

Abstract
Viruses utilize the protein synthetic machinery of their host. Nonetheless, certain features of the synthesis of viral proteins are distinct from those of host-cell translation. Examples include internal ribosome entry sites in some viral mRNAs and ribosomal frameshifting during production of retroviral proteins. Viruses often inhibit host translation and/or possess mechanisms leading to preferential synthesis of viral proteins. In addition, a participant in the cellular antiviral response is the enzyme PKR (protein kinase, RNA activated), which is involved in the control of cellular translation. Thus, viruses and host cells wage war on the battlefield of translation. The distinctive features of protein synthesis in virally infected cells provide potential targets for therapeutic intervention. Translation-targeted therapeutics have precedence in antibiotics like tetracycline and erythromycin. Means for discovery of translation-targeted therapeutics for viral disease are discussed.
Keywords: Virus, Picornavirus, HIV, Protein synthesis, Antivirals, Frameshifting
IN simplistic terms, the ideal therapeutic agent for an infectious disease would be one that targets an event or process critical to the pathogen’s life cycle without affecting the host’s metabolism. Conceivably, this distinction between pathogen and host could be achieved simply because the biochemical target of the therapeutic agent does not exist in the host. An example of such a target is bacterial cell wall biosynthesis. Because human cells do not make a structure comparable to a bacterial cell wall, an inhibitor of a critical step in bacterial cell wall biosynthesis might be expected to have little deleterious effect on human metabolism (with the caveat that seemingly unrelated processes may have biochemical features in common that render the processes susceptible to inhibition by the same agent). Agents that inhibit the synthesis of bacterial cell walls include penicillin and bacitracin (Gale et al., 1981).
Alternatively, the distinction between pathogen and host might arise from biochemical differences between analogous targets that exist in both organisms. In this case, the agent would need to display selectivity for inhibiting the process of the pathogen to possess an acceptable therapeutic index. An example of this type of selectivity is found in antibiotic agents that target bacterial translation. Because translation is the means by which all organisms convert the genetic information of messenger RNA (mRNA) into proteins, a nonspecific inhibition of translation would be harmful to both pathogen and host. However, the machineries of translation in prokaryotes and eukaryotes have differences in addition to similarities, and these differences account for the selectivity of certain antibiotics. For example, a number of antibiotics (e.g., erythromycin and tetracycline) display selectivity for binding to bacterial ribosomal RNAs (rRNAs) or ribosomal proteins (Gale et al., 1981).
The purpose of this article is to discuss translation as a possible target for therapeutic intervention in viral diseases in humans. As is the case with those agents intended for use against bacterial pathogens, it is critical that these “translation-targeted therapeutics” be aimed at some aspect of translation or translational control that is unique to the viral pathogen. This represents a somewhat more daunting challenge for viral pathogens than for bacteria because the proteins of viral pathogens are synthesized by the human cell’s translation apparatus. Nonetheless, as understanding of
tiation Factor 2]. The initiation of translation is a complicated process involving a large number of cellular factors, and considerable evidence exists suggesting that translation initiation is a major site of regulation (Rhoads, 1993; Hershey, 1993).
A major distinction between prokaryotic and eukaryotic translation involves the means by which ribosomes engage the mRNA and select the AUG start codon. In prokaryotes, ribosomes associate with the mRNA through a sequence known as the Shine-Dalgarno site, which resides in the transcript 4–7 nucleotides 5′ of the start codon (McCarthy and Gualerzi, 1990). The Shine-Dalgarno site displays complementarity to the 16S rRNA of the 30S bacterial ribosomal subunit. Base pairing between these complementary sequences positions the ribosome near the start codon. The actual recognition of the start codon also involves base pairing, in this instance between the anticodon loop of the f-met-tRNA and the AUG initiator codon. In prokaryotes, three initiation factors (IF-1, IF-2, and IF-3) are involved in initiating translation at the start codon.
The start of protein synthesis in eukaryotes also requires that the AUG initiator codon be correctly identified (Figs. 2 and 3). This process begins with an association of a complex (consisting of the 40S ribosomal subunit, met-tRNA, and translation factors) with the 5′ end of the mRNA, which is modified with a cap structure consisting of a 7-methylguanosine residue joined by a 5′–5′ triphosphate linkage (Banerjee, 1980; Furuichi and Shatkin, 1989). The 5′ cap structure increases the efficiency of translation, with most translation in vivo being highly cap dependent. Initiation factor eIF-4F, consisting of three interacting subunits (eIF-4A, eIF-4E, and eIF-4γ), binds to the cap structure through eIF-4E and serves as an RNA helicase. The cap dependence of translation may be related to the need to unwind the 5′ untranslated region (UTR) of the mRNA. For an mRNA with a relatively unstructured 5′UTR, translation may be less cap dependent. The movement of the preinitiation complex to the initiator AUG is envisioned in the “scanning model” as a linear migration in the 5′ to 3′ direction until the first AUG codon is encountered. As with prokaryotes, actual recognition of the AUG involves base pairing with the anticodon loop of the initiator tRNA.
FIG. 2.
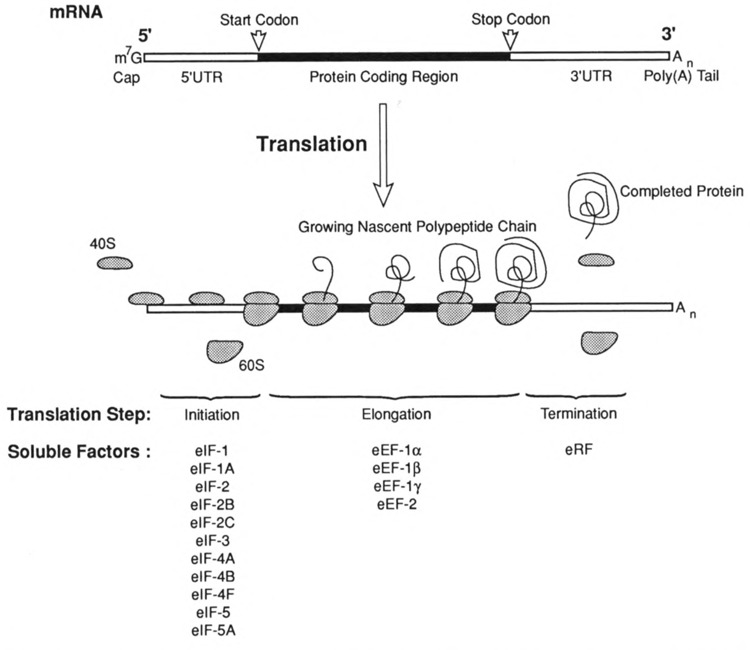
The translation of an mRNA. The “anatomy” of a typical mRNA molecule in higher eukaryotes is shown in the upper portion of the figure. The mature mRNA molecule can be envisioned as consisting of three divisions. Central to the structure and function of the molecule is the open reading frame that codes for protein. The open reading frame is defined by a start codon and an in-frame stop codon. On either side of the protein coding region are the untranslated regions (UTRs). The 5′UTR is modified at its 5′ end by the post- or cotranscriptional addition of a 7-methylguanosine cap that is linked to the remainder of the transcript in 5′-5′ triphosphate linkage. The 3 ′UTR is modified at its 3′ end by the posttranscriptional addition of a poly(A) tail. Translation is divided into the steps of Initiation, Elongation, and Termination as indicated. Initiation involves the binding of 40S ribosomal subunit to the cap, scanning to the start codon, and 60S ribosomal subunit joining. Elongation is a cyclical process by which the triplet nucleotide codons of the mRNA are decoded as aminoacyl tRNAs deliver the appropriate amino acid for the growth of the nascent polypeptide chain. Termination occurs when the ribosome reaches the stop codon where the completed protein and the components of the translation machinery are released. Soluble factors that have been identified as participants in each of the steps of translation factors listed are themselves comprised of multiple polypeptide subunits.
FIG. 3.
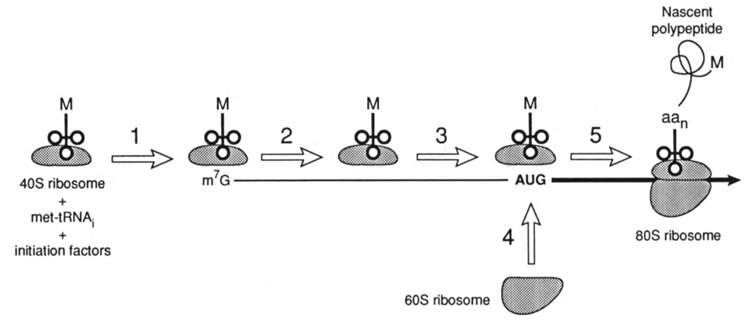
The scanning model for translation initiation. The complex consisting of a 40S ribosome, met-tRNAi, and initiation factors interacts with the mRNA cap (step 1). The complex then scans in a 5′ to 3′ direction (step 2) until the start codon is encountered (step 3). The 60S ribosome joins at the AUG (step 4) and elongation proceeds (step 5). The nascent polypeptide is shown after the nth codon has directed the addition of its amino acid (aan).
Translational elongation that starts after 60S ribosome joining involves a cyclic series of events by which the nucleotide sequence of the mRNA is read in the 5′ to 3′ direction, three nucleotides at a time by tRNAs corresponding to the successive codons. This process results in lengthening of the nascent polypeptide at what will be its carboxy-terminal end. Elongation continues until a termination codon is reached. Whereas many features of translation initiation differ between prokaryotes and eukaryotes, the process of elongation is quite similar. Although most attention has been focused on the regulation of initiation, there is a growing body of evidence suggesting that translation elongation can also be regulated (Ryazanov et al., 1991).
Termination of translation occurs when a stop codon is encountered in frame with the start codon (Tate and Brown, 1992). There is no mammalian tRNA whose anticodon is complementary to the stop codons (UAA, UAG, and UGA). However, stop codons are actively recognized by the eukaryotic release factor (eRF). At the stop codon, peptide elongation ceases and the eRF binds to the site left vacant in the absence of a cognate aminoacyl tRNA. Release of the newly synthesized protein requires GTP hydrolysis and hydrolysis of the peptidyl-tRNA bond. The completed protein is released, and all the components of translation are made available for another round of translation.
INTERNAL RIBOSOME ENTRY IN CERTAIN VIRUSES
The scanning model depicted in Fig. 3 appears to accommodate most mRNAs having caps and 5′UTRs in which upstream AUGs are rare. However, it is more difficult to envision how translation of mRNAs from picornaviruses (e.g., poliovirus) occurs via a scanning mechanism that begins at the 5′ end of the mRNA. The picornavirus mRNAs have many upstream AUGs within an extremely long and highly structured 5′UTR. Over the past 10 years, evidence has accumulated indicating that the picornavirus mRNAs contain internal ribosome entry sites (IRES) (Donahue, 1990; Sonenberg, 1990; Oh and Sarnow, 1993). The picornavirus IRES elements provide a means by which the translation machinery can assemble internal to the 5′ end of the mRNA near the appropriate AUG for the viral protein. The function of the IRES can be envisioned as being analogous to the Shine-Dalgarno sequence, which provides a ribosome binding site near the AUG utilized for translation initiation in prokaryotes. Certain picornaviruses inhibit cellular, cap-dependent initiation by encoding a protease that cleaves eIF-4γ, a critical component of the cap binding initiation factor eIF-4F (Wyckoff, 1993). When cellular cap-dependent translation is inhibited as a result of the action of the picornavirus protease, translation of the viral mRNA is not affected because its translation is initiated in a cap-independent fashion via the IRES element (Fig. 4). In addition to existing in all genera of Picornaviridae family, IRES elements have been implicated in hepatitis C virus (Tsukiyama-Kohara et al., 1992; Wang et al., 1993) and hepatitis B virus (Chang et al., 1990; Ou et al., 1990).
FIG. 4.
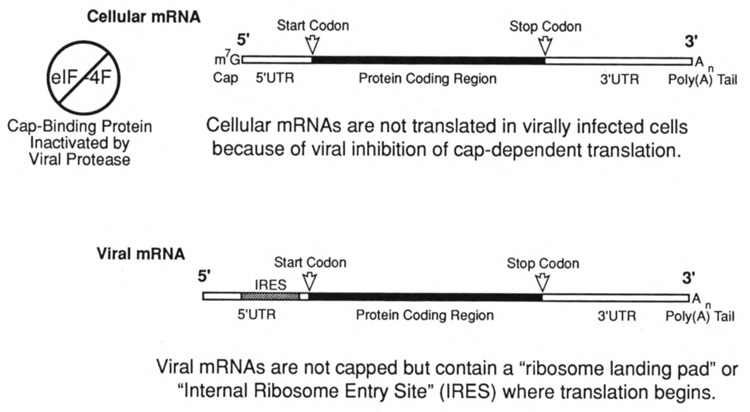
The initiation of translation via a viral IRES element. Certain picornaviruses (e.g., poliovirus, rhinovirus) encode a protease that inactivates the cap binding translation factor eIF-4F resulting in an inhibition of cellular, cap-dependent translation initiation. The 5′UTR of the mRNAs of these viruses contain an Internal Ribosome Entry Site (IRES) that allows translation initiation in the absence of functional eIF-4F.
IRES elements have also been proposed for one human mRNA [immunoglobulin heavy chain binding protein (BiP)] and two Drosophila mRNAs with no known human equivalents (antennapedia and ultrabithorax) (Macejak and Sarnow, 1990). There are several differences between the human BiP IRES and viral IRES elements. Unlike picornaviral and hepatitis C viral mRNAs, BiP mRNA is capped and the IRES element is less than 220 nt in length. In addition, the BiP IRES has no sequence or structural relationship to any known viral IRES element and also appears to function differently than any known viral IRES element. Most significantly, the BiP IRES is not functionally equivalent to a viral IRES; unlike viral IRES elements, insertion of the BiP IRES does not stimulate translation of an uncapped mRNA in poliovirus-infected cells (Macejak et al., 1990). It has also been proposed that BiP mRNA is translated in vivo by both cap-dependent and cap-independent mechanisms, with the former being predominant (Macejak and Sarnow, 1990). Thus, it appears likely that selective inhibition of the cap-independent, IRES-mediated translation of the mRNAs of picornaviruses and hepatitis C would not be harmful to human mRNA translation (including that of BiP mRNA). This would mean that an antiviral agent that targets IRES-mediated translation might be expected to have minimal side effects. This hypothesis, although seemingly sound, remains to be tested because there are currently no agents known to inhibit IRES-mediated translation selectively.
THE IMPACT OF VIRAL INFECTION ON TRANSLATION
The picornavirus shut-off of host cell translation through the proteolytic cleavage of a cellular initiation factor is but one example of the impact of viral infection on translation. Indeed, translation seems to be a major battlefield in the war of virus versus host. Some viruses appear to require inhibition of host cell protein synthesis to replicate, although the precise advantage this affords the virus is the subject of active research (e.g., Zhang and Schneider, 1994). For viruses that do inhibit protein synthesis, the inhibition must be selective if the virus is to replicate. The picornavirus IRES is one means by which that selectivity is achieved. In the case of influenza virus, the 5′UTR of the viral mRNAs appear to confer translatability to these transcripts under circumstances of reduced overall translation (Garfinkel and Katze, 1993, 1994). Similarly, the late phase of adenovirus infection is characterized by an inhibition of cellular protein synthesis. In this case the inhibition involves a dephosphorylation and consequent inactivation of a critical initiation factor (eIF-4E). The adenoviral mRNAs are unusual in that they display a markedly reduced dependence upon this initiation factor (Zhang and Schneider, 1993).
The mammalian cell also attempts to do battle against infecting viruses on the field of translation. The host defense involves an attempt at a universal inhibition of translation. If this shut-off of translation is achieved, the host cell itself perishes, but this cellular suicide would be advantageous in that the spread of the viral infection to other cells of the organism would be blocked (Fig. 5). The means by which cells attempt translational suicide involves the enzyme known as PKR (for Protein Kinase, RNA activated). PKR has been referred to by a number of other names, including p68 (for the human enzyme; p65 for the murine enzyme), dsRNA-PK, eIF-2α kinase, and DAI. PKR is induced by interferon and activated as a kinase by viral double-stranded RNA (Fig. 5). The phosphorylation catalyzed by this kinase results in the shut-off of protein synthesis (Hovanessian, 1993; Katze, 1993). Two of the factors in translation initiation (eIF-2 and eIF-2B) appear to be involved in the antiviral pathway mediated by PKR. For eIF-2 to function in more than one round of initiation, bound GDP must be exchanged for GTP, and this reaction is catalyzed by eIF-2B. eIF-2 contains a site of serine phosphorylation (serine 51 of the α subunit), and when phosphorylated by PKR, eIF-2 binds tightly to eIF-2B (Hershey, 1993; Mathews, 1993). The formation of a complex between eIF-2B and the phosphorylated form of eIF-2 blocks exchange and thereby inhibits translation initiation.
FIG. 5.
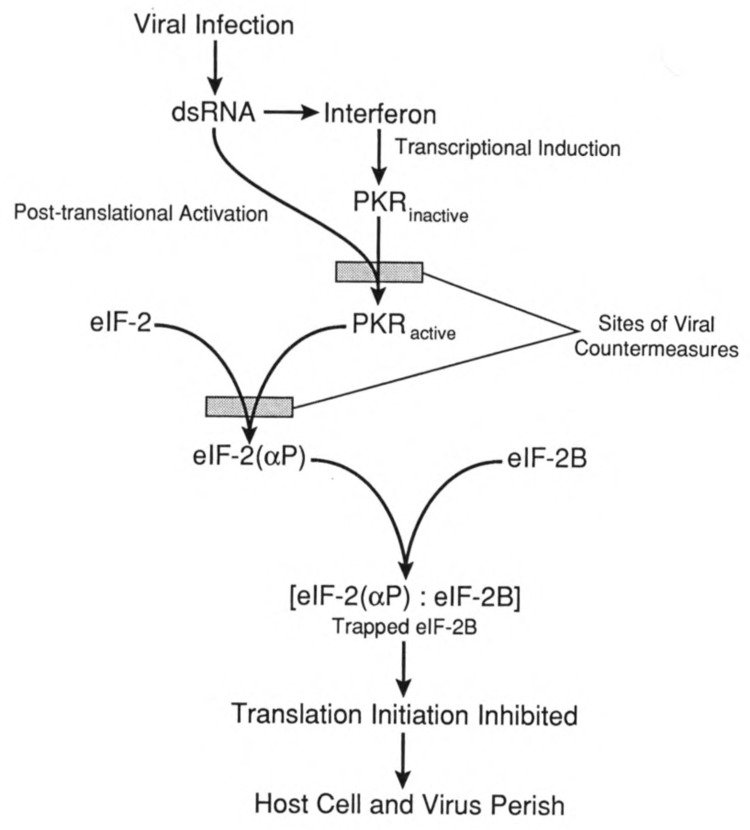
The PKR pathway. Double-stranded RNA (dsRNA) resulting from a viral infection triggers interferon production. Interferon is a transciptional inducer of a number of genes including the protein kinase, RNA activated (PKR). The viral dsRNA also activates PKR through its two RNA binding motifs. Active PKR catalyzes phosphorylation of eIF-2 at serine 51 of its α subunit. The phosphorylated eIF-2 forms a complex with eIF-2B, the guanine nucleotide exchange factor, and the trapped eIF-2B is inactive. Failure to exchange bound GDP for GTP prevents eIF-2 from participating in translation initiation, resulting in a global inhibition of protein synthesis. If this pathway is effective, the infected host cell would perish, but viral replication and spread to other host cells would be aborted. Various viruses possess countermeasures to prevent either PKR activation or the phosphorylation of eIF-2.
A number of viruses attempt to circumvent the PKR antiviral pathway by blocking the activation of the kinase (Katze, 1993; Mathews, 1993). Viruses may reduce the level of PKR by proteolysis (e.g., poliovirus, Black et al., 1989) or by inhibiting its activation via virally encoded RNAs (e.g., adenovirus, Mathews, 1993) or via the unmasking of a cellular inhibitor of PKR (e.g., influenza, Garfinkel and Katze, 1994). Other viral strategies include sequestration of the activating dsRNA (e.g., reovirus, Imani, 1988; vaccinia virus, Chang et al., 1992), or production of a viral protein that mimics the cellular substrate (eIF-2α) (e.g., vaccinia virus, Beattie et al., 1991). Reports have appeared indicating that human immunodeficiency viruses (HIV) tat protein controls PKR-catalyzed phosphorylation (Roy et al., 1990) and that the TAR RNA, which is bound by tat, can activate PKR (Roy et al., 1991). It should be noted that the relationship between TAR and PKR remains controversial (e.g., Geballe and Gray, 1992; Gunnery et al., 1992). The fact that a number of viruses possess a variety of seemingly unrelated countermeasures to prevent the activation of PKR strongly supports the contention that PKR activation plays a central role in the cellular antiviral response.
Physiological regulation of translation initiation also appears to occur via eIF-4 polypeptides (Rhoads, 1993; Hershey, 1993). Much of the evidence implicating eIF-4 polypeptides (notably eIF-4E and eIF-4γ) is based upon genetic manipulation. Nonetheless, it is noteworthy that infection of cells with certain picornaviruses results in proteolytic cleavage of eIF-4γ, concurrent with the cell’s loss of ability to translate capped mRNAs (Wyckoff, 1993). Adenoviruses also inactivate eIF-4F but through an underphosphorylation of its eIF-4E component (Zhang and Schneider, 1993). The adenovirus late mRNAs can be translated under conditions of limited eIF-4F, most likely because the 5′UTR of these mRNAs (referred to as the tripartite leader) is relatively devoid of secondary structure and is therefore less dependent on the RNA-unwinding activity possessed by eIF-4F. Infection of cells with influenza virus also leads to dephosphorylation of eIF-4E (Feigenblum and Schneider, 1993). Influenza virus mRNAs are also selectively translated after host cell protein synthesis is inhibited. It should be noted that adenoviruses (Zhang and Schneider, 1993) and influenza virus (Garfinkel and Katze, 1993) appear to utilize multiple mechanisms to modulate host translation, and these may play distinct roles in various stages of viral life cycle.
As was noted above, if a translation-targeted antiviral is to have an acceptable therapeutic index, it must be directed at some aspect of translation or translational control that is not critical for the host cell. The mechanism(s) by which viruses shut-off and hijack host cell translation would appear to be good candidates. After all, absent the viral infection, normal host cells do not engage in translational suicide via PKR. Moreover, the diverse mechanisms used by various viruses to prevent PKR-mediated cell suicide involve, for the most part, viral gene products. Therefore, it seems likely that therapeutic agents aimed at these virally encoded processes would not be deleterious to the host cell.
FRAMESHIFTING IN RETROVIRUSES INCLUDING HIV
In general, the information that the mRNA carries from the DNA of the nucleus is conveyed into the amino acid sequence of protein with high fidelity. This fidelity is the result of a strict maintenance of the reading frame by the ribosomal machinery of translation. Despite the importance of maintaining the reading frame to the proper translation of mRNA into protein, certain mRNAs are programmed to disregard the reading frame at specific points in the process of translation elongation (Weiss et al., 1990). The process of frame-shifting is capable of producing more than one protein from the same translation start. Examples of this phenomenon occur in retroviruses where the overlap of two reading frames and a frame-shift between the gag and pol genes allows the proteins encoded by the pol gene to be translated as the gag-pol fusion protein.
Frameshifting is an essential component of the life cycle of HIV (Pavlakis and Felber 1990; Wong-Staal, 1990). The HIV-1 frameshift is a representative of the class of frameshifting characterized by a −1 shift of the ribosome during translation; that is, during elongation, the ribosome shifts backward from its initial reading frame (termed frame 0) into the −1 reading frame. Such frameshifting appears to involve a “slippery sequence” characterized by a run of repetitive nucleotides and an element of RNA secondary structure just 3′ of the slippery sequence (Weiss et al., 1990; Brierley et al., 1992) (Fig. 6). The secondary structure element is thought to impede translation elongation, and while the ribosome is stalled over the slippery sequence, a frameshift occurs at some (usually low) frequency. For some retroviruses the secondary structure element is a pseudoknot, whereas for others (e.g., HIV) the RNA secondary structure appears to be a moderately stable stem-loop structure.
FIG. 6.
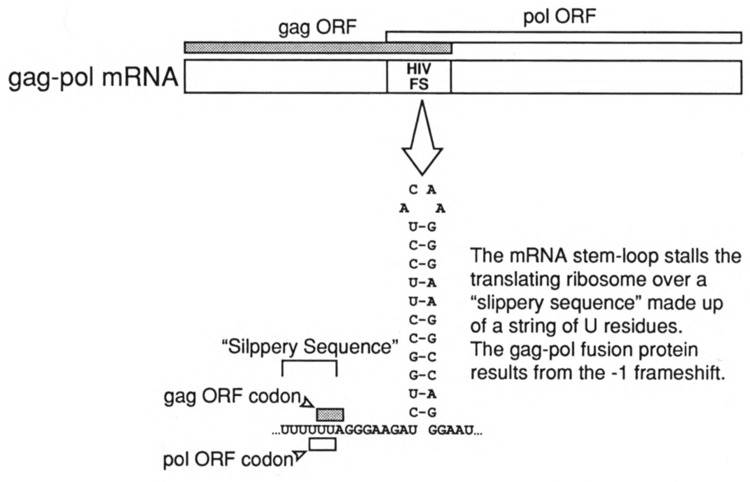
The frameshifting sequence of HIV-1. The gag and pol gene products of HIV-1 are encoded by a single mRNA. The open reading frames (ORFs) of the gag protein and the pol proteins overlap by approximately 200 nucleotides. Within the region of overlap is contained a frame-shifting sequence (HIV FS) consisting of “slippery sequence” (six consecutive U residues) and a moderately stable stem-loop structure. Frameshifting from the gag ORF to the pol ORF (a −1 frameshift) is thought to occur when the elongating ribosome is paused over the slippery sequence as a consequence of being impeded by the RNA secondary structure. Frameshifting in HIV-1 occurs at a frequency of 5–10% and is the only means for producing the pol proteins required for viral replication.
Most importantly from the perspective of translation-targeted therapeutics, there exists no evidence that production of any cellular protein is dependent upon a −1 frameshift analogous to that needed to make the pol gene products of HIV. Thus, an inhibitor of this process would be expected to inhibit the synthesis of essential viral proteins without affecting host cell protein synthesis.
THE DISCOVERY OF TRANSLATION-TARGETED THERAPEUTICS
Much attention has focused on antisense oligonucleotides or their derivatives as antiviral agents (Cohen, 1991; Goodchild, 1991). In addition to mediating the destruction of mRNA by RNase H, it has been suggested that antisense oligonucleotides may decrease viral gene expression by an arrest of translation. Translation arrest is a particularly attractive mechanism for explaining the inhibition of viral protein synthesis by oligonucleotide derivatives (e.g., methylphosphonates) that do not support RNase H cleavage of the mRNA targeted by the antisense molecule (Cohen, 1991). Obstacles to the use of antisense molecules as antiviral agents include issues related to their stability and the ability to deliver the antisense therapeutics effectively. The example provided by antibiotics like tetracycline, which is a small molecule with oral bioavailability, gives encouragement in the search for small molecules directed at translation-related targets as an alternative to the antisense approach.
Several means might be envisioned for the discovery of translation-targeted therapeutics for viral diseases. The first would be serendipity, and this means of discovery would be analogous to that of many known antibacterial agents. For the most part, the drugs now known to be translation-targeted antibiotics were identified as having antibacterial properties prior to an understanding of their mechanism of action. Only subsequently was it discerned that these agents interfered with bacterial translation. The National Cancer Institute conducts a massive screen for anti-HIV agents, testing thousands of defined chemical entities or natural product extracts for antiviral activity (Boyd, 1988). This screening program is conducted initially without concern for their mechanism of action. One might imagine (even suspect) that this program will eventually turn up agents that target some aspect of HIV translation (e.g., frameshifting).
A second means of discovering translation-targeted therapeutics would be rational drug design. This means of drug discovery has long been the dream of pharmaceutical scientists and has recently begun to bear fruit in the area of antivirals with the design of a new influenza hemaglutinin inhibitor based on the crystal structure of the protein (von Itzstein et al., 1993). Unfortunately, the molecular components involved in translation and its control during viral infection are not characterized in sufficient detail to allow for rational targeting of these molecules. This will come in time.
The third approach is, in a sense, a hybrid of the first two. A specific molecular target related to translation can be selected and incorporated into a “smart” screen. Such a screen would incorporate a reporter gene with an easily detectable readout (e.g., luciferase). Once such a smart screen is reduced to practice, testing of large numbers of substances could reveal an agent that interferes with the function of the chosen target. This process does not necessarily depend upon knowledge in exquisite detail of how the translation-related target functions in cells. To illustrate this approach using the examples described above, one could envision screens incorporating picornaviral IRES elements or the HIV frameshifting sequence. Screens could also be designed to assess the ability of chemical compounds or natural product mixtures to interfere with the means by which a given virus attempts to block PKR-mediated suicide. If activity in these smart screens was detected, the putative translation-targeted therapeutic could be examined in more detail, including confirmation of mechanism of action, examination of structure-activity relationships, and testing as an antiviral in cell culture or animal models.
SUMMARY AND PERSPECTIVES
This chapter describes, albeit briefly, some of the connections that are known to exist between the process of translation and viral infections, and suggests that translation-targeted therapeutics may constitute a new class of antivirals. Translation-targeted therapeutics directed at bacterial pathogens are well known, but as yet the paradigm provided by antibiotics such as tetracycline and erthyromycin has not been extended to human viral pathogens. In part, this is because such viral pathogens utilize their host’s translation apparatus, and a safe and effective therapeutic must target some feature of the translation of the viral mRNA that is unique to the pathogen or to some aspect of cellular mRNA translation that only occurs in virally infected cells. It is quite clear that viruses alter cellular translation using viral-specific mechanisms and prevent cellular antiviral responses that relate to translation. It is also known that the translation of certain viral mRNAs involves molecular processes that do not appear to be utilized for host mRNA translation (e.g., frameshifting or IRES-mediated initiation). Thus, there are a number of sites in the course of a viral infection at which translation-targeted therapeutics might be directed.
Translation-targeted therapeutics ought not be thought of as being limited to bacterial and viral pathogens. Fungal pathogens also might be effectively addressed using translation as a target. As eukaryotes, fungi are not as different from humans as are bacteria. Nonetheless, there exist differences between fungal and human translation mechanisms that might be exploited therapeutically. For example, fungi appear to utilize an additional soluble translation factor termed EF-3, which is encoded by an essential gene and which may be involved in translation accuracy (Belfield and Tuite, 1993). Moreover, even where analogous translation factors exist in fungi and humans, their protein sequences are sufficiently distinct to envision antifungal agents capable of distinguishing between the two.
Translation or its control is also altered in non-infectious pathogenic states, and it may be possible to address these diseases with translation-targeted therapeutics. For example, several proteins involved in translation have been shown to be effectors of cell growth and tumorigenesis (Sonenberg, 1993). Interestingly, there is considerable overlap between these proteins and those involved in the translational events accompanying certain viral infections. For example, eIF-4E, the cap binding component of eIF-4F, leads to transformation of cells in which it is overexpressed (Lazaris-Karatzas et al., 1990; De Benedetti and Rhoads, 1990). It is also noteworthy that ras-transformed cells have been shown to display an increased rate of protein synthesis and increased phosphorylation of eIF-4E (Rinker-Schaeffer et al., 1992). Although not a translation factor per se, the enzyme PKR has also been implicated in tumor formation. PKR exhibits properties similar to those of known tumor suppressor proteins (e.g., p53, Rb) in that transfection of a mutated form of PKR cDNA resulted in malignant transformation (Koromilas et al., 1992; Meurs et al., 1993). A cellular 58-kDa protein that is activated as an inhibitor of PKR during influenza virus infection has been demonstrated recently to have oncogenic properties (Barber et al., 1994), and ras-transformed cells express a distinct protein that inhibits PKR (Mundshau and Faller, 1992). The fact that interferon has both antiviral and antitumor activities may be related, at least in part, to its induction of PKR (Lengyel, 1993). Finally, evidence has been presented indicating that constitutive expression of the elongation factor EF-1α causes fibroblasts to become more susceptible to transformation induced by chemical carcinogen and UV light (Tatsuka et al., 1992). Taken together, these findings implicate the regulation of translation as a critical means by which the normal rate of cell growth and division are maintained. It appears that various means of perturbing this translational regulation can result in oncogenesis or increased susceptibility to carcinogenesis. This conclusion raises the possibility that translation-targeted therapeutics may be useful as anticancer agents. Treatment of rapidly dividing cancer cells having a high rate of translation with a therapeutic agent capable of returning translation to its normal rate would be expected to slow cell growth and could cause reversion to the untransformed state.
Modern molecular biology has led to a nearly explosive growth in understanding the components of the eukaryotic translation apparatus. The means by which viruses alter cellular translation are also becoming more clear, as are the processes involved in translation of mRNAs of fungal pathogens. Alteration of translation in oncogenically transformed cells has also emerged as a major area of research activity. It is reasonable to assume that, as our understanding of translation and its control continues to grow, new therapeutic targets related to translation will become apparent.
ACKNOWLEDGEMENTS
Drs. S. Green, V. Miles, C. Moehle, J. Watson, and G. Witherell of RiboGene, Inc. have played major roles in developing the concept of translation-targeted therapeutics for nonbacterial pathogens and pathogenic states. The Scientific Advisory Board of RiboGene, Inc., consisting of Drs. T. Donahue, A. Geballe, J. Hershey, A. Hinnebusch, M. Katze, M. Mathews, D. Morris, and N. Sonenberg, has also contributed significantly to this process.
REFERENCES
- Banerjee A. K. (1980), Microbiol Rev 44, 175–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber G. N., Thompson S., Lee T. G., Strom T., Jagus R., Darveau A., and Katze M. G. (1994), Proc Natl Acad Sci USA 91, 4278–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie E., Tartaglia J., and Paoletti E. (1991), Virology 183, 419–422. [DOI] [PubMed] [Google Scholar]
- Belasco J. G. and Brawerman G. (eds.) (1993), Control of Messenger RNA Stability, Academic Press, San Diego. [Google Scholar]
- Belfield G. P. and Tuite M. F. (1993), Mol Microbiol 9, 411–418. [DOI] [PubMed] [Google Scholar]
- Black T. L., Safer B., Hovanessian A., and Katze M. (1989), J Virol 63, 2244–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd M. R. (1988), in AIDS: Etiology, Diagnosis, Treatment and Prevention (deVita V. T., Hellman S., and Rosenberg S. A., eds.), J. B. Lippincott, Philadelphia, pp. 305–319. [Google Scholar]
- Brierley I., Jenner A. J., and Inglis S. C. (1992), J Mol Biol 227, 463–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L.-J., Ganem D., and Varmus H. E. (1990), Proc Natl Acad Sci USA 87, 5158–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.-W., Watson J. C., and Jacobs B. L. (1992), Proc Natl Acad Sci USA 89, 4825–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. S. (1991), Antiviral Res 16, 121–133. [DOI] [PubMed] [Google Scholar]
- De Benedetti A. and Rhoads R. E. (1990), Proc Natl Acad Sci USA 87, 8212–8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue T. F. (1990), Curr Opin Cell Biol 2, 1087–1091. [DOI] [PubMed] [Google Scholar]
- Feigenblum D. and Schneider R. J. (1993), J Virol 67, 3027–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi Y. and Shatkin A. J. (1989), Methods Enzymol 180, 164–176. [DOI] [PubMed] [Google Scholar]
- Gale E. F., Cundliffe E., Reynolds P. E., Richmond M. H., and Waring M. J. (1981), The Molecular Basis of Antibiotic Action, John Wiley & Sons, London. [Google Scholar]
- Garfinkel M. S. and Katze M. G. (1993), Gene Expr 3, 109–118. [PMC free article] [PubMed] [Google Scholar]
- Garfinkel M. S. and Katze M. G. (1994), Sci Am Sci Med 1, 2–11. [Google Scholar]
- Geballe A. P. and Gray M. K. (1992), Nucleic Acids Res 20, 4291–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild J. (1991), Antisense Res Dev 1, 361–364. [DOI] [PubMed] [Google Scholar]
- Gunnery S., Green S. R., and Mathews M. B. (1992), Proc Natl Acad Sci USA 89, 11557–11561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harford J. (1994), in The Liver: Biology and Pathobiology, Third Edition (Arias I. M. et al. , eds.), Raven Press, New York, pp. 69–84. [Google Scholar]
- Hershey J. W. B. (1991), Annu Rev Biochem 60, 717–755. [DOI] [PubMed] [Google Scholar]
- Hershey J. W. B. (1993), Semin Virol 4, 201–207. [Google Scholar]
- Hovanessian A. G. (1993), Semin Virol 4, 237–245. [Google Scholar]
- Imani F. and Jacobs B. L. (1988), Proc Natl Acad Sci USA 85, 7887–7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katze M. G. (1993), Semin Virol 4, 508.1–508.10. [Google Scholar]
- Koromilas A. E., Roy S., Barber G. N., Katze M. G., and Sonenberg N. (1992), Science 257, 1685–1689. [DOI] [PubMed] [Google Scholar]
- Kozak M. (1992), Annu Rev Cell Biol 8, 197–225. [DOI] [PubMed] [Google Scholar]
- Lazaris-Karatzas A., Smith M. R., Frederickson R. M., Jaramillo M. L., Liu Y. L., Kung H.-F., and Sonenberg N. (1990), Genes Dev 6, 1631–1642. [DOI] [PubMed] [Google Scholar]
- Lengyel P. (1993), Proc Natl Acad Sci USA 90, 5893–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macejak D. G. and Sarnow P. (1991), Nature 353, 90–94. [DOI] [PubMed] [Google Scholar]
- Macejak D. G., Hambidge S. J., Najita L., and Sarnow P. (1990), in New Aspects of Positive-Strand Viruses (Brinton M. A. and Heinz F. X., eds.), American Society for Microbiolgy, Washington, DC, pp. 152–157. [Google Scholar]
- Mathews M. B. (1993), Semin Virol 4, 247–257. [Google Scholar]
- McCarthy J. E. G. and Gualerzi C. (1990), Trends Genet 6, 78–85. [DOI] [PubMed] [Google Scholar]
- Merrick W. C. (1992), Microbiol Rev 56, 291–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurs E. F., Galabru J., Barber G. N., Katze M. G., and Hovanessian A. G. (1993), Proc Natl Acad Sci USA 90, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundshau L. J. and Faller D. V. (1992), J Biol Chem 267, 23092–23098. [PubMed] [Google Scholar]
- Oh S.-K. and Sarnow P. (1993), Curr Opin Genet Dev 3, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S.-K., Scott M. P., and Sarnow P. (1992), Genes Dev 6, 1643–1653. [DOI] [PubMed] [Google Scholar]
- Ou J.-H., Bao H., Shih C., and Tahara S. M. (1990), J Virol 64, 4578–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlakis G. N. and Felber B. K. (1990), New Biol 2, 20–31. [PubMed] [Google Scholar]
- Rinker-Schaeffer C. W., Austin V., Simmer S., and Rhoads R. E. (1992), J Biol Chem 267, 2593–2598. [PubMed] [Google Scholar]
- Roy S., Katze M. G., Parkin N. T., Edery I., Hovanessian A. G., and Sonenberg N. (1990), Science 247, 1216–1219. [DOI] [PubMed] [Google Scholar]
- Roy S., Agy M., Hovanessian A. G., Sonenberg N., and Katze M. G. (1991), J Virol 65, 632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads R. E. (1993), J Biol Chem 268, 3017–3020. [PubMed] [Google Scholar]
- Ryazanov A. G., Rudkin B. B., and Spirin A. S. (1991), FEBS Lett 285, 170–175. [DOI] [PubMed] [Google Scholar]
- Sonenberg N. (1990), Curr Top Microbiol Immunol 161, 23–47. [DOI] [PubMed] [Google Scholar]
- Sonenberg N. (1993), Curr Opin Cell Biol 5, 955–960. [DOI] [PubMed] [Google Scholar]
- Tate W. P. and Brown C. M. (1992), Biochemistry 31, 2443–2450. [DOI] [PubMed] [Google Scholar]
- Tatsuka M., Mitsui H., Wada M., Nagata A., Nojima H., and Okayama H. (1992), Nature 359, 333–336. [DOI] [PubMed] [Google Scholar]
- Tsukiyama-Kohara K., Iizuka N., Kohara M., and Nomoto A. (1992), J Virol 66, 1476–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Itstein M., Wu W.-Y., Kok G. B., Pegg M. S., Dyason J. C., Jin B., Phan T. V., Smythe M. L., White H. F., Oliver S. W., Colman P. M., Varghese J. N., Ryan D. M., Woods J. M., Bethell R. C., Hotham V. J., Cameron J. M., and Penn C. R. (1993), Nature 363, 418–423. [DOI] [PubMed] [Google Scholar]
- Wang C., Sarnow P., and Siddiqui A. (1993), J Virol 67, 3338–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss R. B., Dunn D. M., Atkins J. F., and Gesteland R. F. (1990), Prog Nucleic Acid Res 39, 159–183. [DOI] [PubMed] [Google Scholar]
- Wong-Staal F. (1990), in Virology, 2nd edition (Fields B. N. and Knipe D. M., eds.), Raven Press, New York, pp. 1529–1543. [Google Scholar]
- Wool I. G., Endo Y., Chan Y.-L., and Glück A. (1990), in The Ribosome (Hill W. E. et al. , eds.), American Society for Microbiology, Washington, DC, pp. 203–214. [Google Scholar]
- Wyckoof E. E. (1993), Semin Virol. 4, 209–215. [Google Scholar]
- Zhang Y. and Schneider R. J. (1993), Semin Virol 4, 229–236. [Google Scholar]
- Zhang Y. and Schneider R. J. (1994), J Virol 68, 2544–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]