

Abstract
Thrombotic microangiopathy (TMA) is one of the most devastating sequalae of kidney transplantation. A number of published articles have covered either de novo or recurrent TMA in an isolated manner. We have, hereby, in this article endeavored to address both types of TMA in a comparative mode. We appreciate that de novo TMA is more common and its prognosis is poorer than recurrent TMA; the latter has a genetic background, with mutations that impact disease behavior and, consequently, allograft and patient survival. Post-transplant TMA can occur as a recurrence of the disease involving the native kidney or as de novo disease with no evidence of previous involvement before transplant. While atypical hemolytic uremic syndrome is a rare disease that results from complement dysregulation with alternative pathway overactivity, de novo TMA is a heterogenous set of various etiologies and constitutes the vast majority of post-transplant TMA cases. Management of both diseases varies from simple maneuvers, e.g., plasmapheresis, drug withdrawal or dose modification, to lifelong complement blockade, which is rather costly. Careful donor selection and proper recipient preparation, including complete genetic screening, would be a pragmatic approach. Novel therapies, e.g., purified products of the deficient genes, though promising in theory, are not yet of proven value.
Keywords: Kidney transplantation, De novo thrombotic microangiopathy, Thrombotic microangiopathy, Recurrent thrombotic microangiopathy, Atypical hemolytic uremic syndrome
Core tip: Many articles in the literature have covered either de novo or recurrent thrombotic microangiopathy (TMA) in an isolated manner; we tried here in this article to gather the criteria of both types in one review for comparison. Contrary to what was believed in the past, de novo TMA is more common and its prognosis is poorer. On the other hand, recurrent TMA relies on a wide base of genetic backgrounds, with mutation errors differing in their impact on disease behavior and consequently on allograft and patient survival. This base for instance is rapidly expanding, and ultimately warrants a parallel robust work up regimen.
INTRODUCTION
Thrombotic microangiopathy (TMA) is a debilitating complication of kidney transplantation that is associated with poor patient and graft outcomes. The incidence of post-transplant TMA has been reported to be 5.6 cases per 1000 renal transplant recipients per year with a 50% mortality rate three years after diagnosis[1]. TMA after transplantation can be classified into either: (1) De novo TMA, i.e., developed for the first time without any evidence of the disease before transplant; and (2) Recurrent TMA, i.e., native kidneys failed as a result of TMA and it came back in renal transplantation. Since renal biopsy of native kidney is not performed in many patients with end stage renal disease (ESRD), missed diagnosis of TMA prior to kidney transplantation is likely. With the advent of the drug eculizumab, an anti C5 monoclonal antibody, that is highly effective in prevention as well as treatment of atypical hemolytic uremic syndrome (aHUS), it would be crucial to know the etiology of ESRD in order to differentiate de novo from recurrence. Such distinction will invariably have clear clinical and therapeutic implications. In this review, we shall try to discuss the main differences between the two categories in the pathophysiology, clinical course and available approaches of prevention and treatment.
DE NOVO TMA
In the presence of acquired or genetic dysregulation of the alternative complement pathway (AP), a number of precipitating factors have been identified in the context of renal transplantation that trigger the development of de novo TMA. These factors include the following: (1) Antibody mediated rejection (AMR); (2) Immunosuppressive-associated TMA: Calcineurin inhibitors (CNI) or mTOR inhibitors (mTORi), single or combined; (3) Other medications: e.g., anti-vascular endothelial growth factor inhibitors (anti-VGFI); (4) Viral infection: e.g., HCV, CMV, BK and parvovirus; (5) Genetic abnormalities in the complement cascade; (6) Phenotypical shift of C3 glomerulopathy (with ESRD), to an aHUS post transplantation; and (7) Missed diagnosis of TMA in the native kidney as a cause of ESRD (i.e., recurrent TMA)[2].
Which is more prevalent, de novo or recurrent TMA?
Reynolds et al[1], in a United States Renal Data System (USRDS)-based study, declared that the number of recurrent TMA cases was only 12 compared to 112 patients with de novo TMA, though the risk of post-transplant TMA recurrence was 36.5 times higher in kidney transplant recipients with ESRD due to hemolytic uremic syndrome (HUS) as compared to other etiologies (29.2% vs 0.8%)[1]. Langer et al[3] reported the incidence of de novo TMA to be 1.5%. However, the incidence of de novo TMA is mentioned to be as high as 3%-14%[4,5]. It is clear that de novo TMA is more prevalent after kidney transplantation and presumably underestimated. Graft loss rate of 40% is reported in de novo TMA within a couple of years of diagnosis[5,6].
Etiopathogenesis of de novo TMA
AMR and medications are the two main causes of de novo TMA. In addition, the role of complement abnormalities is becoming more apparent with one study reporting an underlying complement mutational abnormality in one third of patients with de novo TMA[7].
Calcineurin-induced TMA: The link between CNI (CyA and tacrolimus) administration and the evolution of de novo TMA is not a new concept. Three underlying mechanisms could explain the role of CNI in TMA development: (1) Loss of the normal balance between the vasodilator peptides (e.g., prostaglandin (PG) E2 and prostacyclin (PG12)) and the vasoconstrictor peptides (e.g., thromboxane A2 and endothelin), results in arteriolar vasoconstriction[8,9], renal ischemia and establishment of endothelial injury[10]; (2) CNI-induced platelet activation, pro-coagulant and anti-fibrinolytic activity have been shown to be involved in TMA evolution, particularly so, with an injured endothelium due to AMR, ischemia-reperfusion injury or any other etiology[10-12]; and (3) Microparticle production from endothelial cells, a known effect of CyA that can result in activation of the AP, a well-known mechanism that is implicated in TMA evolution[13]. However, three trap points have been speculated to oppose the role of CNI: (1) Patients utilizing CNI to maintain immunosuppression represent more than 95% of kidney transplant recipients (KTR), and only a small percentage can develop TMA, which suggests the presence of another underlying predisposing factor (s)[14]; (2) CNI withdrawal in de novo TMA does not always guarantee a favorable graft outcome[6]; (3) A USRDS-based study demonstrates a significantly higher incidence of TMA in the group of KTR that was not under CNI maintenance therapy (11.9/1000/year), as compared to those on CNI maintenance (5.0/1000/year)[1].
mTOR inhibitor-associated TMA: mTORi can inhibit cell cycle progression and proliferation. Both sirolimus and everolimus have been reported to be implicated in the pathogenesis of de novo TMA. The following explanations have been given: (1) mTORi has antiangiogenic properties, and can decrease renal expression of vascular endothelial growth factor (VEGF) with death of the endothelial progenitor cells. These effects are proven to be implicated in TMA pathogenesis[15,16]; (2) The VEGF inhibition has been recently proven to be associated with reduced renal levels of complement factor H (CFH)[17]. Patients with underlying CFH genetic mutations are more susceptible to develop de novo TMA, particularly with mTORi exposure[7]; (3) Repair of endothelial injury could be hampered by mTORi use[18-20]; and (4) Furthermore, the procoagulant and the antifibrinolytic activity of mTORi might play additional roles in de novo TMA development[21,22].
The exact role of mTORi in the evolution of de novo TMA is not fully understood[3,18,23]. Some authors have suggested that the impact of these medications may exceed that of CNI in the development of de novo TMA[1,24]. However, interpretation of these data may be limited by the fact that mTORi itself, e.g., sirolimus, may be used as a rescue medication in the case of diagnosis of CNI-induced TMA[1,24]. The risk of development of TMA with combined CNI and mTORi protocols is higher than using mTORi alone, an effect that has been documented in several studies. While Fortin et al[18] reported that the highest risk of de novo TMA was in the group using CNI and mTORi, Nava et al[20] studied 396 KTR, 36 (7.3%) developed TMA and 17 of them were drug-related. Not only were the drug levels of CNI and mTORi higher in the TMA group, but the sum of both drug levels in the TMA group was also higher[18-20]. An explanation for this additive risk is that the repair of the endothelial injury induced by CNI is hampered by mTORi[18-20]. Therefore, immunosuppression protocols using drug combinations should be planned cautiously, when high doses of these agents are usually used in the early post-transplant period[7].
AMR-associated de novo TMA: The role of AMR in the development of post-transplant TMA is commonly reported and well-recognized[1]. Endothelial cells are a well-known target of allo-immune response. The peritubular capillary (PTC) C4d staining (a well-recognized surrogate marker of AMR) has been reported to be present in 16.2% of biopsied recipients with TMA[1,25]. Moreover, Satoskar et al[6] reported an incidence of 55% of de novo TMA patients who express diffuse PTC C4d positivity. The observed prevalent administration of CyA in this study argued that it may have an augmenting effect on TMA prevalence. However, the observed difference between TMA in patients with C4d positive biopsy (13.6%) and that in C4d negative biopsies (3.6%) favors a postulated role of humoral rejection in the evolution of post-transplant TMA[2]. Both studies, for instance, demonstrated that clustering of both AMR and TMA would predict much worse graft outcome[6,26].
Other causes: Several less common etiologies have been reported to be involved in TMA pathogenesis and include: Viral infection, e.g., CMV infection[27,28], BK virus[29], parvovirus[30,31], chronic hepatitis C virus (with or without anti-cardiolipin seropositivity)[32,33], and antiviral medications, e.g., ribavirin and interferon[34] and disseminated histoplasmosis[35,36]. Ischemia-reperfusion injury can augment complement-associated injury through complement activation[37]. An acquired disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) deficiency-another rare risk factor- has been shown in one case to represent post-transplant TMA[38,39]. Unfortunately, the role of rare risk factors is rather difficult to evaluate in controlled studies. Living donation, on the other hand, has not been shown to guarantee any protection against graft dysfunction[5]. Interestingly, a C3 glomerulopathy disease in a native kidney can undergo phenotypical shift and present after kidney transplantation as de novo TMA[40].
Complement gene mutations: Chua et al[41] reported that renal complement activation is the common denominator in such a heterogeneous condition. They observed C4d deposits in more than 88% and C4d with localized C5b-9 in about 60% of 42 biopsy samples from patients with histologically confirmed diagnosis of TMA from a heterogenous group of patients[41]. Moreover, Le Quintrec et al[7] reported the presence of genetic mutations in CFH, Complement Factor I(CFI) or both in 29% of their studied de novo TMA patients, 25% showed low Complement Factor B (CFB) and/or low C3, suggesting an AP complement activation. No mutations have been found in healthy controls (100) or in TMA-free KTR controls[7].
Relation to TMA evolution: The AP depends on two main regulators: CFH and CFI. CFH has the ability to inhibit the C3 cleaving enzyme C3bBb. Moreover, it can serve as co-factor for FI, and the latter has the ability to inactivate C3b. Consequently, inactivation of these proteins either due to genetic mutations or development of neutralizing antibodies, can trigger an uncontrolled AP activity, leading to endothelial injury, the pathogenetic basis of TMA. Interpreting the results of the above study may suggest an overlap between aHUS and TMA. However, multiple mutational gene varieties related to complement and the coagulation-fibrinolysis cascades have been recently recognized in TMA patients[42].
Clinical manifestations
Timing: TMA could develop at any time in the post transplantation course[5,43], however this syndrome is mostly encountered in the first 3-6 mo post transplantation. This is probably when the CNI immunosuppressive trough levels are relatively higher[1].
Salient features: TMA manifestations are quite variable and can vary from a limited form confined to the kidney to a full blown systemic variant[4,6,44]. The systemic form of TMA consists of the classic triad of thrombocytopenia, microangiopathic hemolytic anemia (MAHA) and acute kidney injury (AKI). Features of MAHA include raised lactic acid dehydrogenase (LDH), drop in hemoglobin (HB) and decreased haptoglobin with schistocytes on peripheral blood smear. Localized (limited) TMA is usually presented later in TMA course, as compared to the systemic form, which can be explained by the urgency of the systemic type, necessitating the diagnostic allograft biopsy[4]. When a renal transplant recipient has significant renal dysfunction and the biopsy does not show any acute rejection, one must suspect two possibilities: (1) TMA or (2) Renal artery stenosis. The histopathologic changes are usually non-specific but vary in the acute status to the chronic angiopathic changes. In the active stage, there is evidence of endothelial cell injury with platelet aggregation (thrombosis), fibrinoid necrosis and glomerular ischemia. In the chronic stage, the basement membranes undergo duplication and multilayering with increased matrix layers and vessel wall cells, which ultimately ends in the unique onion skin formation (Figure 1)[2,45].
Figure 1.

Acute and chronic thrombotic microangiopathy and calcineurin inhibitors-associated arteriolopathy with severe acute ischemic tubular lesions. A: Advanced interstitial inflammatory fibrosis (Masson trichrome stain); B: Immunofluorescence, diffuse and segmental C3; C: C1q deposits within glomerular capillary walls; D: Diffuse acute and chronic arteriolar and glomerular thrombotic microangiopathy lesions on light microscopy (LM). (Adapted from: Yassine et al[45]).
Once the diagnosis of TMA has been established, a prompt revision of the etiology of the native kidney ESRD should be instituted. In aHUS patients who do not show systemic manifestations, the diagnosis could be obscure. In the absence of renal biopsy, many cases can be misdiagnosed as hypertensive nephrosclerosis[2]. Consequently, a prompt testing for genetic mutations should be accomplished to unmask an underlying complement dysregulation and avoid missing the diagnosis of a recurrent aHUS. This approach has key therapeutic implications, since de novo TMA has limited therapeutic options, in contrast to recurrent aHUS after transplantation, which has a better chance of C-5 blockade through the monoclonal antibody eculizumab, an effective therapeutic agent not only for treatment, but also for prevention of recurrence[2,46].
Prognosis of de novo TMA: The prognosis of post-transplant de novo TMA is quite poor for the patient and as well as the allograft. About one half of the patients loses their graft within the first two years after diagnosis[4,6]. This is supported by the USRDS-based report presented by Reynolds et al[1] that reported a patient mortality rate of 50% after three years of diagnosis. Many studies support these results[4-6,18]. To compare systemic versus localized TMA, Schwimmer et al[4] reported that 54% of systemic TMA develops dialysis-requiring AKI and 38% lost their grafts. On the other hand, none of the patients with localized TMA developed TMA-related early graft loss or required dialysis. Unfortunately, this variation in both types of behavior has not reflected on graft survival, as both types of TMA face poor long-term graft survival[2,4].
RECURRENT TMA AFTER RENAL TRANSPLANTATION
Etiology of recurrent TMA
aHUS; thrombotic thrombocytopenic purpura (TTP); and autoimmune diseases: e.g., scleroderma and systemic lupus erythematosus, with or without anti-phospholipid antibody syndrome[2].
aHUS: Recurrence of TMA in the allograft depends on the underlying type involving the native kidney. Overactivation of the AP is known to be the underlying etiology of aHUS. By far, aHUS is the most common diagnosis in TMA associated with recurrence. Risk of recurrence is greatly dependent on the underlying associated abnormality[47]. For example, mutational abnormality involving CFH and CFI, regulatory complement components produced by the liver, results in aberrant CFH and CFI. After transplant, CFH and CFI have a robust impact in the evolution of aHUS recurrence. The reported rate of aHUS recurrence approached 70%-90%[47,48]. Membrane co-factor protein (MCP), a transmembrane complement regulatory component that is produced by kidney endothelial cells even in post-transplant period, keeps aHUS recurrence lower unless other mutational gene defects have been associated[47-49]. Additional MCP mutations (> 22%), as reported by Bresin et al[50], led to graft loss due to recurrence of aHUS in one third of patients. The global rate of recurrence in aHUS patients is reported to be as high as 60%. Untreated patients, however, ultimately develop graft loss at a rate of 90%, with 80% of them occurring in the first year[50].
TTP: TTP is the second recognized etiology in TMA. Genetic or acquired lack of ADAMTS13 has been recognized. For a long period, differentiation between TTP and HUS relied primarily on the presence of neurologic manifestation in TTP and renal dysfunction in HUS to settle the diagnosis. Serological evaluation of ADAMTS13 activity is now feasible. However, complete distinction between the two clinical entities is not always possible because of overlap in manifestations. Recently, Zafrani et al[51] documented the presence of AKI in more than half of TTP patients (with low ADAMTS13 activity) and 50% progression of CKD and even ESRD. It is reasonable to expect TTP recurrence as long as the underlying defect is present after transplantation[52]. The same explanation can be applied to the autoimmune diseases, e.g., lupus nephritis, wherein patients can develop TMA in 5%-10% with documented recurrence after kidney transplantation[53-57].
Pathology: aHUS is a variety of TMA that represents the tissue response to an ongoing endothelial injury. Thrombotic features, e.g., fibrin/platelet plugging and intraluminal fibrin are not always seen in renal allograft biopsy. Non-thrombotic features can appear as denuded and swollen endothelium, mesangiolysis, glomerular basement membrane double contour, as well as accumulation of electrolucent material in the subendothelium. Arterial and arteriolar intraluminal fibrin, myxoid intimal thickening as well as concentric myointimal proliferation (onion skin appearance) have also been described[58] (Table 1).
Table 1.
Morphological features in microangiopathy
| Active lesions | Chronic lesions |
| Glomeruli: Thrombi - Endothelial swelling or denudation - Fragmented RBCs - Subendothelial flocculent material. EM: Mesangiolysis - Microaneurysms | Glomeruli: LM: Double contours of peripheral capillary walls, with variable mesangial interposition - EM: New subendothelial basement membrane - Widening of the subendothelial zone |
| Arterioles: Thrombi - Endothelial swelling or denudation-Intramural fibrin-Fragmented red blood cells-Intimal swelling-Myocyte necrosis | Arterioles: Hyaline deposits Arteries: Fibrous intimal thickening with concentric lamination (onion skin) |
| Arteries: Thrombi - Myxoid intimal swelling -Intramural fibrin- Fragmented red blood cells |
Adapted from: Goodship et al[58]. EM: Electron microscopy; LM: Light microscopy.
PATHOPHYSIOLOGY OF TMA RECURRENCE
The AP is constitutively active and is, therefore, fine-tuned. The regulatory components exist either in the serum (fluid phase) or attached onto cell membranes. CFH is the main inhibitor of the AP. CFH has the ability to work in fluid phase as well as on cell surfaces. Furthermore, CFH can act as a co-factor to CFI[59,60]. Regulatory components on cell surfaces, or “membrane regulators” include the following: (1) Membrane cofactor protein (MCP/CD46); (2) Complement receptor 1 (CR1/CD35); (3) Decay accelerating factor (DAF/CD55); and (4) Protectin (CD59), which prohibits MAC formation[61,62].
Any disturbance involving any of this protective shield will ultimately lead to complement activation with subsequent endothelial cell derangement[63]. It is increasingly recognized that complement dysregulation is the fundamental etiology involved in TMA evolution. Both genetic aberrations as well as autoantibodies can be involved in this process. Usually, there is (are) an inciting environmental trigger factor(s).
Current classification of TMA includes the following
Primary hereditary TMA: Includes mutations in ADAMTS13, MMACHC (cb1c deficiency), or in genes encoding complement components.
Primary acquired TMA: Autoantibodies to ADAMTS13 or to CFH, which occurs with homozygous CFHR3/1 deletion.
Infection-associated TMA: Shiga toxin-producing Escherichia coli-HUS (STEC-HUS) and pneumococcal HUS have distinct mechanisms that result in TMA; in other infections, the processes are ill-defined and sometimes can trigger manifestations of the primary TMA.
Secondary TMA: Presents in a variety of conditions, and in many conditions the culprit mechanisms are usually multifactorial or unknown. The shown classification (Figure 2) is not unequivocal, i.e., in some secondary forms of TMA, e.g., pregnancy-associated TMA or de novo TMA after transplantation, a significant percentage of cases may be associated with genetic predisposition (Figure 2)[64].
Figure 2.

Spectrum of thrombotic microangiopathy[64]. AAV: ANCA-associated vasculitis; ADAMTS13: A disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; aHUS: Atypical hemolytic uremic syndrome; C3G: C3 glomerulopathy; CAPS: Catastrophic antiphospholipid syndrome; cblC: Cobalamin C type; DGKE: Gene encoding diacylglycerol kinase ε; FH: Factor H; HELLP: Syndrome of hemolysis, elevated liver enzymes, and low platelets; HUS: Hemolytic uremic syndrome; IgAN: IgA nephropathy; MN: Membranous nephropathy; MPGN: Membranoproliferative GN; SRC: Scleroderma renal crisis; STEC: Shiga toxin–producing Escherichia coli; TMA: Thrombotic microangiopathy; TTP: Thrombotic thrombocytopenic purpura.
The most common complement mutation in aHUS is CFH, with 40% of cases inherited and 25% sporadic[65,66]. Furthermore, not only CFH has its impact on TMA evolution, but the CFH-related genes (CFHR1-5) have additional roles. Through deletion, hybrid protein formation and duplication[67] of these genes, the endothelial cell surface becomes denuded from its protective shield, and consequently aHUS may supervene[65,68].
The risk of aHUS recurrence could be four times higher with CFH mutations or with the carriers of CFH/CFHR1 hybrid genes[24]. On the other hand, the impact of CFI mutations is controversial. While early reports about CFI mutations documented a high rate of recurrence and graft loss[69-71], Bienaime et al[72] denied any risk of recurrence associated with CFI mutations. Le Quintrec et al[24] were in agreement with them. As MCP can normally be expressed by the endothelial cell surface of the allograft, aHUS recurrence is seldom influenced by MCP gene mutations. No more than three cases of MCP-associated recurrence have been reported[73,74], where recurrence was attributed either to combined gene mutations[49] or microchimerism related to the recipient’s endothelium[74] (Table 2).
Table 2.
Risk of atypical hemolytic uremic syndrome recurrence according to the implicated genetic abnormality
| Gene mutation | Location | Functional impact | Mutation frequency in aHUS (%) | Recurrence after transplantation (%) |
| CFH | Plasma | Loss | 20-30 | 75-90 |
| CFI | Plasma | Loss | 2-12 | 45-80 |
| CFB | Plasma | Gain | 1-2 | 100 |
| C3 | Plasma | Gain | 5-10 | 40-70 |
| MCP | Membrane | Loss | 10-15 | 15-20 |
| THBD | Membrane | Loss | 5 | One case |
| Homozygous CFHR1 del (3%-8%) | Circulating | Undetermined | 14-23 (> 90% with anti-CHF AB) | NA |
Adapted from Salvadori et al[74]. NA: Not available; CFH: Complement factor H; CFI: Complement factor I; CFB: Complement factor B; C3: Complement component 3; MCP: Membrane cofactor protein; THBD: Thrombomodulin.
There is a paucity of data on the role of thrombomodulin (THBD) gene mutations in aHUS. Like MCP, THBD is membrane-anchored, so the possibility of recurrence is rarely seen. Only a few cases have been reported[75,76]. Gain of function mutation (C3 and CFB) is vulnerable for recurrence. Recurrent aHUS with subsequent graft loss have been reported in up to four cases of CFB carriers[77,78]. On the other hand, data related to C3-asociated recurrence are conflicting. While Le Quintrec et al[24] documented recurrence in four of five allografts, Noris et al[79] reported only two cases out of seven transplants with C3 mutations. Zuber et al[80] postulated that normal C3 supplied by the graft tissues might have a protective effect.
Role of diacylglycerol kinase-ε (DGKE) mutations: Until recently, the vast majority of aHUS patients were thought to be associated with AP dysregulation. On the contrary, most patients with DGKE mutations exhibit no evidence of complement overactivity. Homozygous mutations in the gene encoding for DGKε and DGKε-associated nephropathy have been recently uncovered. Complete loss of function is associated with acute renal failure, thrombocytopenia and hemolytic anemia. Consequently, it has been postulated that the DGKε protein may play a fundamental role in regulating thrombosis in renal tissues, a robust fact that urged expert renal clinicians to include DGKE mutations in the pathophysiology of aHUS[81,82] (see treatment below).
Environmental triggers: The process of aHUS recurrence can be triggered by anti-HLA antibodies[6], viral infection, ischemia-reperfusion injury and immunosuppressive medications[83], either isolated or in clusters, which can initiate the cascade of complement activation in susceptible patients.
Clinical assessment of aHUS: Any HUS that is not due to STEC-HUS has been called aHUS[75]. The recent progress in understanding the pathophysiology and the underlying genetic factors led to the current classification of aHUS[84]. Consequently, the term “primary HUS” has been addressed by some clinicians when there is underlying abnormality in the AP. However, patients with underlying complement abnormality need a trigger factor, e.g., infection, including pneumococcal infection (T-antigen associated TMA), surgery, medications, pregnancy, so that aHUS can clinically manifest[85,86].
Acute vs chronic lesion?
Timing of an aHUS episode is not easily predictable. Many patients are at persistent risk of recurrence. In medical genetics, penetrance of any disease-causing mutation means the percentage of subjects with genetic mutations who can express clinical symptoms[87]. Penetrance in aHUS is age-related, by age 70, penetrance reaches 64%[88], which supports the presence of disease modifiers by the aging process. The fact that certain patients (3%-5%) may express more than one genetic variant supports the postulation that mutation burden determines the magnitude of disease penetrance. The late presentation of aHUS reflects the impact of the environmental triggers. However, dissociation between the pathological entities and the clinical presentation have been reported. For example, TMA can be diagnosed in tissue biopsy without simultaneous decline in platelet count. Moreover, the current use of eculizumab has its impact on the natural history of aHUS[89]. Complement inhibition can improve glomerular perfusion enough to maintain kidney function. Once this biological agent is withdrawn, the renal endothelium may interact with the complement system through an unknown mechanism. More studies are obviously warranted to declare these alterations[58].
Extrarenal manifestation: Twenty percent of aHUS patients can express extrarenal manifestations in the form of digital gangrene, cerebral artery thrombosis, myocardial infarction, in addition to ocular, GIT, pulmonary and neurologic involvement[42,90-98]. Drusen formation is not common in aHUS[99].
Laboratory investigations and differential diagnosis: Once the diagnosis of aHUS is suspected, exclusion of ADAMTS13 activity is urgently mandated to exclude TTP diagnosis. In children, TTP is less common; therefore, eculizumab therapy should be instituted early without waiting for the results of ADAMTS13 activity. In addition, 5% of STEC-HUS patients have no prodromal diarrhea and 30% of complement-mediated aHUS patients can present with a diarrheal prodrome[100].
Complement assessment in aHUS: Before commencing plasma therapy, serum complement component should be thoroughly evaluated. C3 is low in 30% of aHUS patients and, therefore cannot be used as a screening criteria for aHUS[97,101]. CD46 surface expression should be assessed by flow cytometry. Functional parameters as well as activation markers should be also determined. Whether these biological markers can be used to guide therapy requires further investigation[102] (Table 3).
Table 3.
Complement studies for atypical hemolytic uremic syndrome (aHUS)
| Complement test | aHUS |
| Complement protein levels | C3, C4, FB1, C51 |
| Complement regulatory protein levels | FH, FI, Properdin1, CD462 |
| Complement split products | C3c1, C3d1, Bb1, sC5b-91 |
| Complement functional assays | CH50, AH50, hemolytic assays, FH assays1 |
| Autoantibodies | Anti-FH |
| Genetic screening | CFH, CFI, C3, CD46, CFB Genomic rearrangements across the FH-FHR locus (e.g., by MLPA) Sequencing of coding regions and assessment of CNV Non-complement genetic screening includes THBD and DGKE |
Currently available only at specific laboratories; they are research and not clinically validated assays;
CD46 is also known as MCP. Adapted from: Goodship et al[58]. AH50: Alternative pathway hemolytic assay; C3: Complement component 3; C4: Complement component 4; C5: Complement component 5; CFB: Complement factor B gene; CFH: Complement factor H gene; CFHR: Complement factor H related genes; CFI: Complement factor I gene; CH50: Classical pathway hemolytic assay; CNV: Copy number variation; DGKE gene: Diacylglycerol kinase epsilon gene; FB: Complement factor B; FH: Complement factor H; FI: Complement factor I; MLPA: Multiplex ligation-dependent probe amplification; sC5b-9: Soluble C5b-9; THBD: Thrombomodulin; aHUS: Atypical hemolytic uremic syndrome.
Panel of genetic testing: The diagnostic list of genes of aHUS should include at least CFH, CFI, C3, CFB, THBD, CFHR1, CFHR5 and DGKE[48,65,75,103-105]. Genotyping workup should also include CFH-H3 and MCP ggaac haplotypes[106]. Recent advances in genetic surveys addressed the use of copy number variation (CNV), hybrid genes, and the complex genomic rearrangements of CFH/CFHRs genomic region[68,107-111]. The full-detailed genetic mapping, however, allows proper diagnosis and therapeutic plans, and helps in genetic counseling, particularly in living related-donation[112]. The role of living-related kidney donor transplantation in aHUS is that the culprit agent(s), either acquired or genetic, should be well-recognized, and the donor should be free of this factor(s) at the same time. Consequently, the presence of CFH or MCP mutations in the donor is not-per se- a contraindication for donation[58].
Rationale for genetic screening: The current progress in understanding the underlying genetic background of aHUS and its molecular basis makes it paramount to provide a full detailed genetic map before transplant, and the following explanations have been given: (1) Determination of the actual cause of the disease that allows for correct genetic counseling; (2) Drawing the plan of disease management; (3) Evaluating the expected response for therapy; and (4) Defining the prognostic course as well as patient and allograft survival. These studies, however, did not hamper the progress in clinical diagnosis and therapy institution before irreversible sequalae have been established[113]. A schematic presentation for the “genetic drivers” of aHUS is supplied in Figure 3[58].
Figure 3.

Genetic drivers in atypical hemolytic uremic syndrome (Adapted from: Goodship et al[58]). aHUS: Atypical hemolytic uremic syndrome; C3G: C3 glomerulopathy; CNV: Copy number variation; SCR: Short consensus repeat.
Interpretation of the genetic variants: Genetic mutations can be interpreted as: (1) Benign; (2) Likely benign; (3) Variant of uncertain significance; (4) Likely pathogenic; or (5) Pathogenic, according to the international guidelines[114].
The pathogenic mutations in aHUS have the ability to hamper the capacity to protect the endothelial lining and the platelet from the devastating effect of complement or its activation[78,115-121]. It is well-documented now that pathogenic variant combinations as well as clustering of risk factors facilitate the evolution of aHUS[49,88,122-125]. Genetic designation also has its impact on therapeutic plans, response to therapy as well as the chance for aHUS recurrence[79,126] (Table 4).
Table 4.
Genotype-phenotype correlations in atypical hemolytic uremic syndrome (data refer to the period before introduction of eculizumab)
| Gene | Risk of death or ESRD at onset or first yr | Risk of recurrence | Risk of death or ESRD after 3-5 yr | Risk of recurrence in allograft |
| CFH or CFH-CFHR1/3 hybrid genes | 50%-70% | 50% | 75% | 75%-90% |
| CFI | 50% | 10%-30% | 50%-60% | 45%-80% |
| MCP single | 0%-6% | 70%-90% | 6%-38% | < 20% |
| MCP combined1 | 30%-40% | 50% | 50% | 50%-60% |
| C3 | 60% | 50% | 75% | 40%-70% |
| CFB | 50% | 100% | 75% | 100% |
| THBD | 50% | 30% | 54% | ? |
| Anti-FH | 30%-40% | 40%-60% | 35%-60% | Depends on antibody titers |
Combined with CFH or CFI or C3 mutations. Adapted from: Goodship et al[58]. CFB: Complement factor B gene; CFH: Complement factor H gene; CFHR: Complement factor H-related genes; CFI: Complement factor I gene; FH: Factor H protein; THBD: Thrombomodulin gene.
Acquired drivers of aHUS: The FH autoantibodies are the best reported example. It is typically characterized by homozygosity for delCFHR3-CFHR1. Test results need to be confirmed after two weeks if the initial results were positive. According to the consensus guidelines in pediatrics, CFH autoantibodies assessment should be confirmed, if positive, on a regular basis[84]. About a quarter of patients with anti-CFH-associated HUS are vulnerable for relapse.
Diagnosis of aHUS recurrence: A full detailed clinical history is usually warranted. A proven tissue diagnosis with light microscopy (LM), immunofluorescence (IF) and electron microscopy (EM) studies supporting the diagnosis of aHUS in the native kidney should be available. However, once diagnosis of aHUS is suspected, a full battery of biochemical, genetic as well as pathological investigations of the AP should be accomplished[127], including the following: (1) Estimation of the anti-CFH AB; (2) MCP screening on the peripheral blood WBCs; (3) Examination of the recombination in CFHR region; and (4) Screening of the genetic mutations related to CFH, CFI, CFB, C3, and MCP.
The impact of various genetic mutations on allograft survival is not universally quantifiable. Not all of the genetic mutations share the same magnitude of risk on allograft survival. Despite the fact that genetic screening is difficult and complex and the spectrum of gene mutation is a continuously expanding field[102], performing such studies is fundamental to determining the possible outcome of the kidney transplant in the set of aHUS recurrence[128].
THERAPY OF POST-TRANSPLANT TMA
Treatment of de novo TMA
In view of the extreme heterogenicity of the mechanisms related to variable etiologies of TMA, therapeutic maneuvers should be individualized for each patient. Institution of therapeutic options is highly dependent on diagnosis as well as the patient’s response. The following approaches have been suggested: (1) Immunosuppressive medication management: the role of immunosuppressive medications (e.g., CNI or mTORi) has been reported in the literature, with a documented better response after switching from one CNI member to another or to an mTORi)[5,129-134]. However, this was not agreed by Satoskar et al[6], who denied any difference in outcomes between temporary discontinuation, dose modulation, withdrawal or continuation of CyA in management of de novo TMA. Whatever the situation would be, the withdrawal of the offending agent should be the first line in treating de novo TMA, a fundamental step that ultimately results in correction of the hematological profile[2]; (2) Plasmapheresis (PE) and intravenous immunoglobulins (IVIG): The following rationales have been addressed in favor of PE/IVIG therapy: Depending on its efficacy in treating patients with TTP[135,136], and previous choice as a first line therapy for aHUS (replaced now by eculizumab), PE with IVIG has been extrapolated to be used early in treating de novo TMA patients. In 2003, Karthikeyan et al[43] reported a graft salvage rate with PE approaching 80%. Two benefits have been postulated for this type of therapy: Removal of the platelet aggregation factors, e.g., thromboxane A2 and the simultaneous replenishment of the deficient factors, e.g., PGI2-stimulating factor[43]. With the possibility of the presence of underlying complement dysregulation in patients undergoing kidney transplantation due to systemic TMA[7], in the same manner, it is reasonable to speculate that PE can be beneficial for two reasons: Removal of the abnormal mutant complement proteins and supplying normally functioning complement components[7]. In AMR-associated TMA, an improved outcome has been reported, which was attributed to removal of the anti-HLA antibodies[6,137]. A 100% response has been reported to be associated with PE/IVIG therapy in five solid organ transplantation with systemic TMA with no evidence of relapse after withdrawal of the culprit agent (e.g., tacrolimus) in a recent study[2]; (3) Belatacept: A promising alternate option that allows withdrawal of the offending drug incriminated in TMA evolution. Belatacept is an immunosuppressive co-stimulatory blocker against CD80 and CD86 surface ligands and CD28 on T cells. The first case report in 2009 documented TMA resolution after belatacept therapy used for immunosuppression in post-transplantation TMA due to CNI-induced endothelial toxicity[138]. Two case series have followed, thereafter documenting fair graft outcome due to resolution of the CNI-induced TMA[139,140]. Of note, belatacept has nothing to do with the underlying endothelial derangement, its role is only to replace/displace the culprit drug[2]; and (4) Complement inhibition: Eculizumab, an anti-C5 agent, blocks the lytic C5b-9 membrane attack complex generation. This recombinant monoclonal antibody addressed a breakthrough in the management of aHUS, as it was proven to be effective in treatment as well as in prevention of recurrent aHUS after renal transplantation[141]. A large percentage of patients with diagnosed TMA express complement activation, including those patients with unrecognized complement genes[2]. For example, Chua et al[41] reported C4d renal deposition in all histologically documented cases with post-transplantation TMA. These data delineate that complement overactivation can be considered as one of the final common pathways incriminated in TMA evolution[2]. Consequently, anti-complement therapy has been suggested to have a fundamental role in the management of de novo post-transplantation TMA. Efficacy of eculizumab has been documented in several case reports and case series in management of resistant cases of medication-associated TMA, including cases with unrecognized genetic defects[142-147]. This efficacy has been also documented in patients with refractory AMR with TMA[147-156].
On the other hand, Cornell et al[157] reported no difference in death-censored graft survival or biopsy finding at one year when they compared the outcome of eculizumab-treated patients with positive cross matching with controls, even though the incidence of acute AMR was less in the eculizumab group. So, in view of these conflicting results as well as considering the high cost of the drug, the use of this vital biological agent should be confined to a specified subset of de novo TMA patients, presumably: (1) AMR-associated TMA; (2) Patients who became PE-dependent; and (3) Refractory hemolysis persists despite maximum doses of PE therapy. However, more efforts are still warranted to declare the best way to utilize this unique agent and which subset of TMA patients are the best candidates for this costly drug. An urgent need for new biomarkers is also warranted for early detection of complement overactivity[2] (see kidney transplantation without eculizumab prophylaxis below).
Treatment of recurrent TMA
Recommendations for recurrent TMA: First of all, it is worthy to remember that most of the recommendations about recurrence and therapeutic advices relied primarily on case reports (level 4 evidence) as well as experts’ opinions (level 5 evidence) rather than on randomized controlled trials (level 1b evidence). (1) The minimal list of genetic screening should include: CFH, CFI, CFHR, CFB, MCP and C3[158]; (2) All patients with primary or suspected aHUS, should be surveyed for all complement components and its related proteins; (3) Patients with isolated MCP associated mutations (not combined with other mutations) may be safe for kidney donation; (4) Patients with documented aHUS and with lack of definite genetic mutations can proceed in renal transplantation under the umbrella of intensive plasma exchange therapy[159]; and (5) Polygenic pattern for aHUS patients should be handled with extreme caution in case of living donation[80].
Prevention of aHUS: The following strategies are suggested to decrease/prevent aHUS: (1) Complement activity incited by an injury to endothelium, e.g., ischemia-reperfusion injury, viral infection and immunosuppressive medications[127], should be avoided; (2) Certain relations have been reported between CNI use and aHUS recurrence[160], which is not confirmed by other authors[15,112], even the usual substitute in such a case (an mTOR) is not innocent and can induce recurrence[15,112]; (3) We cannot depend solely on PE therapy in management of aHUS recurrence for several reasons: PE failed to prevent aHUS recurrence in many cases[161]; PE cannot guarantee prevention of aHUS recurrence after cessation of therapy; Many cases under PE therapy were proved to develop “subclinical” aHUS recurrence, which means that PE therapy cannot influence complement activity; Prophylactic use of rituximab proved to be efficacious as anti-CFH-antibodies[162], the beneficial effect of rituximab can be enhanced by adding PE therapy[163,164]; and (4) The anti-C5 monoclonal antibiotic eculizumab has been reported to be used successfully to prevent aHUS recurrence in patients with CFH, CFH/CFHR1 hybrid genes as well as with C3 gene mutations[165-168] (see below).
Prophylactic complement blockade: Gene abnormalities have been reported to be associated with aHUS recurrence in 80% of patients[112]. In light of robust evidence of increased complement activity during aHUS episodes[169,170] after exposure to a trigger, e.g., surgery or infection, clinical indication of complement blockade is suggested[171]. However, this explanation lacks enough evidence (Figure 4[58]).
Figure 4.

Prophylaxis against atypical hemolytic uremic syndrome recurrence in allograft based on a risk-assessment strategy[96] (Adapted from: Goodship et al[58]). 1Requires complete screening of all genes implicated in atypical hemolytic uremic syndrome; 2Prophylactic regimens are based on local center protocols; no trial data exist to support superiority of one protocol over another; 3Liver transplantation can be considered for renal transplant recipients with liver-derived complement protein abnormalities, uncontrolled disease activity despite eculizumab therapy or financial considerations regarding cost of long-term eculizumab therapy; 4Decision to perform or not to perform prophylactic plasma exchange or complement inhibition is left to the discretion of the clinician. aHUS: Atypical hemolytic uremic syndrome; CFI: Complement factor I gene; FH: Complement factor H protein; MCP: Membrane cofactor protein gene.
Therapeutic protocols for aHUS recurrence: Once the diagnosis of primary aHUS has been established, complement blockade therapy should be instituted. The available data points to two strategies: (1) Minimal dosage to establish complement blockade; and (2) Dose withdrawal scheme[142]. Both options, however, lack enough evidence and require precise monitoring of complement blockade (Table 5).
Table 5.
Eculizumab dosing in atypical hemolytic uremic syndrome based on dosing goal, one additional monitoring may be required during intercurrent events (e.g., infection, surgery, vaccination) to detect unblocked complement activity
| Minimal dose |
| Desire to continue dosing with the minimal dose required to achieve a pre-identified level of complement blockade 1 Dose reduction or interval extension Goal CH50 < 10% (recommended) Goal AH50 < 10% (recommended) Goal eculizumab trough > 100 μg/mL |
| Discontinuation |
| Desire to discontinue complement blockade: No consensus exists regarding tapering of dose |
Adapted from: Goodship et al[58]. AH50: Alternative pathway hemolytic activity; CH50: Total complement activity.
FH autoantibody-driven aHUS: Anti-cellular therapy is recommended, with close monitoring of the antibody titer (Figure 5). How to monitor complement blockade? Detailed description is shown in Table 6.
Figure 5.

Treatment of complement factor H autoantibody-mediated atypical hemolytic uremic syndrome. There are no prospective controlled studies in patients with atypical hemolytic uremic syndrome (aHUS) due to anti–factor H protein (FH) antibodies, and thus the proposed management is based on a pediatric consensus[84] (Adapted from: Goodship et al[58]). aAbnormal titer depends on the testing laboratory; bThe decision to use plasma therapy versus eculizumab will be based on patient age and local resource availability; cCyclophosphamide, rituximab, or mycophenolate mofetil; dThe decision to continue anticomplement therapy indefinitely is not informed by data; eThe interval may be monthly or quarterly and is based on local resources; fThis recommendation is based on limited retrospective case reviews[172-174].
Table 6.
Monitoring eculizumab therapy
| Description | |
| CH50 (total complement activity) | Measures the combined activity of all of the complement pathways Tests the functional capability of serum complement components to lyse 50% of sheep erythrocytes in a reaction mixture Low in congenital complement deficiency (C1-8) or during complement blockade Normal range is assay dependent Recommended goal during therapeutic complement blockade: < 10% of normal |
| AH50 (alternative pathway hemolytic activity) | Measures combined activity of alternative and terminal complement pathways Tests the functional capability of alternate or terminal pathway complement components to lyse 50% of rabbit erythrocytes in a Mg2+-EGTA buffer Will be low in congenital C3, FI, FB, properdin, FH, and FD deficiencies or during terminal complement blockade Normal range is assay dependent Recommended goal during complement blockade: < 10% of normal |
| Eculizumab trough | May be a free or bound level ELISA: Using C5 coated plates, patient sera, and an anti-human IgG detection system Not affected by complement deficiencies Recommended trough level during complement blockade: 50-100 μg/mL |
| Alternative assays | The following assays are under investigation (or awaiting to be replicated in different laboratories)[83] as a means to monitor therapeutic complement blockade Free C5 In vitro human microvascular endothelial cell test sC5b -9 (also referred to as sMAC and TCC) may remain detectable in aHUS patients in remission and therefore is not recommended as a monitoring tool |
Adapted from: Goodship et al[58]. aHUS: Atypical hemolytic uremic syndrome; C3: Complement component 3; C5: Complement component 5; EGTA: Ethyleneglycol tetraacetic acid; ELISA: Enzyme-linked immunosorbent assay; FB: Complement factor B; FD: Complement factor D; FH: Complement factor H; FI: Complement factor I; sC5b-9: Soluble C5b-9; sMAC: Soluble membrane attack complex; TCC: Terminal complement complex.
Duration of therapy: There is not enough data supporting life-long therapy for aHUS. Cessation of therapy appears to be plausible in certain situations (Figure 6). Enough time, however, should be permitted to optimize renal recovery and satisfy TMA resolution. Early biomarkers of disease relapse due to complement activation or endothelial derangement as well as their inciting triggers should be thoroughly investigated in the future.
Figure 6.
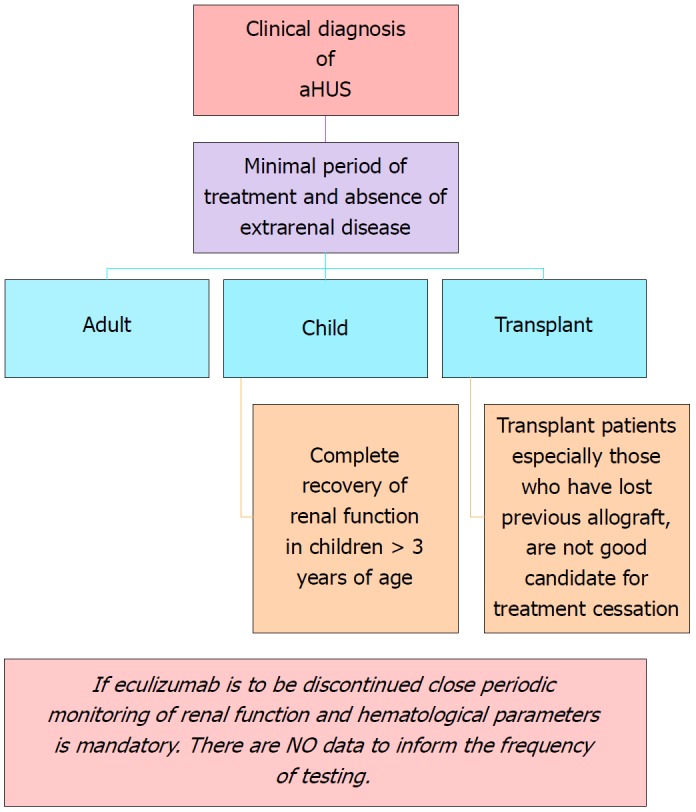
Recommendations for cessation of treatment with complement inhibitors. There are no prospective controlled studies in patients with atypical hemolytic uremic syndrome (aHUS) to define criteria for discontinuation of eculizumab therapy. This flow diagram is based on expert opinion[176-178]. Discontinuation can be considered on a case-by-case basis in patients after at least 6-12 mo of treatment and at least 3 mo of normalization (or stabilization in the case of residual CKD) of kidney function. Earlier cessation (at 3 mo) may be considered in patients (especially children) with pathogenic variants in MCP if there has been rapid remission and recovery of renal function. Patients on dialysis, eculizumab should be maintained for at least 4 to 6 mo before discontinuation. In this setting, assessment of fibrotic changes in kidney biopsy may be helpful. In transplant patients, especially patients who have lost previous allografts, discontinuation is not recommended. Adapted from: Goodship et al[58]. aHUS: Atypical hemolytic uremic syndrome.
Unanswered questions: There is paucity of information about this biological agent, e.g., what is the most optimal dose? What are the ideal dose-intervals? For how long should this kind of costly therapy be continued?[175] What impact does this agent have on the spectrum of renal transplantation[113]?
Cessation of therapy: The following scheme is suggested for withdrawal of complement blockade therapy (Figure 6).
Kidney transplantation without eculizumab prophylaxis: A case series presented by Verhave et al[179] described successful kidney transplantation without recurrence in four high risk aHUS patients. They received living donor kidney with therapeutic protocol consisted of: Basiliximab for induction, tacrolimus in low dose, and prednisone and mycophenolate mofetil as immunosuppressive in addition to a statin. Additional precautions include lowering the blood pressure and minimizing the cold ischemic time. No recurrence or rejection has been observed after 16-21 mo. This case series heralds the possibility of successful kidney transplantation in recurrent aHUS without the need for prophylactic eculizumab through minimizing cold ischemic time, decreasing the risk of rejection and, thereby, providing endothelial protection[179].
Treatment of DGKE mutation associated TMA: The role of complement blockade here is questionable. Many cases experienced disease remission with no specific therapy. Azukaitis[82] and colleagues reported the feasibility of kidney transplantation in five patients with no recurrence after transplantation.
RENAL TRANSPLANTATION
Timing
Renal transplantation should be postponed six months after institution of dialysis, as limited kidney recovery can occur several months after commencing eculizumab therapy[170,180]. Disappearance of the extrarenal manifestations as well as resolution of TMA hematological parameters are the prerequisite for kidney transplantation. The magnitude of risk of recurrence can be utilized to guide the necessity of anti-complement blockade (Table 2).
Risk of kidney donation
Two risks have been reported to be associated with living-related kidney donation: (1) Recurrent disease in the recipient; and (2) De novo disease in the donor, if he/she is a genetic mutation carrier[169]. Any potential donor proved to exhibit alternative pathway dysregulation should be excluded. On the other hand, any potential living-related donor devoid of complement gene abnormalities can be permitted[113]. “Liver transplantation” may be reserved for patients with liver-derived complement protein aberrations, particularly in patients poorly responding to complement blockade[181].
Future therapy
The following future therapeutic agents have been addressed: (1) Purified products of the deficient genes; and (2) C3 convertase inhibitors[182].
Research targets
The following agents are under investigation: (1) The anti-C3b blocker, compstatin analog Cp40[183]; and (2) The anti-C3 convertase monoclonal antibodies[184].
CONCLUSION
The impact of TMA, either de novo or recurrent, on allograft longevity is underestimated. The spectrum of the culprit genes implicated in the evolution of TMA is currently expanding. Despite the landmark breakthrough of immense efficacy of complement blockade therapy, the outlook of this devastating syndrome remains poor if the diagnosis is delayed. In contrast, the recurrent TMA is much more optimistic if there is timely intervention by complement blockade before permanent damage sets in. More efforts targeting genetic mutation management as well as the advent of early predictors of TMA recurrence are warranted for better disease control and, thereby, better patient and allograft outcome.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Transplantation
Country of origin: United Kingdom
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: No conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 11, 2018
First decision: March 30, 2018
Article in press: July 10, 2018
P- Reviewer: Sanchez-Zapardiel E S- Editor: Ji FF L- Editor: Filipodia E- Editor: Yin SY
Contributor Information
Fedaey Abbas, Nephrology Department, Jaber El Ahmed Military Hospital, Safat 13005, Kuwait; Faculty of Health and Science, University of Liverpool, Institute of Learning and Teaching, School of Medicine, Liverpool L69 3GB, United Kingdom.
Mohsen El Kossi, Faculty of Health and Science, University of Liverpool, Institute of Learning and Teaching, School of Medicine, Liverpool L69 3GB, United Kingdom; Doncaster Renal Unit, Doncaster Royal Infirmary, Doncaster DN2 5LT, United Kingdom.
Jon Jin Kim, Faculty of Health and Science, University of Liverpool, Institute of Learning and Teaching, School of Medicine, Liverpool L69 3GB, United Kingdom; Nottingham Children Hospital, Nottingham NG7 2UH, United Kingdom.
Ajay Sharma, Faculty of Health and Science, University of Liverpool, Institute of Learning and Teaching, School of Medicine, Liverpool L69 3GB, United Kingdom; Transplant Surgery, Royal Liverpool University Hospitals, Liverpool UK L7 8XP, United Kingdom.
Ahmed Halawa, Faculty of Health and Science, University of Liverpool, Institute of Learning and Teaching, School of Medicine, Liverpool L69 3GB, United Kingdom; Department of Transplantation Surgery, Sheffield Teaching Hospitals, Sheffield S57AU, United Kingdom. ahmed.halawa@sth.nhs.uk.
References
- 1.Reynolds JC, Agodoa LY, Yuan CM, Abbott KC. Thrombotic microangiopathy after renal transplantation in the United States. Am J Kidney Dis. 2003;42:1058–1068. doi: 10.1016/j.ajkd.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Garg N, Rennke HG, Pavlakis M, Zandi-Nejad K. De novo thrombotic microangiopathy after kidney transplantation. Transplant Rev (Orlando) 2018;32:58–68. doi: 10.1016/j.trre.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Langer RM, Van Buren CT, Katz SM, Kahan BD. De novo hemolytic uremic syndrome after kidney transplantation in patients treated with cyclosporine-sirolimus combination. Transplantation. 2002;73:756–760. doi: 10.1097/00007890-200203150-00017. [DOI] [PubMed] [Google Scholar]
- 4.Schwimmer J, Nadasdy TA, Spitalnik PF, Kaplan KL, Zand MS. De novo thrombotic microangiopathy in renal transplant recipients: a comparison of hemolytic uremic syndrome with localized renal thrombotic microangiopathy. Am J Kidney Dis. 2003;41:471–479. doi: 10.1053/ajkd.2003.50058. [DOI] [PubMed] [Google Scholar]
- 5.Zarifian A, Meleg-Smith S, O’donovan R, Tesi RJ, Batuman V. Cyclosporine-associated thrombotic microangiopathy in renal allografts. Kidney Int. 1999;55:2457–2466. doi: 10.1046/j.1523-1755.1999.00492.x. [DOI] [PubMed] [Google Scholar]
- 6.Satoskar AA, Pelletier R, Adams P, Nadasdy GM, Brodsky S, Pesavento T, Henry M, Nadasdy T. De novo thrombotic microangiopathy in renal allograft biopsies-role of antibody-mediated rejection. Am J Transplant. 2010;10:1804–1811. doi: 10.1111/j.1600-6143.2010.03178.x. [DOI] [PubMed] [Google Scholar]
- 7.Le Quintrec M, Lionet A, Kamar N, Karras A, Barbier S, Buchler M, Fakhouri F, Provost F, Fridman WH, Thervet E, et al. Complement mutation-associated de novo thrombotic microangiopathy following kidney transplantation. Am J Transplant. 2008;8:1694–1701. doi: 10.1111/j.1600-6143.2008.02297.x. [DOI] [PubMed] [Google Scholar]
- 8.Remuzzi G, Bertani T. Renal vascular and thrombotic effects of cyclosporine. Am J Kidney Dis. 1989;13:261–272. doi: 10.1016/s0272-6386(89)80032-0. [DOI] [PubMed] [Google Scholar]
- 9.Ramírez C, Olmo A, O’Valle F, Masseroli M, Aguilar M, Gómez-Morales M, Revelles F, García-Chicano MJ, Arrebola F, Reguero ME, et al. Role of intrarenal endothelin 1, endothelin 3, and angiotensin II expression in chronic cyclosporin A nephrotoxicity in rats. Exp Nephrol. 2000;8:161–172. doi: 10.1159/000020664. [DOI] [PubMed] [Google Scholar]
- 10.Sahin G, Akay OM, Bal C, Yalcin AU, Gulbas Z. The effect of calcineurin inhibitors on endothelial and platelet function in renal transplant patients. Clin Nephrol. 2011;76:218–225. [PubMed] [Google Scholar]
- 11.Tomasiak M, Rusak T, Gacko M, Stelmach H. Cyclosporine enhances platelet procoagulant activity. Nephrol Dial Transplant. 2007;22:1750–1756. doi: 10.1093/ndt/gfl836. [DOI] [PubMed] [Google Scholar]
- 12.Verpooten GA, Cools FJ, Van der Planken MG, Bedert LC, Claes R, Van Gaal LF, De Broe ME. Elevated plasminogen activator inhibitor levels in cyclosporin-treated renal allograft recipients. Nephrol Dial Transplant. 1996;11:347–351. doi: 10.1093/oxfordjournals.ndt.a027265. [DOI] [PubMed] [Google Scholar]
- 13.Renner B, Klawitter J, Goldberg R, McCullough JW, Ferreira VP, Cooper JE, Christians U, Thurman JM. Cyclosporine induces endothelial cell release of complement-activating microparticles. J Am Soc Nephrol. 2013;24:1849–1862. doi: 10.1681/ASN.2012111064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mulgaonkar S, Kaufman DB. Conversion from calcineurin inhibitor-based immunosuppression to mammalian target of rapamycin inhibitors or belatacept in renal transplant recipients. Clin Transplant. 2014;28:1209–1224. doi: 10.1111/ctr.12453. [DOI] [PubMed] [Google Scholar]
- 15.Sartelet H, Toupance O, Lorenzato M, Fadel F, Noel LH, Lagonotte E, Birembaut P, Chanard J, Rieu P. Sirolimus-induced thrombotic microangiopathy is associated with decreased expression of vascular endothelial growth factor in kidneys. Am J Transplant. 2005;5:2441–2447. doi: 10.1111/j.1600-6143.2005.01047.x. [DOI] [PubMed] [Google Scholar]
- 16.Miriuka SG, Rao V, Peterson M, Tumiati L, Delgado DH, Mohan R, Ramzy D, Stewart D, Ross HJ, Waddell TK. mTOR inhibition induces endothelial progenitor cell death. Am J Transplant. 2006;6:2069–2079. doi: 10.1111/j.1600-6143.2006.01433.x. [DOI] [PubMed] [Google Scholar]
- 17.Keir LS, Firth R, Aponik L, Feitelberg D, Sakimoto S, Aguilar E, Welsh GI, Richards A, Usui Y, Satchell SC, et al. VEGF regulates local inhibitory complement proteins in the eye and kidney. J Clin Invest. 2017;127:199–214. doi: 10.1172/JCI86418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fortin MC, Raymond MA, Madore F, Fugère JA, Pâquet M, St-Louis G, Hébert MJ. Increased risk of thrombotic microangiopathy in patients receiving a cyclosporin-sirolimus combination. Am J Transplant. 2004;4:946–952. doi: 10.1111/j.1600-6143.2004.00428.x. [DOI] [PubMed] [Google Scholar]
- 19.Robson M, Côte I, Abbs I, Koffman G, Goldsmith D. Thrombotic micro-angiopathy with sirolimus-based immunosuppression: potentiation of calcineurin-inhibitor-induced endothelial damage? Am J Transplant. 2003;3:324–327. doi: 10.1034/j.1600-6143.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- 20.Nava F, Cappelli G, Mori G, Granito M, Magnoni G, Botta C, Solazzo A, Fontana F, Baisi A, Bonucchi D. Everolimus, cyclosporine, and thrombotic microangiopathy: clinical role and preventive tools in renal transplantation. Transplant Proc. 2014;46:2263–2268. doi: 10.1016/j.transproceed.2014.07.062. [DOI] [PubMed] [Google Scholar]
- 21.Keller K, Daniel C, Schöcklmann H, Endlich KH, Kerjaschki D, Johnson RJ, Hugo C. Everolimus inhibits glomerular endothelial cell proliferation and VEGF, but not long-term recovery in experimental thrombotic microangiopathy. Nephrol Dial Transplant. 2006;21:2724–2735. doi: 10.1093/ndt/gfl340. [DOI] [PubMed] [Google Scholar]
- 22.Baas MC, Gerdes VE, Ten Berge IJ, Heutinck KM, Florquin S, Meijers JC, Bemelman FJ. Treatment with everolimus is associated with a procoagulant state. Thromb Res. 2013;132:307–311. doi: 10.1016/j.thromres.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Crew RJ, Radhakrishnan J, Cohen DJ, Stern L, Goldstein M, Hardy M, D’Agati VD, Markowitz GS. De novo thrombotic microangiopathy following treatment with sirolimus: report of two cases. Nephrol Dial Transplant. 2005;20:203–209. doi: 10.1093/ndt/gfh334. [DOI] [PubMed] [Google Scholar]
- 24.Le Quintrec M, Zuber J, Moulin B, Kamar N, Jablonski M, Lionet A, Chatelet V, Mousson C, Mourad G, Bridoux F, et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant. 2013;13:663–675. doi: 10.1111/ajt.12077. [DOI] [PubMed] [Google Scholar]
- 25.Meehan SM, Kremer J, Ali FN, Curley J, Marino S, Chang A, Kadambi PV. Thrombotic microangiopathy and peritubular capillary C4d expression in renal allograft biopsies. Clin J Am Soc Nephrol. 2011;6:395–403. doi: 10.2215/CJN.05870710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu K, Budde K, Schmidt D, Neumayer HH, Lehner L, Bamoulid J, Rudolph B. The inferior impact of antibody-mediated rejection on the clinical outcome of kidney allografts that develop de novo thrombotic microangiopathy. Clin Transplant. 2016;30:105–117. doi: 10.1111/ctr.12645. [DOI] [PubMed] [Google Scholar]
- 27.Rane S, Nada R, Minz M, Sakhuja V, Joshi K. Spectrum of cytomegalovirus-induced renal pathology in renal allograft recipients. Transplant Proc. 2012;44:713–716. doi: 10.1016/j.transproceed.2011.11.052. [DOI] [PubMed] [Google Scholar]
- 28.De Keyzer K, Van Laecke S, Peeters P, Vanholder R. De novo thrombotic microangiopathy induced by cytomegalovirus infection leading to renal allograft loss. Am J Nephrol. 2010;32:491–496. doi: 10.1159/000321328. [DOI] [PubMed] [Google Scholar]
- 29.Petrogiannis-Haliotis T, Sakoulas G, Kirby J, Koralnik IJ, Dvorak AM, Monahan-Earley R, DE Girolami PC, DE Girolami U, Upton M, Major EO, et al. BK-related polyomavirus vasculopathy in a renal-transplant recipient. N Engl J Med. 2001;345:1250–1255. doi: 10.1056/NEJMoa010319. [DOI] [PubMed] [Google Scholar]
- 30.Ardalan MR, Shoja MM, Tubbs RS, Jayne D. Parvovirus B19 microepidemic in renal transplant recipients with thrombotic microangiopathy and allograft vasculitis. Exp Clin Transplant. 2008;6:137–143. [PubMed] [Google Scholar]
- 31.Waldman M, Kopp JB. Parvovirus-B19-associated complications in renal transplant recipients. Nat Clin Pract Nephrol. 2007;3:540–550. doi: 10.1038/ncpneph0609. [DOI] [PubMed] [Google Scholar]
- 32.Baid S, Pascual M, Williams WW Jr, Tolkoff-Rubin N, Johnson SM, Collins B, Chung RT, Delmonico FL, Cosimi AB, Colvin RB. Renal thrombotic microangiopathy associated with anticardiolipin antibodies in hepatitis C-positive renal allograft recipients. J Am Soc Nephrol. 1999;10:146–153. doi: 10.1681/ASN.V101146. [DOI] [PubMed] [Google Scholar]
- 33.Baid-Agrawal S, Farris AB 3rd, Pascual M, Mauiyyedi S, Farrell ML, Tolkoff-Rubin N, Collins AB, Frei U, Colvin RB. Overlapping pathways to transplant glomerulopathy: chronic humoral rejection, hepatitis C infection, and thrombotic microangiopathy. Kidney Int. 2011;80:879–885. doi: 10.1038/ki.2011.194. [DOI] [PubMed] [Google Scholar]
- 34.Yamazaki S, Takayama T, Inoue K, Higaki T, Makuuchi M. Transplantation-related thrombotic microangiopathy triggered by preemptive therapy for hepatitis C virus infection. Transplantation. 2008;86:1010–1011. doi: 10.1097/TP.0b013e31818747d8. [DOI] [PubMed] [Google Scholar]
- 35.Dwyre DM, Bell AM, Siechen K, Sethi S, Raife TJ. Disseminated histoplasmosis presenting as thrombotic microangiopathy. Transfusion. 2006;46:1221–1225. doi: 10.1111/j.1537-2995.2006.00873.x. [DOI] [PubMed] [Google Scholar]
- 36.Sethi S. Acute renal failure in a renal allograft: an unusual infectious cause of thrombotic microangiopathy. Am J Kidney Dis. 2005;46:159–162. doi: 10.1053/j.ajkd.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 37.de Vries DK, van der Pol P, van Anken GE, van Gijlswijk DJ, Damman J, Lindeman JH, Reinders ME, Schaapherder AF, Kooten Cv. Acute but transient release of terminal complement complex after reperfusion in clinical kidney transplantation. Transplantation. 2013;95:816–820. doi: 10.1097/TP.0b013e31827e31c9. [DOI] [PubMed] [Google Scholar]
- 38.Pham PT, Danovitch GM, Wilkinson AH, Gritsch HA, Pham PC, Eric TM, Kendrick E, Charles LR, Tsai HM. Inhibitors of ADAMTS13: a potential factor in the cause of thrombotic microangiopathy in a renal allograft recipient. Transplantation. 2002;74:1077–1080. doi: 10.1097/00007890-200210270-00003. [DOI] [PubMed] [Google Scholar]
- 39.Ulinski T, Charpentier A, Colombat M, Desconclois C, Mougenot B, Fremaux-Bacchi V, Suberbielle C, Deschênes G, Bensman A, Veyradier A. From humoral rejection to generalized thrombotic microangiopathy--role of acquired ADAMTS13 deficiency in a renal allograft recipient. Am J Transplant. 2006;6:3030–3036. doi: 10.1111/j.1600-6143.2006.01574.x. [DOI] [PubMed] [Google Scholar]
- 40.Lorcy N, Rioux-Leclercq N, Lombard ML, Le Pogamp P, Vigneau C. Three kidneys, two diseases, one antibody? Nephrol Dial Transplant. 2011;26:3811–3813. doi: 10.1093/ndt/gfr436. [DOI] [PubMed] [Google Scholar]
- 41.Chua JS, Baelde HJ, Zandbergen M, Wilhelmus S, van Es LA, de Fijter JW, Bruijn JA, Bajema IM, Cohen D. Complement Factor C4d Is a Common Denominator in Thrombotic Microangiopathy. J Am Soc Nephrol. 2015;26:2239–2247. doi: 10.1681/ASN.2014050429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371:654–666. doi: 10.1056/NEJMra1312353. [DOI] [PubMed] [Google Scholar]
- 43.Karthikeyan V, Parasuraman R, Shah V, Vera E, Venkat KK. Outcome of plasma exchange therapy in thrombotic microangiopathy after renal transplantation. Am J Transplant. 2003;3:1289–1294. doi: 10.1046/j.1600-6143.2003.00222.x. [DOI] [PubMed] [Google Scholar]
- 44.Nadasdy T. Thrombotic microangiopathy in renal allografts: the diagnostic challenge. Curr Opin Organ Transplant. 2014;19:283–292. doi: 10.1097/MOT.0000000000000074. [DOI] [PubMed] [Google Scholar]
- 45.Bouatou Y, Bacchi VF, Villard J, Moll S, Martin PY, Hadaya K. Atypical Hemolytic Uremic Syndrome Recurrence after Renal Transplantation: C3-Glomerulonephritis as an Initial Presentation. Transplant Direct. 2015;1:e9. doi: 10.1097/TXD.0000000000000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matar D, Naqvi F, Racusen LC, Carter-Monroe N, Montgomery RA, Alachkar N. Atypical hemolytic uremic syndrome recurrence after kidney transplantation. Transplantation. 2014;98:1205–1212. doi: 10.1097/TP.0000000000000200. [DOI] [PubMed] [Google Scholar]
- 47.Noris M, Remuzzi G. Thrombotic microangiopathy after kidney transplantation. Am J Transplant. 2010;10:1517–1523. doi: 10.1111/j.1600-6143.2010.03156.x. [DOI] [PubMed] [Google Scholar]
- 48.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676–1687. doi: 10.1056/NEJMra0902814. [DOI] [PubMed] [Google Scholar]
- 49.Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, Pinto S, Goodship TH, Alberti M, Ribes D, et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. 2013;24:475–486. doi: 10.1681/ASN.2012090884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bresin E, Daina E, Noris M, Castelletti F, Stefanov R, Hill P, Goodship TH, Remuzzi G; International Registry of Recurrent and Familial HUS/TTP. Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: prognostic significance of genetic background. Clin J Am Soc Nephrol. 2006;1:88–99. doi: 10.2215/CJN.00050505. [DOI] [PubMed] [Google Scholar]
- 51.Zafrani L, Mariotte E, Darmon M, Canet E, Merceron S, Boutboul D, Veyradier A, Galicier L, Azoulay E. Acute renal failure is prevalent in patients with thrombotic thrombocytopenic purpura associated with low plasma ADAMTS13 activity. J Thromb Haemost. 2015;13:380–389. doi: 10.1111/jth.12826. [DOI] [PubMed] [Google Scholar]
- 52.Mise K, Ubara Y, Matsumoto M, Sumida K, Hiramatsu R, Hasegawa E, Yamanouchi M, Hayami N, Suwabe T, Hoshino J, et al. Long term follow up of congenital thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome) on hemodialysis for 19 years: a case report. BMC Nephrol. 2013;14:156. doi: 10.1186/1471-2369-14-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song D, Wu LH, Wang FM, Yang XW, Zhu D, Chen M, Yu F, Liu G, Zhao MH. The spectrum of renal thrombotic microangiopathy in lupus nephritis. Arthritis Res Ther. 2013;15:R12. doi: 10.1186/ar4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barbour TD, Crosthwaite A, Chow K, Finlay MJ, Better N, Hughes PD, Cohney SJ. Antiphospholipid syndrome in renal transplantation. Nephrology (Carlton) 2014;19:177–185. doi: 10.1111/nep.12217. [DOI] [PubMed] [Google Scholar]
- 55.Canaud G, Kamar N, Anglicheau D, Esposito L, Rabant M, Noël LH, Guilbeau-Frugier C, Sberro-Soussan R, Del Bello A, Martinez F, et al. Eculizumab improves posttransplant thrombotic microangiopathy due to antiphospholipid syndrome recurrence but fails to prevent chronic vascular changes. Am J Transplant. 2013;13:2179–2185. doi: 10.1111/ajt.12319. [DOI] [PubMed] [Google Scholar]
- 56.George JN. The thrombotic thrombocytopenic purpura and hemolytic uremic syndromes: evaluation, management, and long-term outcomes experience of the Oklahoma TTP-HUS Registry, 1989-2007. Kidney Int Suppl. 2009:S52–S54. doi: 10.1038/ki.2008.622. [DOI] [PubMed] [Google Scholar]
- 57.Mariotte E, Blet A, Galicier L, Darmon M, Parquet N, Lengline E, Boutboul D, Canet E, Traineau R, Schlemmer B, et al. Unresponsive thrombotic thrombocytopenic purpura in critically ill adults. Intensive Care Med. 2013;39:1272–1281. doi: 10.1007/s00134-013-2873-4. [DOI] [PubMed] [Google Scholar]
- 58.Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Frémeaux-Bacchi V, Kavanagh D, Nester CM, Noris M, Pickering MC, Rodríguez de Córdoba S, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017;91:539–551. doi: 10.1016/j.kint.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 59.Schmidt CQ, Herbert AP, Hocking HG, Uhrín D, Barlow PN. Translational mini-review series on complement factor H: structural and functional correlations for factor H. Clin Exp Immunol. 2008;151:14–24. doi: 10.1111/j.1365-2249.2007.03553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bettoni S, Bresin E, Remuzzi G, Noris M, Donadelli R. Insights into the Effects of Complement Factor H on the Assembly and Decay of the Alternative Pathway C3 Proconvertase and C3 Convertase. J Biol Chem. 2016;291:8214–8230. doi: 10.1074/jbc.M115.693119. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. 2006;118:127–136. doi: 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 62.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol. 2015;6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frimat M, Tabarin F, Dimitrov JD, Poitou C, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood. 2013;122:282–292. doi: 10.1182/blood-2013-03-489245. [DOI] [PubMed] [Google Scholar]
- 64.Brocklebank V, Wood KM, Kavanagh D. Thrombotic Microangiopathy and the Kidney. Clin J Am Soc Nephrol. 2018;13:300–317. doi: 10.2215/CJN.00620117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaimé F, Dragon-Durey MA, Ngo S, Moulin B, Servais A, Provot F, Rostaing L, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8:554–562. doi: 10.2215/CJN.04760512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maga TK, Meyer NC, Belsha C, Nishimura CJ, Zhang Y, Smith RJ. A novel deletion in the RCA gene cluster causes atypical hemolytic uremic syndrome. Nephrol Dial Transplant. 2011;26:739–741. doi: 10.1093/ndt/gfq658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zipfel PF, Edey M, Heinen S, Józsi M, Richter H, Misselwitz J, Hoppe B, Routledge D, Strain L, Hughes AE, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet. 2007;3:e41. doi: 10.1371/journal.pgen.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Valoti E, Alberti M, Tortajada A, Garcia-Fernandez J, Gastoldi S, Besso L, Bresin E, Remuzzi G, Rodriguez de Cordoba S, Noris M. A novel atypical hemolytic uremic syndrome-associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H-dependent complement regulation. J Am Soc Nephrol. 2015;26:209–219. doi: 10.1681/ASN.2013121339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sethi S, Haas M, Markowitz GS, D’Agati VD, Rennke HG, Jennette JC, Bajema IM, Alpers CE, Chang A, Cornell LD, et al. Mayo Clinic/Renal Pathology Society Consensus Report on Pathologic Classification, Diagnosis, and Reporting of GN. J Am Soc Nephrol. 2016;27:1278–1287. doi: 10.1681/ASN.2015060612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, Vigneau C, Kuypers D, Boudailliez B, Loirat C, Rondeau E, Fridman WH. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. 2004;41:e84. doi: 10.1136/jmg.2004.019083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chan MR, Thomas CP, Torrealba JR, Djamali A, Fernandez LA, Nishimura CJ, Smith RJ, Samaniego MD. Recurrent atypical hemolytic uremic syndrome associated with factor I mutation in a living related renal transplant recipient. Am J Kidney Dis. 2009;53:321–326. doi: 10.1053/j.ajkd.2008.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bienaime F, Dragon-Durey MA, Regnier CH, Nilsson SC, Kwan WH, Blouin J, Jablonski M, Renault N, Rameix-Welti MA, Loirat C, et al. Mutations in components of complement influence the outcome of Factor I-associated atypical hemolytic uremic syndrome. Kidney Int. 2010;77:339–349. doi: 10.1038/ki.2009.472. [DOI] [PubMed] [Google Scholar]
- 73.Fremeaux-Bacchi V, Moulton EA, Kavanagh D, Dragon-Durey MA, Blouin J, Caudy A, Arzouk N, Cleper R, Francois M, Guest G, et al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2006;17:2017–2025. doi: 10.1681/ASN.2005101051. [DOI] [PubMed] [Google Scholar]
- 74.Salvadori M, Bertoni E. Complement related kidney diseases: Recurrence after transplantation. World J Transplant. 2016;6:632–645. doi: 10.5500/wjt.v6.i4.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, Del-Favero J, Plaisance S, Claes B, Lambrechts D, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:345–357. doi: 10.1056/NEJMoa0810739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sinibaldi S, Guzzo I, Piras R, Bresin E, Emma F, Dello Strologo L. Post-transplant recurrence of atypical hemolytic uremic syndrome in a patient with thrombomodulin mutation. Pediatr Transplant. 2013;17:E177–E181. doi: 10.1111/petr.12151. [DOI] [PubMed] [Google Scholar]
- 77.Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, López-Trascasa M, Sánchez-Corral P, Morgan BP, Rodríguez de Córdoba S. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2007;104:240–245. doi: 10.1073/pnas.0603420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roumenina LT, Jablonski M, Hue C, Blouin J, Dimitrov JD, Dragon-Durey MA, Cayla M, Fridman WH, Macher MA, Ribes D, et al. Hyperfunctional C3 convertase leads to complement deposition on endothelial cells and contributes to atypical hemolytic uremic syndrome. Blood. 2009;114:2837–2845. doi: 10.1182/blood-2009-01-197640. [DOI] [PubMed] [Google Scholar]
- 79.Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, Daina E, Fenili C, Castelletti F, Sorosina A, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5:1844–1859. doi: 10.2215/CJN.02210310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zuber J, Le Quintrec M, Sberro-Soussan R, Loirat C, Frémeaux-Bacchi V, Legendre C. New insights into postrenal transplant hemolytic uremic syndrome. Nat Rev Nephrol. 2011;7:23–35. doi: 10.1038/nrneph.2010.155. [DOI] [PubMed] [Google Scholar]
- 81.Epand RM, So V, Jennings W, Khadka B, Gupta RS, Lemaire M. Diacylglycerol Kinase-ε: Properties and Biological Roles. Front Cell Dev Biol. 2016;4:112. doi: 10.3389/fcell.2016.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Azukaitis K, Simkova E, Majid MA, Galiano M, Benz K, Amann K, Bockmeyer C, Gajjar R, Meyers KE, Cheong HI, et al. The Phenotypic Spectrum of Nephropathies Associated with Mutations in Diacylglycerol Kinase ε. J Am Soc Nephrol. 2017;28:3066–3075. doi: 10.1681/ASN.2017010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ruggenenti P. Post-transplant hemolytic-uremic syndrome. Kidney Int. 2002;62:1093–1104. doi: 10.1046/j.1523-1755.2002.00543.x. [DOI] [PubMed] [Google Scholar]
- 84.Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, Coppo R, Emma F, Johnson S, Karpman D, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016;31:15–39. doi: 10.1007/s00467-015-3076-8. [DOI] [PubMed] [Google Scholar]
- 85.Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, Mele C, Bresin E, Cassis L, Gamba S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108:1267–1279. doi: 10.1182/blood-2005-10-007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol. 2013;33:508–530. doi: 10.1016/j.semnephrol.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wikipedia. Penetrance. Available from: https://en.wikipedia.org/wiki/Penetrance.
- 88.Sansbury FH, Cordell HJ, Bingham C, Bromilow G, Nicholls A, Powell R, Shields B, Smyth L, Warwicker P, Strain L, et al. Factors determining penetrance in familial atypical haemolytic uraemic syndrome. J Med Genet. 2014;51:756–764. doi: 10.1136/jmedgenet-2014-102498. [DOI] [PubMed] [Google Scholar]
- 89.Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:1035–1050. doi: 10.1681/ASN.2004100861. [DOI] [PubMed] [Google Scholar]
- 90.Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, Alpers CE, Bajema IM, Bedrosian C, Braun M, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84:1079–1089. doi: 10.1038/ki.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hou J, Markowitz GS, Bomback AS, Appel GB, Herlitz LC, Barry Stokes M, D’Agati VD. Toward a working definition of C3 glomerulopathy by immunofluorescence. Kidney Int. 2014;85:450–456. doi: 10.1038/ki.2013.340. [DOI] [PubMed] [Google Scholar]
- 92.Larsen CP, Ambuzs JM, Bonsib SM, Boils CL, Cossey LN, Messias NC, Silva FG, Wang YH, Gokden N, Walker PD. Membranous-like glomerulopathy with masked IgG kappa deposits. Kidney Int. 2014;86:154–161. doi: 10.1038/ki.2013.548. [DOI] [PubMed] [Google Scholar]
- 93.Larsen CP, Messias NC, Walker PD, Fidler ME, Cornell LD, Hernandez LH, Alexander MP, Sethi S, Nasr SH. Membranoproliferative glomerulonephritis with masked monotypic immunoglobulin deposits. Kidney Int. 2015;88:867–873. doi: 10.1038/ki.2015.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cook HT. C4d Staining in the Diagnosis of C3 Glomerulopathy. J Am Soc Nephrol. 2015;26:2609–2611. doi: 10.1681/ASN.2015040433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sethi S, Nasr SH, De Vriese AS, Fervenza FC. C4d as a Diagnostic Tool in Proliferative GN. J Am Soc Nephrol. 2015;26:2852–2859. doi: 10.1681/ASN.2014040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.West CD, Witte DP, McAdams AJ. Composition of nephritic factor-generated glomerular deposits in membranoproliferative glomerulonephritis type 2. Am J Kidney Dis. 2001;37:1120–1130. doi: 10.1053/ajkd.2001.24511. [DOI] [PubMed] [Google Scholar]
- 97.Sethi S, Fervenza FC, Zhang Y, Zand L, Vrana JA, Nasr SH, Theis JD, Dogan A, Smith RJ. C3 glomerulonephritis: clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int. 2012;82:465–473. doi: 10.1038/ki.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sethi S, Gamez JD, Vrana JA, Theis JD, Bergen HR 3rd, Zipfel PF, Dogan A, Smith RJ. Glomeruli of Dense Deposit Disease contain components of the alternative and terminal complement pathway. Kidney Int. 2009;75:952–960. doi: 10.1038/ki.2008.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Recalde S, Tortajada A, Subias M, Anter J, Blasco M, Maranta R, Coco R, Pinto S, Noris M, García-Layana A, et al. Molecular Basis of Factor H R1210C Association with Ocular and Renal Diseases. J Am Soc Nephrol. 2016;27:1305–1311. doi: 10.1681/ASN.2015050580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gerber A, Karch H, Allerberger F, Verweyen HM, Zimmerhackl LB. Clinical course and the role of shiga toxin-producing Escherichia coli infection in the hemolytic-uremic syndrome in pediatric patients, 1997-2000, in Germany and Austria: a prospective study. J Infect Dis. 2002;186:493–500. doi: 10.1086/341940. [DOI] [PubMed] [Google Scholar]
- 101.Servais A, Noël LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, Macher MA, Zuber J, Karras A, Provot F, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–464. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 102.Angioi A, Fervenza FC, Sethi S, Zhang Y, Smith RJ, Murray D, Van Praet J, Pani A, De Vriese AS. Diagnosis of complement alternative pathway disorders. Kidney Int. 2016;89:278–288. doi: 10.1016/j.kint.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 103.Abarrategui-Garrido C, Martínez-Barricarte R, López-Trascasa M, de Córdoba SR, Sánchez-Corral P. Characterization of complement factor H-related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood. 2009;114:4261–4271. doi: 10.1182/blood-2009-05-223834. [DOI] [PubMed] [Google Scholar]
- 104.Lemaire M, Frémeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, Fakhouri F, Taque S, Nobili F, Martinez F, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet. 2013;45:531–536. doi: 10.1038/ng.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. 2010;31:E1445–E1460. doi: 10.1002/humu.21256. [DOI] [PubMed] [Google Scholar]
- 106.Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, Carreras Berges L, López-Trascasa M, Sánchez-Corral P, Rodríguez de Córdoba S. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet. 2005;14:703–712. doi: 10.1093/hmg/ddi066. [DOI] [PubMed] [Google Scholar]
- 107.Challis RC, Araujo GS, Wong EK, Anderson HE, Awan A, Dorman AM, Waldron M, Wilson V, Brocklebank V, Strain L, et al. A De Novo Deletion in the Regulators of Complement Activation Cluster Producing a Hybrid Complement Factor H/Complement Factor H-Related 3 Gene in Atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol. 2016;27:1617–1624. doi: 10.1681/ASN.2015010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen Q, Wiesener M, Eberhardt HU, Hartmann A, Uzonyi B, Kirschfink M, Amann K, Buettner M, Goodship T, Hugo C, et al. Complement factor H-related hybrid protein deregulates complement in dense deposit disease. J Clin Invest. 2014;124:145–155. doi: 10.1172/JCI71866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gale DP, de Jorge EG, Cook HT, Martinez-Barricarte R, Hadjisavvas A, McLean AG, Pusey CD, Pierides A, Kyriacou K, Athanasiou Y, et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet. 2010;376:794–801. doi: 10.1016/S0140-6736(10)60670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tortajada A, Yébenes H, Abarrategui-Garrido C, Anter J, García-Fernández JM, Martínez-Barricarte R, Alba-Domínguez M, Malik TH, Bedoya R, Cabrera Pérez R, et al. C3 glomerulopathy-associated CFHR1 mutation alters FHR oligomerization and complement regulation. J Clin Invest. 2013;123:2434–2446. doi: 10.1172/JCI68280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Venables JP, Strain L, Routledge D, Bourn D, Powell HM, Warwicker P, Diaz-Torres ML, Sampson A, Mead P, Webb M, et al. Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med. 2006;3:e431. doi: 10.1371/journal.pmed.0030431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Noris M, Remuzzi G. Managing and preventing atypical hemolytic uremic syndrome recurrence after kidney transplantation. Curr Opin Nephrol Hypertens. 2013;22:704–712. doi: 10.1097/MNH.0b013e328365b3fe. [DOI] [PubMed] [Google Scholar]
- 113.Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux-Bacchi V; French Study Group for aHUS/C3G. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8:643–657. doi: 10.1038/nrneph.2012.214. [DOI] [PubMed] [Google Scholar]
- 114.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Manuelian T, Hellwage J, Meri S, Caprioli J, Noris M, Heinen S, Jozsi M, Neumann HP, Remuzzi G, Zipfel PF. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest. 2003;111:1181–1190. doi: 10.1172/JCI16651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Marinozzi MC, Vergoz L, Rybkine T, Ngo S, Bettoni S, Pashov A, Cayla M, Tabarin F, Jablonski M, Hue C, et al. Complement factor B mutations in atypical hemolytic uremic syndrome-disease-relevant or benign? J Am Soc Nephrol. 2014;25:2053–2065. doi: 10.1681/ASN.2013070796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Noris M, Galbusera M, Gastoldi S, Macor P, Banterla F, Bresin E, Tripodo C, Bettoni S, Donadelli R, Valoti E, et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. 2014;124:1715–1726. doi: 10.1182/blood-2014-02-558296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pérez-Caballero D, González-Rubio C, Gallardo ME, Vera M, López-Trascasa M, Rodríguez de Córdoba S, Sánchez-Corral P. Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet. 2001;68:478–484. doi: 10.1086/318201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sánchez-Corral P, Pérez-Caballero D, Huarte O, Simckes AM, Goicoechea E, López-Trascasa M, de Córdoba SR. Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet. 2002;71:1285–1295. doi: 10.1086/344515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schramm EC, Roumenina LT, Rybkine T, Chauvet S, Vieira-Martins P, Hue C, Maga T, Valoti E, Wilson V, Jokiranta S, et al. Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood. 2015;125:2359–2369. doi: 10.1182/blood-2014-10-609073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ståhl AL, Vaziri-Sani F, Heinen S, Kristoffersson AC, Gydell KH, Raafat R, Gutierrez A, Beringer O, Zipfel PF, Karpman D. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood. 2008;111:5307–5315. doi: 10.1182/blood-2007-08-106153. [DOI] [PubMed] [Google Scholar]
- 122.Bernabéu-Herrero ME, Jiménez-Alcázar M, Anter J, Pinto S, Sánchez Chinchilla D, Garrido S, López-Trascasa M, Rodríguez de Córdoba S, Sánchez-Corral P. Complement factor H, FHR-3 and FHR-1 variants associate in an extended haplotype conferring increased risk of atypical hemolytic uremic syndrome. Mol Immunol. 2015;67:276–286. doi: 10.1016/j.molimm.2015.06.021. [DOI] [PubMed] [Google Scholar]
- 123.Caprioli J, Castelletti F, Bucchioni S, Bettinaglio P, Bresin E, Pianetti G, Gamba S, Brioschi S, Daina E, Remuzzi G, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet. 2003;12:3385–3395. doi: 10.1093/hmg/ddg363. [DOI] [PubMed] [Google Scholar]
- 124.Harris CL, Heurich M, Rodriguez de Cordoba S, Morgan BP. The complotype: dictating risk for inflammation and infection. Trends Immunol. 2012;33:513–521. doi: 10.1016/j.it.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pickering MC, de Jorge EG, Martinez-Barricarte R, Recalde S, Garcia-Layana A, Rose KL, Moss J, Walport MJ, Cook HT, de Córdoba SR, et al. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med. 2007;204:1249–1256. doi: 10.1084/jem.20070301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sánchez Chinchilla D, Pinto S, Hoppe B, Adragna M, Lopez L, Justa Roldan ML, Peña A, Lopez Trascasa M, Sánchez-Corral P, Rodríguez de Córdoba S. Complement mutations in diacylglycerol kinase-ε-associated atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2014;9:1611–1619. doi: 10.2215/CJN.01640214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zuber J, Le Quintrec M, Morris H, Frémeaux-Bacchi V, Loirat C, Legendre C. Targeted strategies in the prevention and management of atypical HUS recurrence after kidney transplantation. Transplant Rev (Orlando) 2013;27:117–125. doi: 10.1016/j.trre.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 128.Chua S, Wong G, Lim WH. The importance of genetic mutation screening to determine retransplantation following failed kidney allograft from recurrent atypical haemolytic ureamic syndrome. BMJ Case Rep. 2014;2014:pii: bcr2013202875. doi: 10.1136/bcr-2013-202875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kais H, Nourredine C, Raoudha B, Emira C, Tarek S, Fehmi S, Ezzedine B, Jalel H, Jamel M. Treatment of Tacrolimus-associated thrombotic microangiopathy in renal transplant recipient with fresh frozen plasma: A case report. Saudi J Kidney Dis Transpl. 2006;17:58–61. [PubMed] [Google Scholar]
- 130.Young BA, Marsh CL, Alpers CE, Davis CL. Cyclosporine-associated thrombotic microangiopathy/hemolytic uremic syndrome following kidney and kidney-pancreas transplantation. Am J Kidney Dis. 1996;28:561–571. doi: 10.1016/s0272-6386(96)90468-0. [DOI] [PubMed] [Google Scholar]
- 131.Cortina G, Trojer R, Waldegger S, Schneeberger S, Gut N, Hofer J. De novo tacrolimus-induced thrombotic microangiopathy in the early stage after renal transplantation successfully treated with conversion to everolimus. Pediatr Nephrol. 2015;30:693–697. doi: 10.1007/s00467-014-3036-8. [DOI] [PubMed] [Google Scholar]
- 132.Nwaba A, MacQuillan G, Adams LA, Garas G, Delriviere L, Augustson B, DeBoer B, Moody H, Jeffrey GP. Tacrolimus-induced thrombotic microangiopathy in orthotopic liver transplant patients: case series of four patients. Intern Med J. 2013;43:328–333. doi: 10.1111/imj.12048. [DOI] [PubMed] [Google Scholar]
- 133.Franco A, Hernandez D, Capdevilla L, Errasti P, Gonzalez M, Ruiz JC, Sanchez J; HUS-Sirolimus Spanish Study Group. De novo hemolytic-uremic syndrome/thrombotic microangiopathy in renal transplant patients receiving calcineurin inhibitors: role of sirolimus. Transplant Proc. 2003;35:1764–1766. doi: 10.1016/s0041-1345(03)00614-6. [DOI] [PubMed] [Google Scholar]
- 134.Epperla N, Hemauer K, Hamadani M, Friedman KD, Kreuziger LB. Impact of treatment and outcomes for patients with posttransplant drug-associated thrombotic microangiopathy. Transfusion. 2017;57:2775–2781. doi: 10.1111/trf.14263. [DOI] [PubMed] [Google Scholar]
- 135.Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, Spasoff RA. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325:393–397. doi: 10.1056/NEJM199108083250604. [DOI] [PubMed] [Google Scholar]
- 136.Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med. 1991;325:398–403. doi: 10.1056/NEJM199108083250605. [DOI] [PubMed] [Google Scholar]
- 137.Djamali A, Kaufman DB, Ellis TM, Zhong W, Matas A, Samaniego M. Diagnosis and management of antibody-mediated rejection: current status and novel approaches. Am J Transplant. 2014;14:255–271. doi: 10.1111/ajt.12589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ashman N, Chapagain A, Dobbie H, Raftery MJ, Sheaff MT, Yaqoob MM. Belatacept as maintenance immunosuppression for postrenal transplant de novo drug-induced thrombotic microangiopathy. Am J Transplant. 2009;9:424–427. doi: 10.1111/j.1600-6143.2008.02482.x. [DOI] [PubMed] [Google Scholar]
- 139.Koppula S, Yost SE, Sussman A, Bracamonte ER, Kaplan B. Successful conversion to belatacept after thrombotic microangiopathy in kidney transplant patients. Clin Transplant. 2013;27:591–597. doi: 10.1111/ctr.12170. [DOI] [PubMed] [Google Scholar]
- 140.Cicora F, Paz M, Mos F, Roberti J. Use of belatacept as alternative immunosuppression in three renal transplant patients with de novo drug-induced thrombotic microangiopathy. Case Rep Med. 2013;2013:260254. doi: 10.1155/2013/260254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham C, Cohen DJ, Delmas Y, Douglas K, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368:2169–2181. doi: 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- 142.Shochet L, Kanellis J, Simpson I, Ta J, Mulley W. De novo thrombotic microangiopathy following simultaneous pancreas and kidney transplantation managed with eculizumab. Nephrology (Carlton) 2017;22 Suppl 1:23–27. doi: 10.1111/nep.12936. [DOI] [PubMed] [Google Scholar]
- 143.Dedhia P, Govil A, Mogilishetty G, Alloway RR, Woodle ES, Abu Jawdeh BG. Eculizumab and Belatacept for De Novo Atypical Hemolytic Uremic Syndrome Associated With CFHR3-CFHR1 Deletion in a Kidney Transplant Recipient: A Case Report. Transplant Proc. 2017;49:188–192. doi: 10.1016/j.transproceed.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 144.Ikeda T, Okumi M, Unagami K, Kanzawa T, Sawada A, Kawanishi K, Omoto K, Ishida H, Tanabe K. Two cases of kidney transplantation-associated thrombotic microangiopathy successfully treated with eculizumab. Nephrology (Carlton) 2016;21 Suppl 1:35–40. doi: 10.1111/nep.12768. [DOI] [PubMed] [Google Scholar]
- 145.Safa K, Logan MS, Batal I, Gabardi S, Rennke HG, Abdi R. Eculizumab for drug-induced de novo posttransplantation thrombotic microangiopathy: A case report. Clin Nephrol. 2015;83:125–129. doi: 10.5414/CN108163. [DOI] [PubMed] [Google Scholar]
- 146.Chandran S, Baxter-Lowe L, Olson JL, Tomlanovich SJ, Webber A. Eculizumab for the treatment of de novo thrombotic microangiopathy post simultaneous pancreas-kidney transplantation--a case report. Transplant Proc. 2011;43:2097–2101. doi: 10.1016/j.transproceed.2011.02.064. [DOI] [PubMed] [Google Scholar]
- 147.Wilson CH, Brown AL, White SA, Goodship TH, Sheerin NS, Manas DM. Successful treatment of de novo posttransplant thrombotic microangiopathy with eculizumab. Transplantation. 2011;92:e42–e43. doi: 10.1097/TP.0b013e318230c0bd. [DOI] [PubMed] [Google Scholar]
- 148.Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, Cosio FG, Gandhi MJ, Kremers W, Gloor JM. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant. 2011;11:2405–2413. doi: 10.1111/j.1600-6143.2011.03757.x. [DOI] [PubMed] [Google Scholar]
- 149.González-Roncero F, Suñer M, Bernal G, Cabello V, Toro M, Pereira P, Angel Gentil M. Eculizumab treatment of acute antibody-mediated rejection in renal transplantation: case reports. Transplant Proc. 2012;44:2690–2694. doi: 10.1016/j.transproceed.2012.09.038. [DOI] [PubMed] [Google Scholar]
- 150.Chehade H, Rotman S, Matter M, Girardin E, Aubert V, Pascual M. Eculizumab to treat antibody-mediated rejection in a 7-year-old kidney transplant recipient. Pediatrics. 2015;135:e551–e555. doi: 10.1542/peds.2014-2275. [DOI] [PubMed] [Google Scholar]
- 151.Locke JE, Magro CM, Singer AL, Segev DL, Haas M, Hillel AT, King KE, Kraus E, Lees LM, Melancon JK, et al. The use of antibody to complement protein C5 for salvage treatment of severe antibody-mediated rejection. Am J Transplant. 2009;9:231–235. doi: 10.1111/j.1600-6143.2008.02451.x. [DOI] [PubMed] [Google Scholar]
- 152.Lonze BE, Dagher NN, Simpkins CE, Locke JE, Singer AL, Segev DL, Zachary AA, Montgomery RA. Eculizumab, bortezomib and kidney paired donation facilitate transplantation of a highly sensitized patient without vascular access. Am J Transplant. 2010;10:2154–2160. doi: 10.1111/j.1600-6143.2010.03191.x. [DOI] [PubMed] [Google Scholar]
- 153.Stewart ZA, Collins TE, Schlueter AJ, Raife TI, Holanda DG, Nair R, Reed AI, Thomas CP. Case report: Eculizumab rescue of severe accelerated antibody-mediated rejection after ABO-incompatible kidney transplant. Transplant Proc. 2012;44:3033–3036. doi: 10.1016/j.transproceed.2012.03.053. [DOI] [PubMed] [Google Scholar]
- 154.Tran D, Boucher A, Collette S, Payette A, Royal V, Senécal L. Eculizumab for the Treatment of Severe Antibody-Mediated Rejection: A Case Report and Review of the Literature. Case Rep Transplant. 2016;2016:9874261. doi: 10.1155/2016/9874261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Orandi BJ, Zachary AA, Dagher NN, Bagnasco SM, Garonzik-Wang JM, Van Arendonk KJ, Gupta N, Lonze BE, Alachkar N, Kraus ES, et al. Eculizumab and splenectomy as salvage therapy for severe antibody-mediated rejection after HLA-incompatible kidney transplantation. Transplantation. 2014;98:857–863. doi: 10.1097/TP.0000000000000298. [DOI] [PubMed] [Google Scholar]
- 156.Kulkarni S, Kirkiles-Smith NC, Deng YH, Formica RN, Moeckel G, Broecker V, Bow L, Tomlin R, Pober JS. Eculizumab Therapy for Chronic Antibody-Mediated Injury in Kidney Transplant Recipients: A Pilot Randomized Controlled Trial. Am J Transplant. 2017;17:682–691. doi: 10.1111/ajt.14001. [DOI] [PubMed] [Google Scholar]
- 157.Cornell LD, Schinstock CA, Gandhi MJ, Kremers WK, Stegall MD. Positive crossmatch kidney transplant recipients treated with eculizumab: outcomes beyond 1 year. Am J Transplant. 2015;15:1293–1302. doi: 10.1111/ajt.13168. [DOI] [PubMed] [Google Scholar]
- 158.Vernon KA, Gale DP, de Jorge EG, McLean AG, Galliford J, Pierides A, Maxwell PH, Taube D, Pickering MC, Cook HT. Recurrence of complement factor H-related protein 5 nephropathy in a renal transplant. Am J Transplant. 2011;11:152–155. doi: 10.1111/j.1600-6143.2010.03333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Loirat C, Fremeaux-Bacchi V. Hemolytic uremic syndrome recurrence after renal transplantation. Pediatr Transplant. 2008;12:619–629. doi: 10.1111/j.1399-3046.2008.00910.x. [DOI] [PubMed] [Google Scholar]
- 160.Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. 2013;33:479–492. doi: 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zuber J, Le Quintrec M, Krid S, Bertoye C, Gueutin V, Lahoche A, Heyne N, Ardissino G, Chatelet V, Noël LH, et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant. 2012;12:3337–3354. doi: 10.1111/j.1600-6143.2012.04252.x. [DOI] [PubMed] [Google Scholar]
- 162.Goicoechea de Jorge E, Caesar JJ, Malik TH, Patel M, Colledge M, Johnson S, Hakobyan S, Morgan BP, Harris CL, Pickering MC, et al. Dimerization of complement factor H-related proteins modulates complement activation in vivo. Proc Natl Acad Sci USA. 2013;110:4685–4690. doi: 10.1073/pnas.1219260110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kwon T, Dragon-Durey MA, Macher MA, Baudouin V, Maisin A, Peuchmaur M, Fremeaux-Bacchi V, Loirat C. Successful pre-transplant management of a patient with anti-factor H autoantibodies-associated haemolytic uraemic syndrome. Nephrol Dial Transplant. 2008;23:2088–2090. doi: 10.1093/ndt/gfn063. [DOI] [PubMed] [Google Scholar]
- 164.Waters AM, Pappworth I, Marchbank K, Bockenhauer D, Tullus K, Pickering MC, Strain L, Sebire N, Shroff R, Marks SD, et al. Successful renal transplantation in factor H autoantibody associated HUS with CFHR1 and 3 deficiency and CFH variant G2850T. Am J Transplant. 2010;10:168–172. doi: 10.1111/j.1600-6143.2009.02870.x. [DOI] [PubMed] [Google Scholar]
- 165.Zimmerhackl LB, Hofer J, Cortina G, Mark W, Würzner R, Jungraithmayr TC, Khursigara G, Kliche KO, Radauer W. Prophylactic eculizumab after renal transplantation in atypical hemolytic-uremic syndrome. N Engl J Med. 2010;362:1746–1748. doi: 10.1056/NEJMc1001060. [DOI] [PubMed] [Google Scholar]
- 166.Román-Ortiz E, Mendizabal Oteiza S, Pinto S, López-Trascasa M, Sánchez-Corral P, Rodríguez de Cordoba S. Eculizumab long-term therapy for pediatric renal transplant in aHUS with CFH/CFHR1 hybrid gene. Pediatr Nephrol. 2014;29:149–153. doi: 10.1007/s00467-013-2591-8. [DOI] [PubMed] [Google Scholar]
- 167.Nester C, Stewart Z, Myers D, Jetton J, Nair R, Reed A, Thomas C, Smith R, Brophy P. Pre-emptive eculizumab and plasmapheresis for renal transplant in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2011;6:1488–1494. doi: 10.2215/CJN.10181110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Krid S, Roumenina LT, Beury D, Charbit M, Boyer O, Frémeaux-Bacchi V, Niaudet P. Renal transplantation under prophylactic eculizumab in atypical hemolytic uremic syndrome with CFH/CFHR1 hybrid protein. Am J Transplant. 2012;12:1938–1944. doi: 10.1111/j.1600-6143.2012.04051.x. [DOI] [PubMed] [Google Scholar]
- 169.Donne RL, Abbs I, Barany P, Elinder CG, Little M, Conlon P, Goodship TH. Recurrence of hemolytic uremic syndrome after live related renal transplantation associated with subsequent de novo disease in the donor. Am J Kidney Dis. 2002;40:E22. doi: 10.1053/ajkd.2002.36938. [DOI] [PubMed] [Google Scholar]
- 170.Povey H, Vundru R, Junglee N, Jibani M. Renal recovery with eculizumab in atypical hemolytic uremic syndrome following prolonged dialysis. Clin Nephrol. 2014;82:326–331. doi: 10.5414/CN107958. [DOI] [PubMed] [Google Scholar]
- 171.Licht C, Heinen S, Józsi M, Löschmann I, Saunders RE, Perkins SJ, Waldherr R, Skerka C, Kirschfink M, Hoppe B, et al. Deletion of Lys224 in regulatory domain 4 of Factor H reveals a novel pathomechanism for dense deposit disease (MPGN II) Kidney Int. 2006;70:42–50. doi: 10.1038/sj.ki.5000269. [DOI] [PubMed] [Google Scholar]
- 172.Dragon-Durey MA, Sethi SK, Bagga A, Blanc C, Blouin J, Ranchin B, André JL, Takagi N, Cheong HI, Hari P, et al. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol. 2010;21:2180–2187. doi: 10.1681/ASN.2010030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Khandelwal P, Gupta A, Sinha A, Saini S, Hari P, Dragon Durey MA, Bagga A. Effect of plasma exchange and immunosuppressive medications on antibody titers and outcome in anti-complement factor H antibody-associated hemolytic uremic syndrome. Pediatr Nephrol. 2015;30:451–457. doi: 10.1007/s00467-014-2948-7. [DOI] [PubMed] [Google Scholar]
- 174.Sinha A, Gulati A, Saini S, Blanc C, Gupta A, Gurjar BS, Saini H, Kotresh ST, Ali U, Bhatia D, et al. Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int. 2014;85:1151–1160. doi: 10.1038/ki.2013.373. [DOI] [PubMed] [Google Scholar]
- 175.Tanimoto T, Oshima Y, Kami M. Eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;369:1378–1379. doi: 10.1056/NEJMc1308826. [DOI] [PubMed] [Google Scholar]
- 176.Sheerin NS, Kavanagh D, Goodship TH, Johnson S. A national specialized service in England for atypical haemolytic uraemic syndrome-the first year’s experience. QJM. 2016;109:27–33. doi: 10.1093/qjmed/hcv082. [DOI] [PubMed] [Google Scholar]
- 177.Wetzels JF, van de Kar NC. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome. Am J Kidney Dis. 2015;65:342. doi: 10.1053/j.ajkd.2014.04.039. [DOI] [PubMed] [Google Scholar]
- 178.Ardissino G, Possenti I, Tel F, Testa S, Salardi S, Ladisa V. Discontinuation of eculizumab treatment in atypical hemolytic uremic syndrome: an update. Am J Kidney Dis. 2015;66:172–173. doi: 10.1053/j.ajkd.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 179.Verhave JC, Westra D, van Hamersvelt HW, van Helden M, van de Kar NC, Wetzels JF. Living kidney transplantation in adult patients with atypical haemolytic uraemic syndrome. Neth J Med. 2013;71:342–347. [PubMed] [Google Scholar]
- 180.Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, Delmas Y, Douglas K, Furman RR, Gaber OA, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. 2015;87:1061–1073. doi: 10.1038/ki.2014.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Coppo R, Bonaudo R, Peruzzi RL, Amore A, Brunati A, Romagnoli R, Salizzoni M, Galbusera M, Gotti E, Daina E, et al. Liver transplantation for aHUS: still needed in the eculizumab era? Pediatr Nephrol. 2016;31:759–768. doi: 10.1007/s00467-015-3278-0. [DOI] [PubMed] [Google Scholar]
- 182.Zhang Y, Nester CM, Holanda DG, Marsh HC, Hammond RA, Thomas LJ, Meyer NC, Hunsicker LG, Sethi S, Smith RJ. Soluble CR1 therapy improves complement regulation in C3 glomerulopathy. J Am Soc Nephrol. 2013;24:1820–1829. doi: 10.1681/ASN.2013010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhang Y, Shao D, Ricklin D, Hilkin BM, Nester CM, Lambris JD, Smith RJ. Compstatin analog Cp40 inhibits complement dysregulation in vitro in C3 glomerulopathy. Immunobiology. 2015;220:993–998. doi: 10.1016/j.imbio.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Paixão-Cavalcante D, Torreira E, Lindorfer MA, Rodriguez de Cordoba S, Morgan BP, Taylor RP, Llorca O, Harris CL. A humanized antibody that regulates the alternative pathway convertase: potential for therapy of renal disease associated with nephritic factors. J Immunol. 2014;192:4844–4851. doi: 10.4049/jimmunol.1303131. [DOI] [PMC free article] [PubMed] [Google Scholar]