

Abstract
Local drug delivery to the ear has gained wide clinical acceptance, with the choice of drug and application protocol in humans largely empirically-derived. Here, we review the pharmacokinetics underlying local therapy of the ear using the drugs commonly used in clinical practice as examples. Based on molecular properties and perilymph measurements interpreted through computer simulations we now better understand the principles underlying entry and distribution of these and other drugs in the ear. From our analysis, we have determined that dexamethasone-phosphate, a pro-drug widely-used clinically, has molecular and pharmacokinetic properties that make it ill-suited for use as a local therapy for hearing disorders. This polar form of dexamethasone, used as a more soluble agent in intravenous preparations, passes less readily through lipid membranes, such as those of the epithelia restricting entry at the round window membrane and stapes. Once within the inner ear, dexamethasone-phosphate is cleaved to the active form, dexamethasone, which is less polar, passes more readily through lipid membranes of the blood-perilymph barrier and is rapidly eliminated from perilymph without distributing to apical cochlear regions. Dexamethasone-phosphate therefore provides only a brief exposure of the basal regions of the cochlea to active drug. Other steroids, such as triamcinolone-acetonide, exhibit pharmacokinetic properties more appropriate to the ear and merit more detailed consideration.
Keywords: Intracochlear, intralabyrinthine, perilymph, round window membrane, oval window, drug delivery, inner ear
1. Introduction
It has been known for years that injecting a drug solution through the tympanic membrane into the middle ear allows the drug to reach and influence function of the inner ear (Ersner et al., 1951; Schuknecht, 1956). The field of local drug delivery to the ear took on greater relevance when in the mid-1990’s local delivery of gentamicin became a widely-accepted clinical therapy for the treatment of Meniere’s disease (Lange 1989; Nedzelski et al., 1993; Toth & Parnes, 1995). Since that time, we have learned that the pharmacokinetics of the inner ear with locally-applied drugs is rather complex, involving the interaction of multiple elements. Some share similarities with other systems of the body, such as entry from the vasculature which has some similarities to that in the eye or the brain. Others, such as passage through the round window membrane, distribution through the different fluid and tissue compartments of the ear, and fluid exchange across the cochlear aqueduct, are unique to the ear. Here we review what we know so far about inner ear pharmacokinetics with a primary emphasis on drugs currently used in clinical practice.
The ear consists of a number of interconnected compartments that an applied drug can access.
1) Middle ear
The middle ear is normally gas-filled but becomes a fluid-filled space communicating with perilymph when drug solution is applied there. The middle ear is lined with epithelium that on the ventral surface, leading to the Eustachian tube, is of endodermal origin and is densely ciliated. In contrast, dorsal surfaces of the epithelium and regions in the vicinity of the round window membrane and stapes are of neural crest origins and are not ciliated (Thompson & Tucker 2013). The epithelium is both highly vascularized and includes lymphatic drainage to the retroauricular and junctional lymph nodes (Lim & Hussl; 1975). Fluid and/or drug loss through the Eustachian tube, via the vasculature and via the lymphatics can all contribute to the decline of middle ear concentration with time after drug application, as can fluid or mucus secretion by the epithelium. An initial breakdown (metabolism) of drugs in the middle ear also likely occurs but only limited quantitative data are yet available. The primary function of the middle ear epithelium is to maintain the normal gas-filled state and removal of applied drug solutions by these multiple processes occurs as a result of that specialization.
2) Inner Ear
The inner ear comprises prominent fluid spaces containing endolymph or perilymph, but drugs entering the inner ear do not remain confined to just the fluid spaces. Most of the adjacent tissue spaces are not bounded by tissues with tight junctions so drugs rapidly equilibrate with the extracellular spaces of the spiral ligament, the organ of Corti, the spiral ganglion and of the auditory and vestibular nerves. Depending on permeability properties, drugs may enter the intracellular compartments of these tissues or become membrane-bound if lipophilic. Distribution between endolymph and perilymph depends on where the drug enters the ear, whether by systemic or local application, and whether the drug can pass through the tight, cellular endolymph-perilymph barrier. In the cochlea, distribution of charged molecules between endolymph and perilymph is also influenced by the endocochlear potential. Fluid spaces in the bone of the otic capsule also interact with perilymph, with incomplete bone-lining cells (Chole & Tinling, 1994) and a lacuno-canalicular system in the bone in open fluid communication with perilymph (Zehnder et al., 2005).
3) Cranium
Perilymph is in open fluid communication with cerebrospinal fluid (CSF). The endolymphatic sac also contacts the dura mater in the posterior fossa. These communications raise the possibility that substances applied to perilymph may gain access to the brain. In rodents, where the cochlear aqueduct is relatively large, passage of drugs through the aqueduct is largely mitigated by the high rate of CSF turnover. While the CSF may be providing a sink to which perilymphatic drugs are lost (Salt et al., 2015), drug accumulation in CSF is generally low. Although in humans the aqueduct is longer and narrower, there are instances of hearing loss after intrathecal administration of ototoxic drug (Maarup et al, 2015). The passage of drugs from the ear to the brain via the auditory and vestibular nerves has also been proposed (Praetorius et al. 2007, Zhang et al., 2012).
4) Vasculature
For the inner ear the vasculature represents a large sink to which drugs can be lost (or gained following systemic applications), impeded by the tight blood labyrinth barriers. This includes the blood-perilymph and blood-strial barriers which may have different characteristics. Each of the tissues of the middle and inner ear, including the bone of the otic capsule, has an associated vasculature that may contribute to the overall pharmacokinetics of the inner ear. It should also be borne in mind that any barrier is only as good as its weakest segment, with pharmacokinetics potentially influenced by local defects in the barrier.
A schematic of the main processes and compartments underlying inner ear pharmacokinetics with intratympanic drug applications is shown in Figure 1. The figure shows a drug-containing formulation injected through the tympanic membrane into the middle ear cavity. Drug enters the inner ear through multiple pathways, including through the round window (RW) membrane and the stapes (King et al., 2011, Salt et al., 2012a). Drug is lost from the middle ear by multiple mechanisms, as discussed above. As the drug enters perilymph it initially distributes throughout the fluid and tissue spaces of basal turn and vestibule, with spread along the scalae towards the cochlear apex occurring more slowly. In the basal turn of ST, drug levels are diluted by CSF, either entering through the cochlear aqueduct as a volume flow, or as a CSF-perilymph exchange caused by pressure-induced fluid oscillations across the CA. Drug can be lost from the inner ear fluids by multiple processes, including uptake, buffering or binding to cellular or non-cellular components of the ear, or through elimination by the vasculature across the blood-labyrinth barriers.
Figure 1.
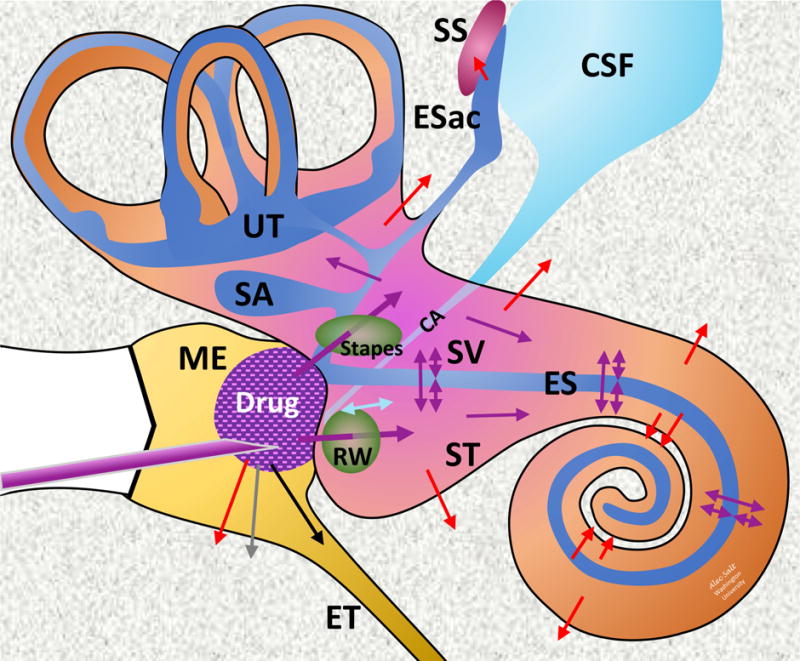
Schematic of drug applied intratympanically to the inner ear. Colored arrows indicate movements of drug; Purple: Distribution; Red: Elimination to blood; Cyan: CSF-Perilymph fluid exchange; Gray: Elimination to lymphatics; Black: Elimination via the Eustachian tube. Abbreviations are: CSF: Cerebrospinal Fluid; CA: Cochlear aqueduct; Esac: Endolymphatic Sac; ES: Endolymphatic Space; ET: Eustachian tube; ME: Middle Ear; RW: Round Window; SA: Saccule; SS: Sigmoid Sinus; ST: Scala Tympani; SV: Scala Vestibuli; UT: Utricle.
2. Pharmacokinetics of the ear
Pharmacokinetics is the science of drug movements in the body, including absorption, distribution, metabolism, and excretion. In practice, however, all the applied drug may not be available so factors related to drug liberation are usually also considered, leading to the acronym LADME: Liberation, Absorption, Distribution, Metabolism and Elimination. In 2009, we first applied this concept to the ear (Salt & Plontke, 2009), providing a structured framework for systematically studying, interpreting, and computer modeling drug movements in the ear.
L: Liberation is used to represent the release of drug from its pharmaceutical or dosage form into the middle or inner ear spaces. This encompasses controlled-release formulations applied to the middle ear or inner ear spaces including polymers (solid or gels), nanoparticles, liposomes, particulate suspensions, etc. It includes fluid injections into the middle ear by implanted osmotic or digital pumps or solutions delivered directly into perilymph by volume injection or oscillation of fluid across an application cannula. It includes delivery from implanted devices specifically for controlled drug release or implanted auditory prostheses with drug delivery cannulas and delivery from electrode carriers through drug-filled compartments, drug-eluting coatings or drug incorporated into the electrode carrier itself. Liberation can also include in situ drug generation as a result of gene or cell based therapy where specific cells are modified to synthesize a therapeutic substance. Various articles have reviewed the approaches with different drug delivery systems (Swan et al., 2008; Ayoob et al. 2015; Gillespie et al. 2014; Hendricks et al. 2008; Nakagawa et al. 2011; Nguyen et al. 2017; Plontke et al. 2017; Pyykkö et al. 2011; Roemer et al. 2017; Senn et al. 2017).
Delivery protocols play a prominent role in influencing liberation, including drug formulations, application procedures and devices, injection pump rates, elution rates etc. These are the main aspects of local drug delivery that are under the control of the experimenter or physician.
A: Absorption at the body level is the process by which the drug reaches the bloodstream from the site of administration, affected by the bioavailability of the drug and the ability to pass through cellular boundaries, such as the gut for oral delivery or the endothelial cells of the capillaries for intramuscular injections. For the ear, we used absorption to represent the passage of drug into the inner ear from an application site outside the inner ear. This is mostly applicable to drugs applied to the middle ear. Pathways from the middle ear include the RW membrane, stapes and in some animals through areas of the otic capsule where the bone is thin (King et al., 2011, Salt et al., 2012a, Mikulec et al. 2009). This inner-ear-focused absorption can therefore also represent drug passage following systemic application from the vascular system to the inner ear through the blood-labyrinth barriers. In contrast, movement from the CSF to the inner ear following intracerebral or intrathecal applications would not be included in absorption as this this does not occur across a cellular boundary.
D: Distribution for the body is the process by which drug diffuses or passes from the systemic circulation into interstitial and intracellular fluids. The distribution of a drug between tissues depends on various factors such as vascular permeability, blood flow, binding of drug to tissue and plasma proteins, its lipid solubility and other factors. For the ear, we use distribution to represent the multitude of processes whereby drugs spread through the fluid and tissue spaces of the inner ear and to adjacent compartments. This differs considerably from the whole-body distribution phenomenon, primarily because the blood is highly stirred so drug distribution in the systemic circulation can be assumed to be homogenous. In contrast, the inner ear fluids are not stirred so substantial drug gradients commonly occur between different regions. Distribution in the ear is dominated by the spread in open fluid spaces containing endolymph and perilymph, primarily by diffusion but also influenced by volume flow of the fluids if present. Volume flow can be extremely slow in the normal, sealed cochlear state but can also be rapid when any part of the inner ear is perforated. Distribution in the ear also includes spread from fluid spaces to extracellular spaces of the tissue compartments (the organ of Corti, spiral ligament, spiral ganglion, nerves, etc.) which occurs readily when boundary cell layers are incomplete or lack tight junctions. Distribution also includes spread into the intracellular spaces of those tissues across cell membranes, together with buffering and binding to extracellular structures (e.g. basilar membrane and tectorial membrane), to intracellular proteins and to lipophilic components to lipid membranes. Distribution also includes the redistribution processes from tissues initially exposed to higher drug concentrations as perilymph drug levels decline with time. Distribution therefore represents all forms of drug spread within the inner ear.
M: Metabolism, also called biotransformation, is the chemical conversion of drugs. For the body, this commonly occurs in the liver where substances are converted to more hydrophilic compounds which are more readily eliminated by the kidneys and by other routes. For the ear, metabolism of drugs can be of great importance to their distribution and action, as we consider extensively below. For dexamethasone-phosphate and triamcinolone-acetonide, metabolism in the ear has great influence. The metabolite may have different affinity for the receptor site and different physical properties that influence permeability properties through cell layers, thus influencing, for example, the rate of elimination. Passage across the RW membrane and stapes, and elimination to the vasculature can be highly influenced by whether drug metabolism occurs in the middle or inner ear, and how fast metabolism occurs.
E: Elimination at the body level is the removal of unchanged drug or metabolite from the body via renal, biliary, or pulmonary processes. For the ear, we use elimination to represent losses of drug from the ear at multiple levels. From the middle ear, it can represent drug losses to the pharynx (Eustachian tube), to vascular system, to the lymphatics. From inner ear, it includes losses to the vascular system, to CSF (fluid oscillation across CA, or even superior canal dehiscence), and to the middle ear (perilymph leaks through round or oval window fistulas). Losses of drug from the inner ear to the middle ear would generally be characterized as elimination, rather than negative or reverse absorption.
3. Molecular Properties of Drugs used in the Ear
At present, only a small number of drugs are widely used in routine clinical practice for therapy of the ear. The aminoglycoside gentamicin is used to treat Meniere’s disease (Lange 1989; Nedzelski et al., 1993) and corticosteroids, mainly dexamethasone and methylprednisolone, are used to treat Meniere’s disease, idiopathic sudden sensorineural hearing loss and other forms of acute hearing loss (Hamid & Trune, 2008). The different forms of these molecules that are used for therapy of the ear are illustrated in Figure 2. For ionized forms, the formula weights given do not include the counter ion. The drug molecule dissociates from the counter-ions in aqueous solution so mobility and transport characteristics are not influenced by them. The differences in chemical structure, such as between the larger dexamethasone-phosphate compared to dexamethasone, confer different physical properties which have a substantial impact on aqueous solubility (given in the figure) and on inner ear pharmacokinetics.
Figure 2.
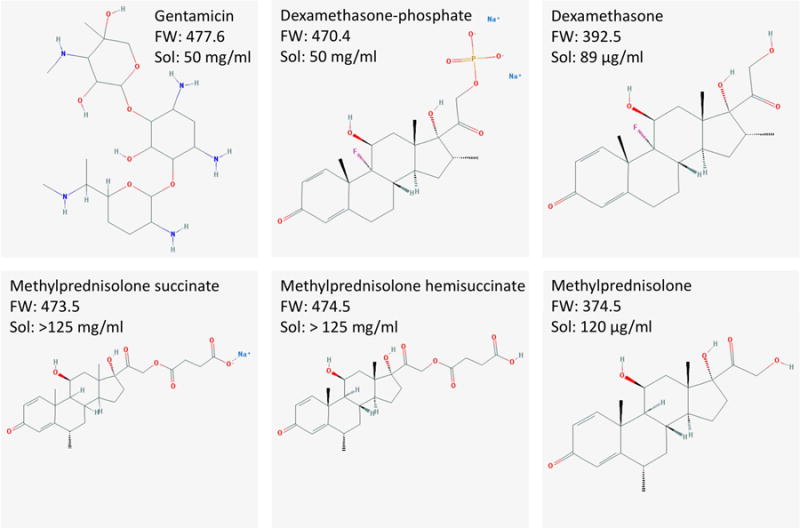
Drugs in common use for intratympanic therapy of the inner ear. Formula weights (FW) given do not include counter ions from which they dissociate in solution. “Sol” indicates the solubility in aqueous solution. Formulae courtesy of the Pubchem website (https://pubchem.ncbi.nlm.nih.gov).
Gentamicin is highly soluble in aqueous solution and is typically available as a 40 mg/ml solution, intended for intravenous (IV) use but used off-label as an intratympanic injection. The pH of the solution is quite low (~4.5) so some groups, primarily in the USA, mix the preparation with sodium bicarbonate solution (Nedzelski et al, 1993) to bring the pH closer to the physiological value. This also results in a lower applied concentration (26.7 mg/ml).
Dexamethasone is typically applied intratympanically as a 4 to 12 mg/ml (sometimes 24 mg/ml) solution of dexamethasone-phosphate (disodium salt) formulated for IV use, and used off-label. Dexamethasone-phosphate is a pro-drug and is inactive until phosphatases cleave the polar phosphate group, converting to the active base form, dexamethasone. Dexamethasone-phosphate is larger (FW 470.4), more polar and substantially more soluble than the base form of dexamethasone (FW 392.5). The less-soluble form has been used as a suspension in Poloxamer gel to provide a more prolonged delivery in animals (Wang et al. 2011) and in humans (Lambert et al. 2016). The base form of methylprednisolone is also poorly soluble in aqueous solution and is typically administered as a more soluble form, either as methylprednisolone succinate or methylprednisolone hemisuccinate (Solu-Medrol). It is used in concentrations from 20 to 125 mg/ml (most commonly 40 mg/ml), again formulated for IV or intramuscular use and used off label. Some formulations of methylprednisonone include ~8.8 mg/ml benzyl alcohol as a preservative, a concentration close to that which has been shown to increase RW membrane permeability (Mikulec et al., 2008).
The ability of molecules to pass passively through biological membranes has been shown to depend strongly on the lipid solubility and on the polar properties of the molecule (Egan et al., 2000). The SwissADME website (http://http://www.swissadme.ch) enables calculation of a number of physical properties for any defined molecule using uniform methodology. Two important parameters are WLOGP, an estimate of the lipid partition coefficient for the molecule, and TPSA/A2, a measure of the topological polar surface area of the molecule. TPSA/A2 takes into account the extent and presence of polar, charged groups (dependent on the size of the molecule) even when the overall molecule is uncharged. Molecules that are lipid soluble (high WLOPG value), small and non-polar (low TPSA/A2) tend to pass through membranes more readily than those that are hydrophilic (low WLOGP), large and polar (high TPSA/A2). Plotting these two variables against each other gives a graphic representation of the passive permeability properties of the molecule (Daina & Zoete, 2016) as shown in Figure 3. The molecular properties of the drugs commonly used for inner ear therapy have been calculated and are shown on the plots. The yellow ellipse (“egg yolk”) on the plot encompasses the parameter range that typically allows a drug to pass through the blood-brain barrier. The ellipse was derived from analysis of a dataset describing the entry of 260 molecules into the human brain. The white ellipse (“egg white”) similarly represents the range of parameters for molecules that permeate the human gut, derived from a dataset describing the entry of 600 molecules in humans (Daina & Zoete, 2016).
Figure 3.
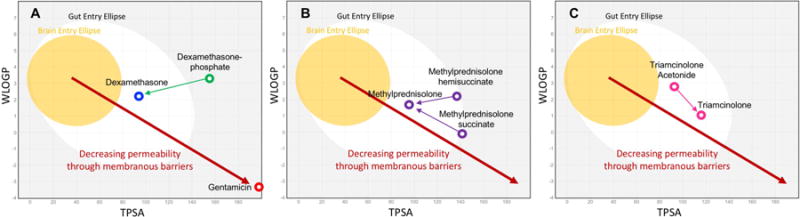
Membrane permeation-related characteristics, WLOGP (lipid solubility, Y-axis) and TPSA (topological polar surface area; X-axis) for a number of drugs calculated by the SwissADME website (http://http://www.swissadme.ch). The yellow ellipse bounds the statistical range for molecules that pass through the blood-brain barrier and the white ellipse bounds the range for molecules that permeate the gut (Daina & Zoete, 2016; Daina et al., 2017). Based on this analysis, dexamethasone and methylprednisolone would be expected to permeate membranes more readily than dexamethasone-phosphate, methylprednisolone-hemisuccinate, and methylprednisolone-succinate. Gentamicin would be expected to be substantially less permeable than all forms of the steroids.
Figure 3A shows the base form of dexamethasone lies substantially to the left of the more polar form, dexamethasone-phosphate, suggesting it would be more permeable through membranous barriers. Gentamicin, in contrast is highly polar and hydrophilic. It lies at the lower right side of the plot, suggesting gentamicin would less readily pass through cellular boundaries. Figures 3B and 3C show similar calculations for the different forms of methylprednisolone and triamcinolone respectively. These calculations show that it is critically important to know the exact form of the drug being used, as small differences in molecular configuration influence polarity and lipophilicity, therefore altering the pharmacokinetic properties of the molecule.
The same physical parameters also play a major role in the aqueous solubility of different drugs. Small, nonpolar lipophilic drugs, such as the base forms of dexamethasone and methylprednisolone, are relatively insoluble in water, as seen by comparing solubility data in Figure 2 with properties plotted in Figure 3. Adding polar groups to the molecule, such as the phosphate or succinate groups for dexamethasone and methylprednisolone respectively, greatly increase aqueous solubility by increasing TPSA/A2. The use of these polar, more soluble forms of steroids was a successful strategy to increase the total drug amount delivered in IV formulations. When given intravenously the soluble formulation is rapidly dispersed by blood flow before the polar groups are cleaved in tissues such as the liver, forming the less-soluble active molecule. Unfortunately, there has been little consideration of how such molecular transformations influence the pharmacokinetics of the drug when used with local applications to the ear.
It should be noted that permeability characteristics inferred from molecular properties all relate to passive movements of small molecules across membranous boundaries. Many small nonpolar lipophilic substances such as steroids which gain access to the brain are actively transported out (Karssen et al., 2001; Löscher & Potschka 2005). Such transport, superimposed on passive movements, can potentially influence and even dominate pharmacokinetics for the substance. Active transport of drugs across the boundaries of the ear, however, has not yet been demonstrated.
4. Middle Ear Kinetics
When drug solution is injected into the middle ear it does not remain “undisturbed”, waiting for the dissolved drug to diffuse into the inner ear. As discussed earlier, the middle ear has a number of powerful mechanisms to remove fluids and drugs. For intratympanic applications in humans, the patient lies in a supine position for 20 – 30 min with the head orientated to keep the Eustachian tube uppermost, so that drug solution applied to the RW niche does not immediately drain towards the Eustachian tube and be swallowed. Also, depending on middle ear and mastoid anatomy (e.g. pneumatization and mucosal state) fluid can easily be disseminated through the mastoid cells or diluted by fluid in the middle ear.
Middle ear drug kinetics has not been extensively studied but even in anesthetized recumbent animals applied substances are rapidly lost from the middle ear. The concentration time course of drug in the middle ear, the so-called “residence time” of the drug critically influences the perilymph drug level achieved with intratympanic applications (Salt & Plontke, 2009.). Mikulec et al., (2009) reported the decline time course of the marker TMPA measured in real time in the RW niche with ion-specific microelectrodes. Within just 30 min, concentration of marker solution in the niche fell to 52% of that initially-measured. Figure 4 shows the normalized decline of middle ear concentration measured for gentamicin (Salt et al., 2016) and for dexamethasone-phosphate in our studies. In both cases, solutions were applied to the round window niche in experiments where perilymph was collected after a 1 hr application period. After perilymph collection, the drug solution remaining in the RW niche was sampled. For gentamicin, a substance that is retained well in the inner ear, the middle ear concentration was found to fall to an average of 46% after 83 min (Salt et al, 2016). For dexamethasone-phosphate the decline in concentration was even more rapid, falling to 10% of that applied at 93 min after application (Salt et al., 2018). It is notable that gentamicin was lost more slowly than dexamethasone-phosphate, in accordance with its molecular properties calculated in Figure 3. These studies show that applied drugs are eliminated rather rapidly from the middle ear when applied as a solution.
Figure 4.
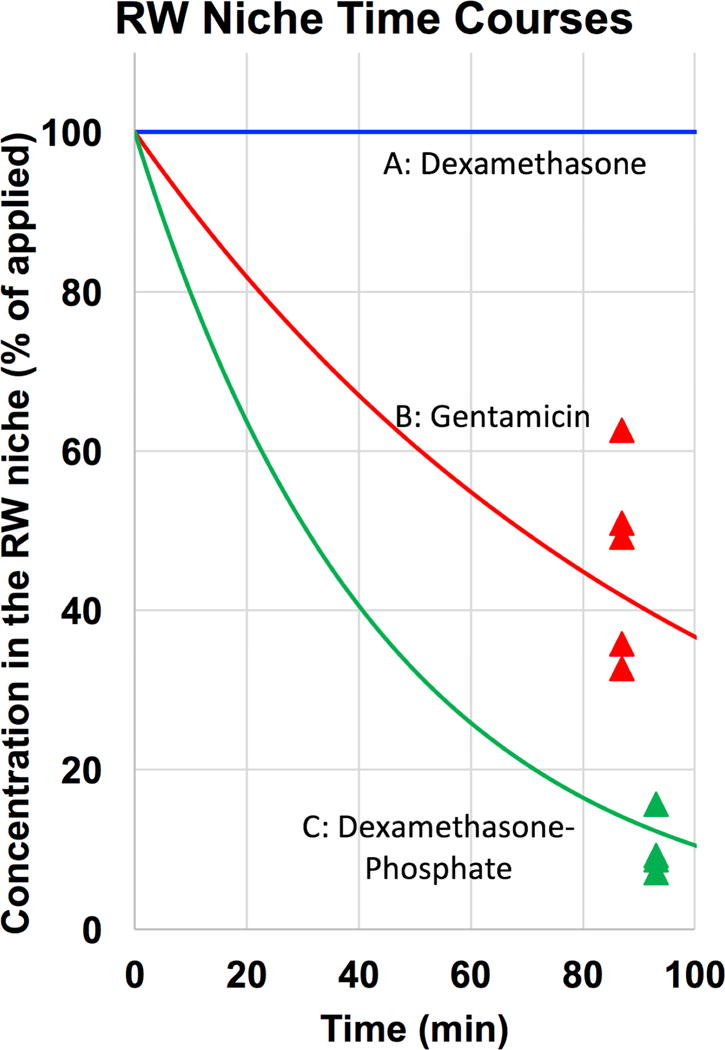
Data points show the measured drug concentration remaining in the RW niche in perilymph sampling experiments that used a single 20 μL application to the RW niche in the guinea pig. For dexamethasone-phosphate the measure was the sum of that metabolized to the base form (72%) and that remaining as the phosphate form (28%). Subsequent calculations have assumed an exponential decline of concentration with time as indicated by the curves. For dexamethasone applied as a suspension of micronized solid drug, a uniform concentration over the application period was assumed in calculations.
In other studies, we applied dexamethasone base in its micronized form to the RW niche as a 4.5% suspension in phosphate-buffered saline. The concentration of dexamethasone in the aqueous supernatant of this formulation was measured to be 94.2 μg/ml, which is close to the solubility published on Pubchem (https://pubchem.ncbi.nlm.nih.gov) of 89 μg/ml. There was insufficient volume in the RW niche to collect uncontaminated supernatant so no measure of middle ear concentration was possible. It is assumed that dexamethasone concentrations remain high during the application period due to the presence of solid drug. This is shown in Figure 4 as an unchanging concentration over this short period. These studies show that for drugs dissolved in a solution, drug concentration in the applied solution falls remarkably rapidly, even though the primary systems for elimination of volume from the ear, specifically the ciliated epithelium and Eustachian tube, are dysfunctional in the anesthetized preparation.
Middle ear kinetics is also potentially influenced by many factors not studied here, including variances in petrous bone pneumatization, and pathologic conditions such as infection or inflammation of the middle ear when substantial vasodilation and fluid effusion may occur. Another important consideration is that with intratympanic drug applications, tissues of the middle ear are exposed to substantially higher drug levels than are tissues of the inner ear and to the composition of the delivery vehicle. Histological examination of the middle ear after intratympanic injection of drugs has shown some inflammation but no major middle ear pathology (Wang et al., 2009; Wang et al, 2014)
5. Inner ear: Entry and elimination kinetics
5.1 Barriers of the ear
The main barriers to drug entry into the inner ear are formed by specialized cell layers, as reviewed elsewhere (Salt & Hirose, 2017). Endothelial cells of blood vessels provide the blood-perilymph and blood-strial barriers. They restrict the entry of systemically-applied drugs into the ear and control the elimination of locally-applied drugs. For drugs applied intratympanically, the primary barrier restricting passage into perilymph is the epithelium of the middle ear, which covers both the round window (RW) and the oval window at the stapes, the main sites for drug entry (Goycoolea, 2001; King et al., 2013). For both endothelial and epithelial cell layers, tight junctions between adjacent cells restrict paracellular solute movements. The lipid membranes of the cells restrict passive solute movements through the cell layer. A plethora of transport proteins within the membranes (ion channels, water channels, pumps and transporters) further influence movements of specific solutes through the layer with about 1500 genes (about 8% of the genome) dedicated to membrane transport functions (Ye et al. 2014).
5.2 Elimination measurement
Interpretation of perilymph concentrations measured following intratympanic drug application is made difficult by the measure also depending on how rapidly the drug is eliminated from perilymph. The perilymph concentration resulting from rapid entry at the round window combined with rapid elimination from scala tympani (ST) could be similar to that with slow entry at the round window combined with slow elimination from ST. Although there have been many pharmacokinetic studies focused on kinetics in ST resulting from drug entry through the round window membrane, it has recently been appreciated that drugs enter perilymph independently through both the RW membrane, into ST, and through the stapes, into scala vestibuli (SV) and the vestibule (King et al., 2011, Salt et al., 2012a). Pharmacokinetic studies therefore often need to consider the entire perilymphatic space, not just ST.
We have pioneered techniques to quantify elimination kinetics throughout the perilymphatic spaces by first loading the ear with drug by injection from a pipette sealed into the lateral SCC. As the cochlear aqueduct provides the outlet for fluid during injection, the procedure fills virtually the entire perilymphatic space with drug solution. After delays of zero to 4 hours, perilymph was then sampled from the fenestration used for injection with a sequential sampling method. This allows the distribution of drug throughout the perilymphatic space to be documented as a function of time. Figure 5 shows the group mean sample curves for our 3 drugs of interest established for gentamicin by Salt et al., (2016) and for dexamethasone and dexamethasone-phosphate by Salt et al. (2012b). A progressive decline of each drug over time is apparent, differing in rate for each substance. The elimination half-times for SV and ST derived from simulations fitted simultaneously to all the delay times for each substance are also shown in the figure. Simulations used consistent perilymph flow and perilymph-CSF interaction parameters derived from recent marker studies with a high molecular weight fluorescent dextran (Salt et al., 2015). The analysis confirms the visual interpretation of the decline curves, showing that gentamicin is best retained in perilymph (with longest half-time) and also showing that the base form of dexamethasone is eliminated most rapidly. The differences in elimination rate are generally in accordance with the molecular properties calculated in Figure 3. Gentamicin, having low WLOGP and high TPSA/A2, was expected to be least permeable while the base form of dexamethasone was expected to be the most permeable, as found here.
Figure 5.
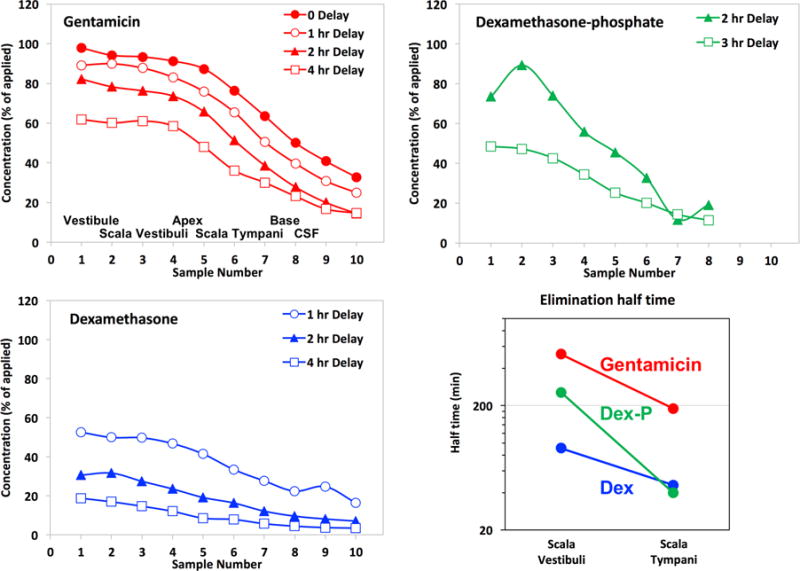
Perilymph elimination compared by perilymph loading followed by sequential sampling from the LSCC after the delay times indicated. The approximate origins of the samples are indicated in the first panel. These curves show the progressive decline of perilymph drug concentration with time. Dexamethasone and dexamethasone-phosphate data are from Salt et al., (2012b) and gentamicin data are from Salt et al., (2016).
5.3 Entry measurements
Entry from the middle ear into perilymph from intratympanic drug applications has also been quantified by sequential perilymph sampling from the lateral semicircular canal. In all studies, the perilymph samples were collected after a 1 hour drug application to the RW niche. The short application time allows entry at the round window membrane and at the stapes to be distinguished before being significantly influenced by local inter-scala communication between ST and SV (Salt et al., 2012a). It also allows the entry measurement to be dominated by the initial period when the middle ear concentration is high, and therefore less influenced by a declining middle ear drug level. Perilymph concentrations measured for the three drugs of interest, normalized with respect to their applied concentrations, are shown in Figure 6. The mean measured perilymph levels (black lines) were highest for dexamethasone, reaching an average across all 10 samples of 2.3 % of the applied concentration. Measured perilymph levels with application of dexamethasone-phosphate were substantially lower averaging just 0.13 % of the applied concentration. This measurement included both forms of the drug, dexamethasone-phosphate and dexamethasone base summed, which in these samples averaged 11% dexamethasone-phosphate and 89% dexamethasone base. Dexamethasone levels averaged across all samples were therefore almost 18× those of dexamethasone-phosphate with RW niche applications. Gentamicin was found at an intermediate concentration, reaching a mean of 0.62 % of the applied concentration.
Figure 6.
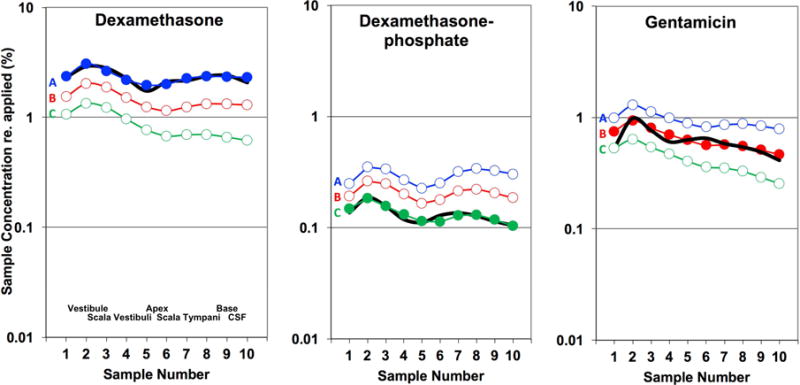
Perilymph concentrations resulting from RW niche applications for 3 drugs. Black lines show the group mean measured perilymph concentrations in 10 sequential samples taken from the lateral semi-circular canal. The number of experiments were 4, 4 and 5 respectively. Concentrations are normalized (as %) of the drug concentration applied to the RW niche. The applied concentration of dexamethasone was 94.2 μg/ml (dissolved concentration in an applied 4.5% suspension), of dexamethasone-phosphate was 4 mg/ml and of gentamicin was 40 mg/ml respectively. The approximate spatial origins of the perilymph samples are given in the first panel. Colored curves are simulations of the experiments calculated for each of the 3 time courses of drug decline in the middle ear. A, blue: unchanging with time, comparable to micronized dexamethasone suspension, B, red: 74.5 min middle ear decline half-time, comparable to gentamicin solution, C: green: 25.7 min middle ear decline half-time, comparable to dexamethasone phosphate. These decline rates correspond to the curves shown in Figure 4, and are detailed in the text. Dexamethasone and dexamethasone-phosphate data are from Salt et al., 2018 and gentamicin data are from Salt et al., 2016.
Interpretation of these measurements needed to take into account the differences in middle ear kinetics, established in Figure 4, and of perilymph kinetics, established in Figure 5, for the 3 drugs. As concentrations were normalized to the applied concentration, the lower measured dexamethasone-phosphate concentrations could partially result from the rapid decline of this drug in the middle ear. The colored curves in Figure 6 with solid symbols show simulations of the experiments for the appropriate, corresponding middle ear drug time course and perilymph kinetics, with RW membrane and stapes permeability characteristics adjusted to best fit the measured curves. In each case, sample concentrations were also calculated for the other two middle ear decline conditions as indicated by the letters. These calculations show that the drug time course in the middle ear does substantially influence the amount of drug in perilymph, but not to the degree necessary to account for the 18× measured difference between dexamethasone and dexamethasone-phosphate. For example, the blue curve “A” for dexamethasone-phosphate indicates the predicted sample concentrations if middle ear concentration had remained constant, rather than declining as measured and shown in curve “C”. Even when middle ear declines for gentamicin and dexamethasone-phosphate are taken into account (e.g. comparing curves A or curves C for the two drugs), entry remains about 8× times greater for dexamethasone than for dexamethasone-phosphate, in accordance with the molecular properties of the two forms. Gentamicin entry was measured to be at an intermediate level, greater than that of dexamethasone-phosphate, which does not follow the expectation based on molecular properties calculated in Figure 3.
6. Comparisons between drugs
The above analysis confirms that dexamethasone enters the ear far more readily than dexamethasone-phosphate, but can only be applied at low concentration due to its limited solubility. An important question is therefore which form of dexamethasone gives the highest perilymph concentration in absolute terms. The measured perilymph concentration, averaged across all 10 samples collected, was 2.18 μg/ml for dexamethasone (applied at a concentration of 94.2 μg/ml) and was 4.15 μg/ml for dexamethasone-phosphate (applied at a concentration of 4000 μg/ml). Measured perilymph concentration was therefore higher in absolute terms when dexamethasone-phosphate was applied compared to the lower solubility base form. However, the comparison of samples taken after 1 hour application is quite misleading and it is also important to consider the time courses resulting from the two forms of drug, as shown in Figure 7.
Figure 7.
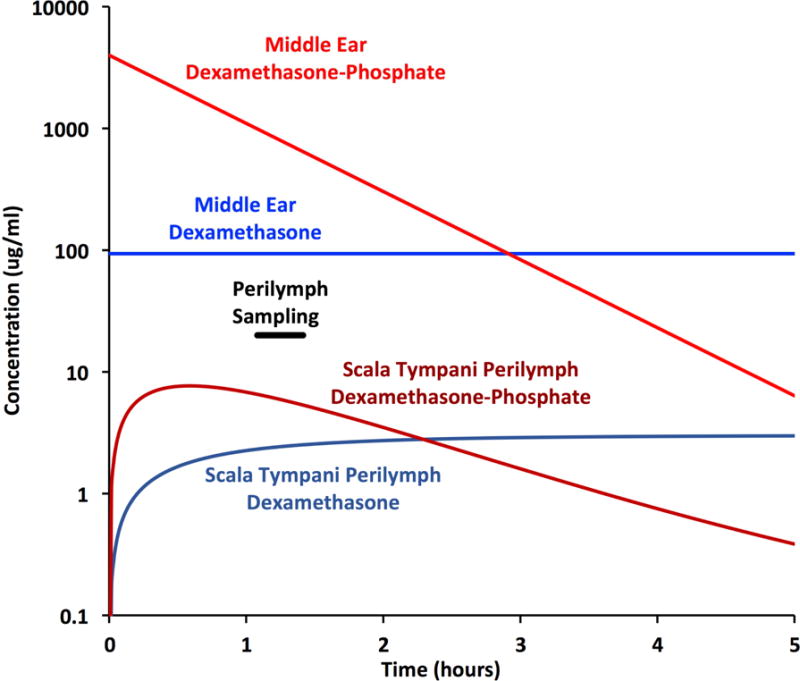
Time courses of dexamethasone-phosphate and dexamethasone base concentrations derived from simulations fitted to the measured sample data. Concentrations are shown for perilymph of the entire scala tympani pooled. Perilymph concentration is initially higher with application of dexamethasone-phosphate, due to the higher applied concentration, and was higher at the time of sampling which commenced after 1 hour as indicated. Nevertheless, the higher dosing is short-lived and after 2.3 hours concentration falls below that when dexamethasone suspension is administered.
On a logarithmic scale, the exponential decline of dexamethasone-phosphate concentration (shown in Figure 4) appears as a declining straight line. At this rate, it takes approximately 3 hours for the middle ear concentration to fall below the free concentration in the applied suspension of micronized dexamethasone. Due to the higher initial concentration, even though dexamethasone-phosphate is less permeable through the RW membrane, ST perilymph levels will initially be higher, in accordance with the sample data on which the simulations are based. However, due to the declining middle ear concentration combined with ongoing elimination from perilymph, perilymph concentration falls rapidly with time for dexamethasone-phosphate applications. Dexamethasone-phosphate thus provides only a brief exposure, largely confined to the basal regions of the cochlea (distribution with location not shown here).
The use of gentamicin as an effective therapy for Meniere’s disease was largely responsible for the initial advance in the field of local drug delivery. Our pharmacokinetic studies show that gentamicin enters the ear at both the RW membrane and the stapes more readily than dexamethasone-phosphate. It is possible that passage of gentamicin through the membranous boundaries at the RW membrane and stapes are influenced by factors other than lipid membrane permeability, such as by some form of transport. Alternatively, it is also possible that other factors related to the commercial gentamicin formulation applied to the middle ear could have influenced permeability of the cell layer to the drug. As the applied formulation is of non-physiologic pH (pH 3 – 4) and osmolarity (Mikulec et al, 2008) and is applied directly to the epithelial layer of the middle ear the non-physiologic composition could disrupt the cell’s properties, thereby influencing permeability to gentamicin.
Although methylprednisolone is also used extensively for inner ear therapy, there are only limited pharmacokinetic data related to its pharmacokinetics in the ear. In an interpretation of human samples from the cochleostomy site after intratympanic application (Bird et al., 2007), the combined clearance half time for methylprednisolone from the middle ear and the inner ear was estimated as 27min (Plontke et al. 2008a). In similarity with the different forms of dexamethasone, the soluble forms of methylprednisolone typically administered would be expected to be less permeable through the membranous boundaries at the RW membrane and stapes compared to the base, less polar form, as suggested by its molecular properties in Figure 3. However, to our knowledge there are still no data showing that the succinate or hemisuccinate forms are metabolized to the base form in the ear, and if they are metabolized, at what rate. These important issues related to the molecular properties and potency of different forms of methylprednisolone need to be added to the long list of uncertain aspects of steroid therapy (Trune & Canlon, 2012).
Triamcinolone has properties quite unlike dexamethasone and methylprednisolone in its common molecular forms and could potentially overcome some of the obstacles to effective steroid use in the ear. As shown in Figure 3, the form most commonly administered is triamcinolone-acetonide, the less-polar and less water-soluble form (aqueous solubility 21-33 μg/ml), which is used because it has higher potency (8 times the potency of prednisone) compared to triamcinolone (1-2 times the potency of prednisone), and because the crystal suspension provides a depot effect, at least in other indications outside the ear. Similar to the base form of dexamethasone, it is given as a suspension of micronized solid drug particles in a physiological background solution of normal pH and osmolarity. Preliminary pharmacokinetic measurements show that triamcinolone-acetonide readily enters the ear at the RW membrane and stapes, but is also, as expected, rapidly eliminated from perilymph (Salt at al. 2018). However, perilymph analysis also showed that triamcinolone-acetonide was metabolized to triamcinolone, the less potent but less permeable form, which exhibits a distribution profile in perilymph consistent with it not being rapidly eliminated. These properties (readily entering the ear, being metabolized there to a form that is eliminated slowly and which therefore becomes widely distributed throughout the ear) could make triamcinolone-acetonide a better candidate for local therapy of hearing disorders. Intratympanic triamcinolone-acetonide suspension (applied form verified with the authors) has been used clinically in patients with Ménierè’s syndrome and reported to be a safe and potentially effective procedure for vertigo control (Jumaily et al., 2017). It has also been used as a standard therapy for secondary (salvage) treatment of idiopathic sudden sensorineural hearing loss in some institutions including in one the authors (SKP), (Loader et al. 2013, Plontke 2017). Animals treated with dexamethasone or triamcinolone-acetonide delivered intratympanically in hydrogels were found to have reduced inflammation but no significant improvement post-implantation hearing (Honeder et al., 2015). However, hearing changes when normally-hearing animals are implanted could arise from trauma or mechanical factors that may not be remedied by steroids.
The dependence of steroid properties on molecular structure further raises the importance of drug metabolism in the middle and inner ear and its influence on pharmacokinetics. For steroids, the potency of each form of the drug is also relevant. Potency is influenced by the affinity of the cytosolic glucocorticoid and/or mineralocorticoid receptors to the drug and on the ability of the drug to penetrate the cell membrane to reach the cytosolic receptors (Grossmann et al., 2004). Dexamethasone-phosphate is metabolized in the middle ear and to an even greater degree in the inner ear, as evidenced by samples from the middle ear and of perilymph showing dexamethasone present as the phosphate and the base forms (Plontke et al. 2008b, Hahn et al. 2012). In the liver, dexamethasone is metabolized by the enzyme CYP3A4 and to a lesser extent by CYP17A. CYP17A has been demonstrated to be present in the inner ear (Lecain et al., 2003), leading to the possibility that local degradation in the ear could contribute to the rapid elimination of dexamethasone from perilymph. Inhibitors of these pathways, specifically using 10 μM ketoconazole and 10 μM abiraterone respectively had no influence dexamethasone elimination with time suggesting that they do not contribute significantly to loss of dexamethasone from perilymph. (Salt et al., 2014).
Little is known about whether the polar, more soluble forms of methylprednisolone, methylprednisolone-succinate and methylprednisolone-hemisuccinate, are metabolized in the ear, or the differences in potency of these different molecular forms. Kinetic studies are lacking with the exception one compromised by technical problems (Parnes et al., 1999; Salt & Plontke, 2005). We would expect that, like dexamethasone, the more soluble, polar forms would less-readily enter the at the RW membrane and stapes than the less polar form, but this has never been measured. More importantly, it has not been shown whether the polar forms are metabolized in the ear to the base form or whether they act on the receptors in the applied form.
7. Delivery protocols compared
7.1 Intratympanic applications
The amount and distribution of drug in perilymph depends both on the substance applied and on the application protocol. Based on the kinetic properties for the 3 drugs presented above we can calculate their likely distribution throughout the human inner ear for the application protocols typically used clinically, as shown in Figure 8. This calculation assumes that pharmacokinetic properties derived from animal experiments are comparable to those in humans when scaled to the larger human ear. For a dexamethasone suspension, concentration in the vestibule will be higher than that in the cochlea, due to the faster rate of elimination from ST compared to SV and the vestibule. Even when a steady-state is achieved, a substantial gradient in concentration will remain along the cochlea, with higher-frequency regions exposed to higher drug levels than low-frequency regions. The 500-Hz region is calculated to be exposed to a 500× lower drug concentration than the 8-kHz region.
Figure 8.
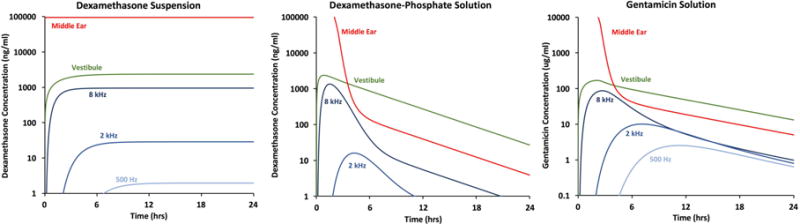
Influence of drug properties and application protocols on calculated drug levels achieved in different regions of the human inner ear using kinetic parameters established in the studies above. Sustained delivery from a suspension of dexamethasone results in the generation of a steady-state, with substantial concentration gradients between the vestibule and the cochlea, and between high-frequency and low-frequency cochlear regions. In contrast, delivery of dexamethasone-phosphate solution results in a transient drug exposure, primarily to vestibular and high-frequency cochlear regions, due to the rapid rate of elimination of dexamethasone from perilymph. Delivery of gentamicin also results in a transient drug exposure, with greater spread to low frequency cochlear regions then dexamethasone-phosphate, due to a lower rate of elimination from cochlear perilymph.
For drug solutions applied intratympanically we have assumed the entire injected volume remains in place for 30 mins, with drug elimination rates comparable to those shown in Figure 4, after which there would be a superimposed decline of drug volume in the middle ear, corresponding to the loss down the Eustachian tube when the patient resumed their normal posture. With dexamethasone-phosphate, concentrations in the vestibule and at the 8-kHz region of the cochlea are calculated to transiently exceed those generated with a suspension, due to the higher solubility of this form of the drug. However, the drug exposure is transient with even larger gradients along the cochlea, so that exposure of low-frequency regions to drug is extremely limited. In this case, the 500-Hz region (not shown as it is below the graph minimum) would be exposed to over 2000× lower peak drug concentration than the 8-kHz region. In comparison, for gentamicin the vestibule is again exposed to higher drug level than the 8-kHz cochlear region but the decline of gentamicin concentration occurs more slowly with distance along the cochlea than with dexamethasone-phosphate so the gradients along the cochlear are considerably smaller. For gentamicin, it is calculated that the 500-Hz region would be exposed to only 34× lower peak drug concentration than the 8-kHz region. A complete interpretation of these curves would also require knowledge of the therapeutic (for steroid) or toxic (for gentamicin) dose of the drug for inner ear tissues, and whether each region of the inner ear must be treated locally or whether treating part of the ear is sufficient. Such detail is not yet available for tissues of the ear.
These calculations present an enigma in that dexamethasone-phosphate administration, which is often given with the intended purpose of providing therapy for hearing disorders, provides far less exposure of middle and apical cochlear regions to drug than does gentamicin, which is given to ablate vestibular hair cells while trying to avoid damage to hearing. When applied in the form of dexamethasone-phosphate solution the expected drug distribution along the cochlea, based on pharmacokinetic studies in animals, appears contrary to its intended purpose.
Although the polar molecular properties of the phosphate form confer higher aqueous solubility, it also makes dexamethasone-phosphate substantially less permeable through the membranes of the RW and stapes than the base form of dexamethasone. When the lower entering amount does reach perilymph of the ear, it is then metabolized to the active base form, dexamethasone, which is both predicted to be more permeable though membranous boundaries of the vasculature and confirmed here by measurements to be rapidly lost from perilymph. An additional consequence of the rapid elimination rate is that dexamethasone does not distribute apically very far along the cochlea, exposing the speech frequency regions of the human cochlea to only low drug levels and for only a brief period of time. These properties are not just “less than ideal” for treating cochlear disorders but represent a highly unfortunate combination of pharmacokinetic properties. Intratympanically-applied dexamethasone-phosphate therefore appears to have physical properties completely inappropriate for treating cochlear disorders. This conclusion may be supported by the absence of improvement of auditory symptoms of Meniere’s disease (Silverstein et al. 1998) and a lack of correlation between outcomes and dexamethasone dosing in patients with SSNL (Liebau et al. 2017). Recent studies have reported that a higher dosing regimen using 24 mg/ml dexamethasone-phosphate may give better recovery of hearing in ISSNHL (Alexander et al., 2015; Kordiš & Battelino, 2017). Nevertheless, our analysis raises serious concerns that when administered as the dexamethasone-phosphate form, the speech regions of the human cochlea will only be exposed to low drug levels and that exposure will be for a brief period. Drugs with physical properties more appropriate for their purpose in the ear should be sought. In addition, there needs to be greater awareness that the applied form of a specific drug can make a substantial difference to perilymphatic drug levels. This is especially problematic as drug nomenclature in many publications is often not precise, with the base form of the drug (e.g. dexamethasone, methylprednisolone or triamcinolone) commonly stated without details of the exact form of the drug used. As shown here, the applied form of drug may have physical properties substantially different from the stated, base form.
7.2 Intracochlear and intralabyrinthine applications
Intracochlear and intralabyrinthine applications have the advantage of bypassing adsorption barriers like the RW, the stapes, and the blood labyrinth barriers. This is of special interest for the application of substances that do not readily cross these barriers (see above), and for gene and cell-based therapies. In addition, it has been demonstrated that intracochlear application of only small amounts e.g. of dexamethasone phosphate by injection through the round window membrane leads to a lower variability of the intracochlear concentration, to an increased absolute perilymph concentration, and to a more uniform distribution of the substance in scala tympani (Hahn et al. 2012). Losses of the substance due to leaks in the round window membrane, which occur by injections through the round window membrane, however, must be taken into consideration. These losses may be reduced by using “sealing” materials such as biopolymer gels or biocompatible tissue glue (Plontke et al. 2016).
For chronic intracochlear delivery of drugs, micropumps have been developed (as for instance with reciprocating infuse-withdraw technology). These reciprocating delivery systems distribute drugs into a volume in the base of the cochlea, while the primary determinant of distribution throughout more distal regions of the cochlea is diffusion. It was observed that over longer time courses maintenance of drug concentration in the basal part of scala tympani may prove more advantageous for extending apical delivery than increases in flow rate, but basal-apical concentrations gradients remain even after longer intracochlear delivery time periods. (Chen et al. 2005, Pararas et al. 2011).
For gene therapy and cell transplantation, intracochlear or intralabyrinthine delivery methods are essential. However, translation of these experimental approaches on the application of biological therapies is presently at an early stage, and pharmacokinetic data have yet to be collected (Roemer et al. 2017).
Another approach on intracochlear drug delivery is the application of substances in combination with auditory implants, specifically cochlear implants, aiming to reduce adverse events of cochlear implantation and improvement of the frequency spectrum, resolution, and the dynamic range of auditory implants (Hendricks et al. 2008). Initial work focused on the use of neurotrophic factors in order to induce afferent fiber growth towards the implant electrodes and preventing or at least diminishing spiral ganglion degeneration (Landry et al., 2013).
Intracochlear applications of neurotrophin-eluting nanoparticles have been shown to improve survival of neurons in the spiral ganglion (Wise et al., 2016). The current focus is more on the use of steroids and anti-apoptotic agents to minimize fibrous tissue and hearing loss due to surgical trauma and reactions to implant with the major goal of preserving hearing for combined electro-acoustic stimulation (EAS) but also to maintain impedance and functions of conventional implants (Faramand et al., 2010; Plontke et al. 2017).
Intracochlear drug application during CI implantation has the advantage that the cochlea is already opened. It possible to inject substance (e.g. glucocorticosteroids) into scala tympani at the time of CI electrode insertion (De Ceulaer et al. 2003, Ye et al. 2007, Paasche et al. 2006, Honeder et al. 2015). It has also been suggested to insert both a conventional cochlear implant and a controlled release, biodegradable drug delivery system, into scala tympani (Plontke et al. 2017).
The main focus of research, however, is the development of drug-device combinations with drug release from cochlear implant electrode carriers. Different options are available for releasing substances by the electrode carrier into the cochlea: the substances may be incorporated in the CI electrode carrier; the electrode carrier may be coated with the substance, or the electrode carrier may contain a delivery channel, which is then connected to a drug reservoir or a pump system (reviewed in: Plontke et al. 2017).
The delivery of glucocorticosteroids from cochlear implants has been shown to reduce fibrous tissue growth after cochlear implantation. It is safe for auditory neurons, increases the neuroprotective effect of chronic electrical stimulation in animal models (Douchement et al. 2015, Scheper et al. 2017, Wilk et al. 2016), and decreases electrode impedances in humans (Briggs et al. 2016).
Initial pharmacokinetic investigations on delivery of drugs from CI electrode carriers show a release kinetic with an initial burst release, followed by stable concentrations over the observed period with the loading concentration influencing the peak and duration of the burst release (Liu et al. 2016).
Another way of applying drugs with CI is to incorporate a cannula into the implant, through which drug solution is driven (Salt et al. 2017). Based on prior pharmacokinetic data, the initially observed drug gradients along the scala were expected to decline over a period of 8-12 hours. However, concentration gradients declining along scala tympani towards the apex persisted after 24 hours of sustained injection and were still present in some animals after 7 days injection. One explanation is that inner ear pharmacokinetics are altered in the period after cochlear implantation, possibly by a permeabilization of the blood-labyrinth barrier as part of the immune response to the implant (Salt et al. 2017).
8. Conclusions and Future Directions
In the past decade, our capability to perform quantitative pharmacokinetic studies of the inner ear has improved dramatically. We now have fluid delivery and sampling techniques that have overcome serious technical artifacts that have plagued the field since its outset. We also have computer simulations of delivery and sampling procedures available for the ear that closely reproduce the measured data, allowing pharmacokinetic studies to be interpreted in detail in terms of amount and distribution of drug in the ear. Enough data have been accumulated with a range of drugs to understand the underlying scientific basis of inner ear pharmacokinetics. Unfortunately, this knowledge is revealing that we still have some serious obstacles to overcome. The assumption that any substance can be applied intratympanically and will distribute throughout the inner ear in therapeutic amounts has been shown to be false.
Nevertheless, as we better understand how pharmacokinetics depends on drug properties we are now able to select from an enormous range of drugs that may have value to the ear. Kinetic data derived from the eye or the brain can be used to guide the selection of potentially suitable molecules to treat the ear. Rather than developing inner ear therapies empirically, we are now in a much better position to proceed on a scientific, rational basis. While inner ear drug therapy is very much in its infancy, the future looks very bright indeed.
Supplementary Material
Highlights.
Quantitative pharmacokinetic studies of the inner ear allow drug therapies to be compared.
Molecular properties determine how readily drugs pass through membranous boundaries of the ear.
The properties of dexamethasone-phosphate make it unsuitable as a therapy for hearing disorders.
Acknowledgments
Part of this work (ANS) was supported by the National Institute on Deafness and Other Communication Disorders (NIDCD) of the National Institutes of Health (NIH) under award number R01 DC001368. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander TH, Harris JP, Nguyen QT, Vorasubin N. Dose Effect of Intratympanic Dexamethasone for Idiopathic Sudden Sensorineural Hearing Loss: 24 mg/mL Is Superior to 10 mg/mL. Otol Neurotol. 2015;36:1321–1327. doi: 10.1097/MAO.0000000000000834. [DOI] [PubMed] [Google Scholar]
- Ayoob AM, Borenstein JT. The role of intracochlear drug delivery devices in the management of inner ear disease. Expert Opin Drug Deliv. 2015;12:465–479. doi: 10.1517/17425247.2015.974548. [DOI] [PubMed] [Google Scholar]
- Bird PA, Begg EJ, Zhang M, Keast AT, Murray DP, Balkany TJ. Intratympanic versus intravenous delivery of methylprednisolone to cochlear perilymph. Otol Neurotol. 2007;28:1124–1130. doi: 10.1097/MAO.0b013e31815aee21. [DOI] [PubMed] [Google Scholar]
- Briggs R, O’leary S, Birman C, et al. A first time in human investigation of a combination device delivering a targeted drug therapy to cochlear implant recipients. 14th International Conference on Cochlear Implants; Toronto. 2016. [Google Scholar]
- Chen Z, Kujawa SG, McKenna MJ, Fiering JO, Mescher MJ, Borenstein JT, Swan EE, Sewell WF. Inner ear drug delivery via a reciprocating perfusion system in the guinea pig. J Control Release. 2005;110:1–19. doi: 10.1016/j.jconrel.2005.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chole RA, Tinling SP. Bone lining cells of the mammalian cochlea. Hear Res. 1994;75:233–243. doi: 10.1016/0378-5955(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Daina A, Zoete V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem. 2016;11:1117–1121. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ceulaer G, Johnson S, Yperman M, Daemers K, Offeciers FE, O’Donoghue GM, Govaerts PJ. Long-term evaluation of the effect of intracochlear steroid deposition on electrode impedance in cochlear implant patients. Otol Neurotol. 2003;24:769–774. doi: 10.1097/00129492-200309000-00014. [DOI] [PubMed] [Google Scholar]
- Douchement D, Terranti A, Lamblin J, Salleron J, Siepmann F, Siepmann J, Vincent C. Dexamethasone eluting electrodes for cochlear implantation: Effect on residual hearing. Cochlear Implants Int. 2015;16:195–200. doi: 10.1179/1754762813Y.0000000053. [DOI] [PubMed] [Google Scholar]
- Egan WJ, Merz KM, Jr, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000;43:3867–7387. doi: 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- Ersner MS, Spiegel EA, Alexander MH. Transtympanic injection of anesthetics for the treatment of Ménière’s syndrome. AMA Arch Otolaryngol. 1951;54:43–52. doi: 10.1001/archotol.1951.03750070060005. [DOI] [PubMed] [Google Scholar]
- Farahmand Ghavi F, Mirzadeh H, Imani M, Jolly C, Farhadi M. Corticosteroid-releasing cochlear implant: a novel hybrid of biomaterial and drug delivery system. J Biomed Mater Res B Appl Biomater. 2010;94:388–398. doi: 10.1002/jbm.b.31666. [DOI] [PubMed] [Google Scholar]
- Gillespie LN, Richardson RT, Nayagam BA, Wise AK. Treating hearing disorders with cell and gene therapy. J Neural Eng. 2014;11:065001. doi: 10.1088/1741-2560/11/6/065001. [DOI] [PubMed] [Google Scholar]
- Goycoolea MV. Clinical aspects of round window membrane permeability under normal and pathological conditions. Acta Otolaryngol. 2001;121:437–447. doi: 10.1080/000164801300366552. [DOI] [PubMed] [Google Scholar]
- Grossmann C, Scholz T, Rochel M, et al. Transactivation via the human glucocorticoid and mineralocorticoid receptor by therapeutically used steroids in CV-1 cells: A comparison of their glucocorticoid and mineralocorticoid properties. Eur J Endocrinol. 2004;151:397–406. doi: 10.1530/eje.0.1510397. [DOI] [PubMed] [Google Scholar]
- Hahn H, Salt AN, Biegner T, Kammerer B, Delabar U, Hartsock JJ, Plontke SK. Dexamethasone levels and base-to-apex concentration gradients in the scala tympani perilymph after intracochlear delivery in the guinea pig. Otol Neurotol. 2012;33:660–665. doi: 10.1097/MAO.0b013e318254501b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid M, Trune D. Issues, indications, and controversies regarding intratympanic steroid perfusion. Curr Opin Otolaryngol Head Neck Surg. 2008;16:434–440. doi: 10.1097/MOO.0b013e32830ce796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks JL, Chikar JA, Crumling MA, Raphael Y, Martin DC. Localized cell and drug delivery for auditory prostheses. Hear Res. 2008;242:117–131. doi: 10.1016/j.heares.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honeder C, Landegger LD, Engleder E, Gabor F, Plasenzotti R, Plenk H, Kaider A, Hirtler L, Gstoettner W, Arnoldner C. Effects of intraoperatively applied glucocorticoid hydrogels on residual hearing and foreign body reaction in a guinea pig model of cochlear implantation. Acta Otolaryngol. 2015;135:313–319. doi: 10.3109/00016489.2014.986758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumaily M, Faraji F, Mikulec AA. Intratympanic Triamcinolone and Dexamethasone in the Treatment of Ménière’s Syndrome. Otol Neurotol. 2017;38:386–391. doi: 10.1097/MAO.0000000000001311. [DOI] [PubMed] [Google Scholar]
- Karssen AM, Meijer OC, van der Sandt IC, Lucassen PJ, de Lange EC, de Boer AG, de Kloet ER. Multidrug resistance P-glycoprotein hampers the access of cortisol but not of corticosterone to mouse and human brain. Endocrinology. 2001;142:2686–2694. doi: 10.1210/endo.142.6.8213. [DOI] [PubMed] [Google Scholar]
- King EB, Salt AN, Eastwood HT, O’Leary SJ. Direct entry of gadolinium into the vestibule following intratympanic applications in Guinea pigs and the influence of cochlear implantation. J Assoc Res Otolaryngol. 2011;12:741–751. doi: 10.1007/s10162-011-0280-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King EB, Salt AN, Kel GE, Eastwood HT, O’Leary SJ. Gentamicin administration on the stapes footplate causes greater hearing loss and vestibulotoxicity than round window administration in guinea pigs. Hear Res. 2013;304:159–166. doi: 10.1016/j.heares.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordiš Š, Battelino S. The Role of High Dose Intratympanic Dexamethasone as Salvage Therapy for Idiopathic Sudden Sensorineural Hearing Loss. J Int Adv Otol. 2017;21 doi: 10.5152/iao.2017.3896. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Lambert PR, Carey J, Mikulec AA, LeBel C. Intratympanic Sustained-Exposure Dexamethasone Thermosensitive Gel for Symptoms of Ménière’s Disease: Randomized Phase 2b Safety and Efficacy Trial. Otol Neurotol. 2016;37:1669–1676. doi: 10.1097/MAO.0000000000001227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry TG, Fallon JB, Wise AK, Shepherd RK. Chronic neurotrophin delivery promotes ectopic neurite growth from the spiral ganglion of deafened cochleae without compromising the spatial selectivity of cochlear implants. J Comp Neurol. 2013;521:2818–2832. doi: 10.1002/cne.23318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange G. Gentamicin and other ototoxic antibiotics for the transtympanic treatment of Menière’s disease. Arch Otorhinolaryngol. 1989;246:269–270. doi: 10.1007/BF00463571. [DOI] [PubMed] [Google Scholar]
- Lecain E, Yang TH, Tran Ba Huy P. Steroidogenic enzyme expression in the rat cochlea. Acta Otolaryngol. 2003;123:187–191. doi: 10.1080/0036554021000028106. [DOI] [PubMed] [Google Scholar]
- Lim DJ, Hussl B. Macromolecular transport by the middle ear and its lymphatic system. Acta Otolaryngol (Stockh) 1975;80:19–31. doi: 10.3109/00016487509121296. [DOI] [PubMed] [Google Scholar]
- Loader B, Atteneder C, Kaider A, Franz P. Tympanotomy with sealing of the round window as surgical salvage option in sudden idiopathic sensorineural hearing loss. Acta Otolaryngol. 2013;133:1285–1291. doi: 10.3109/00016489.2013.829921. [DOI] [PubMed] [Google Scholar]
- Löscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx. 2005;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebau A, Pogorzelski O, Salt AN, Plontke SK. Hearing Changes After Intratympanically Applied Steroids for Primary Therapy of Sudden Hearing Loss: A Meta-analysis Using Mathematical Simulations of Drug Delivery Protocols. Otol Neurotol. 2017;38:19–30. doi: 10.1097/MAO.0000000000001254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jolly C, Braun S, Stark T, Scherer E, Plontke SK, Kiefer J. In vitro and in vivo pharmacokinetic study of a dexamethasone-releasing silicone for cochlear implants. Eur Arch Otorhinolaryngol. 2016;273:1745–1753. doi: 10.1007/s00405-015-3760-0. [DOI] [PubMed] [Google Scholar]
- Maarup TJ, Chen AH, Porter FD, Farhat NY, Ory DS, Sidhu R, Jiang X, Dickson PI. Intrathecal 2-hydroxypropyl-beta-cyclodextrin in a single patient with Niemann-Pick C1. Mol Genet Metab. 2015;116:75–79. doi: 10.1016/j.ymgme.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikulec AA, Hartsock JJ, Salt AN. Permeability of the round window membrane is influenced by the composition of applied drug solutions and by common surgical procedures. Otol Neurotol. 2008;29:1020–1026. doi: 10.1097/MAO.0b013e31818658ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikulec AA, Plontke SK, Hartsock JJ, Salt AN. Entry of substances into perilymph through the bone of the otic capsule after intratympanic applications in guinea pigs: implications for local drug delivery in humans. Otol Neurotol. 2009;30:131–138. doi: 10.1097/mao.0b013e318191bff8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Ito J. Local drug delivery to the inner ear using biodegradable materials. Ther Deliv. 2011;2:807–814. doi: 10.4155/tde.11.43. [DOI] [PubMed] [Google Scholar]
- Nedzelski JM, Chiong CM, Fradet G, Schessel DA, Bryce GE, Pfleiderer AG. Intratympanic gentamicin instillation as treatment of unilateral Menière’s disease: update of an ongoing study. Am J Otol. 1993;14:278–282. [PubMed] [Google Scholar]
- Nguyen K, Kempfle JS, Jung DH, McKenna CE. Recent advances in therapeutics and drug delivery for the treatment of inner ear diseases: a patent review (2011–2015) Expert Opin Ther Pat. 2017;27:191–202. doi: 10.1080/13543776.2017.1252751. [DOI] [PubMed] [Google Scholar]
- Pararas EE, Chen Z, Fiering J, Mescher MJ, Kim ES, McKenna MJ, Kujawa SG, Borenstein JT, Sewell WF. Kinetics of reciprocating drug delivery to the inner ear. J Control Release. 2011;152:270–277. doi: 10.1016/j.jconrel.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paasche G, Bockel F, Tasche C, Lesinski-Schiedat A, Lenarz T. Changes of postoperative impedances in cochlear implant patients: the short-term effects of modified electrode surfaces and intracochlear corticosteroids. Otol Neurotol. 2006;27:639–647. doi: 10.1097/01.mao.0000227662.88840.61. [DOI] [PubMed] [Google Scholar]
- Parnes LS, Sun AH, Freeman DJ. Corticosteroid pharmacokinetics in the inner ear fluids: an animal study followed by clinical application. Laryngoscope. 1999;109:1–17. doi: 10.1097/00005537-199907001-00001. [DOI] [PubMed] [Google Scholar]
- Plontke SK, Mikulec AA, Salt AN. Rapid clearance of methylprednisolone after intratympanic application in humans. Comment on: Bird PA, Begg EJ, Zhang M, et al. Intratympanic versus intravenous delivery of methylprednisolone to cochlear perilymph. Otol Neurotol. 2008;29:732–733. doi: 10.1097/MAO.0b013e318173fcea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plontke SK, Biegner T, Kammerer B, Delabar U, Salt AN. Dexamethasone concentration gradients along scala tympani after application to the round window membrane. Otology & Neurotology. 2008;29:401–406. doi: 10.1097/MAO.0b013e318161aaae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plontke SK, Glien A, Rahne T, Mäder K, Salt AN. Controlled release dexamethasone implants in the round window niche for salvage treatment of idiopathic sudden sensorineural hearing loss. Otol Neurotol. 2014;35:1168–1171. doi: 10.1097/MAO.0000000000000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plontke SK, Hartsock JJ, Gill RM, Salt AN. Intracochlear Drug Injections through the Round Window Membrane: Measures to Improve Drug Retention. Audiol Neurootol. 2016;21:72–79. doi: 10.1159/000442514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plontke SK, Götze G, Rahne T, Liebau A. Intracochlear drug delivery in combination with cochlear implants: Current aspects. HNO. 2017;65(Suppl 1):19–28. doi: 10.1007/s00106-016-0285-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plontke SK. Diagnostics and Therapy of Idiopathic Sudden Sensorineural Hearing Loss. Laryngorhinootologie. 2017;96(S 01):S103–S122. doi: 10.1055/s-0042-122385. [DOI] [PubMed] [Google Scholar]
- Praetorius M, Brunner C, Lehnert B, Klingmann C, Schmidt H, Staecker H, Schick B. Transsynaptic delivery of nanoparticles to the central auditory nervous system. Acta Otolaryngol. 2007;127:486–490. doi: 10.1080/00016480600895102. [DOI] [PubMed] [Google Scholar]
- Pyykkö I, Zou J, Zhang W, Zhang Y. Nanoparticle-based delivery for the treatment of inner ear disorders. Curr Opin Otolaryngol Head Neck Surg. 2011;19:388–396. doi: 10.1097/MOO.0b013e32834aa3a8. [DOI] [PubMed] [Google Scholar]
- Roemer A, Staecker H, Sasse S, Lenarz T, Warnecke A. Biological therapies in otology. HNO. 2017;65(Suppl 2):87–97. doi: 10.1007/s00106-016-0306-8. [DOI] [PubMed] [Google Scholar]
- Salt AN, Plontke SK. Local Inner ear drug delivery and pharmacokinetics. Hearing Research and Drug Discovery. Drug Discovery Today. 2005;10:1299–1306. doi: 10.1016/S1359-6446(05)03574-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt AN, Plontke SK. Principles of local drug delivery to the inner ear. Audiol Neurootol. 2009;14:350–360. doi: 10.1159/000241892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt AN, King EB, Hartsock JJ, Gill RM, O’Leary SJ. Marker entry into vestibular perilymph via the stapes following applications to the round window niche of guinea pigs. Hear Res. 2012a;283:14–23. doi: 10.1016/j.heares.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt AN, Hartsock JJ, Gill RM, Piu F, Plontke SK. Perilymph pharmacokinetics of markers and dexamethasone applied and sampled at the lateral semi-circular canal. J Assoc Res Otolaryngol. 2012b;13:771–783. doi: 10.1007/s10162-012-0347-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt AN, Hartsock J, Gill R, Piu F, Plontke S. Manipulations of Dexamethasone Kinetics in Perilymph. 37th Midwinter Research Meeting of the ARO; San Diego. 2014. p. 467. Abstract. [Google Scholar]
- Salt AN, Gill RM, Hartsock JJ. Perilymph Kinetics of FITC-Dextran Reveals Homeostasis Dominated by the Cochlear Aqueduct and Cerebrospinal Fluid. J Assoc Res Otolaryngol. 2015;16:357–371. doi: 10.1007/s10162-015-0512-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt AN, Hartsock JJ, Gill RM, King E, Kraus FB, Plontke SK. Perilymph pharmacokinetics of locally-applied gentamicin in the guinea pig. Hear Res. 2016;342:101–111. doi: 10.1016/j.heares.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt A, Hartsock J, Gill R, Smyth D, Kirk J, Verhoeven K. Perilymph pharmacokinetics of marker applied through a cochlear implant in guinea pigs. PLoS One. 2017;12(8):e0183374. doi: 10.1371/journal.pone.0183374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt AN, Hirose K. Communication Pathways to and from the Inner Ear and their Contributions to Drug Delivery. Hearing Research. 2017 doi: 10.1016/j.heares.2017.12.010. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt AN, Hartsock JJ, Hou J, Piu F. Perilymph Pharmacokinetics Compared for Dexamethasone-Phosphate, Dexamethasone and Triamcinolone with Local Applications. 41st Midwinter Research Meeting of the ARO; San Diego, CA Feb. 2018. Abstract. [Google Scholar]
- Scheper V, Hessler R, Hütten M, Wilk M, Jolly C, Lenarz T, Paasche G. Local inner ear application of dexamethasone in cochlear implant models is safe for auditory neurons and increases the neuroprotective effect of chronic electrical stimulation. PLoS One. 2017;12(8):e0183820. doi: 10.1371/journal.pone.0183820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuknecht HF. Ablation therapy for the relief of Ménière’s disease. Laryngoscope. 1956;66:859–870. doi: 10.1288/00005537-195607000-00005. [DOI] [PubMed] [Google Scholar]
- Senn P, Roccio M, Hahnewald S, Frick C, Kwiatkowska M, Ishikawa M, Bako P, Li H, Edin F, Liu W, Rask-Andersen H, Pyykkö I, Zou J, Mannerström M, Keppner H, Homsy A, Laux E, Llera M, Lellouche JP, Ostrovsky S, Banin E, Gedanken A, Perkas N, Wank U, Wiesmüller KH, Mistrík P, Benav H, Garnham C, Jolly C, Gander F, Ulrich P, Müller M, Löwenheim H. NANOCI-Nanotechnology Based Cochlear Implant With Gapless Interface to Auditory Neurons. Otol Neurotol. 2017;38:e224–e231. doi: 10.1097/MAO.0000000000001439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein H, Isaacson JE, Olds MJ, Rowan PT, Rosenberg S. Dexamethasone inner ear perfusion for the treatment of Meniere’s disease: a prospective, randomized, double-blind, crossover trial. Am J Otol. 1998;19:196–201. [PubMed] [Google Scholar]
- Swan EE, Mescher MJ, Sewell WF, Tao SL, Borenstein JT. Inner ear drug delivery for auditory applications. Adv Drug Deliv Rev. 2008;60:1583–1599. doi: 10.1016/j.addr.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson H, Tucker AS. Dual origin of the epithelium of the mammalian middle ear. Science. 2013;339:1453–1456. doi: 10.1126/science.1232862. [DOI] [PubMed] [Google Scholar]
- Toth AA, Parnes LS. Intratympanic gentamicin therapy for Menière’s disease: preliminary comparison of two regimens. J Otolaryngol. 1995;24:340–344. [PubMed] [Google Scholar]
- Trune DR, Canlon B. Corticosteroid therapy for hearing and balance disorders. Anat Rec (Hoboken) 2012;295:1928–1943. doi: 10.1002/ar.22576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Dellamary L, Fernandez R, Harrop A, Keithley EM, Harris JP, Ye Q, Lichter J, LeBel C, Piu F. Dose-dependent sustained release of dexamethasone in inner ear cochlear fluids using a novel local delivery approach. Audiol Neurootol. 2009;14:393–401. doi: 10.1159/000241896. [DOI] [PubMed] [Google Scholar]
- Wang X, Dellamary L, Fernandez R, Ye Q, LeBel C, Piu F. Principles of inner ear sustained release following intratympanic administration. Laryngoscope. 2011;121:385–391. doi: 10.1002/lary.21370. [DOI] [PubMed] [Google Scholar]
- Wang X, Fernandez R, Tsivkovskaia N, Harrop-Jones A, Hou HJ, Dellamary L, Dolan DF, Altschuler RA, LeBel C, Piu F. OTO-201 nonclinical assessment of a sustained-release ciprofloxacin hydrogel for the treatment of otitis media. Otol Neurotol. 2014;35:459–469. doi: 10.1097/MAO.0000000000000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk M, Hessler R, Mugridge K, Jolly C, Fehr M, Lenarz T, Scheper V. Impedance Changes and Fibrous Tissue Growth after Cochlear Implantation Are Correlated and Can Be Reduced Using a Dexamethasone Eluting Electrode. PLoS One. 2016;11:e0147552. doi: 10.1371/journal.pone.0147552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye AY, Liu Q-R, Li C-Y, Zhao M, Qu H. Human Transporter Database: Comprehensive Knowledge and Discovery Tools in the Human Transporter Genes. PLoS ONE. 2014;9(2):e88883. doi: 10.1371/journal.pone.0088883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Tillein J, Hartmann R, Gstoettner W, Kiefer J. Application of a corticosteroid (Triamcinolon) protects inner ear function after surgical intervention. Ear Hear. 2007;28:361–369. doi: 10.1097/01.aud.0000261655.30652.62. [DOI] [PubMed] [Google Scholar]
- Zehnder AF, Kristiansen AG, Adams JC, Merchant SN, McKenna MJ. Osteoprotegerin in the inner ear may inhibit bone remodeling in the otic capsule. Laryngoscope. 2005;115:172–177. doi: 10.1097/01.mlg.0000150702.28451.35. [DOI] [PubMed] [Google Scholar]
- Zhang YB, Zhang R, Zhang WF, Steyger PS, Dai CF. Uptake of gentamicin by vestibular efferent neurons and superior olivary complex after transtympanic administration in guinea pigs. Hear Res. 2012;283:169–179. doi: 10.1016/j.heares.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.