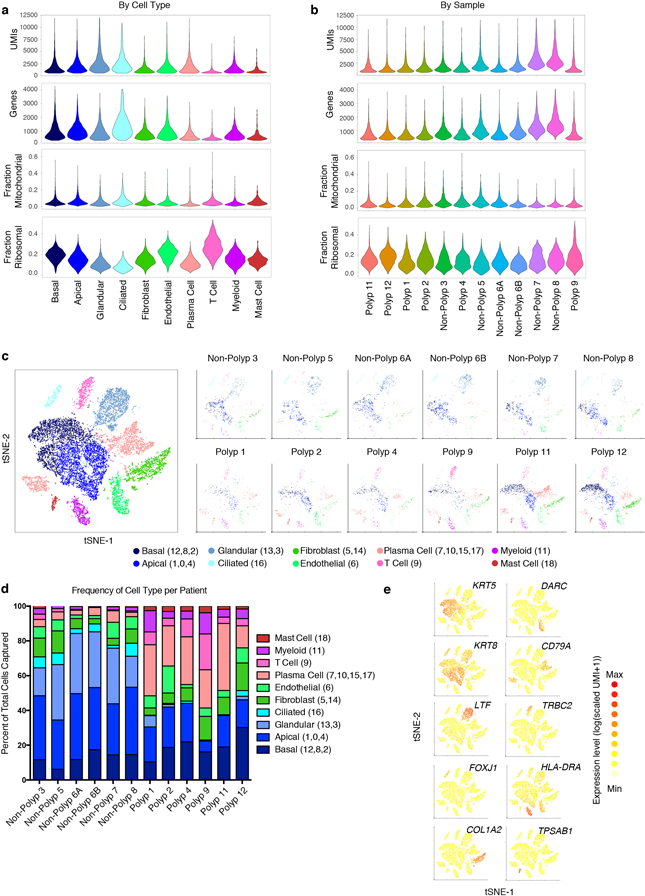
(a) Number of unique molecular identifiers (nUMI) and genes identified, and fraction of reads mapping to mitochondrial or ribosomal genes across recovered cell types; 3,222 basal cells, 4,362 apical cells, 2,192 glandular cells, 498 ciliated cells, 835 T cells, 2,976 plasma cells, 1,724 fibroblasts, 1,143 endothelial cells, 811 myeloid cells, 273 mast cells.
(b) nUMI and genes identified, and fraction of reads mapping to mitochondrial or ribosomal genes across patient samples; 789 Polyp 1 cells, 1,309 Polyp 2 cells, 1,153 Polyp 3 cells, 913 Polyp 4 cells, 1,219 Polyp 5 cells, 1,141 Polyp 6A cells, 1,334 Polyp 6B cells, 1,314 Polyp 7 cells, 1,286 Polyp 8 cells, 1,481 Polyp 9 cells, 2,988 Polyp 11 cells, 3,109 Polyp 12 cells.
(c) tSNE plot as in Fig. 1b colored by cell types across all patients and then separated by sample; 18,036 single cells (n=12 samples).
(d) The percentage of each cell type recovered within each sample.
(e) Select marker gene overlays displaying binned count-based UMI-collapsed expression level (log(scaled UMI+1)) on a tSNE plot from Fig. 1b for key cell types identified (see Supplementary Table 3 for full gene lists); area under the curve (AUC) 0.998 to 0.7 for all markers displayed.
Extended Data Figure 2 |. Top marker genes for cell types by scRNA-seq and bulk tissue RNA-seq from EthSin recovers expected T2I and eosinophilic modules.
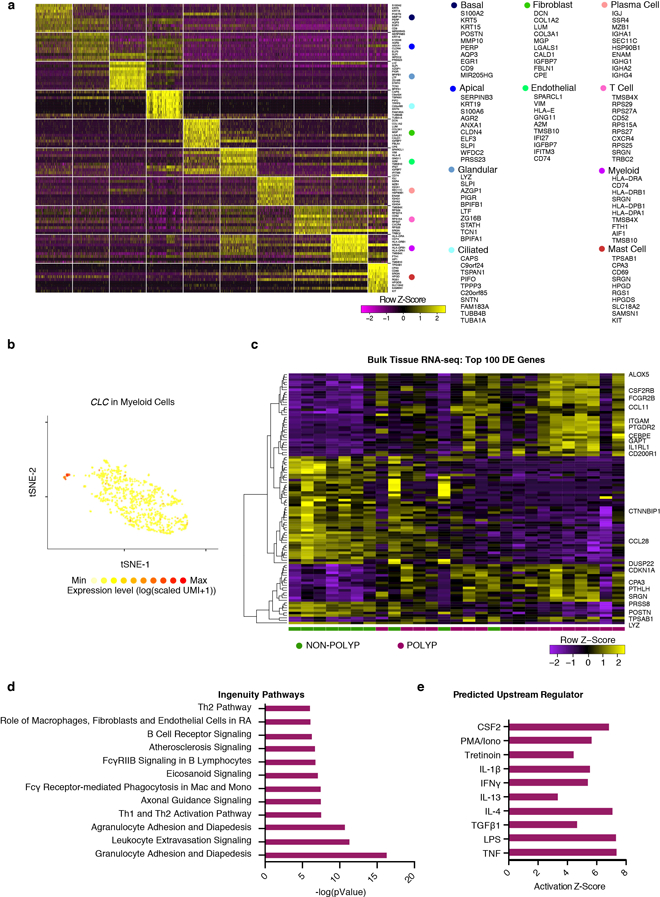
(a) Row-normalized heatmap of the top-10 marker genes identified by ROC-test (AUC>0.73 for all) over all cell types (Fig. 1b,c) with select genes displayed on y-axis and cells on x-axis (see Supplementary Table 3 for full gene lists); maximum 500 cells/type.
(b) An overlay of CLC displaying binned count-based expression level (log(scaled UMI+1)) amongst myeloid cells (a pathognomonic gene for eosinophils); 811 myeloid cells from n=12 samples.
(c) A row-normalized and row-clustered heatmap over the top 100 positively and negatively differentially-expressed genes (50 in each direction) in bulk tissue RNA-seq of 27 samples from non-polyp (n=10) and polyp (n=17) tissue with select genes displayed; DESeq2 Wald Test, all p<9.03×10−5 for genes displayed, corrected for multiple comparisons by Benjamini procedure, samples ordered as in Fig. 3g, (see Supplementary Table 3 for full gene list and associated statistics).
(d) The top differentially regulated pathways identified by Ingenuity Pathway Analysis (see Methods) over the top 1,000 differentially expressed genes, as determined by p<0.05 corrected for multiple comparisons by Benjamini procedure, across polyp and non-polyp tissue.
(e) Predicted upstream regulators based on differentially expressed gene modules in polyp tissue relative to non-polyp determined using Ingenuity Pathway Analysis (see Methods).
Extended Data Figure 3 |. Sub-clustering of myeloid, fibroblast and endothelial cell types from the EthSin T2I inflammatory ecosystem.
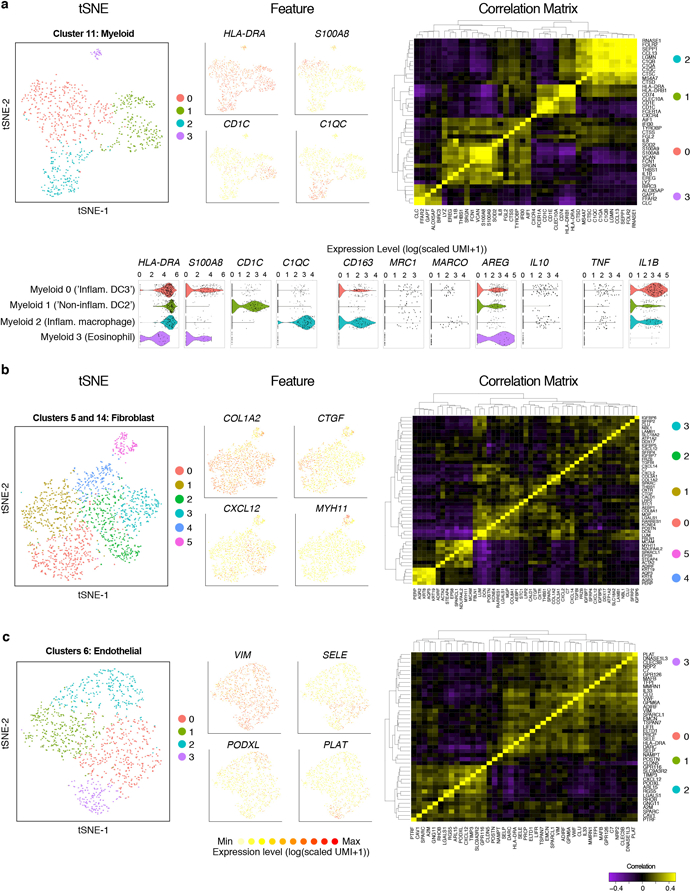
(a) tSNE plot of 811 myeloid cells (n=6 non-polyp, n=6 polyp samples), colored by clusters identified through shared nearest neighbor (SNN) analysis (Supplementary Table 3; Methods), from CRS-EthSin; select marker gene overlays displaying count-based (unique molecular identifier (UMI) collapsed) expression level (log(scaled UMI+1)) on a tSNE plot (see Supplementary Table 3 for full gene lists; genes identified via ROC test with AUC 0.689 for S100A8, 0.763 for CD1C, 0.927 for C1QC); a clustered correlation matrix of marker genes identified in single-cell data from myeloid cells; and violin plots for the expression value (log(scaled UMI+1)) of selected markers of myeloid activation state.
(b) tSNE plot of 1,724 fibroblasts (n=6 non-polyp, n=6 polyp samples), colored by clusters identified through shared nearest neighbor (SNN) analysis (Supplementary Table 3; Methods), from CRS-EthSin; select marker gene overlays displaying count-based (unique molecular identifier (UMI) collapsed) expression level (log(scaled UMI+1)) on a tSNE plot (see Supplementary Table 3 for full gene lists; genes identified via ROC test with AUC 0.691 for CTGF, 0.683 for CXCL12, 0.726 for MYH11); and a clustered correlation matrix of marker genes identified in single-cell data from fibroblasts. NB: Clusters 4 and 5 likely represent doublets with epithelial cells and endothelial cells, respectively. While we exclude these clusters from further formal analyses, we note that there may be interesting biology within pairs of cells found to interact more frequently than by chance.
(c) tSNE plot of 1,143 endothelial cells (n=6 non-polyp, n=6 polyp samples), colored by clusters identified through shared nearest neighbor (SNN) analysis (Supplementary Table 3; Methods), from CRS-EthSin; select marker gene overlays displaying count-based (unique molecular identifier (UMI) collapsed) expression level (log(scaled UMI+1)) on a tSNE plot (see Supplementary Table 3 for full gene lists; genes identified via ROC test with AUC 0.742 for SELE, 0.706 for PODXL, 0.822 for PLAT); and a clustered correlation matrix of marker genes identified in single-cell data from endothelial cells.
Extended Data Figure 4 |. Mapping T2I mediators within EthSin non-polyp or polyp ecosystems and the identities of T cells.
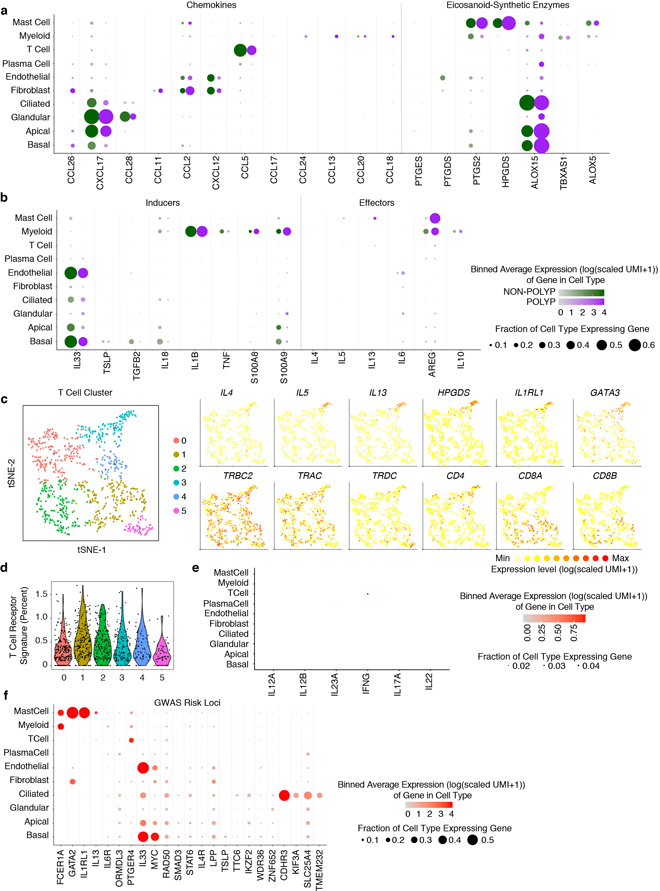
(a) Dot plots of chemokines and lipid mediators with known roles in T2I mapped onto cell types divided by non-polyp or polyp disease state. Dot size represents fraction of cells within that type expressing, and color intensity binned (log(scaled UMI+1)) gene expression amongst expressing cells (related to Figure 1d).
(b) Dot plot of inducers and effectors of T2I mapped onto cell types divided by non-polyp or polyp disease state. Dot size represents fraction of cells within that type expressing, and color intensity binned (log(scaled UMI+1)) gene expression amongst expressing cells (related to Figure 1d).
(c) tSNE plot of re-clustered T cells with select gene overlays displaying binned count-based expression level (log(scaled UMI+1)) for Th2A-specific genes (top row) and canonical T cell markers (bottom row); 835 T cells from n=6 non-polyp and n=6 polyp samples.
(d) Violin plot of five identified T cell clusters scored for expression of T cell receptor complex genes (e.g. TRAC and CD3E, see Methods, Supplementary Table 4); dots represent individual cells, 835 total T cells.
(e) Dot plot of inducers and effectors of Type 1 immunity across all cell types (NB: IL17F not detected).
(f) Dot plot of select GWAS risk alleles41 for allergic disease, mapped onto cell types divided by non-polyp or polyp disease state. Dot size represents fraction of cells within that type expressing, and color intensity binned (log(scaled UMI+1)) gene expression amongst expressing cells (related to Figure 1d).
Extended Data Figure 5 |. Relationship of EthSin epithelial cell clusters and secretory/glandular distinctions.
(a) A phylogenetic tree based on the average cell from each cluster of epithelial cell clusters in gene-space.
(b) Violin plot of expression contribution to a cell’s transcriptome of basal cell genes (see Methods and Supplementary Table 4) across all epithelial cells; 794 cells cluster 12; 924 cells cluster 8; 1,504 cells cluster 2; 1,561 cells cluster 1; 1,600 cells cluster 0; 1,201 cells cluster 4; 725 cells cluster 13; 1,467 cells cluster 3; 498 cells cluster 16; *Mann-Whitney U-test, with Bonferroni correction, p<1.76×10−15, 12, 8 or 2 vs. the mean score of basal/apical epithelial cells; p= 0.5392, 1 vs. the mean score.
(c) Canonical correlation analysis (CCA) displaying our cell type annotations for basal and apical cells derived through clustering and biological curation alongside CCA clusters in tSNE space; 7,584 basal and apical cells.
(d) Violin plots for the count-based expression level (log(scaled UMI+1)) of selected marker genes for each identified epithelial cell subset; cell numbers as in (b).
(e) Row-normalized heatmap of the top marker genes identified by ROC-test (AUC>0.6) within each cell type for each cell cluster with genes displayed on y-axis and cluster annotations on x-axis (see Supplementary Table 3 for full gene lists).
(f) Select overlays on clusters 0 and 4 (differentiating/secretory) and 13 (glandular) displaying binned count-based expression level (log(scaled UMI+1)) in tSNE space for canonical goblet (MUC5B, MUC5AC, SPDEF, FOXA3) and secretory (SCGB1A1) genes; 3,526 cells.
(g) A clustered correlation matrix of glandular, goblet, and secretory cell genes; Pearson’s abs(r)> 0.038 is p<0.05 significant based on asymptotic p-values.
Extended Data Figure 6 |. Glandular cell subsets, their relationship to apical secretory cells, and immune cells recovered through nasal scrapings.
(a) tSNE plots of 5,928 single epithelial cells (n=6 non-polyp samples), and 4,346 single epithelial cells (n=6 polyp samples) colored by clusters identified through (left) shared nearest neighbor (SNN) analysis and (right) original biological curation of cell types (Supplementary Table 3; Methods) as illustrated in Figure 2a. NB: cluster colors in left panels of each disease are not comparable but curated clusters in right are, and glandular cells are highlighted for subsetting in next panel.
(b) Violin plots for the count-based expression level (log(scaled UMI+1)) of selected marker genes identified through marker discovery (ROC test) for each subset of glandular cells; 2,114 total cells (791 cells cluster 1; 709 cells LCN2 cluster; 283 cells SERPINB3 cluster; 209 cells MUC5B cluster; 183 cells PRB1 cluster) with representation of every non-polyp patient in each cluster of cells (e.g. no cluster is unique to one patient) and AUC metric 0.800 for LCN2, 0.736 for SERPINB3, 0.985 for MUC5B, 0.973 for BPIFB2, and 0.908 for PRB1.
(c) Samples were acquired through the two distinct methods of nasal scraping and ethmoid sinus surgical intervention. This allowed for sampling of left: healthy tissue from InfTurb (scraping), middle: of CRS-EthSin-non-polyp tissue (surgery), right: of CRS-EthSin-polyp tissue (surgery), of InfTurb of polyp-bearing individuals (scraping), and of CRS-EthSin-polyp tissue accessible for scraping (scraping). Anatomy of the nasal turbinates (healthy and CRS polyp) and ethmoid sinus (CRS non-polyp and CRS polyp) where samples were acquired is displayed below, highlighting the depth of cells recovered from each site related to Fig. 2. Healthy tissue is annotated with basal and apical cell types, including sub-mucosal glands.
(d) Left: tSNE plot of 18,704 single cells from nasal scrapings (n=9 samples) colored by clusters identified through shared nearest neighbor (SNN) analysis (Supplementary Table 3; Methods); middle: tSNE plot colored by cell types identified through marker discovery (ROC test) and biological curation of identified clusters (Supplementary Table 3; Methods); right: colored by disease and tissue of origin from healthy InfTurb (7,603 cells; n=3 samples), polyp-bearing patient InfTurb (2,298 cells; n=4 samples), and polyp scraping directly from EthSin-polyp (8,803 cells; n=2 samples); with adjacent select marker gene overlays displaying count-based UMI-collapsed expression level (log(scaled UMI+1)) for apical epithelial (KRT8) and hematopoietic (PTPRC) genes.
(e) Select marker gene overlays displaying count-based UMI-collapsed expression level (log(scaled UMI+1)) on a tSNE plot from (a) for key cell types identified (see Supplementary Table 3 for full gene lists); area under the curve (AUC) 0.946 to 0.705 for all markers displayed.
(f) Violin plots for the count-based expression level (log(scaled UMI+1)) for key differentially expressed genes using ROC test within myeloid cells across disease states and tissues identified (Methods); 137 cells, n=3 healthy inferior turbinate; 157 cells, n=4 polyp inferior turbinate; 210 cells, n=2 polyp ethmoid sinus samples; AUC 0.67 for TXNRD1, 0.615 for RALA, 0.647 for TLR2, 0.619 for RIPK2, 0.747 for C1QA, 0.674 for FGL2.
Extended Data Figure 7 |. Changes in cellular composition between EthSin-non-polyp and EthSin-polyp tissue by scRNA-seq and flow cytometric gating and histological strategy for quantification and isolation of basal cells.
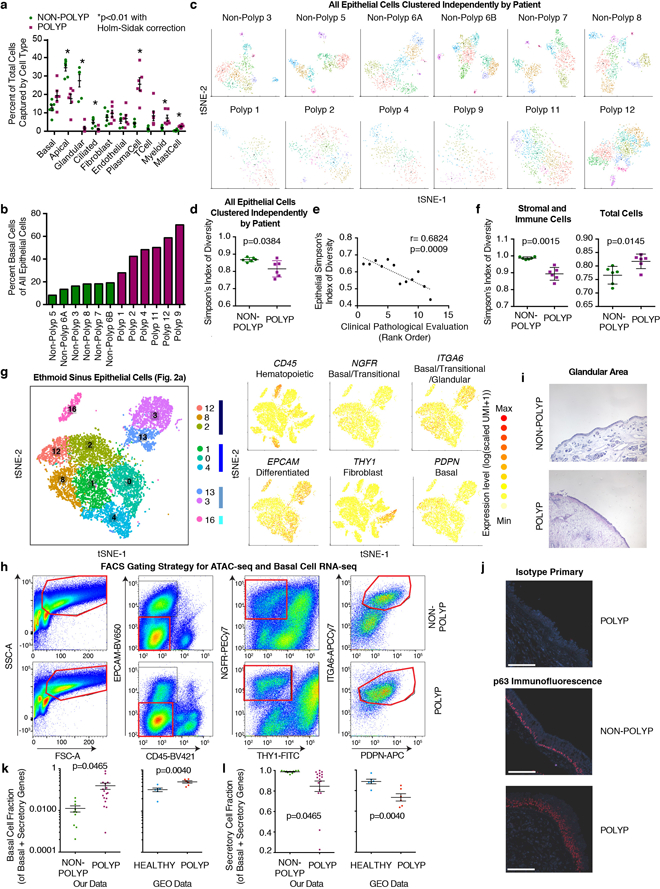
(a) The frequency of each cell type recovered amongst all cells within each patient sample (n=6 non-polyp, n=6 polyp) grouped by disease state; *t-test, two-sided, p=0.0003 for apical, p<0.0001 for glandular, p=0.0047 for ciliated, p=0.00014 for plasma cell, p=0.0098 for myeloid and p=0.00018 for mast cell; all non-polyp vs. polyp with Holm-Sidak correction for multiple comparisons; mean±s.e.m.
(b) The frequency of basal cells amongst epithelial cells captured in scRNA-seq data displayed for each sample and colored by non-polyp or polyp designation.
(c) tSNE plots with each patient’s cells clustered independently over a common list of most variable genes identified from all epithelial cells and with clustering parameters set constant to 12 principal components and resolution set to 1.4; minimum 789 cells in each plot, Extended Data Fig. 1b and Supplementary Table 3 for specific cell numbers.
(d) Simpson’s index of diversity over epithelial cell clusters identified in (c), an indication of the total richness present within an ecosystem, calculated for each patient; n=6 non-polyp and n=6 polyp samples; *t-test, two-tailed, p=0.0384, mean±s.e.m.
(e) Correlation of Simpson’s index of diversity calculated over epithelial cells against the ranked order of samples based on clinical pathological evaluation; n=6 non-polyp and n=6 polyp samples; r=0.6824, p=0.009.
(f) Simpson’s index of diversity over stromal and immune cell types and total cells, an indication of the total richness present within an ecosystem, calculated for each sample (n=6 non-polyp and n=6 polyp); points represent individual samples, *t-test, two-tailed, p=0.0015 stromal and immune, p=0.0145 total cells, non-polp vs. polyp; mean±s.e.m.
(g) Reproduced from Figure 2a: tSNE plot of 10,274 epithelial cells, colored by clusters identified through SNN, with adjacent color bars representing related cell clusters, and overlays displaying binned count-based expression level (log(scaled UMI+1)) of selected genes used to negatively (CD45, EPCAM, THY1) and positively (NGFR, ITGA6, PDPN) identify basal cells.
(h) Full flow cytometric gating strategy for quantification and isolation of basal cells from non-polyp and polyp tissue, (related to Fig. 3c).
(i) Representative histology (5x magnification) of the glandular area detected in haematoxylin and eosin stained tissue sections from non-polyp or polyp patients; quantification in Fig. 3e.
(j) Representative immunofluorescence of p63-staining cells (basal cell marker) relative to isotype control; quantification in Fig. 3d; scale bar 100μm.
(k) Basal cell fraction of transcripts from bulk tissue RNA-seq data of our own data set (related to Fig 3g,h) and two GEO data sets containing healthy and healthy/polyp nasal mucosa biopsies; our data n=10 non-polyp samples, n=17 polyp samples; reference data n=6 healthy, n=6 polyp samples; *t-test, two-tailed p=0.0465 our data and p=0.0040 GEO data; mean±s.e.m.
(l) Secretory cell fraction of transcripts from bulk tissue RNA-seq data of our own data set (related to Fig 3g,h) and two GEO data sets containing healthy and healthy/polyp nasal mucosa biopsies; our data n=10 non-polyp samples, n=17 polyp samples; reference data n=6 healthy, n=6 polyp samples; *t-test, two-tailed p=0.0465 our data and p=0.0040 GEO data; mean±s.e.m
Extended Data Figure 8 |. Epithelial cytokine signatures from CRS-EthSin tissue demonstrate T2I pattern, discovery of gene modules in the fibroblast niche which correlate with basal cell hyperplasia, and differential expression within myeloid and endothelial cells by polyp status.
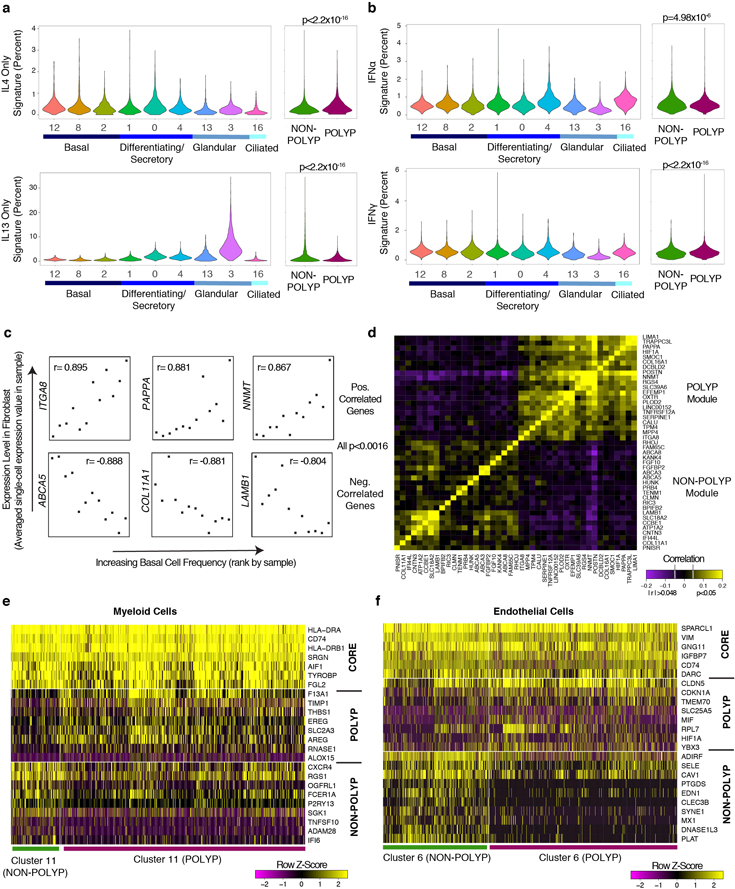
(a) Violin plots of IL-4- or IL-13-uniquely induced gene signatures in respiratory epithelial cell clusters or grouped by disease state presented as expression contribution to a cell’s transcriptome (see Methods, Figure 4b for shared genes, and Supplementary Table 4); 794 cells cluster 12; 924 cells cluster 8; 1,504 cells cluster 2; 1,561 cells cluster 1; 1,600 cells cluster 0; 1,201 cells cluster 4; 725 cells cluster 13; 1,467 cells cluster 3; 498 cells cluster 16; *Mann-Whitney U-test, p<2.2×10−16, 0.305 effect size IL-4 polyp vs. non-polyp and −0.448 effect size IL-13 polyp vs non-polyp.
(b) Violin plots of IFNα- or IFNγ-induced gene signatures in respiratory epithelial cell clusters or grouped by disease state presented as expression contribution to a cell’s transcriptome (see Methods, and Supplementary Table 4); cell numbers as in (a); *Mann-Whitney U-test, p=4.98×10−6, −0.156 effect size IFNα polyp vs. non-polyp; *Mann-Whitney U-test, p<2.2×10−16, 0.161 effect size IFNγ polyp vs non-polyp.
(c) Selected genes detected in fibroblasts from single-cell data which correlate with the samples ranked by basal cell frequency detected within each ecosystem; n=6 non-polyp, 6 polyp samples, all genes used: Spearman correlation, abs(r)>0.7651, p<0.0037. NB: to determine genes correlated in specific cell types (e.g. fibroblasts) with the frequency of basal cells present in a cellular ecosystem, we correlated the average log-normalized single-cell count data for each gene to the rank of samples determined by increasing frequency of basal cells in each ecosystem (8.2% to 19.1% for non-polyp and 27.9% to 70.1% for polyp samples, Extended Data Fig. 7b).
(d) A clustered correlation matrix of genes identified as per (c) in single-cell data from fibroblasts; Pearson’s abs(r)>0.048 is p<0.05 significant based on asymptotic p-values.
(e) Row-normalized heatmap for myeloid cells from ethmoid sinus with select genes displayed on y-axis including a core myeloid signature (ROC-test myeloid cells vs. rest of cells, AUC>0.8), and genes differentially expressed (bimodal test) by disease state, with disease state annotations on x-axis; bimodal test, all non-core genes p<0.0002 or less with Bonferroni correction for multiple hypothesis testing based on number of genes tested.
(f) Row-normalized heatmap for endothelial cells from ethmoid sinus with select genes displayed on y-axis including a core basal signature (ROC-test endothelial cells vs. rest of cells, AUC>0.75), and genes differentially expressed (bimodal test) by disease state, with disease state annotations on x-axis; bimodal test, all non-core genes p<2.43×10−6 or less with Bonferroni correction for multiple hypothesis testing based on number of genes tested.
Extended Data Figure 9 |. Pseudotime analysis on basal and differentiating/secretory cell clusters from EthSin, transcriptional motif enrichments in non-polyp and polyp basal cells, and the identity of cell types in air-liquid interface cultures.
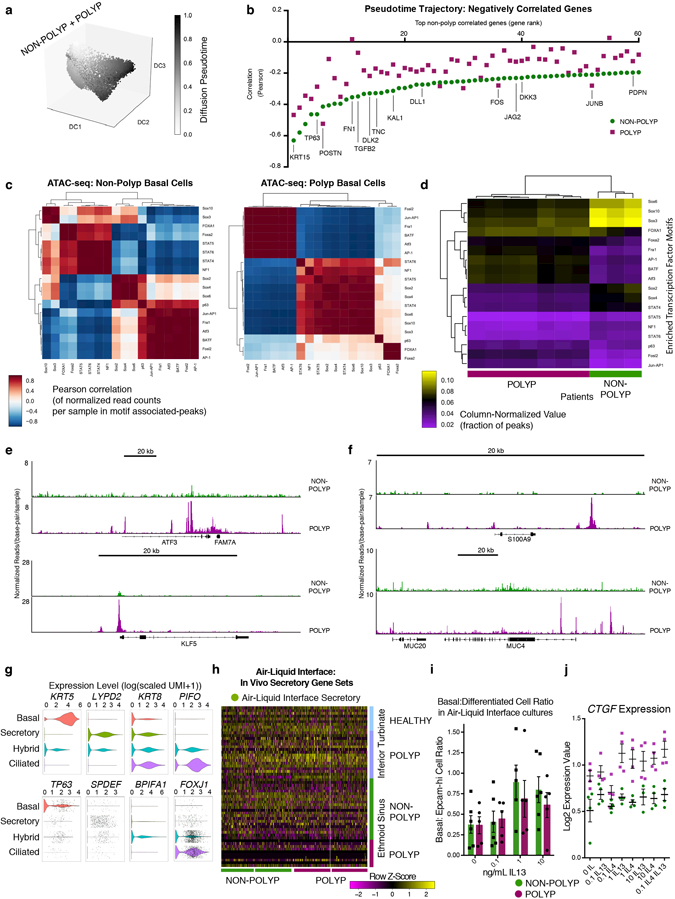
(a) Pseudotime analysis using diffusion mapping (see Methods) of selected clusters of epithelial cells, here displaying diffusion pseudotime (related to Fig. 4d); 3,516 cells (clusters 8/1/4); 4,064 cells (clusters 12/2/0); and n=6 non-polyp, n=6 polyp samples; diffusion map and diffusion coefficients (DC) are calculated over the set of basal and apical marker genes identified in Fig. 1a, see Supplementary Table 3.
(b) The top 60 negatively correlated genes expressed in non-polyp cells with pseudotime trajectory and Pearson correlation values for genes in polyp cells also displayed; differential correlation coefficient analysis using Fisher’s Z-statistic, accounting for number of cells in each group (specific genes highlighted all > 2 Z, full results including Bonferroni corrected p-values in Supplementary Table 3).
(c) Correlation matrices (row and column clustered) of the normalized read counts per sample in motif associated-peaks for non-polyp or polyp samples; Pearson correlation, n=3 non-polyp, n=7 polyp.
(d) A column-normalized heatmap (row and column clustered) for the fraction of peaks with a motif corresponding to accessibility of the respective transcription factor displayed by patient; n=3 non-polyp, n=7 polyp.
(e) IGV tracks for ATF3 and KLF5 based on peaks detected and averaged by non-polyp and polyp samples from ATAC-seq profiling.
(f) IGV tracks for S100A9 and MUC4 based on peaks detected and averaged by non-polyp and polyp samples from ATAC-seq profiling.
(g) Violin plots for the count-based expression level (log(scaled UMI+1)) for key marker genes using ROC test across cell types identified in (Fig. 5a; Supplementary Table 3); 1,345 basal; 6,420 secretory; 6,381 hybrid; and 2,027 ciliated cells; from n=2 non-polyp and 2 polyp patients; AUC 0.943 for KRT5, 0.667 for TP63, 0.644 for LYPD2, <0.55 for SPDEF, <0.55 for KRT8, 0.602 for BPIFA1, 0.813 for PIFO, 0.73 for FOXJ1.
(h) Row-normalized heatmap for ALI-secretory cells (subsampled to 300 cells per donor) as in Fig. 2f of the top in vivo secretory marker genes identified by ROC-test (AUC>0.662) with select genes displayed on y-axis including a core secretory signature (ROC-test secretory cells vs. rest of cells), and then within secretory cells a ROC-test used to identify marker genes within each disease/location category, and basal-cell derived annotations on x-axis (see Supplementary Table 3 for full gene lists, all AUC>0.65 for markers displayed in Fig. 2f).
(i) Quantification of flow cytometry for the ratio of basal to Epcamhi cells (gating as in Extended Data Fig. 7h) from ALI cultures at 21 days stimulated with IL-13 over the indicated doses; points represent individual biological replicates; n=6 non-polyp, n=5 polyp samples for each dose; *2-way ANOVA, n.s. between disease groups at any dose tested; *2-way ANOVA, p=0.0224 for IL-13 dose; mean±s.e.m.
(j) Expression levels for CTGF (Log2 expression value of log-normalized count data) in basal cells from non-polyp or polyp individuals across doses of cytokines displayed; n=4 samples each dose; 2-way ANOVA p<0.0260 for CTGF; all conditions non-polyp vs. polyp except 0.1 ng/mL IL-4 dose for CTGF.
Extended Data Figure 10 |. In vivo blockade with an anti-IL-4Rα monoclonal antibody shifts secretory cell state towards healthy-associated genes.
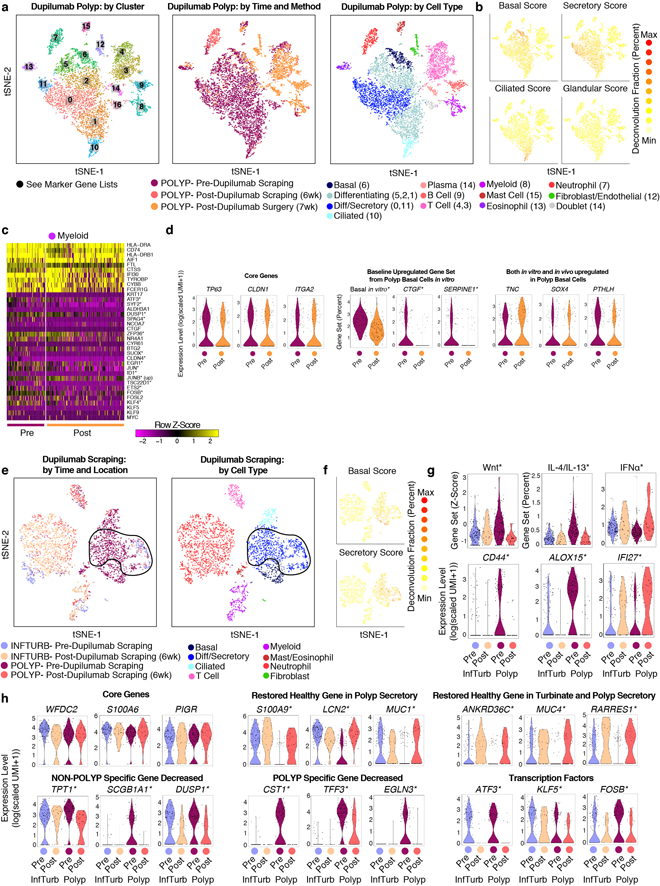
(a) left: tSNE plot of 8,764 single cells (related to Fig. 6g) from the nasal polyps of an anti-IL-4Rα (dupilumab) treated individual (1 patient, sampled at n=3 timepoints) colored by clusters identified through SNN analysis (Supplementary Table 3; Methods); middle: tSNE plot colored by timepoint and tissue of origin from polyp pre-dupilumab scraping (5,731 cells), from polyp post-dupilumab scraping (647 cells), and polyp post-dupilumab surgical sample (2,386 cells); and right: tSNE plot colored by cell types identified through marker discovery (ROC test) and biological curation of identified clusters (Supplementary Table 3; Methods)).
(b) Select cell-type specific score overlays for cell types indicated in original core data set (see Supplementary Table 3 for full gene list).
(c) Row-normalized heatmap for myeloid cells of the top marker genes identified by ROC-test (AUC>0.8) with select genes displayed on y-axis including a core myeloid signature (ROC-test myeloid cells vs. rest of cells), and then genes found to be differentially expressed from (Fig. 5f) in basal cells, and treatment annotations on x-axis; bimodal test, * denotes differential genes in both basal cells and myeloid cells pre- vs post-treatment p<0.003 or less with Bonferroni correction for multiple hypothesis testing based on number of genes tested.
(d) Violin plots for basal cells (200 cells pre-dupilumab and 151 cells post-dupilumab, noted in (a)) for the count-based expression level (log(scaled UMI+1)), except where indicated for gene scores, fraction of transcriptome and z-score (see Methods, Supplementary Table 4 for gene set used) for key basal cell genes for selected biological processes, or from the baseline upregulated gene set from polyp basal cells in vitro (Fig. 5c); differential expression testing for decreased expression post-treatment using bimodal test n.s. unless denoted by * for p<0.00087 or less with Bonferroni correction for multiple hypothesis testing based on number of genes tested; see Supplementary Table 3 for full list; Basal in vitro score Pre vs Post: *t-test, two-tailed, p<3.897×10−15, effect size 0.822.
(e) tSNE plot of 4,486 single cells (related to Fig. 2e, and Fig. 5e) from the inferior turbinate or nasal polyps of an anti-IL-4Rα (dupilumab) treated individual (n=4 samples) colored by timepoint and tissue of origin from inferior turbinate pre-dupilumab scraping (643 cells), from inferior turbinate post-dupilumab scraping (1,596 cells), polyp pre-dupilumab scraping (1,600 cells), and polyp post-dupilumab scraping (647 cells); and tSNE plot colored by cell types identified through marker discovery (ROC test) and biological curation of identified clusters (Supplementary Table 3; Methods)); black outline indicates cells considered in (g).
(f) Select deconvolution score overlays for cell types indicated in original core data set (see Supplementary Table 3 for full gene list).
(g) Violin plot for the gene set score over Wnt pathway (z-score) and expression contribution to a cell’s transcriptome over IFNα- and IL-4/IL-13-commonly induced gene signature in secretory cells grouped as in (e) and sub-sampled to a maximum of 150 cells from each disease/location category from inferior turbinate pre-dupilumab scraping (150 cells), from inferior turbinate post-dupilumab scraping (23 cells), polyp pre-dupilumab scraping (150 cells), and polyp post-dupilumab scraping (38 cells); see Methods, Supplementary Table 3, Supplementary Table 4 for gene lists used; *t-test, two-tailed, Wnt score Pre vs Post Polyp Tissue: effect size 1.02, p=1.091×10−14; Wnt score Pre vs Post Inferior Turbinate Tissue: effect size −0.17, p=0.3706; IL-4/IL-13 score Pre vs Post Polyp Tissue: effect size 1.17, p<2.2×10−16; IL-4/IL-13 score Pre vs Post Inferior Turbinate Tissue: effect size −0.17, p=0.163; IFNα score Pre vs Post Polyp Tissue: effect size −1.25, p=4.254×10−05; IFNα score Pre vs Post Inferior Turbinate Tissue: effect size −0.304, p=0.2766; differential expression testing for decreased expression post-treatment using bimodal test denoted by * and p<7.81×10−06 or less between pre- and post-treated polyp.
(h) Violin plots of secretory cells grouped as in (e) and sub-sampled to a maximum of 150 cells from each disease/location category from inferior turbinate pre-dupilumab scraping (150 cells), inferior turbinate post-dupilumab scraping (23 cells), polyp pre-dupilumab scraping (150 cells), and polyp post-dupilumab scraping (38 cells) for the count-based expression level (log(scaled UMI+1)) and for secretory cell genes from the gene set used in Fig. 2f affected by treatment within anatomical regions indicated by heading; differential expression testing for decreased expression post-treatment using bimodal test n.s. unless denoted by *, all p<6.36×10−5 or less except KLF5 (p=0.0033) and FOSB (p=0.0053) with Bonferroni correction for multiple hypothesis testing based on number of genes tested, see Supplementary Table 3 for all genes tested.
Supplementary Material
ACKNOWLEDGEMENTS
To all the scientists whose foundational work could not be cited in the main text. We thank S.L. Carroll for technical support in Seq-Well experiments; H. Raff for RNA extraction; J. Lai for histology; A. Chicoine of the Brigham and Women’s Human Immunology Flow Core for assistance with isolating cells; L. Ludwig, J. Hammelman, and J. Buenrostro for reagents/analysis advice for ATAC-Seq; D. Lingwood, U.H. von Andrian, B. Walker, S. Pillai, N. Yosef, S. Rakoff-Nahoum, S. Beyaz, C. Borges, M.B. Cole, N. Yosef, R. Satija, C. Bingle, for discussions and comments on the manuscript; Shalek Lab members for experimental and computational advice; and M. Morrison for administrative support. A.K.S. was supported by the Searle Scholars Program, the Beckman Young Investigator Program, the Pew-Stewart Scholars, a Sloan Fellowship in Chemistry, NIH grants 1DP2OD020839, 2U19AI089992, 1U54CA217377, P01AI039671, 5U24AI118672, 2RM1HG006193, 1R33CA202820, 2R01HL095791, 1R01AI138546, 1R01HL126554, 1R01DA046277, 2R01HL095791, and Bill and Melinda Gates Foundation grants OPP1139972 and BMGF OPP1116944; N.A.B. by NIH R01HL120952 and Steven and Judy Kaye Young Innovators Award; T.M.L. by NIH R01HL128241; J.A.B. by NIH U19AI095219; D.F.D. by T32AI007306 (to J.A.B.); K.M.B. by NIH AADCRC Opportunity Fund Award U19AI070535; K.N.C by NIH K23AI118804; S.K.N. was supported by NIH 2R01GM081871–09 to B.B. Support was also provided from the Koch Institute Support (core) Grant P30-CA14051 from the NCI, and Ragon Institute NIH-funded Centers for AIDS Research (P30 AI060354, Harvard University Center for AIDS Research), supported by NIH co-funding and participating Institutes and Centers: NIAID, NCI, NICHD, NHLBI, NIDA, NIMH, NIA, FIC, and OAR. J.O.M. is an HHMI Damon Runyon Cancer Research Foundation Fellow (DRG-2274-16), who would like to thank S. Montanes-Ordovas for encouraging him to work on human allergic disease.
Footnotes
AUTHOR INFORMATION
Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper. Aligned and quality-filtered data and complete statistical outputs for manuscript figures are included in manuscript as Supplementary Tables, with further information at http://shaleklab.com/resources/. Correspondence and requests for materials should be addressed to A.K.S. (shalek@mit.edu) and/or N.A.B. (nbarrett@bwh.harvard.edu).
SUPPLEMENTARY INFORMATION
Extended Data display items, Methods, and Supplementary Information containing tables and additional discussion are available in the online version of the paper at http://www.nature.com/nature.
REFERENCES
- 1.Schleimer RP & Berdnikovs S Etiology of epithelial barrier dysfunction in patients with type 2 inflammatory diseases. J Allergy Clin Immunol 139, 1752–1761, 10.1016/j.jaci.2017.04.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hogan BL et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 15, 123–138, 10.1016/j.stem.2014.07.012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitsett JA & Alenghat T Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol 16, 27–35, 10.1038/ni.3045 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwasaki A, Foxman EF & Molony RD Early local immune defences in the respiratory tract. Nat Rev Immunol 17, 7–20, 10.1038/nri.2016.117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holtzman MJ, Byers DE, Alexander-Brett J & Wang X The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nat Rev Immunol 14, 686–698, 10.1038/nri3739 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwasaki A & Medzhitov R Control of adaptive immunity by the innate immune system. Nat Immunol 16, 343–353, 10.1038/ni.3123 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schleimer RP Immunopathogenesis of Chronic Rhinosinusitis and Nasal Polyposis. Annu Rev Pathol 12, 331–357, 10.1146/annurev-pathol-052016-100401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao L et al. Increase of poorly proliferated p63(+) /Ki67(+) basal cells forming multiple layers in the aberrant remodeled epithelium in nasal polyps. Allergy 72, 975–984, 10.1111/all.13074 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Hansel FK Clinical and Histopathological Studies of the Nose and Sinuses in Allergy Journal of Allergy 1, 43–70 (1929). [Google Scholar]
- 10.Rock JR et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci U S A 106, 12771–12775, 10.1073/pnas.0906850106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karin M & Clevers H Reparative inflammation takes charge of tissue regeneration. Nature 529, 307–315, 10.1038/nature17039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gierahn TM et al. Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat Methods 14, 395–398, 10.1038/nmeth.4179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naik S et al. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature, 10.1038/nature24271 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Netea MG et al. Trained immunity: A program of innate immune memory in health and disease. Science 352, aaf1098, 10.1126/science.aaf1098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rochman M et al. Neurotrophic tyrosine kinase receptor 1 is a direct transcriptional and epigenetic target of IL-13 involved in allergic inflammation. Mucosal Immunol 8, 785–798, 10.1038/mi.2014.109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.von Moltke J, Ji M, Liang HE & Locksley RM Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 529, 221–225, 10.1038/nature16161 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindemans CA et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528, 560–564, 10.1038/nature16460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng LE & Locksley RM Allergic inflammation--innately homeostatic. Cold Spring Harb Perspect Biol 7, a016352, 10.1101/cshperspect.a016352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palm NW, Rosenstein RK & Medzhitov R Allergic host defences. Nature 484, 465–472, 10.1038/nature11047 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gieseck RL 3rd, Wilson MS & Wynn TA Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol, 10.1038/nri.2017.90 (2017). [DOI] [PubMed] [Google Scholar]
- 21.von Andrian UH & Mackay CR T-cell function and migration. Two sides of the same coin. N Engl J Med 343, 1020–1034, 10.1056/NEJM200010053431407 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Allakhverdi Z et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med 204, 253–258, 10.1084/jem.20062211 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wambre E et al. A phenotypically and functionally distinct human TH2 cell subpopulation is associated with allergic disorders. Sci Transl Med 9, 10.1126/scitranslmed.aam9171 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Portelli MA, Hodge E & Sayers I Genetic risk factors for the development of allergic disease identified by genome-wide association. Clin Exp Allergy 45, 21–31, 10.1111/cea.12327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuo WL et al. Ontogeny and Biology of Human Small Airway Epithelial Club Cells. Am J Respir Crit Care Med, 10.1164/rccm.201710-2107OC (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boscke R et al. Wnt Signaling in Chronic Rhinosinusitis with Nasal Polyps. Am J Respir Cell Mol Biol 56, 575–584, 10.1165/rcmb.2016-0024OC (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nusse R & Clevers H Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 169, 985–999, 10.1016/j.cell.2017.05.016 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Ostuni R et al. Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171, 10.1016/j.cell.2012.12.018 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Lambrecht BN & Hammad H The immunology of the allergy epidemic and the hygiene hypothesis. Nat Immunol 18, 1076–1083, 10.1038/ni.3829 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Beyaz S et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58, 10.1038/nature17173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meltzer EO et al. Rhinosinusitis: establishing definitions for clinical research and patient care. J Allergy Clin Immunol 114, 155–212, 10.1016/j.jaci.2004.09.029 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dhariwal J et al. Mucosal Type 2 Innate Lymphoid Cells Are a Key Component of the Allergic Response to Aeroallergens. Am J Respir Crit Care Med 195, 1586–1596, 10.1164/rccm.201609-1846OC (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Proud D, Sanders SP & Wiehler S Human rhinovirus infection induces airway epithelial cell production of human beta-defensin 2 both in vitro and in vivo. J Immunol 172, 4637–4645 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Pipkorn U & Karlsson G Methods for obtaining specimens from the nasal mucosa for morphological and biochemical analysis. Eur Respir J 1, 856–862 (1988). [PubMed] [Google Scholar]
- 35.Wenzel S et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med 368, 2455–2466, 10.1056/NEJMoa1304048 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Beck LA et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med 371, 130–139, 10.1056/NEJMoa1314768 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Bachert C et al. Effect of Subcutaneous Dupilumab on Nasal Polyp Burden in Patients With Chronic Sinusitis and Nasal Polyposis: A Randomized Clinical Trial. JAMA 315, 469–479, 10.1001/jama.2015.19330 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Dwyer DF, Barrett NA, Austen KF & Immunological Genome Project C Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat Immunol 17, 878–887, 10.1038/ni.3445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214, 10.1016/j.cell.2015.05.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Satija R, Farrell JA, Gennert D, Schier AF & Regev A Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502, 10.1038/nbt.3192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poposki JA et al. Group 2 innate lymphoid cells are elevated and activated in chronic rhinosinusitis with nasal polyps. Immun Inflamm Dis 5, 233–243, 10.1002/iid3.161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol, 10.1038/nbt.4096 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDavid A et al. Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinformatics 29, 461–467, 10.1093/bioinformatics/bts714 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hackett NR et al. The human airway epithelial basal cell transcriptome. PLoS One 6, e18378, 10.1371/journal.pone.0018378 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giovannini-Chami L et al. Distinct epithelial gene expression phenotypes in childhood respiratory allergy. Eur Respir J 39, 1197–1205, 10.1183/09031936.00070511 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Naeem S, Thompson LJ, Lawler SP, Lawton JH, Woodfin RM . Declining biodiversity can alter the performance of ecosystems. Nature 368, 734–737, doi: 10.1038/368734a0 (1994). [DOI] [Google Scholar]
- 47.Simpson EH Measurement of Diversity. Nature 163, 688–688, doi: 10.1038/163688a0 (1949). [DOI] [Google Scholar]
- 48.Trombetta JJ et al. Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing. Curr Protoc Mol Biol 107, 4 22 21–24 22 17, 10.1002/0471142727.mb0422s107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323, 10.1186/1471-2105-12-323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olender T et al. The human olfactory transcriptome. BMC Genomics 17, 619, 10.1186/s12864-016-2960-3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang W et al. Transcriptome Analysis Reveals Distinct Gene Expression Profiles in Eosinophilic and Noneosinophilic Chronic Rhinosinusitis with Nasal Polyps. Sci Rep 6, 26604, 10.1038/srep26604 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haghverdi L, Buttner M, Wolf FA, Buettner F & Theis FJ Diffusion pseudotime robustly reconstructs lineage branching. Nat Methods 13, 845–848, 10.1038/nmeth.3971 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490, 10.1038/nature14590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods, 10.1038/nmeth.4396 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589, 10.1016/j.molcel.2010.05.004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fulcher ML, Gabriel S, Burns KA, Yankaskas JR & Randell SH Well-differentiated human airway epithelial cell cultures. Methods Mol Med 107, 183–206 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Mead BE et al. Harnessing single-cell genomics to improve the physiological fidelity of organoid-derived cell types. BMC Biol 16, 62, 10.1186/s12915-018-0527-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gosselin D et al. An environment-dependent transcriptional network specifies human microglia identity. Science 356, 10.1126/science.aal3222 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cells-by-genes matrix generated from ethmoid sinus surgical resections and analyzed during the current study is available along with the manuscript as Supplementary Table 2 along with R code for standard implementation of Seurat. A cells-by-genes matrix from inferior turbinate and polyp scraping data is also available as Supplementary Table 6. Dupilumab treatment cells-by-genes matrices as Supplementary Tables 7 and 8. A metadata table encompassing all scRNA-seq samples is provided as Supplementary Table 9. The count and TPM matrices and associated metadata from bulk tissue RNA-seq are available as Supplementary Tables 10, 11, and 12. FASTQ file format data will be available through dbGaP under accession number XXXX. Marker gene lists for cell types identified in Fig. 1a,b, and from resultant analyses in Fig. 2b, for frequencies of cell clusters and types in Fig. 2c, for cell types identified in Fig. 2e, Fig. 2f, Fig. 3g, Fig. 5a, Fig. 5e, Extended Data Fig. 3a,b,c, Extended Data Fig. 4c, Extended Data Fig. 5e, Extended Data Fig. 6b,d, Extended Data Fig. 10a, selected comparisons of differential expression in Fig. 2d, Fig. 4a, Fig. 5c, Fig. 5f, Extended Data Fig. 2c, Extended Data Fig. 10h, and pseudotime correlation Extended Data Fig. 9b, are available as tabs in Supplementary Table 3. Differential peak calling from epigenetic profiling available in Supplementary Table 5. Additional R code for analyses available on http://shaleklab.com/resources/.