

Abstract
Objective
Cigarette smoking is associated with immune-mediated disorders. We explored the contribution of smoking to polymyositis (PM) and dermatomyositis (DM) phenotypes and attempted to determine whether cigarette smoking effects differ by race and genotype.
Methods
Associations of tobacco smoking with disease features, autoantibodies, HLA types and race were evaluated using multiple logistic regressions in 465 patients.
Results
Caucasian ever-smokers (n=140) were more likely to have PM (adjusted OR=2.24,95%CI:1.41–3.57), anti-synthetase (adjusted OR=1.93, 95%CI:1.12–3.34) and anti-Jo-1 autoantibodies (adjusted OR=1.94,95%CI:1.08–3.46) and less likely to have anti-p155/140 autoantibodies (adjusted OR=0.36,95%CI:0.14–0.92). In Caucasians, ever-smokers had a greater interstitial lung disease (ILD) frequency than never-smokers, while in African-Americans this relationship was inverted, but neither trend reached statistical significance. Pack-years of cigarette smoking showed significant positive associations with PM (adjusted OR=1.02, 95%CI:1.002–1.04) and ILD (adjusted OR=1.02, 95%CI:1.001–1.03) and was inversely associated with anti-p155/140 autoantibodies (adjusted OR=0.93, 95%CI:0.87–0.99) in Caucasians. Caucasians heavy smokers (≥20 pack-years) were more likely to have PM (adjusted OR=2.52, 95%CI:1.25–5.09), ILD (adjusted OR=2.48, 95%CI:1.23–5.00) and anti-Jo-1 autoantibodies (adjusted OR=2.65, 95%CI:1.16–6.08) than never-smokers.
In Caucasians, compared to never-smokers without HLA-DRB1*03:01 allele, ever-smokers with HLA-DRB1*03:01 allele had the highest odds of PM, ILD, ASA and anti-Jo-1 autoantibodies. Risks for those with only one of these two factors were intermediate. An inverse pattern was observed regarding anti-p155/140 autoantibodies.
Conclusion
Tobacco smoking was associated with clinical and autoantibody phenotypes in Caucasians. Our findings also suggest possible interactions among HLA-DRB1*03:01 and smoking on the risk of PM and ILD, as well as, anti-synthetase, anti-Jo-1 and anti-p155/140 autoantibodies in Caucasians.
Keywords: Dermatomyositis, Polymyositis, Smoking, Autoantibody, Cigarette smoking, Autoimmunity
INTRODUCTION
The idiopathic inflammatory myopathies (IIM) are rare autoimmune conditions, characterized by chronic muscle inflammation leading to progressive muscle weakness. They are often associated with other systemic features, including involvement of pulmonary, gastrointestinal, dermatologic and cardiac systems (1). Polymyositis (PM) and dermatomyositis (DM) are two major phenotypes of IIM in adults, affecting about 14–32 per 100,000 of the North American population (2–4). DM is clinically distinguished from other types of IIM by the presence of pathognomonic rashes. Circulating myositis-specific autoantibodies (MSA) and myositis-associated autoantibodies (MAA) have been linked to specific clinical features in both PM and DM and have been shown to have diagnostic and prognostic value in these patients (5, 6). Although the pathogenesis of IIM is unclear, it is thought that a complex interaction of multiple genetic and environmental factors contributes to the disease onset, phenotype and progression (7).
Genetic markers have been linked to disease phenotype and prognosis in IIM patients (8, 9), however, little is known about the effect of environmental agents on these disorders (10, 11). Increasing evidence suggests that cigarette smoking impacts disease risk for many immune-mediated diseases such as rheumatoid arthritis (RA) (12), autoimmune thyroid diseases (13), Crohn’s disease and ulcerative colitis (14, 15). While smoking is associated with an increased risk of Crohn’s disease and Graves’ disease, smoking is reported to be protective for ulcerative colitis and the development of anti-thyroid peroxidase autoantibodies (14, 16–19). A positive association between cigarette smoking and anti-Jo-1 autoantibody frequency was reported in European PM and DM patients (20). The influence of tobacco smoking on other MSA, MAA and different clinical phenotypes, as well as in other racial or ethnic groups, has not been previously investigated. Prior studies have reported that there are differences in HLA allele risk factors between Caucasians and African-Americans with IIM (9, 21–23). Given that race has been identified as a risk modifier in the burden of tobacco-related diseases (24) and the documented racial disparity in genetic susceptibility to IIM (9, 22), we stratified our patient population by race to investigate the potential impact of cigarette smoking on PM and DM. In addition, we investigated the combined impact of HLA-DRB1*03:01 and cigarette smoking on the frequencies of clinical and serologic phenotypes in patients with PM and DM.
PATIENTS AND METHODS
Patients
All patients with adult-onset PM or DM (age at onset ≥18 years) who had available smoking data and were enrolled in Institutional Review Board–approved protocols involving pathogenesis or treatment of IIM at the National Institutes of Health Clinical Center between January 1981 and September 2015 were identified for the present investigation. The protocols included studies on the risk factors, pathogenesis and natural history of IIM. All participants provided written informed consent according to the Declaration of Helsinki for their participation. Diagnosis was made according to Bohan and Peter criteria for probable or definite PM or DM (25) and individuals with cancer-associated myositis (CAM), inclusion body myositis, familial muscle disease, metabolic myopathy, infectious myopathies, or other causes of muscle disease were excluded. CAM was defined as detection of cancer (excluding basal cell carcinoma of the skin) within 5 years of PM or DM diagnosis. Patients who had less than five years of follow-up at the time of evaluation were considered not to have CAM if cancer was not detected by the time of evaluation. Patients with missing tobacco smoking data were not included in this study (Figure 1).
Figure 1.

Flow chart presenting the study subjects selection. * of these patients, 245 had serologic data.
CAM=cancer-associated myositis; MSA= myositis-specific autoantibodies.
Clinical data assessed included demographic features (age at the time of evaluation, gender and race by self-identification), cigarette smoking history, myositis clinical phenotype (PM or DM), and disease course. Blood samples obtained at the time of evaluation were frozen for further analyses. A rheumatologist reviewed the available medical and imaging records to confirm the presence of interstitial lung disease (ILD). In order of preference, diagnosis of ILD was based on lung biopsy consistent with ILD (n=42), chest CT-scan consistent with ILD (n=61), chest x-ray consistent with ILD (n=52) and restrictive pattern observed on pulmonary function test defined as total lung capacity < 80% predicted in conjunction with medical records stating diagnosis of ILD from a non-study physician (n=6). Patients were divided into ever-smoker and never-smoker categories. A positive ever-smoking history was defined as smoking at least 100 cigarettes in a lifetime (24, 26). The quantity of tobacco smoking was expressed as numbers of pack-years, one pack-year being equivalent to an average of 20 cigarettes consumption per day for one year. Patients with cigarette smoking history of <10, 10–20 and ≥20 pack-years were identified as light, moderate and heavy smokers, respectively (27) and were compared with never-smokers.
Patients self-reported their race and ethnicity and were categorized into three different groups: African-American (n=99), Caucasian (n=330) and Other or mixed race (n=36). The Other or mixed race group included participants with Hispanic ethnicity, American Indians, Alaska Natives, Asians, Native Hawaiians or other Pacific Islanders and patients with mixed race.
Laboratory methods
Sera were assessed for the presence of MSA, MAA, and other autoantibodies using previous validated immunoprecipitation methods (28, 29). The MSA group included anti-synthetases (ASA), anti-Mi-2, anti-MJ, anti-signal recognition particle (SRP) and anti-p155/140 autoantibodies. The ASA group was comprised of anti-Jo-1 (histidyl tRNA-synthetase), anti-PL-7 (threonyl), anti-PL-12 (alanyl), anti-OJ (isoleucyl), anti-EJ (glycyl) and anti-KS (asparaginyl) autoantibodies. The MAA category included anti-PM/Scl, anti-Ku, anti-U1RNP, anti-U2RNP, anti-Ro/SSA and anti-La/SSB autoantibodies.
Purified DNA samples were genotyped at the HLA-DRB1 locus using a combination of laboratory designed and commercial reagents for PCR-mediated sequence-specific oligonucleotide probe hybridization, sequence-specific priming techniques and Sanger method sequence based typing (8, 30).
Statistical analyses
Analyses were performed using SAS Enterprise Guide V.4.3 (SAS Institute, Cary, NC, USA). Chi-square and when appropriate Fisher’s exact tests were utilized to analyze categorical variables. Continuous variables were analyzed using Mann–Whitney–Wilcoxon tests. Participants whose data for a variable were not available were excluded from the analysis of that variable and frequency proportions were calculated using the number of non-missing values.
Smoking frequency was adjusted according to age, gender and race as appropriate. Chi-square and Fisher’s exact tests, as appropriate, were performed using the adjusted smoking frequency to assess the association between tobacco use with the clinical phenotype, presence of ILD and autoantibody status. Logistic regression analyses were used to examine the potential effect of smoking on the clinical and serologic status. In addition, unadjusted and adjusted analyses were done separately for Caucasians and African-Americans. All analyses were adjusted for age, gender, and race, as appropriate, and all values reported are adjusted values unless specifically stated otherwise. We did not evaluate the impact of MSA/MAA autoantibodies that were found in fewer than ten subjects.
We hypothesized that cigarette smoking is a risk factor for ILD in PM and DM patients and the possible association between tobacco smoking and ILD is not uniformly distributed across different races, thus a p-value of <0.05 was considered statistically significant. The remaining evaluations, including the association of cigarette smoking with clinical and serologic phenotypes were exploratory analyses, therefore, p-values were not corrected for multiple comparisons.
RESULTS
Demographic characteristics of the patients
There were 465 patients with available smoking history who met the inclusion criteria in the study. The main characteristics of the total population are summarized in Table 1. In terms of demographic features, our data are consistent with other large myositis cohorts (31, 32). Most patients were female (71%) and had PM (58%). The study population was racially diverse with 135 (29%) non-Caucasians. Overall, 180 (39%) were ever-smokers and 147 had available pack-year information. There were significant differences between African-Americans and Caucasians in terms of frequency of PM, ILD status and smoking (Table 1). PM was more frequent in African-Americans (72%) than Caucasians (54%) (P=0.002). African-American patients were also more likely to have ILD (46%) compared to Caucasians (32%) (P=0.018). Caucasians had a higher frequency of ever-smoking (42%) than African-Americans (29%) (P=0.019). The unadjusted and adjusted smoking frequencies are detailed in Supplementary Table 1. An evaluation for selection bias showed that patients with missing values for smoking (n=46), who were not included in this study, did not differ in terms of gender, race, clinical phenotype, autoantibody status (except for anti-PL-12 autoantibodies that were more frequent in patients with missing smoking data) or HLA-DRB1 allele frequency compared to individuals with smoking data (n=465) (data not shown). Patients with missing smoking data were older than those who had available smoking information (mean age of 50.5 and 45.5 years, respectively, P=0.026). In addition, we assessed the possibility of selection bias by excluding CAM patients (n=26). The inclusion of CAM patients in the study population did not change the pattern of associations between cigarette smoking and clinical and serologic status in PM and DM patients described below. Also, comparisons of patients with HLA-DRB1 data with those without HLA-DRB1 data showed that smoking, age, gender, ILD and anti-Jo-1 autoantibody frequencies did not differ between these populations.
Table 1.
Demographic and clinical features of participants in the total myositis population and categorized by race.
| Characteristics | Caucasian (n=330) | African-American (n=99) | Other or Mixed-race (n=36) | Total (n=465) |
|---|---|---|---|---|
| Age, years | ||||
| Mean ± SD | 47.0 ± 13.6 | 43.3 ± 12.8 | 44.4 ± 15.6 | 46.0 ± 13.7 |
| Median (IQR) | 47 (37 – 57) | 43 (32 – 54) | 49 (28.5 – 56) | 46 (35 – 57) |
| Disease duration, months | ||||
| Median (IQR) | 11 (4 – 28) | 12 (4 – 32) | 8.5 (2 – 28.5) | 11 (4 – 28.5) |
| Gender | ||||
| Female (%) | 230 (70%) | 77 (78%) | 23 (64%) | 330 (71%) |
| Clinical Diagnosis | ||||
| Polymyositis (%) | 176 (54%) | 71 (72%) | 20 (56%) | 267 (58%) |
| ILD | ||||
| ILD-positive (%) | 103 (32%) | 43 (46%) | 15 (42%) | 161 (36%) |
| Smoking history | ||||
| Ever-smokers (%) | 140 (42%) | 29 (29%) | 11 (31%) | 180 (39%) |
| Smoking history, pack-years | ||||
| Mean ± SD | 23.0 ± 22.7 | 19.0 ± 15.8 | 8.4 ± 5.6 | 21.2 ± 21.2 |
| Median (IQR) | 15 (7.5 – 35) | 19 (5 – 30) | 6 (4 – 13) | 15 (5 – 30) |
| Pack-years of smoking in ever-smokers | ||||
| <10 (light smokers) | 35 (31%) | 8 (35%) | 6 (55%) | 49 (33%) |
| 10–20 (moderate smokers) | 28 (25%) | 5 (22%) | 5 (45%) | 38 (26%) |
| ≥20 (heavy smokers) | 50 (44%) | 10 (43%) | 0 (0%) | 60 (41%) |
| HLA-DRB1*03:01 | ||||
| Positive/total (%) | 111/215 (52%) | 14/72 (19%) | 3/18 (17%) | 128/305 (42%) |
| Autoantibody status | ||||
| Positive/total (%) | 144/266 (54%) | 43/71 (61%) | 8/24 (33%) | 195/361 (54%) |
| MSA | ||||
| Anti-Synthetase | 83/266 (31%) | 26/71 (37%) | 4/24 (17%) | 113/361 (31%) |
| Anti-Jo-1 | 68/265 (26%) | 17 (71 (24%) | 4/24 (17%) | 89/360 (25%) |
| Anti-PL-7 | 6/261 (2%) | 2/71 (3%) | 0/24 (0%) | 8/356 (2%) |
| Anti-PL-12 | 2/261 (1%) | 5/71 (7%) | 0/24 (0%) | 7/356 (2%) |
| Anti-OJ | 6/235 (2%) | 0/66 (0%) | 0/23 (0%) | 5/324 (2%) |
| Anti-EJ | 0/233 (0%) | 2/66 (3%) | 0/23 (0%) | 2/322 (1%) |
| Anti-KS | 3/236 (1%) | 0/66 (0%) | 0/23 (0%) | 3/325 (1%) |
| Anti-Mi-2 | 14/261 (5%) | 4/71 (6%) | 2/24 (8%) | 20/356 (6%) |
| Anti-MJ | 15/237 (6%) | 1/66 (2%) | 0/24 (0%) | 16/327 (5%) |
| Anti-SRP | 13/261 (5%) | 10/71 (14%) | 1/24 (4%) | 24/356 (7%) |
| Anti-p155/P140 | 29/235 (12%) | 3/66 (5%) | 3/24 (12.5%) | 35/325 (11%) |
For gender, clinical diagnosis, ILD, smoking history, pack-years categories, HLA-DRB1*03:01 and autoantibody status data are listed as numbers of patients in each category and percent in parentheses. Disease duration reflects the time interval between the appearance of the first Bohan and Peter element (proximal muscle weakness, heliotrope rash, Gottron’s sign, Gottron’s papules, elevation of serum levels of skeletal muscle enzymes, muscle biopsy evidence of myopathy or positive EMG) and their evaluation date.
IQR= interquartile range; ILD= interstitial lung disease; MSA= myositis-specific autoantibodies.
Clinical phenotype and smoking history
Adjusted and unadjusted logistic regression analyses are summarized in Table 2 and Supplementary Table 2, respectively. In the total population, ever-smokers were more likely to have PM than never-smokers (adjusted odds ratio [OR]=2.25, 95% confidence interval [CI]: 1.49–3.41, P<0.001). The frequency of PM increased in the total population as the number of pack-years increased (adjusted OR=1.02, 95%CI: 1.004–1.04, P=0.014), indicating a two percent increase in odds of PM diagnosis for each unit increase in number of pack years exposed. In Caucasians, tobacco smoking was positively associated with PM (adjusted OR=2.24, 95%CI: 1.41–3.57, P=0.001). A significant increase in the odds of PM diagnosis was observed with increasing cigarette smoking quantity in Caucasian patients (adjusted OR=1.02, 95%CI: 1.002 – 1.04, P=0.025), with a two percent increased likelihood of PM with each one unit of increase in pack-years. In African-Americans, a similar trend was observed, but the adjusted smoking frequency did not differ statistically between PM and DM patients (37% vs. 20%, respectively, adjusted OR=2.37, 95%CI: 0.79 – 7.16, P=0.12) (Figure 2-A). There was no significant association between the number of pack-years and diagnosis in African-American patients (adjusted OR=1.04, 95%CI: 0.98 – 1.10, P=0.22).
Table 2.
Adjusted logistic regression analyses assessing risks of different clinical and serologic features in patients with polymyositis and dermatomyositis based on cigarette smoking history
| Outcome | Total population (n=465) | Caucasians (n=330) | African-Americans (n=99) | |||
|---|---|---|---|---|---|---|
| OR (95% CI) | P-value | OR (95% CI) | P-value | OR (95% CI) | P-value | |
| Ever-smokers vs. never-smoker | ||||||
| Diagnosis, Polymyositis vs. dermatomyositis | 2.25 (1.49 – 3.41) | <0.001 | 2.24 (1.41 – 3.57) | 0.001 | 2.37 (0.79 – 7.16) | 0.12 |
| Interstitial lung disease | 1.52 (1.002 – 2.29) | 0.049 | 1.61 (0.99 – 2.63) | 0.056 | 0.94 (0.37 – 2.35) | 0.89 |
| Anti-synthetase autoantibodies | 1.60 (0.99 – 2.57) | 0.052 | 1.93 (1.12 – 3.34) | 0.019 | 0.70 (0.22 – 2.20) | 0.54 |
| Anti-Jo-1 autoantibodies | 1.71 (1.03 – 2.83) | 0.037 | 1.94 (1.08 – 3.46) | 0.025 | 0.90 (0.24 – 3.32) | 0.87 |
| Anti-p155/140 autoantibodies | 0.45 (0.20 – 1.04) | 0.061 | 0.36 (0.14 – 0.92) | 0.033 | N/A | N/A |
| Cigarette smoking, pack-years | ||||||
| Diagnosis, Polymyositis vs. dermatomyositis | 1.02 (1.004 – 1.04) | 0.014 | 1.02 (1.002 – 1.04) | 0.025 | 1.04 (0.98 – 1.10) | 0.22 |
| Interstitial lung disease | 1.02 (1.004 – 1.03) | 0.010 | 1.02 (1.001 – 1.03) | 0.037 | 1.04 (0.99 – 1.08) | 0.12 |
| Anti-synthetase autoantibodies | 1.01 (0.99 – 1.02) | 0.23 | 1.01 (0.99 – 1.03) | 0.17 | 1.001 (0.96 – 1.05) | 0.97 |
| Anti-Jo-1 autoantibodies | 1.01 (0.997 – 1.03) | 0.10 | 1.02 (0.999 – 1.03) | 0.059 | 0.99 (0.95 – 1.05) | 0.85 |
| Anti-p155/140 autoantibodies | 0.93 (0.87 – 0.99) | 0.029 | 0.93 (0.87 – 0.99) | 0.037 | N/A | N/A |
Analyses are adjusted for age, gender and race as appropriate. N/A= not applicable. There were only three African-American patients with anti-p155/140 autoantibodies and none of them had a positive ever-smoking history.
Figure 2.
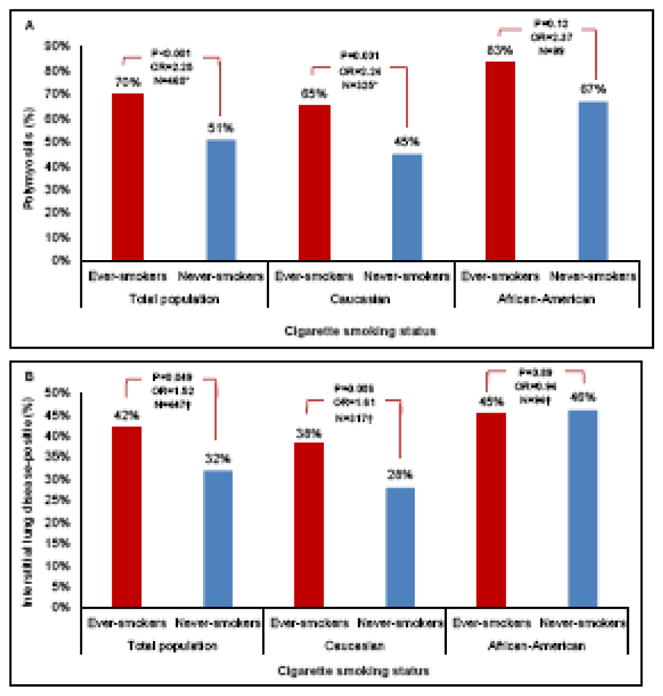
Tobacco smoking frequency by diagnosis (PM, polymyositis; DM, dermatomyositis) (2-A) and interstitial lung disease (ILD) status (2-B) subdivided by race. All values in the figure are adjusted for age, gender, and race as appropriate. Total population includes patients with Caucasian, African-American and Other or mixed races. * Patients with missing values for diagnosis were excluded from this analysis. † Patients with missing values for ILD were excluded from this analysis. The odds ratios and p-values are calculated using adjusted logistic regression analyses shown in Table 2.
ILD was observed in 161 patients (36%) and was evaluated for an association with the adjusted smoking history (Figure 2-B). In the total population, ever smokers were more likely to have ILD than never-smokers (adjusted OR= 1.52, 95%CI: 1.002 – 2.29, P=0.049). A two percent increased odds of ILD was found with each pack-year of cigarette smoking in the total population (adjusted OR=1.02, 95%CI: 1.004–1.03, P=0.010). In Caucasians, ever-smokers were more likely to have ILD compared to never-smokers (unadjusted OR=1.77, 95%CI: 1.10–2.85, P=0.018). Nevertheless, after adjusting for age at the time of evaluation and gender, smoking history was no longer significantly associated with ILD in Caucasian patients (adjusted OR=1.61, 95%CI: 0.99–2.63, P=0.056). A significant association was observed between pack-years and ILD in Caucasians (adjusted OR=1.02, 95%CI: 1.001 – 1.03, P=0.037), indicating a two percent increase in odds of ILD with every unit increase in pack-years. In African-Americans, ILD frequency was not found to be statistically associated with ever-smoker status or pack-years.
Serologic phenotype and smoking history
MSA and MAA serotypes were determined for 361 (78%) and 357 (77%) patients, respectively (Table 1 and Supplementary Table 1). Overall, 195 (54%) patients had a MSA and 105 (29%) had a MAA. Significant differences were noted in the frequency of anti-EJ, anti-PL-12, anti-SRP, MAA, anti-U1RNP and anti-Ro/SSA between African-Americans and Caucasians (P<0.05). Significant differences in other MSA and MAA autoantibodies frequencies were not observed between Caucasian and African-American patients.
The presence of ASA was not significantly associated with the adjusted smoking frequency in the total population (Figure 3-A). In Caucasians, however, ever-smokers were more likely to be ASA-positive compared to never-smokers (adjusted OR=1.93, 95%CI: 1.12 – 3.34, P=0.019). African-American patients showed an opposite trend, as never-smokers had a greater number of ASA-positivity (40%) compared to ever-smokers (31%), but did not reach statistical significance (adjusted OR=0.70, 95%CI: 0.22 – 2.20, P=0.54). No significant associations were observed between the number of pack-years and the presence of ASA in the total population, Caucasian or African-American groups (Table 2).
Figure 3.
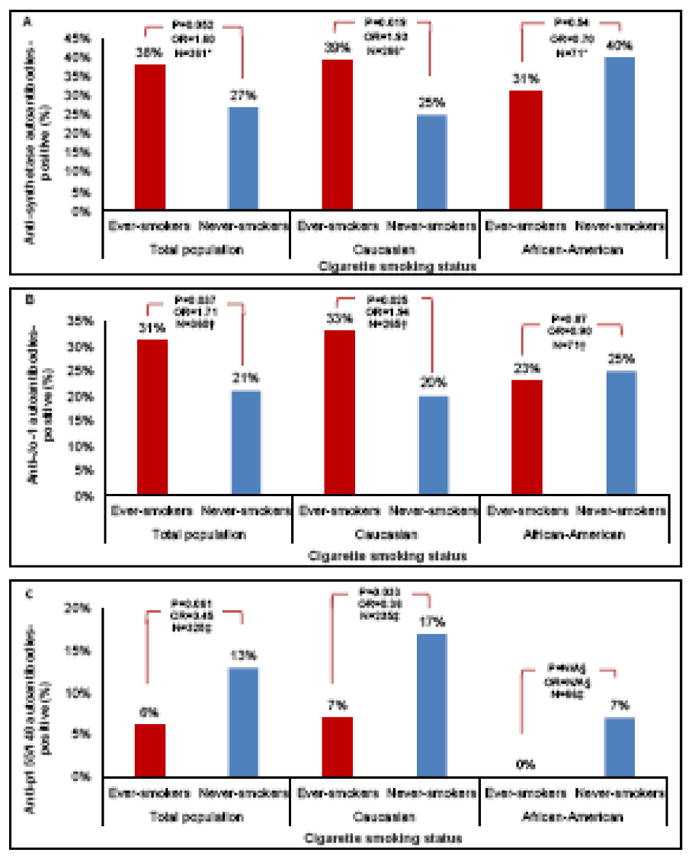
Tobacco smoking frequency based on autoantibody status. Frequency of tobacco smoking based on anti-synthetase autoantibody (ASA) status (3-A). Frequency of tobacco smoking based on anti-Jo-1 autoantibody status (3-B). Frequency of tobacco smoking based on anti-p155/140 autoantibody status (3-C). All values in the figure are adjusted for age, gender and race as appropriate. Total population includes patients with Caucasian, African-American and Other or mixed races. * Patients with missing values for anti-synthetase autoantibodies were excluded from this analysis. † Patients with missing values for anti-Jo-1 autoantibodies were excluded from this analysis. ‡ Patients with missing values for anti-p155/140 autoantibodies were excluded from this analysis. § N/A= not applicable; there were only three African-American patients with anti-p155/140 autoantibodies and none of them had a positive ever-smoking history. The odds ratios and p-values are calculated using adjusted logistic regression analyses shown in Table 2.
The presence of anti-Jo-1 autoantibodies in the total population was positively associated with the adjusted smoking history (adjusted OR=1.71, 95%CI: 1.03 – 2.83, P=0.037) (Figure 3-B). In Caucasian patients, ever-smokers were more likely to be positive for anti-Jo-1 autoantibodies compared to never-smokers (adjusted OR=1.94, 95%CI: 1.08 – 3.46, P=0.025). This was not demonstrated in African-American patients, in whom anti-Jo-1 autoantibodies frequency was 25% in never-smokers and 23% in ever-smokers, however, this finding was not significant (adjusted OR=0.90, 95%CI: 0.24 – 3.32, P=0.87). No significant associations were observed between the number of pack-years and the presence of anti-Jo-1 autoantibodies in the total population, Caucasian or African-American groups.
Anti-p155/140 autoantibodies were not significantly associated with the adjusted smoking history in the total population (adjusted OR=0.45, 95%CI: 0.20 – 1.04, P=0.061), however, the presence of anti-p155/140 autoantibodies was negatively associated with increasing numbers of pack-years in the total population (adjusted OR=0.93, 95%CI: 0.87 – 0.99, P=0.029). In Caucasians, ever-smokers were less likely to be positive for anti-p155/140 autoantibodies than never-smokers (adjusted OR=0.36, 95%CI: 0.14 – 0.92, P=0.033). Increasing numbers of pack-years showed a significant association with decreased likelihood of anti-p155/140 autoantibody positivity in Caucasians (adjusted OR=0.93, 95%CI: 0.87 – 0.99, P=0.037). No association was observed between the adjusted smoking history and anti-p155/140 autoantibodies in African-Americans (Figure 3-C).
Caucasian heavy smokers were more likely to have PM (adjusted OR=2.52, 95%CI: 1.25 – 5.09, P=0.010) and ILD (adjusted OR=2.48, 95%CI: 1.23 – 5.00, P=0.011) compared to never-smokers. Caucasian heavy smokers were also 2.6 times more likely to be positive for anti-Jo-1 autoantibodies compared to never-smokers (adjusted OR=2.65, 95%CI: 1.16 – 6.08, P=0.021). We did not observe any statistically significant associations between light or moderate smoker status compared to never-smoker status in terms of clinical and serologic phenotypes in Caucasians. No significant associations were found between light/moderate/heavy smoking status compared to never-smoking status in African-Americans (data not shown).
Considering the significant positive association between cigarette smoking and PM in this study, separate regression analyses were performed to evaluate cigarette smoking associations with ILD and autoantibody status in PM and DM patients while adjusting for diagnosis (Supplementary Table 3). No autoantibodies were significantly associated with the adjusted smoking frequency in the Other or mixed race groups. No significant associations between the adjusted smoking history and the presence of MSA, anti-Mi-2, anti-MJ, anti-SRP, anti-PM/Scl, anti-U1RNP, anti-Ro/SSA and anti-La/SSB autoantibodies were found in the total population, Caucasian or African-American subgroups.
Combined effects of HLA-DRB1*03:01 and smoking on frequency of clinical and autoantibody phenotypes in Caucasian patients
Two-thirds of all patients were genotyped at the HLA-DRB1 locus (Table 1) and 42% of those were positive for the HLA-DRB1*03:01 allele. The frequency of HLA-DRB1*03:01 was 52% in Caucasians, 19% in African-Americans and 17% in the Other or mixed race group (P<0.001). The frequency of DRB1*03:01 in this Caucasian population is consistent with previous reports (22, 32). The frequency of smoking was similar between patients with and without the HLA-DRB1*03:01 allele (39% vs. 39%, P=0.99, respectively).
Caucasian patients with HLA-DRB1*03:01 were more likely to have ILD (unadjusted OR=2.86, 95%CI: 1.54 – 5.32, P=0.001) and to be positive for anti-synthetase (unadjusted OR=3.99, 95%CI: 2.04 – 7.78, P<0.001) and anti-Jo-1 autoantibodies (unadjusted OR= 7.43, 95%CI: 3.25 – 17.00, P<0.001) and less likely to be positive for anti-p155/140 autoantibodies (unadjusted OR= 0.21, 95%CI: 0.08 – 0.57, P=0.002) compared to those without the allele. Cigarette smoking frequency did not differ significantly between HLA-DRB1*03:01-positive and -negative Caucasian patients (P=0.57).
To examine the combined effect of cigarette smoking and HLA-DRB1*03:01 on the frequencies of clinical and serologic phenotypes in Caucasians with PM and DM, patients were divided into four groups based on their smoking history and HLA-DRB1*03:01 allele status (Table 3). Never-smokers who were negative for HLA-DRB1*03:01 were used as the reference group (never-smokers/03:01-negative) and frequency of different clinical and serologic features were compared among patients based on their cigarette smoking history and HLA-DRB1*03:01 allele status. Multiple logistic regression analyses were performed to explore the association of different levels of smoking and HLA-DRB1*03:01 allele status with outcomes while adjusting for the effect of gender and age.
Table 3.
Adjusted logistic regression analyses assessing risks of different clinical and serologic features by ever-smoking and the presence of HLA-DRB1*03:01 alleles in Caucasian patients with polymyositis and dermatomyositis
| Anti-Jo-1 autoantibodies | |||||
|---|---|---|---|---|---|
| Ever smoking | HLA-DRB1*03:01 allele | Anti-Jo-1 negative, N (%) | Anti-Jo-1 positive, N (%) | Adjusted OR (95%CI) | P value |
| Negative | Negative | 51 (94%) | 3 (6%) | 1.00 | - |
| Positive | Negative | 26 (84%) | 5 (16%) | 4.10 (0.88 – 19.04) | 0.072 |
| Negative | Positive | 35 (60%) | 23 (40%) | 10.85 (2.98 – 39.48) | <0.001 |
| Positive | Positive | 22 (51%) | 21 (49%) | 16.13 (4.29 – 60.62) | <0.001 |
| Anti-synthetase autoantibodies (ASA) | |||||
| Ever smoking | HLA-DRB1*03:01 allele | ASA negative, N (%) | ASA positive, N (%) | Adjusted OR (95%CI) | P value |
| Negative | Negative | 46 (85%) | 8 (15%) | 1.00 | - |
| Positive | Negative | 23 (74%) | 8 (26%) | 2.65 (0.85 – 8.29) | 0.094 |
| Negative | Positive | 33 (57%) | 25 (43%) | 4.03 (1.58 – 10.24) | 0.003 |
| Positive | Positive | 20 (45%) | 24 (55%) | 7.41 (2.76 – 19.86) | <0.001 |
| Interstitial lung disease (ILD) | |||||
| Ever smoking | HLA-DRB1*03:01 allele | ILD negative, N (%) | ILD positive, N (%) | Adjusted OR (95%CI) | P value |
| Negative | Negative | 47 (80%) | 12 (20%) | 1.00 | - |
| Positive | Negative | 31 (79%) | 8 (21%) | 0.92 (0.33 – 2.56) | 0.88 |
| Negative | Positive | 40 (63%) | 23 (37%) | 2.38 (1.04 – 5.44) | 0.039 |
| Positive | Positive | 24 (50%) | 24 (50%) | 3.81 (1.61 – 9.03) | 0.002 |
| Diagnosis, polymyositis vs. dermatomyositis | |||||
| Ever smoking | HLA-DRB1*03:01 allele | Dermatomyositis, N (%) | Polymyositis, N (%) | Adjusted OR (95%CI) | P value |
| Negative | Negative | 43 (70%) | 18 (30%) | 1.00 | - |
| Positive | Negative | 18 (45%) | 22 (55%) | 2.60 (1.11 – 6.05) | 0.027 |
| Negative | Positive | 33 (52%) | 30 (48%) | 2.41 (1.13 – 5.14) | 0.023 |
| Positive | Positive | 15 (31%) | 33 (69%) | 5.24 (2.27 – 12.09) | <0.001 |
| Anti-p155/140 autoantibodies | |||||
| Ever smoking | HLA-DRB1*03:01 allele | Anti-p155/140 negative, N (%) | Anti-p155/140 positive, N (%) | Adjusted OR (95%CI) | P value |
| Negative | Negative | 34 (69%) | 15 (31%) | 1.00 | - |
| Positive | Negative | 22 (85%) | 4 (15%) | 0.37 (0.10 – 1.37) | 0.14 |
| Negative | Positive | 44 (90%) | 5 (10%) | 0.24 (0.08 – 0.74) | 0.014 |
| Positive | Positive | 39 (97.5%) | 1 (2.5%) | 0.07 (0.01 – 0.54) | 0.011 |
Odds ratio are adjusted for the impact of age and gender
Compared to never-smokers who were HLA-DRB1*03:01-negative, ever-smokers with HLA-DRB1*03:01 allele had 16 times increased odds of anti-Jo-1 autoantibody positivity (Table 3). Risks for patients with only one of these two risk factors were intermediate. A similar pattern was observed for ASA, ILD and PM (Supplementary Figure 1).
An opposite pattern was observed when evaluating anti-p155/140 autoantibody frequencies. In this case, never-smokers and smokers with the HLA-DRB1*03:01 allele had lower frequencies of anti-p155/140 autoantibodies compared to the never-smokers without the HLA-DRB1*03:01 allele (adjusted OR=0.24, 95%CI: 0.08 – 0.74 and adjusted OR=0.07, 95%CI: 0.01 – 0.54, respectively). When using an interaction term between HLA-DRB1*03:01 status and smoking history, no significant multiplicative effect was seen regarding the presence of PM, ILD, ASA, anti-Jo-1 and anti-p155/140 autoantibodies.
The same analyses were performed for African-American patients to examine the interrelationships between smoking and HLA-DRB1*03:01 status on the frequency of clinical phenotype, ILD, anti-synthetase, anti-Jo-1 and anti-p155/140 autoantibodies. The frequencies of the clinical and serologic phenotypes did not differ significantly between patients in different smoking and HLA-DRB1*03:01 categories (Supplementary Table 4).
DISCUSSION
The current study is the first to explore the influence of cigarette smoking and cumulative quantity of tobacco exposure on clinical features and autoantibody status in PM and DM across different racial groups. To our knowledge, only two previous studies have investigated the potential effect of tobacco smoking on the risk of IIM (20, 33). In one study, maternal smoking during pregnancy was correlated with the presence of juvenile DM in offspring (33). The other study proposed a gene-environment interaction between HLA-DRB1*03 and cigarette smoking on autoantibody phenotype in European PM and DM patients (20).
Our data support an association between smoking and clinical phenotype. Our results showed that tobacco smoking has a strong association with PM in Caucasians. African-Americans showed a similar trend, although it was not statistically significant after statistical correction. The relative small number of African-American patients might in part explain our inability to detect a statistically significant association.
A smoker’s lung tissue is repeatedly exposed to the potentially toxic chemicals of tobacco smoke, which contributes over time to increased risk of immune system dysregulation, inflammation and epithelial cell damage (34, 35). Based on this, we suspected a possible association between ILD and tobacco smoking in this population. We observed a statistically significant association between ever-smoking and presence of ILD in the total population. Furthermore, pack-years of cigarette smoking was significantly associated with an increased frequency of ILD in Caucasians as each one pack-year increase in smoking consumption increased the risk of ILD by two percent. These data suggest a possible dose-response relationship between cigarette smoking and increasing risk of ILD in Caucasian patients with PM and DM. Although we could not establish a statistically significant association between ever-smoking history and ILD status in the individual Caucasian or African-American racial groups in the adjusted models, we demonstrated that heavy smoking history (cigarette consumption greater than 20 pack-years) was significantly associated with an increased frequency of ILD in Caucasians compared to never-smokers after adjusting for age and gender.
Cigarette smoking has been correlated with autoantibody production as well as the development and course of many rheumatic disorders (16). Tobacco smoke contributes to citrullination and autoantigen modification in RA patients carrying certain HLA-DRB1 shared epitopes, revealing an important gene-environment interaction (14, 36). Smoking has been associated with anti-dsDNA autoantibodies in patients with systemic lupus erythematous (SLE) (37). In addition, active smoking has been linked with greater disease activity, increased organ damage and more severe cutaneous involvement in SLE (38, 39). In our study, associations between cigarette smoking and the presence of certain autoantibodies were observed in Caucasians with PM and DM. In a prior study, Chinoy et al. retrospectively assessed 557 patients with adult-onset PM or DM from four European countries and found that smokers had a higher frequency of anti-Jo-1 autoantibodies (unadjusted OR=1.83, 95%CI 1.18 – 2.83) (20). We found similar results in our US Caucasian population after correcting for the confounding impact of gender and age. In addition, we have demonstrated that Caucasian smokers were more likely to have anti-synthetase autoantibodies, but in contrast, were less likely to have anti-p155/140 autoantibodies. We also observed that pack-years of cigarette smoking was negatively associated with the frequency of anti-p155/140 autoantibodies in Caucasians, which suggests a dose-response relationship between cumulative tobacco smoking and the risk of anti-p155/140 autoantibody positivity in this population. In addition, greater than 20 pack-years of cigarette smoking was significantly associated with increased risk of PM, ILD and anti-Jo-1 autoantibodies in our Caucasian population.
Recent data describe a strong genetic link between HLA-DRB1*03:01, as part of the HLA 8.1 ancestral haplotype, with the development of IIM and the presence of anti-synthetase and anti-Jo-1 autoantibodies in Caucasians with PM and DM (8, 23, 40). To evaluate the possible interrelationship between cigarette smoking and HLA-DRB1*03:01 on the presence of clinical and serologic phenotypes of PM and DM, Caucasian patients were categorized into four groups based upon their HLA-DRB1*03:01 and smoking status. Our findings demonstrate a gradient of increasing frequency of PM clinical phenotype, ILD, anti-synthetase and anti-Jo-1 autoantibody positivity as never-smokers who were also negative for HLA-DRB1*03:01 had the lowest frequencies of PM, ILD, ASA and anti-Jo-1 autoantibodies. The frequencies of these factors increased in ever-smokers who were negative for HLA-DRB1*03:01 and increased further in never-smokers positive for HLA-DRB1*03:01 and finally the highest frequencies were observed in patients that were both positive for the HLA-DRB1*03:01 and were ever-smokers. An opposite pattern was observed in regard to the presence of anti-p155/140 autoantibodies as frequency of this autoantibody decreased in never-smokers/DRB1*03:01-positive and ever-smokers/DRB1*03:01-positive patients compared to never-smokers/DRB1*03:01-negative patients. These findings are consistent with those of Chinoy et al. who have reported a similar pattern of anti-Jo-1 autoantibody frequency for HLA-DRB1*03 and cigarette smoking interaction in a European population; however, HLA-DRB1 locus was not typed to high resolution in that study (20). These findings suggest that genetic background might alter the effect of cigarette smoking on clinical and serologic phenotypes of PM and DM in Caucasians. A similar analysis was performed in African-Americans; however, the sample size was too small to examine associations between cigarette smoking/HLA-DRB1*03:01 status and different clinical and serologic phenotypes in this population.
In the case of the autoantibody phenotypes, we discovered novel and interesting differences based on race. We observed opposite trends of association between smoking history and anti-Jo-1 and anti-synthetase autoantibodies in African-Americans and Caucasians, however, the associations in African-Americans did not reach statistical significance. In contrast to Caucasians, African-American ever-smokers had a lower frequency of anti-Jo-1 and anti-synthetase autoantibodies compared to never-smokers. Different racial and ethnic backgrounds have been associated with different genetic risk factors and phenotype frequencies in patients with IIM (21, 41, 42). The racial variations in tobacco smoking associations with autoantibody status in PM and DM patients suggest that genetic background also affects the impact of this environmental agent. Yet, whether this is attributable to largely genetic differences, or if additional environmental factors also play a role is unclear. There are ethnic differences in regard to smoking behavior, choice of cigarette brand and smoking cessation in the US population that may influence tobacco smoking effect on serologic and clinical phenotypes in PM and DM patients (43).
The strengths of our study include the relatively large sample sizes for a rare disease, inclusion of non-Caucasian patients and the availability of autoantibody and high-resolution HLA data. Some study limitations include smaller numbers of African-American patients, potential selection and referral bias, self-identification of race, detection of ILD solely by chest x-ray or pulmonary function test in a subset of patients as well as some missing data, which reduced our statistical power. Selection bias may have been introduced by excluding CAM patients which could have altered clinical phenotype and autoantibody frequencies. Nevertheless, we did not observe any changes in patterns of association or significance after adding CAM patients to the study population (data not shown). Another potential source of bias in the present study is the impact of factors, such as socioeconomic status and geographic location that were not available for analysis and may potentially influence smoking and outcome frequencies.
The current study suggests that in a US-based population, tobacco smoking is associated with an increased likelihood of developing PM, anti-Jo-1 autoantibodies and anti-synthetase autoantibodies, as well as a decreased likelihood of developing anti-p155/140 autoantibodies, in Caucasian PM and DM patients. Our study also provides further evidence for possible gene-environment interactions between HLA-DRB1*03:01 and cigarette smoking in predicting the presence of PM clinical phenotype, ILD, anti-synthetase, anti-Jo-1 and anti-p155/140 autoantibodies in Caucasians. Thus, both HLA genetics and racial background might modulate the impact of cigarette smoking on PM and DM patients. Future prospective clinical and genetic studies, especially in larger cohorts of African-American and Hispanic patients with genetic ancestry markers will provide more through descriptions of inter-ethnic differences in disease susceptibility as influenced by cigarette smoking history. Studies are warranted to examine possible biologic mechanisms underlying the influence of cigarette smoke on immune system dysregulation and autoantibody formation in the IIM.
Supplementary Material
Acknowledgments
Funding: This research was supported in part by the Intramural Research Programs of the NIH, National Institute of Environmental Health Sciences, National Institute of Arthritis and Musculoskeletal and Skin Diseases and NIH Clinical Center (project ID Z99 CL999999)
We thank Drs. Michael Ward, Peter Grayson and Laura Lewandowski for critical comments on the manuscript. The authors thank the patients who participated in this study, the co-investigators for their contributions to the study, staff at the participating centers and the following contributors for providing support: Natalie Smith, John McGrath, Paul Cacioppo, Anna Jansen, Christine Johnson and Beverly Sellers-Robinson, for assistance in coordination and execution of the study.
Footnotes
Contributors: AS and FWM were involved in the conception and design of the study. All were involved in data acquisition. AS, SFK, WAF, SDA, FWM were involved in data analysis and interpretation. AS, SFK and FWM were involved in manuscript drafting. All were involved in critically revising the manuscript and approved the final version.
Ethics approval: The study was approved by the National Institutes of Health Institutional Review Boards.
Disclaimer: The views expressed in this article are the authors own and are not an official position of the institution or funder.
Competing interests: Ira Targoff reports grants and non-financial support from Oklahoma Medical Research Foundation during the conduct of the study. Rohit Aggarwal reports personal fees from BMS, personal fees from Novartis, personal fees from Octapharma, personal fees from Mallinckrodt, grants from BMS, grants from Mallinckrodt, grants from Pfizer and grants from Momenta outside the submitted work. Lisa Christopher-Stine reports personal fees from Novartis, personal fees from Option Care, personal fees from Idera, grants from CSL Behring, personal fees from Mallinckrodt, and other from Inova Diagnostics outside the submitted work. In addition, Lisa Christopher-Stine has a patent on Detection of HMGCoA reductase (HMGCR) Antibodies in Patient Sera with royalties paid outside the submitted work. Paul Dellaripa reports non-financial support from Boehringer Ingelheim, other from UpToDate and other from Genentech outside the submitted work. The other authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Miller FW. Idiopathic Inflammatory Myopathies. In: Nabel E, editor. American College of Physicians ACP Medicine online. New York: WebMD Professional Publishing; 2013. [Google Scholar]
- 2.Bernatsky S, Joseph L, Pineau CA, Belisle P, Boivin JF, Banerjee D, et al. Estimating the prevalence of polymyositis and dermatomyositis from administrative data: age, sex and regional differences. Ann Rheum Dis. 2009;68:1192–6. doi: 10.1136/ard.2008.093161. [DOI] [PubMed] [Google Scholar]
- 3.Smoyer-Tomic KE, Amato AA, Fernandes AW. Incidence and prevalence of idiopathic inflammatory myopathies among commercially insured, Medicare supplemental insured, and Medicaid enrolled populations: an administrative claims analysis. BMC Musculoskelet Disord. 2012;13:103. doi: 10.1186/1471-2474-13-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furst DE, Amato AA, Iorga SR, Gajria K, Fernandes AW. Epidemiology of adult idiopathic inflammatory myopathies in a U.S. managed care plan. Muscle Nerve. 2012;45:676–83. doi: 10.1002/mus.23302. [DOI] [PubMed] [Google Scholar]
- 5.Tansley SL, McHugh NJ. Myositis specific and associated autoantibodies in the diagnosis and management of juvenile and adult idiopathic inflammatory myopathies. Curr Rheumatol Rep. 2014;16:464. doi: 10.1007/s11926-014-0464-1. [DOI] [PubMed] [Google Scholar]
- 6.Mammen AL. Autoimmune myopathies: autoantibodies, phenotypes and pathogenesis. Nat Rev Neurol. 2011;7:343–54. doi: 10.1038/nrneurol.2011.63. [DOI] [PubMed] [Google Scholar]
- 7.Haq SA, Tournadre A. Idiopathic inflammatory myopathies: from immunopathogenesis to new therapeutic targets. Int J Rheum Dis. 2015;18:818–25. doi: 10.1111/1756-185X.12736. [DOI] [PubMed] [Google Scholar]
- 8.O’Hanlon TP, Carrick DM, Arnett FC, Reveille JD, Carrington M, Gao X, et al. Immunogenetic risk and protective factors for the idiopathic inflammatory myopathies: distinct HLA-A, -B, -Cw, -DRB1 and -DQA1 allelic profiles and motifs define clinicopathologic groups in caucasians. Medicine (Baltimore) 2005;84:338–49. doi: 10.1097/01.md.0000189818.63141.8c. [DOI] [PubMed] [Google Scholar]
- 9.O’Hanlon TP, Rider LG, Mamyrova G, Targoff IN, Arnett FC, Reveille JD, et al. HLA polymorphisms in African Americans with idiopathic inflammatory myopathy: allelic profiles distinguish patients with different clinical phenotypes and myositis autoantibodies. Arthritis Rheum. 2006;54:3670–81. doi: 10.1002/art.22205. [DOI] [PubMed] [Google Scholar]
- 10.Love LA, Weinberg CR, McConnaughey DR, Oddis CV, Medsger TA, Jr, Reveille JD, et al. Ultraviolet radiation intensity predicts the relative distribution of dermatomyositis and anti-Mi-2 autoantibodies in women. Arthritis Rheum. 2009;60:2499–504. doi: 10.1002/art.24702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rider LG, Wu L, Mamyrova G, Targoff IN, Miller FW. Environmental factors preceding illness onset differ in phenotypes of the juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford) 2010;49:2381–90. doi: 10.1093/rheumatology/keq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baka Z, Buzas E, Nagy G. Rheumatoid arthritis and smoking: putting the pieces together. Arthritis Res Ther. 2009;11:238. doi: 10.1186/ar2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krassas GE, Wiersinga W. Smoking and autoimmune thyroid disease: the plot thickens. Eur J Endocrinol. 2006;154:777–80. doi: 10.1530/eje.1.02157. [DOI] [PubMed] [Google Scholar]
- 14.Perricone C, Versini M, Ben-Ami D, Gertel S, Watad A, Segel MJ, et al. Smoke and autoimmunity: The fire behind the disease. Autoimmun Rev. 2016;15:354–74. doi: 10.1016/j.autrev.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Lakatos PL. Environmental factors affecting inflammatory bowel disease: have we made progress? Dig Dis. 2009;27:215–25. doi: 10.1159/000228553. [DOI] [PubMed] [Google Scholar]
- 16.Miller FW. Non-infectious Environmental Agents and Autoimmunity. In: Rose N, Mackay I, editors. The Autoimmune Diseases. 5. Boston: Academic Press; 2014. pp. 283–95. [Google Scholar]
- 17.Belin RM, Astor BC, Powe NR, Ladenson PW. Smoke exposure is associated with a lower prevalence of serum thyroid autoantibodies and thyrotropin concentration elevation and a higher prevalence of mild thyrotropin concentration suppression in the third National Health and Nutrition Examination Survey (NHANES III) J Clin Endocrinol Metab. 2004;89:6077–86. doi: 10.1210/jc.2004-0431. [DOI] [PubMed] [Google Scholar]
- 18.Vestergaard P, Rejnmark L, Weeke J, Hoeck HC, Nielsen HK, Rungby J, et al. Smoking as a risk factor for Graves’ disease, toxic nodular goiter, and autoimmune hypothyroidism. Thyroid. 2002;12:69–75. doi: 10.1089/105072502753451995. [DOI] [PubMed] [Google Scholar]
- 19.Mahid SS, Minor KS, Soto RE, Hornung CA, Galandiuk S. Smoking and inflammatory bowel disease: a meta-analysis. Mayo Clin Proc. 2006;81:1462–71. doi: 10.4065/81.11.1462. [DOI] [PubMed] [Google Scholar]
- 20.Chinoy H, Adimulam S, Marriage F, New P, Vincze M, Zilahi E, et al. Interaction of HLA-DRB1*03 and smoking for the development of anti-Jo-1 antibodies in adult idiopathic inflammatory myopathies: a European-wide case study. Ann Rheum Dis. 2012;71:961–5. doi: 10.1136/annrheumdis-2011-200182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Hanlon TP, Miller FW. Genetic risk and protective factors for the idiopathic inflammatory myopathies. Curr Rheumatol Rep. 2009;11:287–94. doi: 10.1007/s11926-009-0040-2. [DOI] [PubMed] [Google Scholar]
- 22.O’Hanlon TP, Carrick DM, Targoff IN, Arnett FC, Reveille JD, Carrington M, et al. Immunogenetic risk and protective factors for the idiopathic inflammatory myopathies: distinct HLA-A, -B, -Cw, -DRB1, and -DQA1 allelic profiles distinguish European American patients with different myositis autoantibodies. Medicine (Baltimore) 2006;85:111–27. doi: 10.1097/01.md.0000217525.82287.eb. [DOI] [PubMed] [Google Scholar]
- 23.Miller FW, Chen W, O’Hanlon TP, Cooper RG, Vencovsky J, Rider LG, et al. Genome-wide association study identifies HLA 8.1 ancestral haplotype alleles as major genetic risk factors for myositis phenotypes. Genes Immun. 2015;16:470–80. doi: 10.1038/gene.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones MR, Tellez-Plaza M, Navas-Acien A. Smoking, menthol cigarettes and all-cause, cancer and cardiovascular mortality: evidence from the National Health and Nutrition Examination Survey (NHANES) and a meta-analysis. PLoS One. 2013;8:e77941. doi: 10.1371/journal.pone.0077941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts) N Engl J Med. 1975;292:344–7. doi: 10.1056/nejm197502132920706. [DOI] [PubMed] [Google Scholar]
- 26.Neuberger JS, Lai SM. Cigarette Consumption and Cigarette Smoking Prevalence Among Adults in Kansas. Prev Chronic Dis. 2015;12:E93. doi: 10.5888/pcd12.150011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westhoff G, Rau R, Zink A. Rheumatoid arthritis patients who smoke have a higher need for DMARDs and feel worse, but they do not have more joint damage than non-smokers of the same serological group. Rheumatology (Oxford) 2008;47:849–54. doi: 10.1093/rheumatology/ken057. [DOI] [PubMed] [Google Scholar]
- 28.Targoff IN, Mamyrova G, Trieu EP, Perurena O, Koneru B, O’Hanlon TP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006;54:3682–9. doi: 10.1002/art.22164. [DOI] [PubMed] [Google Scholar]
- 29.Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 2004;50:209–15. doi: 10.1002/art.11484. [DOI] [PubMed] [Google Scholar]
- 30.Arons E, Adams S, Venzon DJ, Pastan I, Kreitman RJ. Class II human leucocyte antigen DRB1*11 in hairy cell leukaemia patients with and without haemolytic uraemic syndrome. Br J Haematol. 2014;166:729–38. doi: 10.1111/bjh.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer A, Meyer N, Schaeffer M, Gottenberg JE, Geny B, Sibilia J. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology (Oxford) 2015;54:50–63. doi: 10.1093/rheumatology/keu289. [DOI] [PubMed] [Google Scholar]
- 32.Arnett FC, Targoff IN, Mimori T, Goldstein R, Warner NB, Reveille JD. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum. 1996;39:1507–18. doi: 10.1002/art.1780390910. [DOI] [PubMed] [Google Scholar]
- 33.Orione MA, Silva CA, Sallum AM, Campos LM, Omori CH, Braga AL, et al. Risk factors for juvenile dermatomyositis: exposure to tobacco and air pollutants during pregnancy. Arthritis Care Res (Hoboken) 2014;66:1571–5. doi: 10.1002/acr.22358. [DOI] [PubMed] [Google Scholar]
- 34.Goncalves RB, Coletta RD, Silverio KG, Benevides L, Casati MZ, da Silva JS, et al. Impact of smoking on inflammation: overview of molecular mechanisms. Inflamm Res. 2011;60:409–24. doi: 10.1007/s00011-011-0308-7. [DOI] [PubMed] [Google Scholar]
- 35.Margaritopoulos GA, Vasarmidi E, Jacob J, Wells AU, Antoniou KM. Smoking and interstitial lung diseases. Eur Respir Rev. 2015;24:428–35. doi: 10.1183/16000617.0050-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conigliaro P, Chimenti MS, Triggianese P, Sunzini F, Novelli L, Perricone C, et al. Autoantibodies in inflammatory arthritis. Autoimmun Rev. 2016;15:673–83. doi: 10.1016/j.autrev.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 37.Freemer MM, King TE, Jr, Criswell LA. Association of smoking with dsDNA autoantibody production in systemic lupus erythematosus. Ann Rheum Dis. 2006;65:581–4. doi: 10.1136/ard.2005.039438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turchin I, Bernatsky S, Clarke AE, St-Pierre Y, Pineau CA. Cigarette smoking and cutaneous damage in systemic lupus erythematosus. J Rheumatol. 2009;36:2691–3. doi: 10.3899/jrheum.090403. [DOI] [PubMed] [Google Scholar]
- 39.Ghaussy NO, Sibbitt W, Jr, Bankhurst AD, Qualls CR. Cigarette smoking and disease activity in systemic lupus erythematosus. J Rheumatol. 2003;30:1215–21. [PubMed] [Google Scholar]
- 40.Rothwell S, Cooper RG, Lundberg IE, Miller FW, Gregersen PK, Bowes J, et al. Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann Rheum Dis. 2016;75:1558–66. doi: 10.1136/annrheumdis-2015-208119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reed AM, Ytterberg SR. Genetic and environmental risk factors for idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 2002;28:891–916. doi: 10.1016/s0889-857x(02)00029-7. [DOI] [PubMed] [Google Scholar]
- 42.Schiopu E, Phillips K, MacDonald PM, Crofford LJ, Somers EC. Predictors of survival in a cohort of patients with polymyositis and dermatomyositis: effect of corticosteroids, methotrexate and azathioprine. Arthritis Res Ther. 2012;14:R22. doi: 10.1186/ar3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trinidad DR, Perez-Stable EJ, White MM, Emery SL, Messer K. A nationwide analysis of US racial/ethnic disparities in smoking behaviors, smoking cessation, and cessation-related factors. Am J Public Health. 2011;101:699–706. doi: 10.2105/ajph.2010.191668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.