

Abstract
Aminoacyl-tRNA synthetases (ARSs) are ubiquitously expressed enzymes responsible for charging tRNA with cognate amino acids during protein translation. Non-canonical functions are increasingly recognized, and include transcription and translation control and extracellular signaling. Monoallelic mutations in genes encoding several ARSs have been identified in axonal Charcot-Marie-Tooth (CMT2) disease, whereas biallelic mutations in ARS loci have been associated with multi-tissue syndromes, variably involving the central nervous system, lung, and liver. We report a male infant of non-consanguineous origin, presenting with successive onset of transfusion-dependent anemia, hypothyroidism, cholestasis, interstitial lung disease, and developmental delay. Whole-exome sequencing (WES) revealed compound heterozygosity for two variants (p.Tyr307Cys and p.Arg618Cys) in MARS, encoding methionyl-tRNA synthetase. Biallelic MARS mutations are associated with interstitial lung and liver disease (ILLD). Interestingly, the p.Arg618Cys variant, inherited from an unaffected father, was previously reported in a family with autosomal dominant late-onset CMT2. Yeast complementation assays confirmed pathogenicity of p.Arg618Cys, yet suggested retained function of p.Tyr307Cys. Our findings underscore the phenotypic variability associated with ARS mutations, and suggest genetic or environmental modifying factors in the onset of monoallelic MARS-associated CMT2.
Keywords: aminoacyl-tRNA synthetases, MARS, whole exome sequencing, interstitial lung and liver disease
INTRODUCTION
Aminoacyl-tRNA synthetases (ARSs) function at the first step of protein translation, catalyzing the ligation of amino acids to their cognate tRNAs. Each enzyme catalyzes the esterification of a specific amino acid to a hydroxyl group at the 3′-end of a cognate transfer RNA (Schimmel 1987). ARSs are mostly specific to either the cytoplasm or the mitochondria, the latter appended with the number “2” as in MARS2; three are bifunctional (Antonellis and Green 2008). A subset of ARSs form macromolecular complexes with ARS-interacting multifunctional proteins (AIMP1, AIMP2, and AIMP3) (Lee et al. 2004). Apart from their traditional canonical functions, non-canonical functions of ARSs and ARS macromolecular complexes are becoming increasingly recognized. These include roles in transcription and translation control, signal transduction, cell migration, angiogenesis, inflammation, and tumorigenesis (Castranova et al. 2016, Young, Lee, and Kim 2016).
Monoallelic mutations in cytoplasmic and bifunctional ARSs have been identified in axonal peripheral neuropathies (i.e., AARS [MIM 601065], YARS [MIM 603623], GARS [MIM 600287], KARS [MIM 601421], WARS [MIM 191050]) (Antonellis et al. 2003, Jordanova et al. 2006, Latour et al. 2010, McLaughlin et al. 2010, Tsai et al. 2017), whereas biallelic mutations in mitochondrial ARSs have classically been associated with a wider variety of syndromes, with multi-tissue involvement (i.e., SARS2 [MIM 612804], MARS2 [MIM 609728]; and RARS2 [MIM 611524]). However, recent publications have challenged the dichotomous disease classification associated with cytoplasmic versus mitochondrial ARSs. It is now recognized that biallelic mutations in cytoplasmic ARSs can affect multiple tissues (Oprescu et al. 2017). Examples include biallelic variants in LARS with infantile liver failure syndrome [MIM 615438] (Casey et al. 2012); in AARS with early infantile epileptic encephalopathy-29 [MIM 616339] (Simons et al. 2015); and in KARS with autosomal recessive deafness [MIM 613916] (Santos-Cortez et al. 2013).
Here, we report compound heterozygosity for two MARS variants in an infant with interstitial lung and liver disease [MIM 615486]. Interestingly, one of the mutations, inherited from an unaffected father, was previously reported in a family with autosomal dominant late-onset CMT2 [MIM 616280], highlighting the complexity of genotype-phenotype correlations.
PATIENT DATA
The patient (II-3 in Fig. 1A) was the youngest among 3 siblings born to healthy unrelated parents of Jewish Moroccan/Tunisian/Persian descent. The patient was delivered at term by repeat Cesarean-section at birthweight of 2600 grams, following an unremarkable pregnancy. Soon after birth he developed Coombs-negative transfusion-dependent anemia, necessitating frequent red blood cell (RBC) transfusions. At approximately age 3 months the infant presented with elevated liver enzymes, direct hyperbilirubinemia and hypoalbuminemia. Urine organic acids demonstrated markedly elevated tyrosine metabolites, indicative of liver dysfunction. Liver biopsy showed swollen hepatocytes, diffuse macrovascular and sinusoidal steatosis, and pericentral, portal and sinusoidal fibrosis (Fig. 2). Additional endocrine and metabolic investigations revealed peripheral hypothyroidism, and were otherwise within normal limits. Brain magnetic resonance imaging (MRI) was normal. At four months of age the patient exhibited respiratory deterioration and subsequently became oxygen-dependent. A chest radiograph followed by high-resolution chest computed tomography (CT) revealed advanced interstitial disease (Fig. 2). Bronchoalveolar lavage (BAL) showed multiple foamy macrophages, without evidence of alveolar proteinosis. The patient was fed via a nasogastric tube followed by insertion of a gastrostomy tube at 8 months. He was slow to gain weight, below the 3rd percentile for both weight and length. At 8 months, the patient had significant motor developmental delay: he could not support his head and was unable to roll over. However, he did track objects, had a social smile, and was able to reach for and grasp objects.
Figure 1. Pedigree and identified MARS variants.
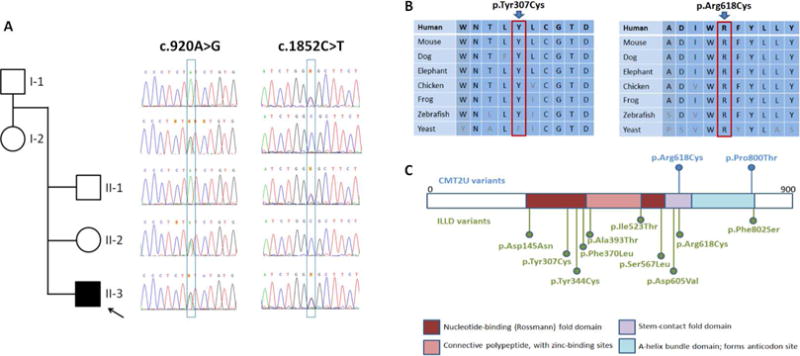
(A) Sanger segregation of the variants in the family. (B) Conservation of affected residues. (C) Position of MARS residues associated with Charcot-Marie-Tooth disease type 2U (CMT2U; in blue) and interstitial lung and liver disease (ILLD; in green).
Figure 2. Liver biopsy and high resolution chest computed tomography.
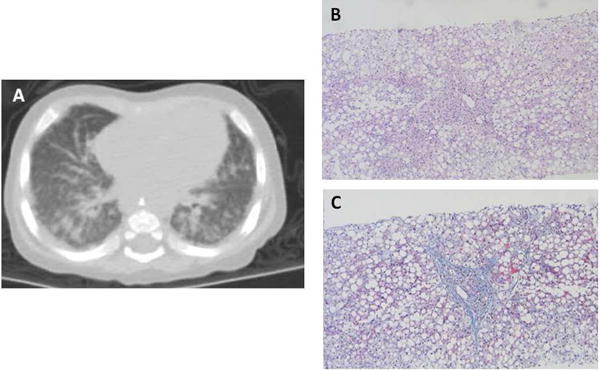
(A) Diffuse infiltrative opacification in the lung periphery indicating severe interstitial lung disease. (B) H&E stain of liver section showing portal and sinusoidal fibrosis, cholangiolar proliferation and diffuse macrovesicular steatosis with ballooning of hepatocytes. (C) Masson trichome stain demonstrating portal and sinusoidal fibrosis.
The patient was treated with hydroxychloroquine in an effort to ameliorate the ongoing inflammatory process and to slow down the progression of interstitial lung disease. Following molecular diagnosis, exogenous methionine supplementation was initiated due to plasma methionine levels in the lower limit of the normal range (8 uM, normal range 7–47 uM) and based on studies in yeast where attenuated MARS activity could be rescued with methionine supplementation (Hadchouel et al. 2015). Approaching his first birthday, the infant’s condition improved. He was weaned off daytime oxygen, hospitalizations became less frequent, and steady weight gain was documented. Head control and motor strength improved with intensive physical and occupational therapy, and the patient learned to support his head and roll from abdomen to back.
MATERIALS AND METHODS
Genetic analysis
The family consented for genetic testing according to a local IRB approved protocol. Exonic sequences from DNA extracted from whole blood of individual II-3 were enriched with the SureSelect Human All Exon 50 Mb V4 Kit (Agilent Technologies, Santa Clara, California, USA). Sequences were generated on a HiSeq2500 (Illumina, San Diego, California, USA) as 125-bp paired-end runs. Read alignment and variant calling were performed with DNAnexus (Palo Alto, California, USA) using default parameters with the human genome assembly hg19 (GRCh37) as reference. Exome analysis of the proband yielded 50.4 million mapped reads with a mean coverage of 74X. Following alignment to the reference genome [hg19] and variant calling, variants were filtered out if the total read depth was less than 8X, and if they were off-target (>8bp from splice junction), synonymous, were predicted benign by MutationTaster, or had minor allele frequency (MAF) >0.01 in the ExAC database (Exome Aggregation Consortium, Cambridge, MA, URL: http://exac.broadinstitute.org).
Segregation analysis
Amplicons containing the potential pathogenic variants in MARS were amplified from genomic DNA of available family members by conventional PCR. PCR products were purified and analyzed by Sanger di-deoxynucleotide sequencing, according to standard procedures.
Yeast complementation assays
Yeast complementation assays were performed using a haploid S. cerevisiae strain with a deleted endogenous MES1 (the yeast ortholog of MARS) as previously described (Gonzalez et al. 2013); viability of this strain is maintained by a vector (pRS316) harboring wild-type MES1. This strain was transformed with a pYY1 construct (Chien et al. 2014) bearing either the wild-type human MARS open-reading frame or the p.Tyr307Cys or p.Arg618Cys MARS open-reading frame, or constructs without a MARS insert (‘empty’ in Fig. 3). Transformed yeast were selected for the presence of the maintenance and experimental vectors on media lacking leucine and uracil. Colonies were grown to saturation in 2mL of selective liquid medium for 48 hours at 30°C, 275rpm. One mL of each saturated culture was spun down and re-suspended in 50ul water, then diluted 1:10 and 1:100. Cultures were spotted on 0.1% 5-FOA complete solid medium (Teknova, Hollister CA), which selected for cells that spontaneously lost the maintenance vector. Yeast were imaged after three days of incubation at 30°C. Two independent transformations were performed with independently generated pYY1 constructs, and two colonies per transformation were tested.
Figure 3. The effects of MARS variants on yeast cell growth.

Representative cultures of the indicated yeast strains were plated on solid growth medium containing 5-FOA. Each strain was previously transformed with a vector containing no insert (Empty pYY1), or one containing either wild-type or mutant (p.Tyr307Cys or p.Arg618Cys) human MARS. Before inoculating on solid growth medium, each strain was either undiluted, or diluted 1:10 or 1:100 in water.
RESULTS
Following filtering of exome data, we identified three genes with rare compound heterozygous variants (Supplemental Table 1). No rare homozygous or de novo variants survived filtering. Two variants identified in MARS provided a sound phenotypic overlap with the clinical presentation of the affected individual (II-3 in Fig. 1A) and segregated with the disease: NM_004990.3:c.920A>G; p.Tyr307Cys (inherited from the mother), and NM_004990.3:c.1852C>T; p.Arg618Cys (inherited from the father). The two unaffected siblings carried either one or the other MARS variant, but not both (Fig. 1A). The p.Tyr307Cys and p.Arg618Cys variants both affect conserved residues (Fig. 1B), are predicted deleterious by various bioinformatic algorithms, and are only present twice in the heterozygous state in the GnomAD database (Table 1). The p.Tyr307Cys variant affects a residue in the catalytic domain, and p.Arg618Cys has been previously reported in association with autosomal dominant, late-onset CMT type 2U (Fig. 1C), with functional studies in yeast supporting pathogenicity (Gonzalez et al. 2013).
Table 1.
Bioinformatic predictions of the MARS variants
| Position | Nucleotide | Amino acid | GERP | SIFT | Mutation Taster | GnomAD frequency |
|---|---|---|---|---|---|---|
| g.chr12:57892235A>G | c.920A>G | p.Tyr307Cys | 4.7 | D | D | 0.000008 |
| g.chr12:57906632C>T | c.1852C>T | p.Arg618Cys | 5.4 | D | D | 0.000008 |
Abbreviations: D – damaging (SIFT)/disease causing (MutationTaster)
The wild-type human MARS open-reading frame complements deletion of the endogenous MES1 locus (Fig. 3). In this assay, yeast growth is a readout for MARS function, because yeast require MARS to survive (an empty vector with no MARS insert cannot support yeast growth; Fig. 3). As was previously demonstrated in MES1, p.Arg618Cys MARS does not support yeast growth, indicating that it is a functionally null allele (Fig. 3). However, yeast expressing p.Tyr307Cys MARS grow similarly to yeast expressing wild-type MARS, indicating that p.Tyr307Cys MARS does not dramatically reduce gene function in our yeast model system.
DISCUSSION
MARS encodes the cytoplasmic methionyl-tRNA synthetase; monoallelic mutations in this gene have been associated with autosomal dominant late-onset CMT2U [MIM 616280] (Gonzalez et al. 2013, Hyun et al. 2014), whereas biallelic mutations have been associated with interstitial lung and liver disease [ILLD, MIM 615486] (van Meel et al. 2013, Hadchouel et al. 2015, Sun et al. 2017). The clinical presentation of the patient studied here closely overlaps that of the patient described by van Meel et al. (2013) (van Meel et al. 2013). Notably, pulmonary alveolar proteinosis, which was a hallmark of the cohort described by Hadchouel et al. (2015), was not suggested by the CT findings of the studied patient nor was it found in BAL.
Most ILLD-associated MARS mutations map to the catalytic domain, whereas the two CMT2U mutations identified to date map to the stem-contact fold domain and the α-helix bundle domain. This has led to the proposal of allele-specific genotype-phenotype correlations (Sun et al. 2017). However, the p.Arg618Cys MARS mutation identified herein was previously reported in CMT2U, with functional support from yeast complementation assays showing an inability of the p.Arg618Cys MES1 to rescue deletion of endogenous MES1 (Gonzalez et al. 2013). When modeled in the human MARS open reading frame, p.Arg618Cys showed similar loss of function (Fig. 3). The p.Arg618Cys mutation in the current study was inherited from an apparently healthy father; nerve conduction velocity (NCV) studies were not available. Clinical reassessment of the father and NCV studies in the sixth decade, corresponding to the reported age of onset of CMT2U, will be of value. Notably, explicit questioning failed to reveal any family history of a late-onset neuropathy in elderly individuals in previous generations.
Interestingly, the second variant p.Tyr307Cys was predicted to be pathogenic by several bioinformatic algorithms (Table 1), yet appeared fully functional in our yeast complementation assay (Fig. 3). It is important to note that the yeast complementation assay may not be sensitive enough to detect minor to moderate decreases in enzyme function, and does not assess for tissue-specific impairments in tRNA charging that might be relevant to the patient phenotype. Furthermore, it is not surprising that p.Tyr307Cys retains some function, based on the non-lethal nature of the phenotype; compound heterozygosity for two functionally null alleles would presumably be embryonic lethal (Seburn et al. 2006, Motley et al. 2011). Thus, a more careful enzymatic analysis is needed to fully characterize the functional consequences of p.Tyr307Cys MARS (Oprescu et al. 2017).
While dominant and recessive inheritance at the same locus is a recognized phenomenon, it can often be traced to the type of variant (i.e., nonsense versus missense), localization of the variant in a specific protein domain, mechanism of action (i.e., loss-of-function versus gain-of-function), or the trigger or escape of nonsense-mediated decay. Controversies in the literature have stemmed from discordant reports concerning pathogenicity of a particular variant (Shy et al. 2006, Seeman et al. 1999). In the current case, based on the clinical data and the functional evidence from yeast studies, we propose that the p.Arg618Cys mutation can cause autosomal dominant CMT2U or can contribute to biallelic ILLD, dependent upon the genetic background of the individual. Possible contributions from other loci and from environmental factors cannot be ruled out. This hypothesis is consistent with the incomplete penetrance of p.Arg618Cys-associated CMT2U (Gonzalez et al. 2013) as well as with the proposed contribution of mutation burden to neuropathies (Gonzaga-Jauregui et al. 2015).
There has been growing attention in recent years to the phenotypic consequences of monoallelic and biallelic mutations in both cytoplasmic and mitochondrial ARSs (Oprescu et al. 2017). In correlation with previous studies, we show that compound heterozygosity for mutations in the gene encoding MARS causes a multi-organ disease involving the liver, lungs, bone marrow, and thyroid function. Moreover, we discuss clinical interventions that may benefit patients with ILLD and highlight the complexity of genotype-phenotype correlations.
Supplementary Material
Acknowledgments
The authors wish to thank the family for their participation in this study. We thank Dr. Stephanie Ben-Shushan for images of the liver biopsy. R.M. is supported by the Michigan Pre-doctoral Training in Genetics Program (GM007544). A.A. is supported by funding from the National Institute of General Medical Sciences (GM110184 and GM118647).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
The ClinVar accession numbers for the DNA variant data reported in this paper are SCV000692448 and SCV000692464.
CONFLICT OF INTEREST: The authors declare that they have no conflict of interest.
References
- Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, Sivakumar K, Ionasescu V, Funalot B, Vance JM, Goldfarb LG, Fischbeck KH, Green ED. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72(5):1293–9. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonellis A, Green ED. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet. 2008;9:87–107. doi: 10.1146/annurev.genom.9.081307.164204. [DOI] [PubMed] [Google Scholar]
- Casey JP, McGettigan P, Lynam-Lennon N, McDermott M, Regan R, Conroy J, Bourke B, O’Sullivan J, Crushell E, Lynch S, Ennis S. Identification of a mutation in LARS as a novel cause of infantile hepatopathy. Mol Genet Metab. 2012;106(3):351–8. doi: 10.1016/j.ymgme.2012.04.017. [DOI] [PubMed] [Google Scholar]
- Castranova D, Davis AE, Lo BD, Miller MF, Paukstelis PJ, Swift MR, Pham VN, Torres-Vázquez J, Bell K, Shaw KM, Kamei M, Weinstein BM. Aminoacyl-Transfer RNA Synthetase Deficiency Promotes Angiogenesis via the Unfolded Protein Response Pathway. Arterioscler Thromb Vasc Biol. 2016;36(4):655–62. doi: 10.1161/ATVBAHA.115.307087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien CI, Chen YW, Wu YH, Chang CY, Wang TL, Wang CC. Functional substitution of a eukaryotic glycyl-tRNA synthetase with an evolutionarily unrelated bacterial cognate enzyme. PLoS One. 2014;9(4):e94659. doi: 10.1371/journal.pone.0094659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzaga-Jauregui C, Harel T, Gambin T, Kousi M, Griffin LB, Francescatto L, Ozes B, Karaca E, Jhangiani SN, Bainbridge MN, Lawson KS, Pehlivan D, Okamoto Y, Withers M, Mancias P, Slavotinek A, Reitnauer PJ, Goksungur MT, Shy M, Crawford TO, Koenig M, Willer J, Flores BN, Pediaditrakis I, Us O, Wiszniewski W, Parman Y, Antonellis A, Muzny DM, Katsanis N, Battaloglu E, Boerwinkle E, Gibbs RA, Lupski JR, Baylor-Hopkins Center for Mendelian Genomics Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Rep. 2015;12(7):1169–83. doi: 10.1016/j.celrep.2015.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M, McLaughlin H, Houlden H, Guo M, Yo-Tsen L, Hadjivassilious M, Speziani F, Yang XL, Antonellis A, Reilly MM, Züchner S, Inherited Neuropathy Consortium Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J Neurol Neurosurg Psychiatry. 2013;84(11):1247–9. doi: 10.1136/jnnp-2013-305049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadchouel A, Wieland T, Griese M, Baruffini E, Lorenz-Depiereux B, Enaud L, Graf E, Dubus JC, Halioui-Louhaichi S, Coulomb A, Delacourt C, Eckstein G, Zarbock R, Schwarzmayr T, Cartault F, Meitinger T, Lodi T, de Blic J, Strom TM. Biallelic Mutations of Methionyl-tRNA Synthetase Cause a Specific Type of Pulmonary Alveolar Proteinosis Prevalent on Réunion Island. Am J Hum Genet. 2015;96(5):826–31. doi: 10.1016/j.ajhg.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun YS, Park HJ, Heo SH, Yoon BR, Nam SH, Kim SB, Park CI, Choi BO, Chung KW. Rare variants in methionyl- and tyrosyl-tRNA synthetase genes in late-onset autosomal dominant Charcot-Marie-Tooth neuropathy. Clin Genet. 2014;86(6):592–4. doi: 10.1111/cge.12327. [DOI] [PubMed] [Google Scholar]
- Jordanova A, Irobi J, Thomas FP, Van Dijck P, Meerschaert K, Dewil M, Dierick I, Jacobs A, De Vriendt E, Guergueltcheva V, Rao CV, Tournev I, Gondim FA, D’Hooghe M, Van Gerwen V, Callaerts P, Van Den Bosch L, Timmermans JP, Robberecht W, Gettemans J, Thevelein JM, De Jonghe P, Kremensky I, Timmerman V. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet. 2006;38(2):197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- Latour P, Thauvin-Robinet C, Baudelet-Méry C, Soichot P, Cusin V, Faivre L, Locatelli MC, Mayençon M, Sarcey A, Broussolle E, Camu W, David A, Rousson R. A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2010;86(1):77–82. doi: 10.1016/j.ajhg.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Cho BH, Park SG, Kim S. Aminoacyl-tRNA synthetase complexes: beyond translation. J Cell Sci. 2004;117(Pt 17):3725–34. doi: 10.1242/jcs.01342. [DOI] [PubMed] [Google Scholar]
- McLaughlin HM, Sakaguchi R, Liu C, Igarashi T, Pehlivan D, Chu K, Iyer R, Cruz P, Cherukuri PF, Hansen NF, Mullikin JC, Biesecker LG, Wilson TE, Ionasescu V, Nicholson G, Searby C, Talbot K, Vance JM, Züchner S, Szigeti K, Lupski JR, Hou YM, Green ED, Antonellis A, NISC Comparative Sequencing Program Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet. 2010;87(4):560–6. doi: 10.1016/j.ajhg.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley WW, Seburn KL, Nawaz MH, Miers KE, Cheng J, Antonellis A, Green ED, Talbot K, Yang XL, Fischbeck KH, Burgess RW. Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels. PLoS Genet. 2011;7(12):e1002399. doi: 10.1371/journal.pgen.1002399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprescu SN, Griffin LB, Beg AA, Antonellis A. Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods. 2017;113:139–151. doi: 10.1016/j.ymeth.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Cortez RL, Lee K, Azeem Z, Antonellis PJ, Pollock LM, Khan Irfanullah S, Andrade-Elizondo PB, Chiu I, Adams MD, Basit S, Smith JD, Nickerson DA, McDermott BM, Ahmad W, Leal SM, University of Washington Center for Mendelian Genomics Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am J Hum Genet. 2013;93(1):132–40. doi: 10.1016/j.ajhg.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmel P. Aminoacyl tRNA synthetases: general scheme of structure-function relationships in the polypeptides and recognition of transfer RNAs. Annu Rev Biochem. 1987;56:125–58. doi: 10.1146/annurev.bi.56.070187.001013. [DOI] [PubMed] [Google Scholar]
- Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW. An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron. 2006;51(6):715–26. doi: 10.1016/j.neuron.2006.08.027. [DOI] [PubMed] [Google Scholar]
- Seeman P, Mazanec R, Marikova T, Rautenstrauss B. Charcot-Marie-Tooth 1A: heterozygous T118M mutation over a CMT1A duplication has no influence on the phenotype. Ann N Y Acad Sci. 1999;883:485–9. [PubMed] [Google Scholar]
- Shy ME, Scavina MT, Clark A, Krajewski KM, Li J, Kamholz J, Kolodny E, Szigeti K, Fischer RA, Saifi GM, Scherer SS, Lupski JR. T118M PMP22 mutation causes partial loss of function and HNPP-like neuropathy. Ann Neurol. 2006;59(2):358–64. doi: 10.1002/ana.20777. [DOI] [PubMed] [Google Scholar]
- Simons C, Griffin LB, Helman G, Golas G, Pizzino A, Bloom M, Murphy JL, Crawford J, Evans SH, Topper S, Whitehead MT, Schreiber JM, Chapman KA, Tifft C, Lu KB, Gamper H, Shigematsu M, Taft RJ, Antonellis A, Hou YM, Vanderver A. Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am J Hum Genet. 2015;96(4):675–81. doi: 10.1016/j.ajhg.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Hu G, Luo J, Fang D, Yu Y, Wang X, Chen J, Qiu W. Mutations in methionyl-tRNA synthetase gene in a Chinese family with interstitial lung and liver disease, postnatal growth failure and anemia. J Hum Genet. 2017 doi: 10.1038/jhg.2017.10. [DOI] [PubMed] [Google Scholar]
- Tsai PC, Soong BW, Mademan I, Huang YH, Liu CR, Hsiao CT, Wu HT, Liu TT, Liu YT, Tseng YT, Lin KP, Yang UC, Chung KW, Choi BO, Nicholson GA, Kennerson ML, Chan CC, De Jonghe P, Cheng TH, Liao YC, Züchner S, Baets J, Lee YC. A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain. 2017 doi: 10.1093/brain/awx058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meel E, Wegner DJ, Cliften P, Willing MC, White FV, Kornfeld S, Cole FS. Rare recessive loss-of-function methionyl-tRNA synthetase mutations presenting as a multi-organ phenotype. BMC Med Genet. 2013;14:106. doi: 10.1186/1471-2350-14-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young HJ, Lee JW, Kim S. Function of membranous lysyl-tRNA synthetase and its implication for tumorigenesis. Biochim Biophys Acta. 2016;1864(12):1707–1713. doi: 10.1016/j.bbapap.2016.09.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.