

Key Points
Ravulizumab provided rapid and sustained reduction in complement-mediated hemolysis at dosing intervals up to 12 weeks.
Patients receiving higher trough exposures experienced greater frequency of LDH normalization, with no episodes of breakthrough hemolysis.
Abstract
Ravulizumab (ALXN1210), a humanized monoclonal antibody to complement component C5, was engineered from eculizumab to have a substantially longer terminal half-life, permitting longer dosing intervals for paroxysmal nocturnal hemoglobinuria (PNH) treatment. Two phase 1b/2 multicenter open-label studies evaluated efficacy and safety of multiple doses and regimens of ravulizumab in PNH patients naive to complement-inhibitor treatment. Patients in study 103 (n = 13) received ravulizumab 900 mg (lower trough exposure) or 1800 mg every 4 weeks (higher trough exposure); those in study 201 (n = 26) received 1000 mg every 4, 1600 mg every 6, 2400 mg every 8, or 5400 mg every 12 weeks. Trough exposure levels with study 201 dosing regimens were similar to the study 103 900-mg every-4-weeks regimen. Rapid sustained reduction of plasma lactate dehydrogenase (LDH) occurred across all cohorts (73%-90% at end point vs baseline). A greater proportion of patients had normalized LDH (<234 U/L) at least once from days 29 to 253 in the higher- (85.7%) vs lower-trough-exposure (50.0%-83.3%) cohorts; the weighted average of the proportion of instances of LDH normalization from days 29 to 253 was highest in higher- vs lower-trough-exposure cohorts (62.3% vs 31.4%-54.5%). No patients in the higher-trough-exposure cohort, but 1 to 2 patients in all lower-trough-exposure cohorts, experienced breakthrough hemolysis. Ravulizumab improved quality of life (QoL) measures in all cohorts. Two patients experienced meningococcal infections; both recovered and continued in the study. In summary, ravulizumab provided rapid and sustained reduction in complement-mediated hemolysis and improved QoL at dosing intervals up to 12 weeks. This trial was registered at www.clinicaltrials.gov as #NCT02598583 (study 103) and NCT02605993 (study 201).
Visual Abstract
Introduction
Prior to the introduction of eculizumab (Soliris; Alexion Pharmaceuticals, Inc, New Haven, CT),1,2 treatment of paroxysmal nocturnal hemoglobinuria (PNH) consisted of supportive therapies such as red blood cell (RBC) transfusions to manage anemia, anticoagulants to manage thrombophilia, and immunosuppressive therapy to manage bone marrow failure, all aiming to control clinical manifestations of the disease.3 Since its approval in 2007,1,2 eculizumab transformed the management of patients with PNH. Eculizumab decreases intravascular hemolysis and thrombotic events, improves anemia, reduces need for transfusions, improves quality of life (QoL), and reduces mortality in patients with PNH.4-9
Eculizumab has a terminal half-life of ∼11 days that necessitates dosing intervals of every 2 weeks.10 Data compiled from patients with PNH have shown that 11% to 27% of patients treated with eculizumab experience breakthrough hemolysis and may require shortened dosing intervals or higher doses.6,11,12 Importantly, breakthrough hemolysis causes adverse clinical signs and symptoms and increases the risk of thrombosis,13,14 the leading cause of death in patients with PNH.15,16 In addition, a high-frequency IV dosing regimen may challenge patient adherence and impair QoL.17
To address these unmet medical needs, ravulizumab (Alexion Pharmaceuticals, Inc), an innovative, humanized monoclonal antibody to complement component C5, was engineered to achieve an extended duration of complement inhibition while retaining the efficacy and safety of eculizumab.18,19 Ravulizumab differs from eculizumab by the substitution of 4 amino acids, which alters the pharmacokinetics and pharmacodynamics of the molecule. Mechanistically, these amino acid substitutions promote endosomal dissociation of the ravulizumab-C5 complex and result in lysosomal degradation of C5, while allowing recycling of ravulizumab to the vascular space due to enhanced affinity of ravulizumab for the neonatal Fc receptor (FcRn) (supplemental Figure 1).20 These modifications resulted in a novel antibody against C5 with a terminal half-life that is 4 times longer than that of eculizumab. The molecule is thus designed to provide highly specific C5 inhibition over extended dosing intervals.18,19,21
The primary objective of studies ALXN1210-PNH-103 (study 103) and ALXN1210-PNH-201 (study 201) was to assess the efficacy and safety of multiple IV dosing regimens of ravulizumab in patients with PNH who were naive to complement inhibitor therapy. Post hoc analyses were performed to evaluate the proportion of patients achieving lactate dehydrogenase (LDH) normalization at least once after day 29 through day 253, as well as the weighted average of the proportion of instances of LDH normalization for the same time period. Additional post hoc analysis evaluated criteria for and identification of breakthrough hemolysis to further inform phase 3 studies. Preliminary reports of these studies demonstrated that ravulizumab provided rapid, sustained inhibition of complement-mediated hemolysis.21,22 Here, we report the protocol-specified end point data for both studies.
Materials and methods
Trial design and oversight
All patients provided written informed consent before study participation. The institutional review board/independent ethics committee at each site approved the protocol and informed consent form. These studies were conducted in accordance with the Declaration of Helsinki and the International Council for Harmonisation Good Clinical Practice Guidelines. The sponsor (Alexion Pharmaceuticals, Inc), in collaboration with investigators and subinvestigators, oversaw the trial design and its conduct with assistance from an independent data and safety monitoring committee. The sponsor collected and analyzed the data, to which all authors had access. All authors meet International Committee of Medical Journal Editors authorship criteria and assume responsibility for the accuracy and completeness of the reported data.
An overview of the study designs is presented in Figure 1. Study 103 was an intrapatient dose-escalation study where patients received ravulizumab once weekly or every 2 weeks during an induction phase and every 4 weeks during the maintenance period (total duration of treatment was up to 169 days). Dose escalation was staggered in successive cohorts to enable safety reviews before initiating higher doses of ravulizumab (Figure 1A). During the maintenance period, patients in cohort 1 received ravulizumab at 900 mg every 4 weeks, resulting in a lower trough exposure, whereas patients in cohort 2 received 1800 mg every 4 weeks, resulting in a higher trough exposure, thereby providing dose ranging to facilitate phase 3 dose selection. Before completion of the 2-year extension period, all patients were transitioned to the phase 3 weight-based dosing regimen of 3000 to 3600 mg every 8 weeks (NCT02946463); results from the extension period will be reported separately.
Figure 1.

Overview of study designs. (A) Study 103. Patients in cohort 1a (n = 2) received ravulizumab 400 mg on days 1 and 8, 600 mg on day 15, and 900 mg administered every 28 days beginning on day 29 for a total of 5 maintenance doses. Patients in cohort 1b (n = 4) received ravulizumab 600 mg on days 1 and 15, and then 900 mg administered every 28 days beginning on day 29 for a total of 5 maintenance doses. Patients in cohort 2 (n = 7) received ravulizumab 600 mg on day 1, 900 mg on day 15, and then 1800 mg administered every 28 days beginning on day 29 for a total of 5 maintenance doses. After receiving 5 maintenance doses, each cohort entered a 2-year extension period at the same dosage and frequency as during the maintenance period. *Enrollment into cohort 1b began after the first two 400-mg doses were confirmed safe and tolerable in the first 2 patients. **Enrollment into cohort 2 began 14 days after the first maintenance dose of 900 mg was administered to the second patient in cohort 1a and a data monitoring committee reviewed cumulative safety and tolerability data and confirmed no identified safety risks. (B) Study 201. Patients in cohort 1 (n = 6) received ravulizumab1400 mg on day 1, 1000 mg on day 15, and then 1000 mg administered every 28 days beginning on day 29 for a total of 8 maintenance doses. Patients in cohort 2 (n = 6) received ravulizumab 2000 mg on day 1, 1600 mg on day 22, and then 1600 mg every 42 days beginning on day 43 for a total of 5 maintenance doses. Patients in cohort 3 (n = 7) received ravulizumab 1600 mg on days 1 and 15, and then 2400 mg every 56 days beginning on day 29 for a total of 4 maintenance doses. Patients in cohort 4 (n = 7) received ravulizumab 3000 mg on day 1, and then 5400 mg every 84 days for a total of 3 maintenance doses. After the treatment period, each cohort entered a 2-year extension period at the same dosage and frequency that was received during the treatment period. D, day; q8w, every 8 weeks; wks, weeks.
Study 201 was conducted in parallel and tested longer dosing intervals as compared with study 103, with all cohorts achieving ravulizumab trough exposures approximating that of the study 103 lower-trough-exposure cohort. Patients received ravulizumab every 2, 3, or 4 weeks during the induction phase, which was staggered to enable safety evaluations during escalation, and every 4, 6, 8, or 12 weeks during the maintenance period (total duration of treatment was up to 253 days; Figure 1B). Before completion of the 2-year extension period, patients in cohorts 1, 2, and 3 were transitioned to the phase 3 every-8-weeks dosing regimen (NCT02946463); results from the extension period will be reported separately.
Eligibility criteria were identical for both studies, with the aim of enrolling a representative population of hemolytic PNH patients, including those who were transfusion-dependent. Men and nonpregnant women ≥18 years of age who had a diagnosis of PNH confirmed by high-sensitivity flow cytometry and were naive to complement inhibitor therapy were eligible for inclusion in the studies. Patients were required to have a baseline LDH ≥3 times the upper limit of normal (ULN) at screening and documented meningococcal vaccination <3 years prior to dosing. Key exclusion criteria included platelet count <30 × 109/L or absolute neutrophil count <0.5 × 109/L; history of bone marrow transplantation; history of major surgery <90 days prior to dosing on study day 1; or history of Neisseria meningitidis infection. All entry criteria are provided in supplemental Appendix I.
Outcome measures
The end points were similar for both studies. The primary efficacy end point was change in mean plasma LDH levels from baseline to day 169 (study 103), day 253 (study 201, cohorts 1-3), or day 281 (study 201, cohort 4). Secondary end points included changes from baseline in free hemoglobin, haptoglobin, reticulocyte levels, and clinical manifestations of PNH, including fatigue, abdominal pain, dyspnea, dysphagia, chest pain, and erectile dysfunction. Exploratory end points included change from baseline in blood transfusion requirements, major adverse vascular events (MAVEs), and estimated glomerular filtration rate (eGFR; using the Modification of Diet in Renal Disease formula); impact on QoL was measured using the Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue version 4.023,24 and the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire-Core 30 scale (EORTC QLQ-C30) version 3.0.25,26 Total scores for the FACIT-Fatigue questionnaire range from 0 to 52, with a higher score indicating less fatigue. Each subscale on the EORTC QLQ-C30 questionnaire has a range of 0 to 100, with a high score representing a higher response level; a high score on functional scales represents a high level of functioning and a high score on symptom scales represents a high level of symptomatology. Both the EORTC QLQ-C30 and FACIT-Fatigue questionnaires have been validated for use in patients with PNH.25
Post hoc analyses assessed proportions of patients reaching LDH normalization (LDH level of <234 U/L at least once after days 29 to 253, as well as the weighted average of the proportion of instances of normalized LDH from days 29 to 253) and incidence of breakthrough hemolysis during the maintenance period. A definition of breakthrough hemolysis was established for this analysis based upon thorough review of published reports and consultation with experts in PNH therapy. Breakthrough hemolysis was defined as ≥1 symptom or sign of intravascular hemolysis (fatigue, abdominal pain, shortness of breath [dyspnea], anemia [hemoglobin <100 g/L and hemoglobin less than the level at baseline], MAVEs [including thrombosis], dysphagia, or erectile dysfunction) within ±7 days of an elevated LDH ≥2 times ULN after prior LDH reduction to <1.5 times ULN on therapy.
Safety end points included adverse events (AEs) and immunogenicity, reflected by development of anti-drug antibodies.
Statistical analysis
The efficacy and safety data were analyzed using descriptive statistics. LDH levels, measured by a central laboratory, were assessed at each visit and compared with baseline, which was the average of all measurements prior to study drug initiation. The safety population included all patients who received ≥1 dose of ravulizumab. The efficacy population included all patients in the safety population who had a baseline LDH measurement and ≥1 postbaseline LDH measurement. For study 103, a sample size of 12 patients was expected to provide ∼80% power to detect a mean paired difference in LDH from baseline of −40% with an estimated standard deviation (SD) of 45%. For study 201, a sample size of 20 patients was expected to provide ∼95% power to detect a mean (SD) paired difference of −40% (45%).
Results
Patients
Of 50 patients with PNH who were screened and provided informed consent, 39 were treated with ravulizumab (study 103, n = 13; study 201, n = 26). The most common reason for screen failure was LDH levels <3 times ULN (supplemental Figure 2). All patients who fulfilled screening requirements were evaluable through the primary completion date. Patient demographics and clinical characteristics at baseline are summarized in Table 1. Most patients in study 103 were Asian and tended to have a lower baseline weight than patients in study 201. The majority of patients in study 201 cohorts 1 and 2 were white, whereas cohorts 3 and 4 included Asian patients. Patients in cohorts 3 and 4 had higher mean baseline LDH levels than patients in cohorts 1 and 2. Mean baseline free hemoglobin levels (reference range, 0-152 mg/L) and reticulocyte counts (reference range, 0.03 × 1012/L to 0.13 × 1012/L) were above the ULN in all cohorts and mean plasma haptoglobin levels were below the lower limit of normal (LLN; lower limit of detection, 0.1 g/L; reference range, 0.3-2.0 g/L) in all study cohorts. Mean PNH granulocyte clone size ranged from 78.3% to 91.1% at baseline and mean PNH type III erythrocyte clone size ranged from 23.9% to 35.1% across all study cohorts. Disease histories were similar across both studies except for higher incidences of hepatobiliary disorders in patients in study 103, and vascular disorders and history of MAVEs in patients in study 201.
Table 1.
Demographic and baseline clinical characteristics
| Characteristic | Study 201 | Study 103 | ||||||
|---|---|---|---|---|---|---|---|---|
| Cohort 1: | Cohort 2: | Cohort 3: | Cohort 4: | Overall, | Cohort 1: | Cohort 2: | Overall, | |
| 1000 mg q4w, | 1600 mg q6w, | 2400 mg q8w, | 5400 mg q12w, | 900 mg q4w, | 1800 mg q4w, | |||
| n = 6 | n = 6 | n = 7 | n = 7 | N = 26 | n = 6 | n = 7 | N = 13 | |
| Race, n (%) | ||||||||
| White | 5 (83.3) | 4 (66.7) | 3 (42.9) | 3 (42.9) | 15 (57.7) | 0 | 1 (14.3) | 1 (7.7) |
| Asian | 0 | 0 | 4 (57.1) | 3 (42.9) | 7 (26.9) | 6 (100) | 6 (85.7) | 12 (92.3) |
| Not reported | 1 (16.7) | 2 (33.3) | 0 | 0 | 3 (11.5) | 0 | 0 | 0 |
| Other | 0 | 0 | 0 | 1 (14.3) | 1 (3.8) | 0 | 0 | 0 |
| Sex, female, n (%) | 2 (33.3) | 1 (16.7) | 1 (14.3) | 2 (28.6) | 6 (23.1) | 2 (33.3) | 5 (71.4) | 7 (53.8) |
| Age at first infusion, mean (SD), y | 43.1 (14.57) | 48.6 (23.48) | 37.3 (14.03) | 48.5 (13.43) | 44.3 (16.33) | 41.1 (10.87) | 43.6 (13.48) | 42.4 (11.91) |
| Weight, mean (SD), kg | 73.3 (7.91) | 90.0 (15.58) | 73.6 (15.90) | 75.3 (13.72) | 77.8 (14.62) | 71.3 (11.06) | 64.7 (9.81) | 67.7 (10.51) |
| LDH, mean (SD), U/L | 1026.88 (547.843) | 1223.55 (149.693) | 2127.57 (815.875) | 2142.24 (366.511) | 1668.90 (724.341) | 1709.95 (582.118) | 1532.71 (355.050) | 1614.52 (461.172) |
| PNH granulocyte clone size, mean (SD), % | 85.75 (14.582) | 78.30 (24.323) | 79.42 (14.395)* | 91.06 (13.315) | 84.12 (16.914)† | 89.95 (6.981) | 91.10 (6.512) | 90.57 (6.470) |
| PNH type III erythrocyte clone size, mean (SD), % | 23.92 (20.002) | 28.25 (13.213) | 35.13 (18.331) | 32.39 (13.292) | 30.22 (16.016) | 27.08 (9.678) | 28.26 (12.813) | 27.72 (11.022) |
| Free hemoglobin, ‡ mean (SD), mg/L | 273.0 (172.29) | 234.5 (109.65) | 371.0 (278.16) | 507.0 (288.97) | 353.5 (242.05) | 237.2 (83.43) | 264.4 (109.05) | 251.8 (95.11) |
| Plasma haptoglobin,§ mean, g/L | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 |
| Reticulocytes,|| mean (SD), ×1012/L | 0.1508 (0.06227) | 0.1685 (0.06071) | 0.2214 (0.03892) | 0.2259 (0.05690) | 0.1941 (0.06125) | 0.1970 (0.07080) | 0.1549 (0.05066) | 0.1743 (0.06205) |
| Platelets, mean (SD), ×109/L | 176.0 (141.45) | 184.3 (36.11) | 178.6 (92.45) | 215.3 (136.92) | 189.2 (105.28) | 246.2 (72.93) | 224.3 (129.21) | 234.4 (103.41) |
| Neutrophils, mean (SD), ×109/L | 3.282 (2.0649) | 1.783 (0.4950) | 2.291 (1.6252) | 2.401 (1.6134) | 2.432 (1.5622) | 2.885 (1.3742) | 2.724 (1.7062) | 2.798 (1.4998) |
| eGFR, mean (SD), mL/min/1.73 m2 | 88.58 (15.474) | 100.55 (23.351) | 119.73 (32.158) | 91.46 (21.372) | 100.50 (25.986) | 112.82 (21.691) | 115.81 (34.953) | 114.43 (28.448) |
| Patients receiving transfusions in 12 mo prior to ravulizumab dosing, n (%) | 0 | 2 (33.3) | 3 (42.9) | 2 (28.6) | 7 (26.9) | 2 (33.3) | 3 (42.9) | 5 (38.5) |
| FACIT-Fatigue score, mean (SD) | 32.0 (11.02) | 29.7 (17.32) | 28.7 (15.20) | 34.4 (10.08) | 31.2 (13.09) | 35.5 (10.03) | 25.4 (14.35) | 30.1 (13.12) |
| Disease history, n (%) | ||||||||
| Blood/lymphatic disorders | 2 (33.3) | 3 (50.0) | 3 (42.9) | 4 (57.1) | 12 (46.2) | 3 (50.0) | 3 (42.9) | 6 (46.2) |
| History of aplastic anemia | 2 (33.3) | 1 (16.7) | 0 | 2 (28.6) | 5 (19.2) | 1 (16.7) | 1 (14.3) | 2 (15.4) |
| Hepatobiliary disorders | 0 | 0 | 3 (42.9) | 1 (14.3) | 4 (15.4) | 2 (33.3) | 2 (28.6) | 4 (30.8) |
| Infections/infestations | 2 (33.3) | 3 (50.0) | 3 (42.9) | 0 | 8 (30.8) | 1 (16.7) | 2 (28.6) | 3 (23.1) |
| Renal/urinary disorders | 0 | 2 (33.3) | 1 (14.3) | 2 (28.6) | 5 (19.2) | 2 (33.3) | 1 (14.3) | 3 (23.1) |
| Cardiac disorders | 1 (16.7) | 1 (16.7) | 1 (14.3) | 0 | 3 (11.5) | 0 | 2 (28.6) | 2 (15.4) |
| Vascular disorders | 2 (33.3) | 2 (33.3) | 1 (14.3) | 4 (57.1) | 9 (34.6) | 1 (16.7) | 1 (14.3) | 2 (15.4) |
| History of MAVEs, n (%) | 0 | 1 (16.7) | 0 | 2 (28.6) | 3 (11.5) | 0 | 0 | 0 |
| History of antithrombotic medication use, n (%) | 1 (16.7) | 3 (50.0) | 3 (42.9) | 3 (42.9) | 10 (38.5) | 2 (33.3) | 1 (14.3) | 3 (23.1) |
| History of systemic corticosteroid use, n (%) | 2 (33.3) | 0 | 2 (28.6) | 3 (42.9) | 7 (26.9) | 3 (50.0) | 1 (14.3) | 4 (30.8) |
eGFR, estimated glomerular filtration rate; FACIT, Functional Analysis of Chronic Illness Therapy; MAVE, major adverse vascular event; ND, no data; q4w, every 4 weeks; q6w, every 6 weeks; q8w, every 8 weeks; q12w, every 12 weeks; SD, standard deviation.
n = 5.
n = 24.
Upper limit of normal = 152 mg/L.
Reference range, 0.3 to 2.0 g/L; lower limit of detection, 0.1 g/L.
Reference range, 0.03 × 1012/L to 0.13 × 1012/L.
Efficacy
Change in LDH.
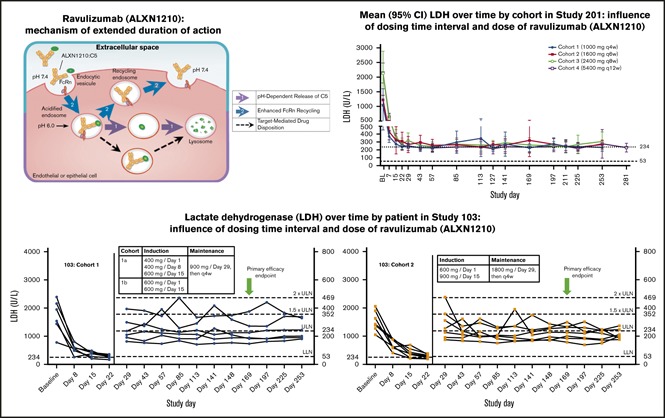
Compared with baseline, treatment with ravulizumab resulted in significant reduction in mean LDH levels in all cohorts and across all time points assessed during induction and maintenance dosing in both studies. At the primary efficacy end point, mean LDH percent reduction from baseline ranged from 72.9% in study 201/cohort 1% to 89.6% in study 201/cohort 4 (Table 2). Time-course studies of patient data from study 103 revealed that treatment with ravulizumab produced rapid, sustained reductions in plasma LDH levels as early as day 8 across both cohorts; however, a greater, more consistent reduction was observed with the higher-trough-exposure dose (cohort 2, 1800 mg every 4 weeks) than with the lower-trough-exposure dose (cohort 1, 900 mg every 4 weeks). Specifically, almost all LDH values in the higher-trough-exposure group were <1.5× ULN at all visits (except for 2 patients on day 29 and 1 patient on day 113). In contrast, there were multiple episodes in which LDH levels exceeded 1.5× ULN in the lower-trough-exposure group (Figure 2A-B). Consistent with study 103, rapid and sustained reductions in LDH were also observed, as early as day 8, in all cohorts in study 201 (Figure 2C).
Table 2.
Hemolysis, LDH normalization, and breakthrough hemolysis
| Study 201 | Study 103 | |||||
|---|---|---|---|---|---|---|
| Cohort 1: | Cohort 2: | Cohort 3: | Cohort 4: | Cohort 1: | Cohort 2: | |
| 1000 mg q4w, | 1600 mg q6w, | 2400 mg q8w, | 5400 mg q12w, | 900 mg q4w, | 1800 mg q4w, | |
| n = 6 | n = 6 | n = 7 | n = 7 | n = 6 | n = 7 | |
| LDH reduction from baseline, mean (SD),* % | 72.85 (12.082) | 77.82 (6.474) | 84.96 (4.423) | 89.58 (3.037)† | 85.95 (3.190) | 84.74 (3.774) |
| LDH at end point,‡ mean (SD), U/L | 230.00 (44.018) | 266.00 (54.262) | 306.14 (130.664) | 228.33 (52.997)† | 232.00 (82.292) | 227.86 (50.581) |
| LDH normalization achievement, days 29-253,§ n (%) | ||||||
| LDH normalized | 5 (83.3) | 3 (50.0) | 4 (57.1) | 5 (71) | 4 (66.7) | 6 (85.7) |
| LDH ≥1.5× ULN | 4 (66.7) | 3 (50.0) | 2 (28.6) | 3 (43) | 2 (33.3) | 1 (14.3) |
| LDH >2× ULN | 2 (33.3) | 1 (16.7) | 2 (28.6) | 1 (14) | 1 (16.7) | 0 |
| No., weighted average proportion of instances of LDH normalization, days 29-253, n (%) | ||||||
| LDH normalized | 38 (54.3) | 22 (31.4) | 37 (44.6) | 43 (52.4) | 36 (54.5) | 48 (62.3) |
| Breakthrough hemolysis, days 29-253,|| n (%) | ||||||
| Incidence of breakthrough hemolysis through end point‡ | 2 (33.3) | 1 (16.7) | 2 (28.6) | 1 (14.3) | 1 (16.7) | 0 (0) |
Primary efficacy end point.
n = 6 for this parameter.
LDH parameters at a protocol-specified end point: Study 103, day 169/24 weeks. Study 201: cohorts 1-3, day 253/36 weeks; cohort 4, day 281/40 weeks.
Patients meeting each parameter at least once after day 29 through day 253. Reference range, 53 to 234 U/L.
Defined as at least 1 symptom or sign of intravascular hemolysis (fatigue, abdominal pain, shortness of breath [dyspnea], anemia [hemoglobin <100 g/L and hemoglobin less than baseline hemoglobin], MAVE [including thrombosis], dysphagia, or erectile dysfunction) within ±7 days of an elevated LDH ≥2× ULN after prior LDH reduction to <1.5× ULN on therapy.
Figure 2.

Influence of dosing time interval and dose by cohort. (A-B) Study 103. LDH over time by patient. Dotted lines delineate the following LDH categories: normal, 53 to <234; 1× to 1.5× ULN, 234 to 351; >1.5× to 2× ULN, 352 to 468; >2× ULN, > 469. (C) Study 201. Mean (95% confidence interval) LDH over time. Doses shown for each cohort are maintenance doses. Green arrows represent the primary efficacy end point.
The greatest proportion of patients achieving LDH normalization at least once after days 29 to 253 (85.7%) occurred in study 103/cohort 2, which received the highest trough exposure of ravulizumab (1800 mg every 4 weeks). In the lower ravulizumab exposure cohorts, 50.0% to 83.3% of patients achieved LDH normalization at least once after days 29 to 253 (Table 2). LDH levels did not reach >2 times ULN during treatment in any patient in study 103/cohort 2 compared with at least 1 patient in every other cohort across the studies. When examined by the weighted average of the proportion of instances of LDH normalization from days 29 to 253, the average percentage of patients reaching LDH normalization was greatest in the higher-trough-exposure group (62.3%) compared with the lower-trough-exposure groups (range, 31.4%-54.5%; Table 2). No patients in study 103/cohort 2 had breakthrough hemolysis, compared with 1 to 2 patients experiencing breakthrough hemolysis in all other cohorts (Table 2).
Key secondary end points.
Mean free hemoglobin levels were above normal in all cohorts in both studies at baseline, and decreased from baseline by ≥10% with ravulizumab treatment in all cohorts except study 201/cohort 1 (supplemental Table 1); mean free hemoglobin levels decreased to the normal range (reference range, 0-152 mg/L) in the higher-trough-exposure cohort (study 103/cohort 2) at study end point. Improvement in total hemoglobin was also observed across all study cohorts (supplemental Table 1). Mean haptoglobin levels were below the LLN at baseline (reference range, 0.3-2.0 g/L) and remained below LLN throughout the maintenance period in all treatment cohorts (supplemental Table 1). Mean reticulocyte counts were above the normal range (reference range, 0.03 × 1012/L to 0.13 × 1012/L) in all cohorts at baseline and remained above ULN during maintenance treatment (supplemental Table 1).
The most common PNH-related symptom reported was fatigue (27 of 39 patients [69.2%] at baseline; 13 of 39 [33.3%] at end of maintenance treatment). Among patients who experienced PNH symptoms at baseline, symptom burden decreased during treatment with ravulizumab. The proportions of patients reporting fatigue, dyspnea, abdominal pain, chest pain, erectile dysfunction, and dysphagia all decreased during the treatment period (Table 3).
Table 3.
Patients reporting PNH-related symptoms
| Symptom | Study 201, n (%), N = 26 | Study 103, n (%), N = 13 | ||
|---|---|---|---|---|
| Baseline | Day 253 | Baseline | Day 169 | |
| Fatigue | 18 (69.2) | 6 (23.1) | 9 (69.2) | 7 (53.8) |
| Dyspnea | 9 (34.6) | 1 (3.8) | 7 (53.8) | 4 (30.8) |
| Abdominal pain | 3 (11.5) | 0 | 3 (23.1) | 0 |
| Chest pain | 3 (11.5) | 0 | 3 (23.1) | 1 (7.7) |
| Erectile dysfunction | 5 (19.2) | 1 (3.8) | 2 (15.4) | 1 (7.7) |
| Dysphagia | 3 (11.5) | 0 | 1 (7.7) | 0 |
Exploratory end points.
Clinically meaningful absolute improvements from baseline in mean FACIT-Fatigue scores (≥3) occurred in all cohorts, ranging from 6.3 to 13.9 (Table 4). Mean scores for the global health status/QoL scale on EORTC QLQ-C30 assessment improved from baseline by 8 to 28 points across the cohorts with ravulizumab treatment. Improvement in mean EORTC QLQ-C30 functional subscale scores was also observed across the study cohorts in the assessments of physical, role, emotional, and social functioning; mean scores for cognitive scale improved in 5 of 6 treatment cohorts (supplemental Table 2). Results for EORTC QLQ-C30 symptom scales are summarized in supplemental Table 3. Mean scores for the fatigue, dyspnea, appetite loss, constipation, and diarrhea symptom scales showed improvement from baseline across all cohorts in both studies. Mean scores on the nausea and vomiting, insomnia, and financial difficulty scales improved in 5 of 6 treatment cohorts; mean scores on the pain scale improved in 4 of 6 treatment cohorts.
Table 4.
FACIT-Fatigue scores
| Parameter | Study 201, mean (SD) | Study 103, mean (SD) | ||||
|---|---|---|---|---|---|---|
| Cohort 1: | Cohort 2: | Cohort 3: | Cohort 4: | Cohort 1: | Cohort 2: | |
| 1000 mg q4w, | 1600 mg q6w, | 2400 mg q8w, | 5400 mg q12w, | 900 mg q4w, | 1800 mg q4w, | |
| n = 6 | n = 6 | n = 7 | n = 7 | n = 6 | n = 7 | |
| Baseline | 32.0 (11.02)* | 29.7 (17.32) | 28.7 (15.20) | 34.4 (10.08) | 35.5 (10.03) | 25.4 (14.35) |
| End point† | 42.0 (6.45) | 39.4 (13.48)* | 40.8 (10.19)‡ | 43.3 (5.57)‡ | 41.8 (8.77) | 39.3 (10.48) |
| Absolute change from baseline | 9.8 (8.70)* | 13.6 (11.01)* | 12.3 (16.87)‡ | 11.2 (8.64)‡ | 6.3 (11.38) | 13.9 (10.42) |
Total scores range from 0 to 52, with higher score indicating less fatigue.
n = 5.
Study 103, day 169. Study 201: cohorts 1-3, day 253; cohort 4, day 281.
n = 6.
Treatment with ravulizumab was associated with a reduction in transfusions. In study 201, 7 of the 26 patients (26.9%) had received at least 1 RBC transfusion within 1 year prior to entering the study; of these 7 patients, 3 (42.8%) remained transfusion-free through the maintenance period. One patient in study 201, who had not previously received an RBC transfusion, required transfusions after initiating treatment with ravulizumab due to the new development of aplastic anemia (supplemental Appendix II). In study 103, 5 of the 13 patients (38.5%) had received at least 1 transfusion within 1 year prior to the study; of these, 3 patients (60.0%) remained transfusion-free through the ravulizumab maintenance period. Further details on the 6 patients who continued to require transfusion are provided in supplemental Appendix II.
No MAVEs were reported in either study. In study 201, mean (SD) eGFR values showed small and inconsistent changes from baseline during the maintenance period, which were not clinically meaningful. In study 103, small decreases in mean (SD) eGFR were observed in both cohorts during the maintenance period, which were not regarded as clinically meaningful.
Safety/adverse events.
Ravulizumab was well tolerated in these studies. AEs are summarized in Table 5. Headache was the most frequently reported AE overall (17 of 39 patients [43.6%]), and the most frequently reported treatment-related AE (6 of 39 patients [15.4%]), occurring most commonly at initiation of treatment with ravulizumab. Aside from meningococcal infections, all other treatment-related AEs occurred in 1 patient each.
Table 5.
Treatment-emergent AEs
| Characteristic | Study 201,* n (%) | Study 103,† n (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| Cohort 1: | Cohort 2: | Cohort 3: | Cohort 4: | Overall, | Cohort 1: | Cohort 2: | Overall, | |
| 1000 mg q4w, | 1600 mg q6w, | 2400 mg q8w, | 5400 mg q12w, | 900 mg q4w, | 1800 mg q4w, | |||
| n = 6 | n = 6 | n = 7 | n = 7 | N = 26 | n = 6 | n = 7 | N = 13 | |
| Patients with TEAEs | 6 (100) | 6 (100) | 7 (100) | 7 (100) | 26 (100) | 5 (83.3) | 7 (100) | 12 (92.3) |
| Patients with related TEAEs‡ | 2 (33.3) | 2 (33.3) | 3 (42.9) | 2 (28.6) | 9 (34.6) | 1 (16.7) | 2 (28.6) | 3 (23.1) |
| Headache | 1 (16.7) | 1 (16.7) | 1 (14.3) | 2 (28.6) | 5 (19.2) | 0 | 1 (14.3) | 1 (7.7) |
| Pain | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14.3) | 1 (7.7) |
| Nausea | 1 (16.7) | 0 | 0 | 0 | 1 (3.8) | 0 | 0 | 0 |
| Abnormal GI sounds | 0 | 0 | 1 (14.3) | 0 | 1 (3.8) | 0 | 0 | 0 |
| Asthenia | 0 | 1 (16.7) | 0 | 0 | 1 (3.8) | 0 | 0 | 0 |
| Infusion site swelling | 0 | 0 | 1 (14.3) | 0 | 1 (3.8) | 0 | 0 | 0 |
| Anemia | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 1 (7.7) |
| Atrial flutter | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14.3) | 1 (7.7) |
| Meningococcal sepsis/infection | 0 | 1 (16.7)§ | 1 (14.3)‡,§ | 0 | 2 (7.7) | 0 | 0 | 0 |
| Myalgia | 0 | 1 (16.7) | 0 | 0 | 1 (3.8) | 0 | 0 | 0 |
| Neutropenia | 0 | 1 (16.7) | 0 | 0 | 1 (3.8) | 0 | 0 | 0 |
| Skin lesion | 0 | 0 | 1 (14.3) | 0 | 1 (3.8) | 0 | 0 | 0 |
| Thrombophlebitis, superficial | 1 (16.7) | 0 | 0 | 0 | 1 (3.8) | 0 | 0 | 0 |
| Patients with serious TEAEs | 2 (33.3) | 2 (33.3) | 3 (42.9) | 1 (14.3) | 8 (30.8) | 0 | 0 | 0 |
| Patients with related serious TEAEs | 0 | 0 | 1 (14.3) | 0 | 1 (3.8) | 0 | 0 | 0 |
GI, gastrointestinal; TEAE, treatment-emergent AE.
Coded using the Medical Dictionary for Regulatory Activities v20.0.
Coded using the Medical Dictionary for Regulatory Activities v18.1.
Judged by the investigator to be possibly, probably, or definitely related to ravulizumab treatment.
This was a serious TEAE.
Two patients in study 201 experienced meningococcal infections presenting as sepsis. One patient was a 28-year-old man receiving penicillin prophylaxis who presented with symptoms on day 57. The blood culture grew serogroup Y/W135 meningococcus that showed intermediate sensitivity to penicillin. The second patient was an 18-year-old man who was not receiving penicillin prophylaxis, and who presented with symptoms on day 222; his blood culture grew penicillin-resistant serogroup Y meningococcus. Both patients had been vaccinated with ACWY and B vaccines as per the risk mitigation protocol for eculizumab; both completely recovered following treatment with IV ceftriaxone and continued receiving ravulizumab. Additional information is provided in supplemental Appendix II.
There were no discontinuations of ravulizumab during the maintenance period, no patient withdrew due to an AE, and no patients developed anti-drug antibodies. No clinically meaningful changes from baseline were observed in physical examination assessments, vital signs, electrocardiogram parameters, or laboratory assessments.
Discussion
The approved maintenance regimen of eculizumab 900 mg given every 2 weeks has been transformative for patients with PNH.4-7 However, breakthrough hemolysis has been observed in some patients especially in the context of complement-activating conditions such as infection, and particularly during the last 1 to 2 days of the 2-week dosing interval5,6,11 and in those who have received eculizumab beyond the indicated 2-week dosing interval.11 In some cases, the burden of the frequency of eculizumab infusions every 2 weeks may result in the use of unapproved dosing intervals to mitigate patients’ concerns regarding QoL and/or convenience.11
In these studies, we show that in patients with PNH who were naive to complement inhibitor therapy, treatment with ravulizumab leads to a rapid reduction in hemolysis (as reflected in LDH levels) beginning at the first evaluable time point (week 1) and continuing for ≤24 to 40 weeks of treatment at dosing intervals up to every 12 weeks. Mean reductions from baseline in LDH levels ranged from 72.9% to 89.6%, and the proportions of patients experiencing LDH normalization at least once after days 29 to 253 ranged from 50.0% to 85.7% across the cohorts. The group with higher ravulizumab trough exposure dosing had the greatest proportion of patients with normal LDH levels at least once after days 29 to 253 (6 of 7 [85.7%]). In addition, the weighted average of the proportion of instances of LDH normalization was greatest in the higher-trough-exposure group (62.3% of patients) compared with the lower-trough-exposure groups (31.4%-54.5% achieved normalization), and the higher-trough-exposure cohort was the only group where there were no incidences of breakthrough hemolysis during the entire study period. The higher-trough-exposure cohort was the only group that reached normal mean free hemoglobin levels. Total hemoglobin levels were also elevated in all cohorts.
Clinically meaningful reductions in hemolysis following treatment with ravulizumab were also associated with improvement of PNH-related symptomatology and QoL. First, the proportion of patients who reported PNH-related symptoms (fatigue [noted to be the most frequent], dyspnea, abdominal pain, chest pain, erectile dysfunction, and dysphagia) at baseline was notably reduced at end point. Second, all ravulizumab study cohorts reported improvement in FACIT-Fatigue scores (ranging from 6.3 to 13.9 points), exceeding the threshold that delineates clinically meaningful improvement (>3 points). Third, improvements in all EORTC QLQ-C30 domains were observed in study 103 and consistent improvements were observed across most of the EORTC QLQ-C30 domains in study 201.
Ravulizumab was well tolerated in these studies and exhibited a safety profile consistent with that observed for eculizumab.4-6 The most frequently reported AE was headache (17 of 39 [43.6%]), which is consistent with previously reported rates for eculizumab, ranging from 44% to 55%.4-6 Headaches that were considered related to ravulizumab commonly occurred early after the initiation of treatment, which may be associated with a transient surge in plasma nitric oxide levels induced by the cessation of the depletion of nitric oxide by free hemoglobin.5 This phenomenon is believed to be the mechanism underlying transient headaches observed during induction of treatment with eculizumab.4-6 Two patients experienced meningococcal infections; however, both fully recovered following antibiotic treatment and continued to receive ravulizumab. The risk of meningococcal infections in patients receiving eculizumab and patients with congenital deficiencies in terminal complement activity is well established4,5,27; therefore, the occurrence of meningococcal infection in patients receiving ravulizumab is not unexpected. For this reason, risk mitigation measures, including meningococcal vaccination, safety cards, and educational materials for patients and physicians have been put in place as part of the risk evaluation and mitigation strategies and risk management plans for patients receiving eculizumab.1,2 Finally, 1 patient developed moderate aplastic anemia during the course of ravulizumab treatment. This patient’s clinical history and treatment course mirrors the natural history of PNH and aplastic anemia. PNH may occur in the setting of aplastic anemia, and both diseases can be successfully treated through complement C5 inhibition and immunosuppressive therapy.8 There is no evidence that complement inhibition may be a precipitant for the development of aplastic anemia.
Meaningful interpretation of these results is facilitated by the use of identical inclusion criteria, which allowed for enrollment of a demographically broad patient population, regardless of transfusion history. The combined size of the patient population across these 2 studies is similar to the 2 original registration trials of eculizumab (TRIUMPH [N = 43]4 and AEGIS [N = 29]28), allowing comparisons to be made among patients naive to C5 antibody treatment. Finally, rigorous criteria were used to define breakthrough hemolysis and LDH normalization, which allowed for in-depth, objective assessment of dose response; use of these criteria may also be applicable in clinical settings.
Interpretation of these results may be limited by several features. First, the number of patients who received the higher-trough-exposure dose of ravulizumab (n = 7) was small compared with the number of patients who received lower-exposure doses of ravulizumab (n = 32). Second, although the duration of treatment for the primary end point was shorter in study 103 (169 days) than in study 201 (253 days for cohorts 1, 2, and 3; 281 days for cohort 4), examination of the time-course data reveals that reductions in LDH in both studies were sustained over time. Third, estimation for LDH normalization in these studies was derived from 2 post hoc analyses that tabulated the number of patients whose LDH achieved normalization at least once at any time between days 29 and 253, or when more stringently evaluated on a per-visit basis. The latter methodology revealed that 62.3% of patients receiving ravulizumab at the higher-trough-exposure levels achieved LDH normalization compared with 31.4% to 54.5% of patients across the lower-trough-exposure cohorts. In contrast, estimation of LDH normalization in the ravulizumab phase 3 studies will use methods that account for the repeated measures of LDH normalization data acquired on a per-visit basis over the 26-week primary analysis period. Finally, variability in racial demographics and mean LDH levels at baseline across the cohorts may limit comparisons between the cohorts. However, the diverse patient population and consistent response to ravulizumab therapy suggest that results here may be generalizable.
In conclusion, in these phase 1b/2 studies, treatment with ravulizumab, administered at extended dosing intervals, achieved a rapid and sustained reduction in complement-mediated hemolysis in patients with PNH. These studies demonstrated drug/exposure vs response relationships, with higher-ravulizumab-trough exposure (1800-mg every-4-weeks cohort) being associated with a greater proportion of patients reaching plasma LDH levels considered to be of low risk for hemolysis (normalization [or below 1.5× ULN]), reductions in free hemoglobin, and lack of breakthrough hemolysis relative to all other cohorts. The prolonged duration of action of ravulizumab will reduce the number of infusions required for the management of PNH, which may have a positive impact on patients’ QoL. In the phase 3 studies, patients randomized to ravulizumab receive a loading dose (2400 mg for patients ≥40 kg to <60 kg, 2700 mg for patients ≥60 kg to <100 kg, and 3000 mg for patients ≥100 kg) on day 1, followed by maintenance doses of ravulizumab (3000 mg for patients ≥40 kg to <60 kg, 3300 mg for patients ≥60 to <100 kg, and 3600 mg for patients ≥100 kg) on day 15 and every 8 weeks thereafter. The 2 phase 3 studies of ravulizumab include >440 patients with PNH who were either naive to (NCT02946463) or receiving prior complement inhibitor therapy (NCT03056040). Additionally, we continue to evaluate the every-12-weeks dosing regimen in study 201/cohort 4.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors, investigators, and sponsor thank the patients and their families for their participation in, and support for, these clinical studies. Medical writing/editorial support was provided by Michael D. Morren of Peloton Advantage, LLC (Parsippany, NJ). Editorial review was provided by Kenneth Pomerantz of Alexion Pharmaceuticals, Inc.
This work, including medical writing/editorial support, was supported by Alexion Pharmaceuticals, Inc.
A complete list of the study investigators is provided in supplemental Appendix III.
Footnotes
Portions of this work were presented at the 59th annual meeting of the American Society of Hematology, 9-12 December 2017, Atlanta, GA.
Authorship
Contribution: A.R., J.W.L., A.H., and H.S. created and designed the study and developed the protocol, recruited patients and collected data, analyzed and interpreted the data, contributed to the manuscript, and approved the final version; S.T.R. and E.S.B. created and designed the study and developed the protocol, analyzed and interpreted the data, wrote the manuscript, and approved the final version; J.S.K., L.T., A.U.-I., R.A.W., J.H.J., and A.G.K. recruited patients and collected data, analyzed and interpreted the data, contributed to the manuscript, and approved the final version; J.S. analyzed and interpreted the data, contributed to the manuscript, and approved the final version; and R.A., A.I.D. and L.S. created and designed the study and developed the protocol, analyzed and interpreted the data, contributed to the manuscript, and approved the final version.
Conflict-of-interest disclosure: A.R. has received honoraria, consulting fees, and research support from Alexion Pharmaceuticals, Inc. S.T.R., R.A., A.I.D., and L.S. are employees and stockholders of Alexion Pharmaceuticals, Inc. E.S.B. was an employee of Alexion Pharmaceuticals, Inc. at the time of the study and is a stockholder in the company. A.H. has received honoraria and consulting fees from Alexion Pharmaceuticals, Inc. H.S. has received travel support, honoraria, and research support (to University of Ulm) from Alexion Pharmaceuticals, Inc. L.T. has received travel support from Alexion Pharmaceuticals, Inc. A.G.K. has received honoraria, travel support, and consulting fees from Alexion Pharmaceuticals, Inc. R.A.W. has received honoraria, research support, and travel support from Alexion Pharmaceuticals, Inc. J.S. has received research support (to Royal Melbourne Hospital), honoraria, consulting fees, and travel support from Alexion Pharmaceuticals, Inc. J.W.L. has received honoraria, travel support, and research support (to Seoul St. Mary’s Hospital) from Alexion Pharmaceuticals, Inc. The remaining authors declare no competing financial interests.
Correspondence: Alexander Röth, Department of Hematology, West German Cancer Center, University Hospital Essen, Hufelandstr 55, D-45122 Essen, Germany; e-mail: alexander.roeth@uk-essen.de; and Jong Wook Lee, Department of Hematology, Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, 222 Banpo-daero, Seocho-gu, Seoul 06591, Republic of Korea; e-mail: jwlee@catholic.ac.kr.
References
- 1.US Food and Drug Administration. Soliris (eculizumab) [prescribing information]. New Haven, CT: Alexion Pharmaceuticals, Inc.; 2018. [Google Scholar]
- 2.European Medicines Agency. Soliris (eculizumab) [summary of product characteristics]. Paris, France: Alexion Europe SAS; 2017. [Google Scholar]
- 3.Risitano AM, Rotoli B. Paroxysmal nocturnal hemoglobinuria: pathophysiology, natural history and treatment options in the era of biological agents. Biologics. 2008;2(2):205-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hillmen P, Young NS, Schubert J, et al. . The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233-1243. [DOI] [PubMed] [Google Scholar]
- 5.Brodsky RA, Young NS, Antonioli E, et al. . Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111(4):1840-1847. [DOI] [PubMed] [Google Scholar]
- 6.Hillmen P, Muus P, Röth A, et al. . Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2013;162(1):62-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loschi M, Porcher R, Barraco F, et al. . Impact of eculizumab treatment on paroxysmal nocturnal hemoglobinuria: a treatment versus no-treatment study. Am J Hematol. 2016;91(4):366-370. [DOI] [PubMed] [Google Scholar]
- 8.Kelly RJ, Hill A, Arnold LM, et al. . Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011;117(25):6786-6792. [DOI] [PubMed] [Google Scholar]
- 9.Hillmen P, Muus P, Dührsen U, et al. . Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110(12):4123-4128. [DOI] [PubMed] [Google Scholar]
- 10.Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria [published correction appears in Nat Biotechnol. 2007;25(12):1488]. Nat Biotechnol. 2007;25(11):1256-1264. [DOI] [PubMed] [Google Scholar]
- 11.Nakayama H, Usuki K, Echizen H, Ogawa R, Orii T. Eculizumab dosing intervals longer than 17 days may be associated with greater risk of breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria. Biol Pharm Bull. 2016;39(2):285-288. [DOI] [PubMed] [Google Scholar]
- 12.Peffault de Latour R, Fremeaux-Bacchi V, Porcher R, et al. . Assessing complement blockade in patients with paroxysmal nocturnal hemoglobinuria receiving eculizumab. Blood. 2015;125(5):775-783. [DOI] [PubMed] [Google Scholar]
- 13.Lee JW, Jang JH, Kim JS, et al. . Clinical signs and symptoms associated with increased risk for thrombosis in patients with paroxysmal nocturnal hemoglobinuria from a Korean Registry. Int J Hematol. 2013;97(6):749-757. [DOI] [PubMed] [Google Scholar]
- 14.Yenerel MN, Muus P, Wilson A, Szer J. Clinical course and disease burden in patients with paroxysmal nocturnal hemoglobinuria by hemolytic status. Blood Cells Mol Dis. 2017;65:29-34. [DOI] [PubMed] [Google Scholar]
- 15.Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood. 2013;121(25):4985-4996. [DOI] [PubMed] [Google Scholar]
- 16.Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253-1258. [DOI] [PubMed] [Google Scholar]
- 17.Richter A, Anton SF, Koch P, Dennett SL. The impact of reducing dose frequency on health outcomes [published correction appears in Clin Ther. 2015;37(8):1870]. Clin Ther. 2003;25(8):2307-2335. [DOI] [PubMed] [Google Scholar]
- 18.Sheridan D, Yu Z, Zhang Y, et al. . Design and preclinical characterization of ALXN1210: a next generation anti-C5 monoclonal antibody with improved pharmacokinetics and duration of action [abstract]. Immunobiology. 2016;221(10):1158 Abstract 63. [Google Scholar]
- 19.Sahelijo L, Mujeebuddin A, Mitchell D, et al. . First in human single-ascending dose study: safety, biomarker, pharmacokinetics and exposure-response relationships of ALXN1210, a humanized monoclonal antibody to C5, with marked half-life extension and potential for significantly longer dosing intervals [abstract]. Blood. 2015;126(23). Abstract 4777. [Google Scholar]
- 20.Sheridan D, Yu ZX, Zhang Y, et al. . Design and preclinical characterization of ALXN1210: a novel anti-C5 antibody with extended duration of action. PLoS One. 2018;13(4):e0195909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JW, Bachman ES, Aguzzi R, et al. . Immediate, complete, and sustained inhibition of C5 with ALXN1210 reduces complement-mediated hemolysis in patients with paroxysmal nocturnal hemoglobinuria (PNH): interim analysis of a dose-escalation study [abstract]. Blood. 2016;128(22). Abstract 2428. [Google Scholar]
- 22.Roeth A, Rottinghaus ST, Hill A, et al. . Optimization of dose regimen for ALXN1210, a novel complement C5 inhibitor, in patients with paroxysmal nocturnal hemoglobinuria (PNH): results of 2 phase 1b/2 studies [abstract]. Blood. 2017;130(suppl 1). Abstract 3482. [Google Scholar]
- 23.Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74. [DOI] [PubMed] [Google Scholar]
- 24.Cella D, Lai JS, Chang CH, Peterman A, Slavin M. Fatigue in cancer patients compared with fatigue in the general United States population. Cancer. 2002;94(2):528-538. [DOI] [PubMed] [Google Scholar]
- 25.Weitz I, Meyers G, Lamy T, et al. . Cross-sectional validation study of patient-reported outcomes in patients with paroxysmal nocturnal haemoglobinuria. Intern Med J. 2013;43(3):298-307. [DOI] [PubMed] [Google Scholar]
- 26.Aaronson NK, Ahmedzai S, Bergman B, et al. . The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. [DOI] [PubMed] [Google Scholar]
- 27.Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev. 1991;4(3):359-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanakura Y, Ohyashiki K, Shichishima T, et al. . Safety and efficacy of the terminal complement inhibitor eculizumab in Japanese patients with paroxysmal nocturnal hemoglobinuria: the AEGIS clinical trial. Int J Hematol. 2011;93(1):36-46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.