

Abstract
Intracellular trafficking is a tightly regulated cellular process, mediated in part by Rab GTPases and their corresponding effector proteins. Viruses have evolved mechanisms to hijack these processes to promote their lifecycles. Here we describe a mechanism by which cleavage of the Rab7 adaptor protein, RILP (Rab interacting lysosomal protein) is induced by viral infection. We report that RILP is directly cleaved by caspase-1 and we have identified a novel caspase-1 recognition site at aspartic acid 75 within the RILP sequence. Alanine substitution at D75 blocks caspase-1-mediated RILP cleavage. Full-length RILP localizes in a tight vesicular structure near the perinuclear region while the cleaved form of RILP re-distributes throughout the cytoplasm. However, cleavage alone was insufficient to re-localize RILP to the cellular periphery and re-localization required specific phosphorylation events near the caspase-1 recognition site. The combination of cleavage and phosphorylation were both needed for release from the dynein component p150Glued and redistribution of CD63+ve intracellular vesicles.
Keywords: Trafficking, Rab, Kinesin, Caspase, p150Glued
INTRODUCTION
Rab-GTPase proteins are central to intracellular trafficking events as they regulate a variety of cellular functions including vesicle translocation and docking at specific and precise fusion sites. Rabs achieve their distinct subcellular localization through conformation-dependent targeting mechanisms that are initiated by GTP binding. One such mechanism is via binding to, or associating with, effector proteins. Rab effectors are highly specialized molecules whose activities are exclusively tailored for individual organelles and transport systems.
RILP, the Rab interacting lysosomal protein, is a Rab7 effector protein that regulates late endocytic transport to degradative compartments. It also plays a direct role in lysosomal morphology and distribution [1]. RILP interacts with Rab7 through its C-terminal domain while its N-terminal domain recruits the dynein-dynactin motor complex. RILP is thus responsible for driving minus-end directed microtubule transport of Rab7-containing vesicles towards the microtubule organizing center [2]. The Rab7-RILP complex acts on many Rab7-containing compartments including phagosomes, melanosomes, and major histocompatibility complex class II-containing compartments [3–5]. These dynein-linked complexes are in a constant state of “tug-of-war” for vesicular trafficking, acting in opposition to kinesin-linked motor complexes [6].
Due to their organelle specificity, the Rab/effector complex is an ideal target for infectious agents to manipulate for their advantage. Chlamydia releases proteins to recruit specific Rabs which redirect them to the Golgi [7] and Salmonella interacts with the GTPase effector SKIP to assure its localization in the perinuclear region [8]. We have recently discovered that both Hepatitis C Virus (HCV) and Sendai virus (SeV) induce the cleavage of RILP. Cleaved RILP still binds to Rab7 but is incapable of making the link to the dynein motor complex. This results in the redirection of Rab7-containing cargo vesicles towards the cell surface. For HCV, this promotes virion secretion [9]. However, the mechanism of how RILP is cleaved remains unknown.
Caspase-1 is typically regarded as a key mediator of inflammatory processes however there is extensive evidence supporting a direct role for caspase-1 in many other cellular processes such as cell survival, metabolic pathways, and protein secretion [10–12]. During these cellular processes, caspase-1 activity is elaborately controlled, both at the duration and timing of activation as well as the subcellular localization of the enzyme. Here we describe a novel mechanism linking caspase-1 to RILP cleavage. This cleavage generates a C-terminal fragment of the protein that no longer mediates inward trafficking but instead, enhances the outward trafficking of Rab7-containing vesicles. These results reveal yet another layer of cellular control regulated by both caspases and trafficking adaptor proteins.
MATERIALS & METHODS
Materials and Antibodies.
General materials were purchased from Sigma-Aldrich (St. Louis, MO) or VWR (Radnor, PA). Opti-MEM, and Lipofectamine 3000 were purchased from Fisher Scientific. Protease inhibitor cocktail (Sigma-Aldrich, P8340) was used at 1:100 dilution.
Plasmids.
RILP-GFP was generated by excising RILP from pTRE2-Bla [9] and inserting into pEGFP-N1. All RILP mutants (RILP(D75A)-GFP, RILP(∆1–75)-GFP, RILP(S76/80A)-GFP, RILP(S76/80D)-GFP, RILP(∆1–75,S76/80A)-GFP, RILP(∆1–75,S76/80D)-GFP, RILP(D75A, S76/80D)-GFP were generated using Q5 Site-Directed Mutagenesis Kit (NEB). Sequences were confirmed by DNA sequencing analysis. HA-RILP-FLAG has been described elsewhere [9]. pCI-Capsase-1 was a gift from Kate Fitzgerald [13] (Addgene, 41552). shRNA Caspase-1 was a gift from Warner Greene [14] (Addgene, 53575). pSicoR-Ef1a-mCh was a gift from Bruce Conklin [15] (Addgene, 31847). pEGFP-p150Glued was a gift from David Stephens [16] (Addgene, 36154).
Cell Culture and Transfection.
HeLa cells were cultured in DMEM supplemented with 10% fetal bovine serum and 1x MEM nonessential amino acids. Transfections were carried out using Lipofectamine 3000 according to manufacturer’s instructions. Sendai virus (SeV, Cantell strain) was used at 400 HAU/ml for 24 h. Drugs were used at the following concentrations: caspase-1 inhibitor VI (Z-YVAD-FMK, 100 µM), caspase-3 inhibitor (DEVD-FMK, 100 µM), JNK inhibitor SP600125 (25 µM), p38 inhibitor V (40 µM), or ERK inhibitor II (100 µM) for 24 hours prior to collection. These concentrations were not associated with toxicity.
Immunofluorescence.
For RILP-GFP vectors, cells grown on coverslips were fixed in ice-cold methanol-acetone (1:1) at room temperature for 10 min and incubated in IF buffer (1% BSA, 2.5 mM EDTA in PBS) for 1 hr at room temperature. The coverslips were mounted in ProLong Gold (Fisher, P36931). For the localization of CD63, cells were fixed in ice-cold methanol for 5 min at room temperature. After blocking in IF buffer, the cells were incubated in primary antibody diluted in IF buffer for 1 h at room temperature. Primary antibodies used were rabbit anti-FLAG antibody (1 µg/ml, Sigma-Aldrich, F7425) and mouse anti-CD63 (Abcam ab8219, 10µg/ml). After washing with PBS, the coverslips were incubated with AlexaFluor conjugated goat secondary antibody (1: 500) for 1 h in the dark at room temperature and mounted as above. Images were acquired by using Nikon Eclipse Ti microscope (Nikon Americas Inc., Melville, NY,).
Western Blotting.
Whole cell lysates were prepared from cells lysed in RIPA buffer and western blotting was performed as stated previously [9]. Anti-GFP tag (G10362) was purchased from Fisher. RILP (H85) was purchased from Santa Cruz Biotechnology. Rab7 (D95F2) and β-tubulin were purchased from Cell Signaling Technology. Caspase-1 [EPR19675] and kinesin heavy chain (ab187680) were purchased from Abcam. To detect phospho-serine, membranes were blocked in BSA blocking buffer (5% BSA, 0.1% Tween-20 in TBS) for 1 h at room temperature. After an overnight incubation at 4°C with phospho-serine Q5 antibody (Qiagen, 1:100 diluted in TBST), rabbit anti-mouse IgG/IgM-HRP (Jackson ImmunoResearch, 315–035-048) was added at a concentration of 1:10,000, diluted in TBS containing 10% milk and 0.1% Tween-20, for 1 h at room temperature. Detection of the HRP-conjugated secondary was performed as above [9].
In Vitro Cleavage Assay.
RILP was cloned into pTNT vector (Promega, L5610) and aspartic acid mutants were generated using the Q5 Site-Directed Mutagenesis Kit. RILP protein was then synthesized using the TNT Quick Coupled Transcription/Translation System (Promega, L1170) according to manufacturer instructions. To assess caspase cleavage, 15µl of the TNT reaction was incubated with 2U recombinant, active caspase-1 (Sigma, C5482) or caspase-3 (Sigma, CC119) enzyme and 5 µl of a 5x caspase assay buffer (250 mM HEPES, pH7.2, 250 mM Nacl, 0.5% CHAPS, 50 mM EDTA, 50 mM DTT, 25% glycerol) in a total volume of 25 µl. The sample was incubated at 37°C for 2 h and 2.5 µl was used to assess for cleavage by western blot using an antibody towards RILP (Sigma, SAB2108446).
Statistics.
Results are expressed as mean ± SE. The Student t test was used for statistical analyses. P ≤ 0.05 was considered significant.
RESULTS
RILP is a direct substrate of caspase-1
We have previously demonstrated that viral infection causes the cleavage of RILP, but the protease(s) involved are unknown. As viral infection typically induces an innate immune inflammatory response we first tested whether caspases were involved. HeLa cells were infected with SeV and treated with inhibitors specific for caspase-1 or caspase-3. Whole cell lysates were then analyzed for RILP by western blot. Treatment with caspase-1 inhibitor significantly blocked the formation of the RILP cleavage product after SeV as well as baseline RILP cleavage (Fig. 1A). Inhibition of caspase-3 had no effect. To confirm the involvement of caspase-1, cells were transfected with shRNA to caspase-1 or a non-targeted sequence. Efficient knockdown of caspase-1 resulted in an inhibition of RILP cleavage (Fig. 1B). Conversely, caspase-1 overexpression increased RILP cleavage in the absence of viral infection (Fig. 1C).
Figure 1. RILP is a direct substrate of caspase-1.
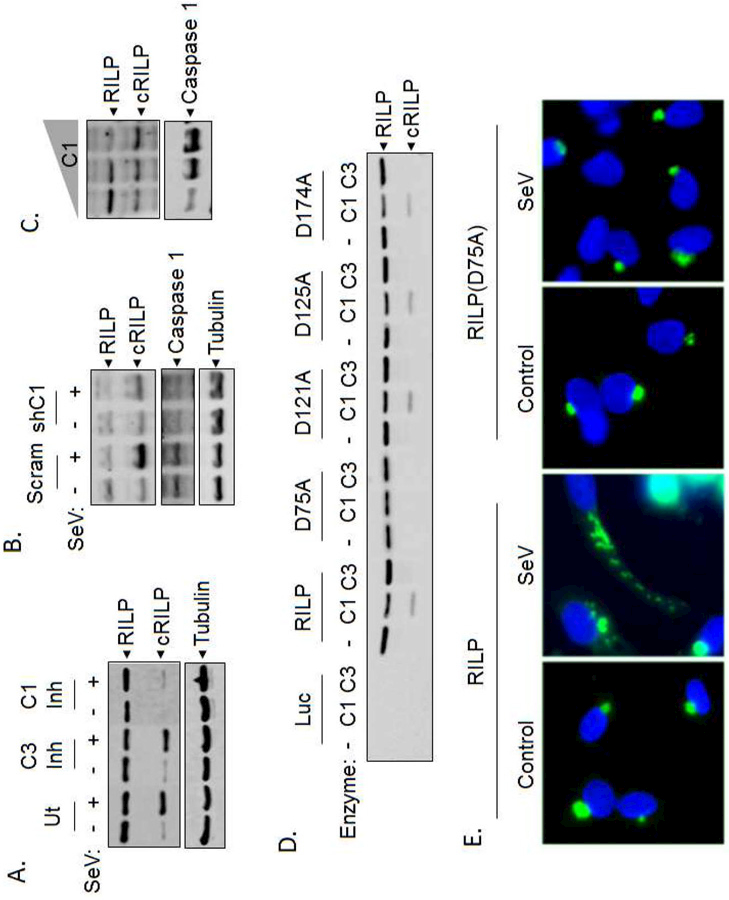
(A) SeV-induced RILP cleavage can be blocked by pharmacological inhibition of caspase-1. (B) Silencing of caspase-1 by shRNA blocks the cleavage of RILP. (C) Titration of caspase-1 overexpression (0, 1.25, or 2.5 µg) shows increased RILP cleavage corresponding with an increase in caspase-1 expression. (D) Purified RILP protein is cleaved at D75 by caspase-1 but not caspase-3. (E) Sendai virus causes cRILP to re-localize to the cellular periphery. Mutation of the caspase cleavage site D75 to alanine blocks this.
We next determined whether RILP is a direct substrate for caspase-1. Incubation of in vitro transcribed and translated RILP with active caspase-1 (C1) produced a single cleavage fragment. In contrast, no caspase 3-induced cleavage products were detected (Fig. 1D). Although all caspases cleave after aspartic acid, RILP does not contain a traditional consensus site for caspase-1 cleavage recognition. Therefore, to identify the cleavage site, we prepared mutant RILP proteins in which candidate aspartic acid residues were converted to alanine, effectively eliminating possible cleavage sites. As shown in Fig. 1D, mutation of D75 to alanine renders RILP completely resistant to caspase-1 cleavage while mutations at positions D121, D125, or D174 had no effect. This confirms that RILP is a direct caspase-1 substrate with cleavage occurring at position D75.
Viral infection is known to activate inflammasome/caspase-1 [17]. To assess whether the virus-mediated redistribution of RILP is caspase-1-dependent, we used a tagged RILP construct containing a C-terminal GFP tag. This allows for the visualization of both full-length RILP as well as cleaved RILP (cRILP). RILP-GFP was expressed in HeLa cells and, after 24 h of SeV infection, the cells were fixed, and localization was assessed. Confirming previous results [9], RILP localized near the perinuclear region (Fig. 1E, control). After SeV infection, this perinuclear structure remained, however smaller punctate structures were observed representing cRILP extending towards the cell periphery. RILP(D75A) had a similar baseline localization when compared to full-length RILP; localizing to the perinuclear region forming a distinct, tight vesicular structure. Its localization, however, was unchanged after SeV infection. Taken together, the data confirms that RILP is specifically cleaved by caspase-1 at D75, liberating a cleaved form of RILP that localizes to structures extending towards the cell periphery.
Efficient RILP cleavage requires phosphorylation
We previously reported that truncation of the N-terminal 80 amino acids of RILP does not alter its localization suggesting that this mutant is still capable of binding dynein complexes [9]. This seemed to contradict our finding that cRILP is formed from a specific cleavage at D75. To examine this discrepancy, we created an N-terminal truncation mutant mimicking cleavage at D75. Like full-length RILP, the cleavage mimic RILP (∆1–75) localizes to the perinuclear region, forming a tight vesicular structure (Fig. 2A). However, after infection with SeV, RILP (∆1–75) almost completely redistributes, the perinuclear structure disappears and smaller punctuate vesicles appear towards the cell periphery. This suggests that cleavage at D75, while necessary, is not sufficient to re-localize cRILP and that a secondary event must occur to see full peripheral localization.
Figure 2. Efficient RILP cleavage requires phosphorylation.
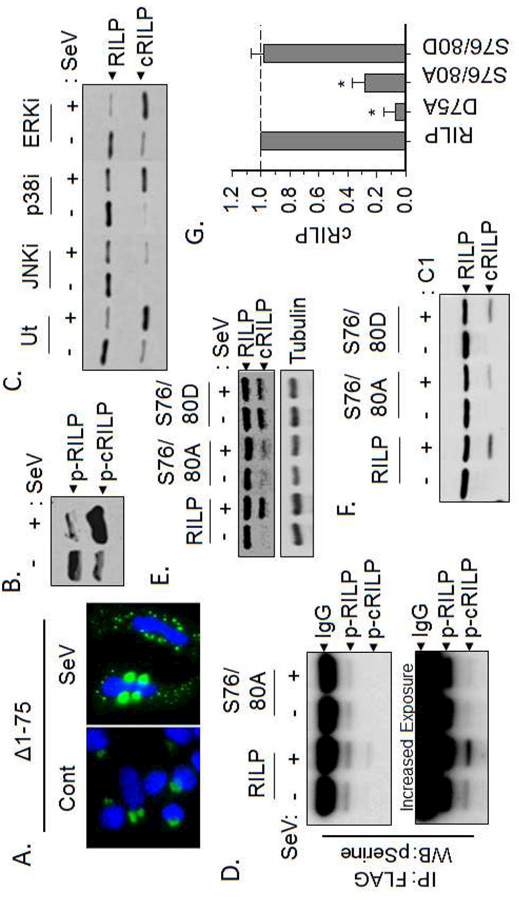
(A) The cleavage mimic pEGFP-N1(RILP(∆1–75)) re-localizes only after Sendai virus infection. (B) Western blot analysis of total phospho-proteins showing that both total and cleaved RILP are phosphorylated however, the amount of phospho-RILP is greatly increased after SeV infection. (C) Inhibition of JNK, but neither p38 nor ERK, blocks RILP cleavage. (D) Western blot analysis of FLAG eluates for phospho-serine shows that cRILP is heavily phosphorylated after SeV infection. Mutation of the serines at positions 76 and 80 within RILP abolish this. (E) SeV induces the cleavage of wild-type RILP. The phospho-deficient RILP(S76/80A) blocks SeV-induced cleavage of RILP while the phosphomimetic RILP(S076/80D) efficiently and spontaneously cleaves. (F-G) Purified RILP protein was incubated with active caspase-1 as stated previously. Western blot analysis for RILP shows that phosphorylation-deficient RILP, S76/80A, is cleaved with less efficiency than wild-type or the phospho-mimic, S76/80D RILP.
Post-translational modifications of proteins regulate and/or reprogram cellular signaling processes. Examination of the RILP sequence suggests that RILP contains a JNK-SAPK phosphorylation motif. Therefore, we next examined the involvement of phosphorylation as a mediator of RILP cleavage and re-localization. We first assessed whether RILP is phosphorylated. Total phosphoproteins were isolated from the lysates of either uninfected or SeV-infected HeLa cells using the PhosphoProtein purification system from Qiagen. Western blot analysis of these phospho-lysates for RILP showed that both full-length and cleaved RILP are phosphorylated (Fig 2B). We next tested the effect of kinase inhibition on RILP cleavage. As seen previously [9], treatment with the JNK inhibitor SP600125 significantly blocked cleavage while inhibition of either p38 or ERK had no effect on the formation of a RILP cleavage product (Fig. 2C). This suggests a possible role of JNK-mediated phosphorylation in the induction of RILP cleavage.
The RILP sequence surrounding the D75 cleavage site contains two serines at positions 76 and 80. We hypothesized that these serines may be phosphosites that play a role in caspase-1-mediated cleavage of RILP. To examine this, we mutated these two residues to alanine, thus creating a phosphorylation-deficient mutant. Both wild-type and the phosphorylation-deficient mutant RILP(S76/80A) were expressed in HeLa cells. After infection with SeV, lysates were collected and the RILP constructs were immunoprecipitated with anti-FLAG magnetic beads. Western blot analysis of the immunoprecipitates for pSerine again confirmed that the cleaved form of RILP is serine phosphorylated (Fig. 2D). Mutation of S76 and S80 to alanine not only abolished the band representing phospho-cRILP, (Fig. 2D), but also blocked SeV-induced cleavage (Fig. 2E). A phospho-mimetic version of RILP, RILP(S76/80D), spontaneously cleaved regardless of virus infection suggesting a role for these two residues in both the phosphorylation and cleavage of RILP.
We next assessed whether mutation of serines 76 and 80 alters caspase-1-induced cleavage of RILP. Incubation of in vitro transcribed/translated RILP with active caspase-1 as performed in Fig. 1, yielded a cleaved RILP band. The phosphorylation-deficient RILP(S76/80A) displayed approximately 65% less cleaved RILP than wild-type (Fig. 2F-G) while the phospho-mimetic version of RILP, RILP(S76/80D), cleaves as efficiently as wild-type RILP when treated with active caspase-1. Taken together, the data suggests that RILP cleavage is enhanced by phosphorylation.
Both phosphorylation and cleavage are required for full re-localization of RILP
To determine whether both cleavage and phosphorylation are required for efficient RILP re-localization, we assessed the localization of several mutants which mimic cleavage and/or phosphorylation. We first looked at the phosphorylation-deficient mutant of full-length RILP, S76/80A. Figure 3A shows that this mutant is unable to re-localize and remains in the perinuclear region even after SeV infection. The phosphomimetic form, S76/80D, spontaneously localizes to the cell periphery with or without the addition of virus. This data confirms that phosphorylation plays a key role in RILP movement. To examine the requirement for cleavage and the interaction between cleavage and phosphorylation on inducing re-localization, we next looked at a phosphorylation-deficient mutant of cleaved RILP, ∆1–75(S76/80A). Unlike the situation with wild-type RILP or ∆1–75 that both redistribute after SeV (Figs. 1E, 2A), ∆1–75(S76/80A) completely failed to re-localize after SeV infection (Fig. 3A) despite its already having the cleavage event. We then used a phosphomimetic form of cleaved RILP, ∆1–75(S76/80D). This mutant spontaneously and efficiently re-localized to the cell periphery irrespective of SeV infection. Furthermore, both spontaneous and SeV-induced re-localization could be blocked by simply removing the caspase-1 cleavage site from the phosphomimetic form of the protein (D75A(S76/80D)). This demonstrates that both cleavage and phosphorylation are required for RILP re-localization. The spontaneous re-localization of full length RILP(S76/80D) thus results from its cleavage even in the absence of virus (Fig. 2E).
Figure 3. Phosphorylation and cleavage are required for full-re-localization of RILP.
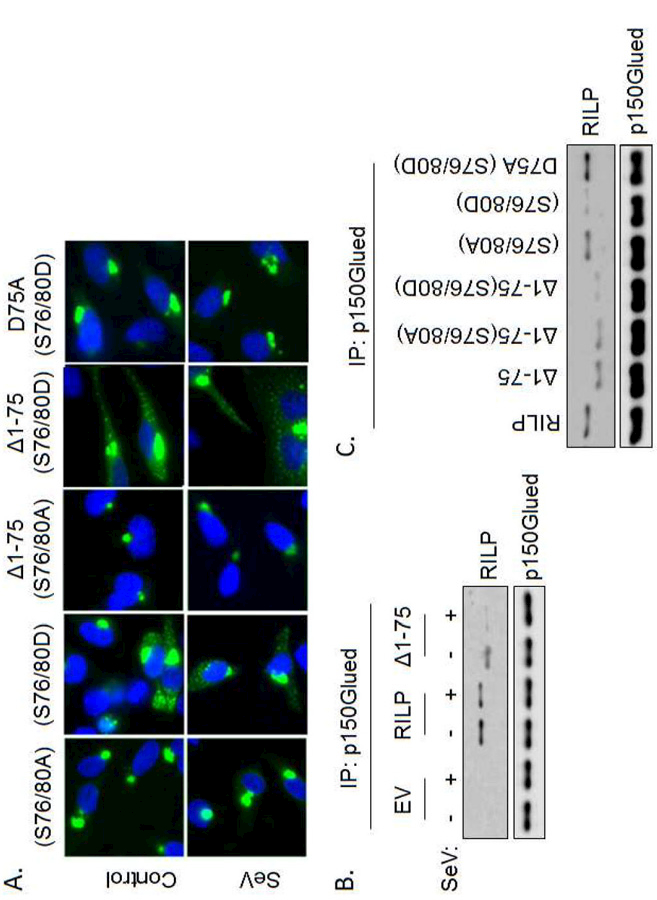
(A) Full RILP re-localization requires both cleavage and phosphorylation. (B) RILP and p150Glued co-immunoprecipitate under basal conditions. SeV decreases this association. The cleavage mimic, RILP(∆1–75) also complexes with p150Glued, however after infection with SeV these two fail to bind. EV = empty vector. (C) Immunoprecipitation of p150Glued and western blot analysis for RILP shows that both cleavage and phosphorylation are required for RILP to bind p150Glued.
RILP cleavage and trafficking motors
The p150Glued subunit of dynactin binds the N-terminal region of RILP and regulates dynein-mediated retrograde transport of cargo vesicles towards the mitotic center (MTOC) [2]. We hypothesized that RILP cleavage results in the removal of the N-terminal portion of RILP that binds p150Glued. This would favor cRILP-induced vesicular movement toward the cell periphery. To test this, HeLa cells were co-transfected with EGFP-p150Glued and HA-RILP-FLAG and were then infected with SeV for 24 h. EGFP-p150Glued was immunoprecipitated from whole cell lysates using an antibody to GFP and its association with RILP was assessed by western blot for FLAG. We found that SeV infection decreased the interaction between p150Glued and RILP (Fig. 3B). The cleavage mimic, ∆1–75 bound p150Glued under control conditions however binding was almost completely eliminated after SeV infection. We next assessed the requirement of phosphorylation in cRILP re-localization. The phosphorylation-deficient mutant of cleaved RILP, ∆1–75(S76/80A) efficiently bound p150Glued while the phosphomimetic ∆1–75(S76/80D) was unable to bind. The full-length phosphomimetic RILP (S76/80D) does not bind p150Glued, yet simply blocking cleavage in this mutant (D75A(S76/80D)) restored the association. This correlates with our hypothesis that cleavage alone is not sufficient to induce RILP re-localization and another secondary event must occur for full re-localization. The data shows that cRILP moves to a peripheral localization more efficiently after SeV as result of loss of binding to p150Glued. It further confirms our findings that both cleavage and phosphorylation are necessary to prevent RILP interaction with the dynein complex and re-localize Rab7-containing vesicles.
RILP cleavage and phosphorylation promote kinesin association and anterograde movement.
Previous data from our lab showed that HCV-induced RILP cleavage increases the secretion of infectious virions in a kinesin-dependent manner [9]. To test whether the cleavage of RILP was directly promoting this outward trafficking, HA-RILP-FLAG or the cleavage mimic HA-RILP(∆1–75)-FLAG were expressed in HeLa cells. After SeV infection, the cells were homogenized in a hypotonic lysis buffer, in the absence of detergents to preserve vesicle structure, and total homogenates were subjected to immunoprecipitation with FLAG-M2 magnetic beads as stated previously. Under control conditions, there was no association between RILP and the kinesin heavy chain. However, SeV infection induced the association of RILP-containing vesicles with the kinesin heavy chain. Similar results were seen with the cleavage mimic RILP(∆1–75) whereby association with kinesin heavy chain was only seen after SeV infection.
We next examined a population of intracellular vesicles known to be under the control of Rab7-mediated trafficking. Various RILP constructs were expressed in HeLa cells and after 24 h, the cells were methanol fixed and immunostained for FLAG (to detect RILP) and endogenous CD63, a marker of the multi-vesicular body. Under control conditions (Fig. 4B, asterisks), CD63 localizes loosely in a perinuclear cluster. As shown previously [9], cRILP, in contrast, localizes in vesicular structures that extend towards the cellular periphery. Expression of cRILP is able to completely redistribute CD63 throughout the cytoplasm. This is in complete contrast to the non-cleavable form of RILP. D75A is tightly associated with the microtubule organizing center and completely sequesters CD63 in this location.
Figure 4. RILP cleavage and phosphorylation promote kinesin association and anterograde movement.
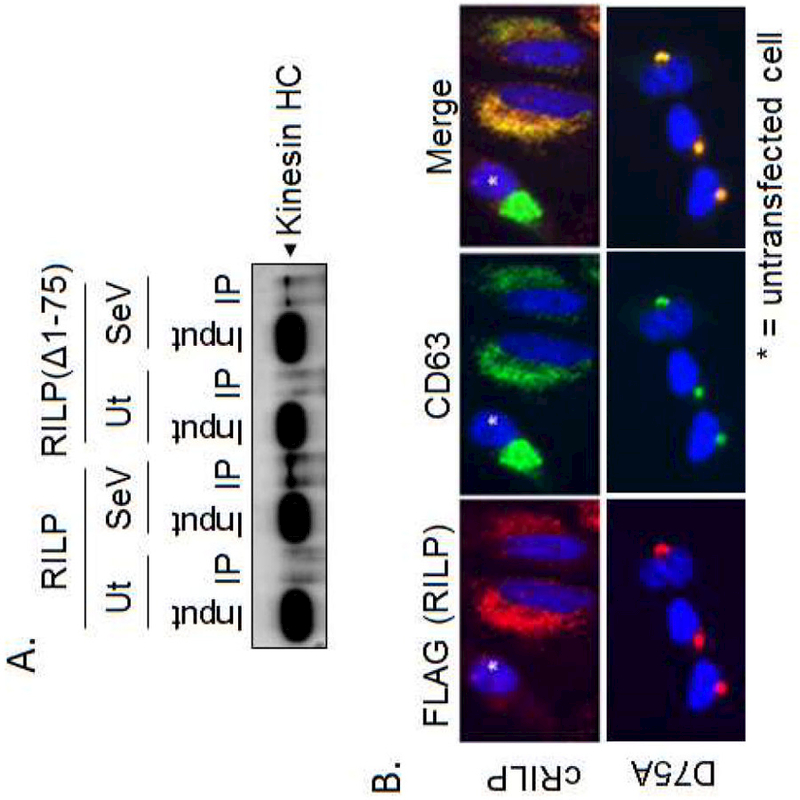
(A) RILP constructs were expressed in HeLa cells and infected with SeV for 24 h. Cells were lysed in hypotonic lysis buffer and RILP was immunoprecipitated using FLAG M2 magnetic beads as described previously. Eluates were assessed by western blot analysis for kinesin heavy chain. (B) The localization of the MVB marker CD63 was assessed in HeLa cells expressing either cRILP-FLAG or the cleavage mutant RILP(D75A)-FLAG.
DISCUSSION
In this study, we demonstrate a novel finding linking caspase activation to the cleavage of a trafficking adaptor protein, RILP. However, the mechanism of how RILP is cleaved and how this cleavage results in a complete re-routing of these intracellular vesicles remained unclear.
This study showed that RILP is directly cleaved by caspase-1 even though RILP does not contain the traditional, and preferred, (W/L)EHD motif. Caspase motifs are most often influenced by the 4 residues surrounding the cleavage site. However, for caspase-1 the flanking 20 amino acids surrounding the P1 Asp contain important determinants of specificity that dramatically affect cleavage efficiency [18]. This is particularly true for the caspase-1 target, IL-1β whose YVHD recognition site is a deviation of the preferred [19].
The potential for crosstalk between phosphorylation and proteolysis in cellular processes has long been recognized. Our results show that both cleavage and phosphorylation of RILP are required for cellular redistribution. First, cleavage is necessary but not sufficient for RILP redistribution. Viral infection causes the redistribution of wild-type RILP but the cleavage deficient mutant D75A was incapable of cellular re-localization with or without virus. In every case, prevention of cleavage blocked RILP redistribution regardless of the phosphorylation state. Further support for the necessity of cleavage was obtained in our previous data using a dually tagged RILP construct which showed that redistributed RILP always represented a cleaved RILP fragment comprising the C-terminal portion of the and never full-length RILP [9].
Phosphorylation was also necessary but not sufficient for RILP redistribution. Preventing phosphorylation blocked redistribution of even the pre-cleaved mutant yet, phosphorylation alone was not sufficient since the uncleavable phosphomimetic mutant (D75A, S76/80D) did not re-localize. Curiously, the full length RILP(S76/80D) phosphomimetic mutant spontaneously cleaved without the addition of virus thus suggesting that phosphorylation might serve two purposes, promoting cleavage as well as being part of the changes that result in loss of dynein binding and redistribution.
Because Rab proteins are specific as to which organelles they reside, they (and their effectors) are commonly exploited by intracellular pathogens [20]. Previous data from our lab has shown that although RILP cleavage occurs at a basal level, pathogen infection (HCV or SeV) can hijack this mechanism to increase cleavage resulting in enhanced vesicular trafficking towards the plasma membrane and thus virus secretion [9]. A consequence of such a rearrangement is the re-localization of Rab7-containing intracellular vesicles including a redisposition of components of the endocytic system such as the multivesicular body shown here. Yet, the mechanism responsible for linking these vesicles to kinesin remains unclear. We demonstrate that the phosphorylation of cleaved RILP acts as a key regulator of its cellular localization. We show that a RILP cleavage mutant missing the N-terminal 75 amino acids (∆1–75), did not spontaneously re-localize and required either virus or an additional mutation mimicking phosphorylation (∆1–75, S76/80D) to do so. This correlated with the fact that both cleavage and phosphorylation are required for RILP to lose its association with p150Glued, a component of the dynein motor complex that promotes inward, anterograde trafficking. Altogether, the data shows that RILP cleavage and phosphorylation essentially liberate the cargo vesicle from the control of dynein, allowing kinesin-based motor complexes to take over and efficiently promote outward trafficking.
In conclusion, viruses depend on host cells for propagation and as such, they have evolved specific mechanisms to modulate or hijack Rab-mediated trafficking functions to perform essential steps in their lifecycles. In this study, we demonstrate that RILP cleavage is specifically up-regulated by viral infection. RILP-mediated trafficking alteration is regulated by both proteolysis and phosphorylation, suggesting that kinase and caspase pathways work in concert to regulate vesicular trafficking.
Supplementary Material
HIGHLIGHTS.
Viral infection causes cleavage of the trafficking adapter RILP.
RILP cleavage promotes secretion by re-localizing Rab7 vesicles to the cell periphery.
RILP is cleaved by caspase 1 and this prevents its binding to dynein.
Both RILP cleavage and phosphorylation are required for trafficking effects.
RILP is a mechanism of inflammation-induced trafficking changes.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health P30 GM118247 and the Lied Pilot Grant Program from the KU Medical Center Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Cantalupo G, Alifano P, Roberti V, Bruni CB, Bucci C , Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes, The EMBO journal, 20 (2001) 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Johansson M, Rocha N, Zwart W, Jordens I, Janssen L, Kuijl C, Olkkonen VM, Neefjes J, Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin, The Journal of cell biology, 176 (2007) 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bains M, Zaegel V, Mize-Berge J, Heidenreich KA, IGF-I stimulates Rab7-RILP interaction during neuronal autophagy, Neurosci Lett, 488 (2011) 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Daniele T, Hackmann Y, Ritter AT, Wenham M, Booth S, Bossi G, Schintler M, Auer-Grumbach M, Griffiths GM, A role for Rab7 in the movement of secretory granules in cytotoxic T lymphocytes, Traffic (Copenhagen, Denmark: ), 12 (2011) 902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ohbayashi N, Maruta Y, Ishida M, Fukuda M, Melanoregulin regulates retrograde melanosome transport through interaction with the RILP-p150Glued complex in melanocytes, Journal of cell science, 125 (2012) 1508–1518. [DOI] [PubMed] [Google Scholar]
- [6].Hancock WO, Bidirectional cargo transport: moving beyond tug of war, Nat Rev Mol Cell Biol, 15 (2014) 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Grieshaber SS, Grieshaber NA, Hackstadt T, Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process, Journal of cell science, 116 (2003) 3793–3802. [DOI] [PubMed] [Google Scholar]
- [8].Harrison RE, Brumell JH, Khandani A, Bucci C, Scott CC, Jiang X, Finlay BB, Grinstein S, Salmonella impairs RILP recruitment to Rab7 during maturation of invasion vacuoles, Molecular biology of the cell, 15 (2004) 3146–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wozniak AL, Long A, Jones-Jamtgaard KN, Weinman SA, Hepatitis C virus promotes virion secretion through cleavage of the Rab7 adaptor protein RILP, Proceedings of the National Academy of Sciences of the United States of America, 113 (2016) 12484–12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Keller M, Ruegg A, Werner S, Beer HD, Active caspase-1 is a regulator of unconventional protein secretion, Cell, 132 (2008) 818–831. [DOI] [PubMed] [Google Scholar]
- [11].Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman J Salfeld, et al. , Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock, Cell, 80 (1995) 401–411. [DOI] [PubMed] [Google Scholar]
- [12].Saleh M, Caspase-1 builds a new barrier to infection, Cell, 126 (2006) 1028–1030. [DOI] [PubMed] [Google Scholar]
- [13].Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA, AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC, Nature, 458 (2009) 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, Greene WC, Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection, Nature, 505 (2014) 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Salomonis N, Schlieve CR, Pereira L, Wahlquist C, Colas A, Zambon AC, Vranizan K, Spindler MJ, Pico AR, Cline MS, Clark TA, Williams A, Blume JE, Samal E, Mercola M, Merrill BJ, Conklin BR, Alternative splicing regulates mouse embryonic stem cell pluripotency and differentiation, Proceedings of the National Academy of Sciences of the United States of America, 107 (2010) 10514–10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Watson P, Stephens DJ, Microtubule plus-end loading of p150(Glued) is mediated by EB1 and CLIP-170 but is not required for intracellular membrane traffic in mammalian cells, Journal of cell science, 119 (2006) 2758–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shrivastava G, Leon-Juarez M, Garcia-Cordero J, Meza-Sanchez DE, Cedillo-Barron L, Inflammasomes and its importance in viral infections, Immunologic research, 64 (2016) 1101–1117. [DOI] [PubMed] [Google Scholar]
- [18].Shen J, Yin Y, Mai J, Xiong X, Pansuria M, Liu J, Maley E, Saqib NU, Wang H, Yang XF, Caspase-1 recognizes extended cleavage sites in its natural substrates, Atherosclerosis, 210 (2010) 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Thornberry NA, Lazebnik Y, Caspases: enemies within, Science (New York, N.Y.), 281 (1998) 1312–1316. [DOI] [PubMed] [Google Scholar]
- [20].Spearman P, Viral interactions with host cell Rab GTPases, Small GTPases, 9 (2018) 192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.