

Abstract
Cyclin D1, DNA topoisomerase I, and proliferating cell nuclear antigen (PCNA) are three important cell cycle regulatory proteins. Recently, their promoters have been isolated, thus facilitating molecular analysis of transcriptional control mechanisms of these genes. Transcription of these three promoters in stable K562 transfectants during different cell cycle phases was analyzed after cell cycle synchronization. About 1 kb of 5′ flanking region from either cyclin D1 or DNA topoisomerase I gene is sufficient to confer G1 or S-phase-specific transcription activity to chloramphenicol acetyltransferase (CAT) reporter genes, respectively. In contrast, 2.8 kb of 5′ flanking sequences from the PCNA gene led to constitutive transcription, but the inclusion of a segment of the PCNA gene first intron, which contains evolutionarily conserved sequences, could enhance transcription in G1/S-enriched nuclei. This PCNA intron region contains a binding site recognized by the transcription factor E2F. To test whether this site is functional, we cotransfected PCNA-CAT genes with E2F-1 and DP-1 expression plasmids. Expression of the E2F-1/DP-1 heterodimer activated the CAT gene with the PCNA intron. Therefore, this intron region, involved in transcriptional activation at the cell cycle G1/S boundary, is also E2F inducible.
Keywords: Cell cycle, PCNA gene, Transcription factor E2F, Transcription regulation
THE cellular processes of DNA replication and cell division, carried out during the S- and M-phases of the cell cycle, involve a large number of proteins. Expression of those genes encoding many of these proteins is regulated in a cell cycle-dependent manner. Studies of molecular mechanisms involved in cell cycle transcriptional control have been limited by the small number of promoters that have so far been isolated. We investigated the transcriptional regulation of three important cell cycle proteins: cyclin Dl, DNA topoisomerase I, and proliferating cell nuclear antigen (PCNA).
Cyclin D1 is one of the mammalian homologs of yeast G1 cyclins (reviewed in Sherr, 1993). Although the cell cycle function(s) of this protein has not been well defined, it is believed to be important for cell cycle progression (Baldin et al., 1993; Quelle et al., 1993). Cyclin D1 protein interacts with cyclin-dependent kinases (Xiong et al., 1992b) and the retinoblastoma protein (Dowdy et al., 1993). Expression of mouse cyclin D1 is stimulated by colony-stimulating factor 1 (CSF-1) in macrophages (reviewed in Sherr, 1991). Overexpression of cyclin D1 is observed in many tumors, due to chromosomal translocation or gene amplification, thus linking it to the bcl-1 proto-oncogene (Motokura and Arnold, 1993). Recently, the human cyclin D1 promoter has been isolated (Inaba et al., 1992; Xiong et al., 1992a; Herber et al., 1994). Here we show that a reporter gene under the control of this promoter is transcribed actively during G1.
Both DNA topoisomerases are DNA replication enzymes, and are probably involved in the relaxation of supercoils generated during the replication process (Wang, 1991). In yeast, at least 17 replication enzyme genes, including a topoisomerase gene, are coordinately activated at START by the Mlu I cell cycle box (MCB) binding factor (MBF) complex (reviewed in Andrews and Herskowitz, 1990; Johnston, 1992). The existence of a similar subset of cell cycle-regulated mammalian genes is likely (McKinney and Heintz, 1991). The recent isolation of human type I and type II DNA topoisomerase promoters will facilitate such studies (Kunze et al., 1991; Hochhauser et al., 1992). In this article, we show that the DNA topoisomerase I promoter is transcriptionally most active during S-phase.
PCNA, as the DNA polymerase delta auxiliary protein, is also required for DNA replication (Bravo et al., 1987; Prelich and Stillman, 1988; Waga and Stillman, 1994). In addition, PCNA is associated with cyclin-dependent kinases (Zhang et al., 1993). The yeast PCNA gene is regulated by MBF at START (reviewed in Andrews and Herskowitz, 1990; Johnston, 1992). Previously, the mammalian PCNA gene was thought to be growth dependent but not cell cycle regulated (reviewed in Baserga, 1991). Recently, the human and rodent PCNA genes were isolated (Travali et al., 1989; Yamaguchi et al., 1991; Liu, 1992). We found evolutionarily conserved sequences in both the promoter regions and within the first intron. However, previous analyses of the PCNA promoter were focused on the 5′ flanking region (Charollais et al., 1992; Labrie et al., 1993), with the exception of negative regions in intron 1 (Alder et al., 1992) and intron 4 (Ottavio et al., 1990). We constructed two PCNA reporter genes, both containing 2.8 kb of 5′ flanking sequences, but only one with 413 bp of conserved intron sequences. We show that intron 1 sequences can enhance transcription in G1/S-enriched nuclei. This conserved PCNA intron region contains a binding site recognized by the transcription factor E2F, which is known to regulate expression of S-phase genes at the G1/S cell cycle transition (Nevins, 1992; Helin and Harlow, 1993; La Thangue, 1994). To investigate whether this E2F site is functional, we cotransfected E2F-1 (Helin et al., 1992; Kaelin et al., 1992; Shan et al., 1992) and DP-1 (Girling et al., 1993; Helin et al., 1993b) expression plasmids with the two PCNA reporter genes and found that only the PCNA intron reporter gene was activated.
MATERIALS AND METHODS
Plasmid Constructions
Cyclin D1-CAT plasmid was constructed from 1.3-kb Pvu II fragment of pDl-G0650 (Xiong et al., 1992a) inserted into the Sma I site of pUMSV-0CAT (Salier and Kurachi, 1989). DNA topoisomerase I-CAT plasmid was constructed from 990-bp EcoRI-XhoI fragment of pTOPOI (Kunze et al., 1991) inserted into the Sma I site of pUMSV-0CAT. PCNA-HN-CAT and PCNA-HH-CAT plasmids were constructed from 2.8-kb Hinc II-Nru I or 3.6-kb Hinc II fragment of p3BS2 (Travali et al., 1989) inserted into the Sma I site of pUMSV0CAT. A 2.6-kb Xba I-Eag I fragment was excised from PCNA-HH-CAT to yield PCNA-EH-CAT. A 320-bp Hinc II-Nae I fragment was inserted into PCNA-EH-CAT to produce PCNA-EN-CAT. A 74-bp Eco 47 III-Apa I fragment was excised from PCNA-EH-CAT to generate PCNA-EHΔ60/133-CAT. A 319-bp Eco 47 III-Pfl M I fragment was removed from PCNA-EH-CAT to generate PCNA-EHΔ60/378-CAT. An oligonucleotide containing the PCNA intron E2F sequence (CGCGTTTGTGGCTTTGGCGCGAAAAAAGAGGGGAC) was inserted between Asc I and Ppu MI sites of PCNA-EHΔ60/378-CAT to yield PCNA-E2F-CAT.
Cell Culture
Human K562 erythroleukemia cells (ATCC CCL 243) were cultured in RPMI 1640 medium supplemented with 10% calf serum. Human Saos-2 osteosarcoma cells (ATCC HTB 85) were cultured in McCoy’s 5a medium supplemented with 15% fetal bovine serum (FBS).
Stable DNA Transfection by Electroporation
Electroporation of K562 cells was performed as described (Spandidos et al., 1987). Stable K562 transfectants were selected with 300 μg/ml hygromycin. A mixed population of stable transfectants was expanded for cell cycle synchronization.
Synchronization of K562 Cells by Drug Treatment
Mitotic K562 cells were first synchronized with nocodazole as described (Liu et al., 1994) and were later G2-arrested with mimosine. S-phase K562 cells were obtained by thymidine block.
Counterflow Centrifugal Elutriation and Flow Cytometry
Counterflow centrifugal elutriation of K562 cells was performed as described (Kauffman et al., 1990) with Beckman JE 5.0 rotor. We routinely used 12–15 1 cultures (3 ∼ 5 × 109 cells). The overall cell cycle distribution of a population of approximately 5 × 105 cells was determined as described (Vindelov and Christensen, 1990). Fluorescence was measured using a Becton Dickinson FACScan and was analyzed on a Hewlett Packard computer with CellFIT software.
Nuclear Transcription Reactions
Nuclear run on transcription was performed as described (Greenberg and Ziff, 1984) with the following modifications. K562 cells were harvested at 1500 rpm for 5 min, then washed with cold PBS three times. The cell pellet (5 × 107 cells) was resuspended in 4 ml 0.1% NP40 lysis buffer [10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.1% (v/v) NP40] by pipetting up-and-down three to five times, followed by centrifugation. The nuclear pellet was washed once with 4 ml storage buffer (50 mM Tris-HCl, pH 8.0, 0.1 mM EDTA, 5 mM MgCl2, 40% glycerol), and then resuspended in 200 μl storage buffer for storage in liquid N2. For run-on transcription assay, the nuclei were thawed and mixed with 25 μl 10× reaction buffer (100 mM Tris-HCl, pH 8.0, 10 mM EDTA, 3 mM MgCl2, 25 mM DTT, 700 mM KCI), 5 μl of 25 mM each of ATP, CTP, and GTP, 20 μl of [α-32P]UTP (200 μCi), and 1 μl RNase inhibitor to react at 30°C for 30 min. To terminate transcription reaction, 1 μl 250 mM CaCl2 and 20 μl of 1 unit/μl DNase I were added and incubated at 30°C for 10 min. To remove proteins, 30 μl 10 × SET buffer (5% SDS, 50 mM EDTA, 100 mM Tris-HCl, pH 7.4) and 2 μl proteinase K (10 mg/ml) were added to react at 42 °C for 30 min. The guanidinium thiocyanate phenol chloroform procedure was used to isolate RNA as described (Chomczynski and Sacchi, 1987). After ethanol precipitation, 32P-labeled RNA was dissolved in 100 μl of TES (10 mM N-Tris[hydroxymethyl]-methyl 2-aminoethane sulphonic acid, pH7.4, 0.2% SDS, and 10 mM EDTA) and then 1 μl was used for scintillation counting. The value of counting should be more than 1 × 105 CPM. NaOH was added to give a final concentration of 0.2 M to denature 32P-labeled RNA for 10 min on ice. The solution was neutralized by the addition of HEPES to a final concentration of 0.5 M and NaCl concentration was adjusted to 0.7 M. In a given experiment, each set of filters was hybridized with the same number of CPM of 32P-labeled RNA in 1 ml of TES buffer containing 0.2% SDS at 65°C for 36 h. After hybridization, the filters were washed with several changes of 2 × SSC (1 × SSC = 0.15 M NaCl, 0.0125 M Na citrate, pH 7.0) twice for 1 h at 65°C and then incubated at 37°C in 2 × SSC with 10 μg/ml RNase A for 30 min. The filters were then washed again in 2 × SSC at 37°C for 1 h, air dried, and exposed in a Phospholmager cassette (Molecular Dynamics). For binding to nitrocellulose, plasmid DNA was linearized by restriction enzyme digestion and the DNA was denatured by incubation with 0.2 M NaOH for 30 min at room temperature, followed by neutralization with 10 vol of 6 × SSC. The DNA was loaded onto nitrocellulose using the Schleicher and Schuell slot blot apparatus; 20 μg DNA was applied per slot.
Transient DNA Transfections and CAT Assays
Human Saos-2 osteosarcoma cells were transfected by calcium phosphate-mediated precipitation, with a total of 25 μg of DNA per 100-mm dish, including internal control plasmid pβJ3, which contains the human β-actin promoter (Ng et al., 1989)-directed β-galactosidase gene. Cell extracts were prepared, and CAT assays were performed as previously described (Liu et al., 1993).
RESULTS
Transcription of Cyclin D1 and DNA Topoisomerase I Genes in Cell Cycle Synchronized Nuclei
Cyclin D1-CAT plasmid was constructed as described in the Materials and Methods section. We transfected this plasmid, together with pSV2-hph (Margolskee et al., 1988), into human K562 cells by electroporation. Hygromycin-resistant stable transfectants were assayed for CAT activities and expanded into 1-1 suspension culture. To examine transcription of the reporter gene during the cell cycle, we used a two-step procedure for synchronization of cells in mitosis and G2 by nocodazole- or mimosine-arrest, respectively (Liu et al., 1994). Thymidine block was used to obtain S-phase-arrested cells. Flow cytometric analysis of DNA content showed that drug-arrested cells were about 85% synchronized (Fig. 1A). About 2 × 108 cells were harvested for nuclear transcription reaction, and labeled RNA was hybridized to CAT, cyclin D1, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) DNA probes (Fig. 1B). Phospholmager quantitation of nascent transcripts showed that, relative to the constitutive level of GAPDH transcription, transcription of both endogenous cyclin D1 and transfected cyclin D1-CAT genes is active during G2 only (Fig. 1C).
FIG. 1.
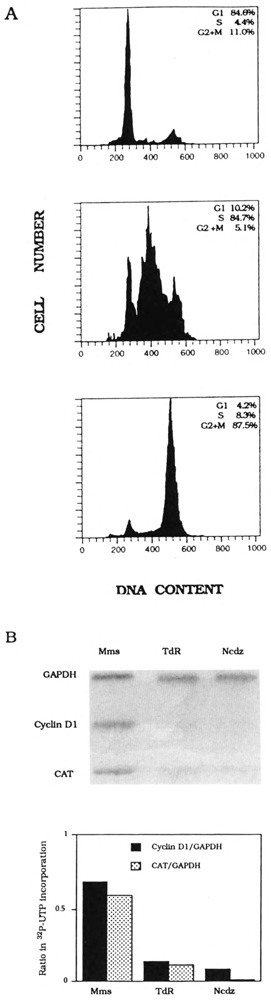
K562 cyclin Dl-CAT stable transfectant cell cycle synchronization and transcription analysis. (A) Flow cytometry analysis of mimosine-arrested cells (top), thymidine-blocked cells (middle), and nocodazole-inhibited cells (bottom). The distribution of DNA contents was determined by FACScan. Percentage of G1 S-phase, and G2 + M cells in each synchronized cell fraction is shown in each inset. (B) Hybridization of nascent nuclear transcripts from mimosine-arrested (Mms), thymidine-blocked (TdR), or nocodazole-inhibited (Ncdz) cells to cyclin D1, CAT, and GAPDH DNA probes. (C) Quantitation of nascent cyclin D1 and CAT transcripts normalized with nascent GAPDH transcripts, respectively, in mimosine-arrested (Mms), thymidine-blocked (TdR), or nocodazole-inhibited (Ncdz) cells.
DNA topoisomerase I-CAT plasmid was constructed as described in the Materials and Methods section. Transfection, selection, cell cycle synchronization, and nuclear transcription were carried out as described earlier for cyclin D1-CAT. Flow cytometric analysis of DNA content showed that these cells were synchronized from 74% to 91% (Fig. 2A). RNA labeled in nuclear transcription reactions were hybridized to CAT, DNA topoisomerase I, and GAPDH DNA probes (Fig. 2B), and quantitation of nascent transcripts showed that, relative to the constitutive level of GAPDH transcription, transcription of both endogenous DNA topoisomerase I and transfected DNA topoisomerase I-CAT genes is most active during S-phase (Fig. 2C).
FIG. 2.
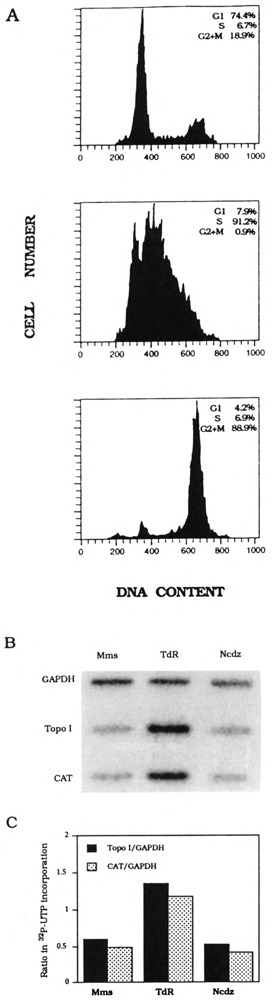
K562 DNA topoisomerase I-CAT stable transfectant cell cycle synchronization and transcription analysis. (A) Flow cytometry analysis of mimosine-arrested cells (top), thymidine-blocked cells (middle), and nocodazole-inhibited cells (bottom). The distribution of DNA contents was determined by FACScan. Percentage of G1 S-phase, and G2 × M cells in each synchronized cell fraction is shown in each inset. (B) Hybridization of nascent nuclear transcripts from mimosine-arrested (Mms), thymidine-blocked (TdR), or nocodazole-inhibited (Ncdz) cells to DNA topoisomerase I, CAT, and GAPDH DNA probes. (C) Quantitation of nascent DNA topoisomerase I and CAT transcripts normalized with nascent GAPDH transcripts, respectively, in mimosine-arrested (Mms), thymidine-blocked (TdR), or nocodazole inhibited (Ncdz) cells.
Transcription of PCNA Gene in Cell Cycle Synchronized Nuclei
Two PCNA-CAT plasmids were constructed as described in the Materials and Methods section, one containing 413 bp of conserved intron sequences (PCNA-HH-CAT) and the other containing an SV40 intron only (PCNA-HN-CAT). Transfection, selection, cell cycle synchronization, and nuclear transcription were carried out as described earlier for cyclin D1-CAT. Flow cytometric analysis of DNA content showed that these cells were synchronized from 74% to 96% (Fig. 3A,B). RNA labeled in nuclear transcription reactions was hybridized to CAT, PCNA, and GAPDH DNA probes, and quantitation of nascent transcripts showed that, relative to the constitutive level of GAPDH transcription, transcription of the endogenous PCNA and transfected intron-containing CAT genes during S-phase is slightly higher than the intronless CAT gene (Fig. 3E,F).
FIG. 3.
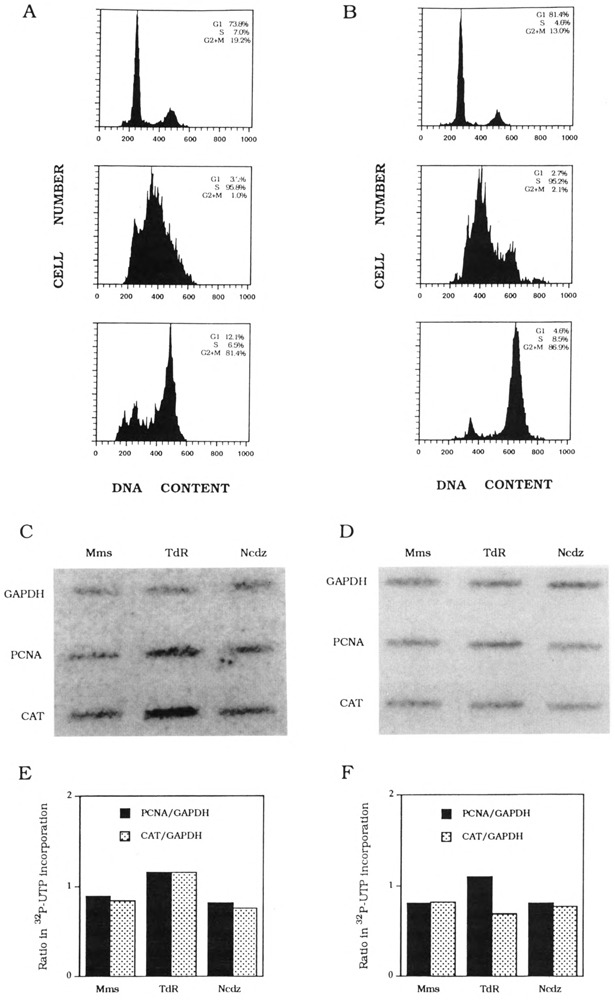
K562 PCNA-HH-CAT and PCNA-HN-CAT stable transfectant cell cycle synchronization and transcription analysis. (A) Flow cytometry analysis of mimosine-arrested K562 PCNA-HH-CAT cells (top), thymidine-blocked cells (middle), and nocodazole-inhibited cells (bottom). The distribution of DNA contents was determined by FACScan. Percentage of G1, S-phase, and G2 × M cells in each synchronized cell fraction is shown in each inset. (B) Flow cytometry analysis of mimosine-arrested K562 PCNA-HNCAT cells (top), thymidine-blocked cells (middle), and nocodazole-inhibited cells (bottom). The distribution of DNA contents was determined by FACScan. Percentage of Gl, S-phase, and G2 × M cells in each synchronized cell fraction is shown in each inset. (C) Hybridization of K562 PCNA-HH-CAT nascent nuclear transcripts to PCNA, CAT, and GAPDH DNA probes. (D) Hybridization of K562 PCNA-HN-CAT nascent nuclear transcripts to PCNA, CAT, and GAPDH DNA probes. (E) Quantitation of K562 PCNA-HH-CAT nascent PCNA and CAT transcripts normalized with nascent GAPDH transcripts, respectively. (F) Quantitation of K562 PCNA-HN-CAT nascent PCNA and CAT transcripts normalized with nascent GAPDH transcripts, respectively.
Due to possible problems of drug-induced alterations of gene expression (reviewed in Pardee and Keyomarsi, 1992), we repeated nuclear transcription analyses of cyclin D1-CAT and PCNA-CAT genes with nuclei fractionated by counterflow centrifugal elutriation (Kauffman et al., 1990). Cell populations were fractionated on the basis of sedimentation properties; therefore, counterflow centrifugal elutriation currently is the best method to obtain physiologically unperturbated cell cycle phase-specific subpopulations. In fact, we chose to work with human K562 cells because it is the cell line of choice for counterflow centrifugal elutriation (Kauffman et al., 1990). Results from a nuclear transcription experiment with elutriation-synchronized cyclin Dl-CAT cells confirm our previous conclusion that this promoter is G1 inducible (Fig. 4).
FIG. 4.
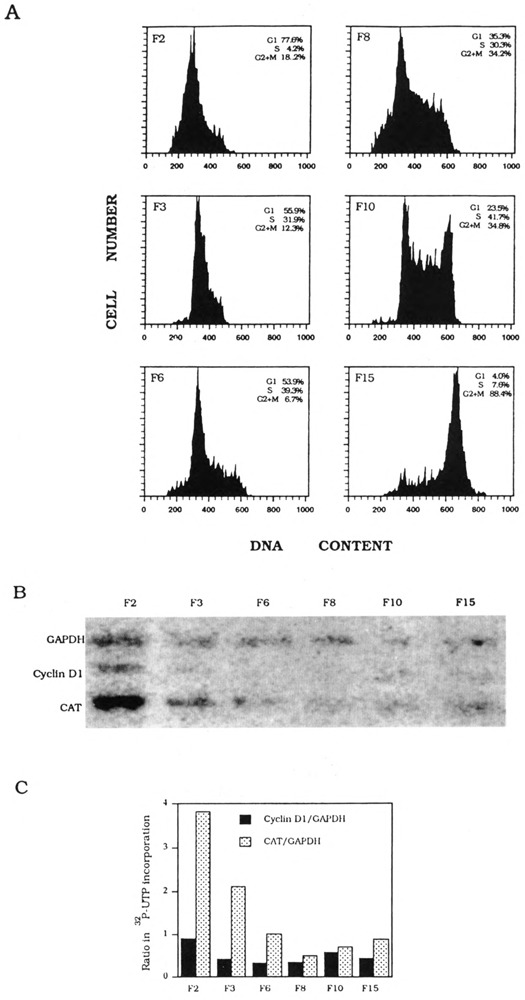
K562 cyclin D1-CAT stable transfectant cell cycle synchronization and transcription analysis. (A) Flow cytometry analysis of DNA contents of counterflow centrifugal elutriated cells (FI-FI5) as determined by FACScan. Percentage of G1 S-phase, and G2 × M cells in each synchronized cell fraction is shown in each inset. (B) Hybridization of nascent nuclear transcripts to cyclin D1, CAT, and GAPDH DNA probes. (C) Quantitation of nascent cyclin D1 and CAT transcripts normalized with nascent GAPDH transcripts, respectively.
To examine whether transcription of PCNA would be transiently activated during G1/S in the cell cycle, we fractionated K562 PCNA-HH-CAT cells into eight subpopulations, labeled as F1 to F8. Unlike drug-synchronized cell populations, counterflow centrifugal elutriation can fractionate K562 cells to yield a G1/S subpopulation. Flow cytometric analysis of DNA content showed that F5 cells were 50% G1 and 41% S-phase (Fig. 5A). Nuclei were prepared from F5 and five other cell fractions for nuclear transcription reactions: labeled RNA was hybridized to CAT, PCNA, and GAPDH DNA probes (Fig. 5B), and quantitation of nascent transcripts showed that, relative to the constitutive level of GAPDH transcription, transcription of both endogenous PCNA and transfected intron-containing CAT genes is clearly more active in F5 nuclei (Fig. 5C). In contrast, transcription reactions carried out with nuclei derived from counterflow centrifugal elutriation-fractionated K562 PCNA-HN-CAT cells showed that the endogenous PCNA gene, but not the transfected intronless CAT gene, is transcriptionally more active in G1/S (F6) nuclei (Fig. 6). Therefore, element(s) within the PCNA gene first intron is (are) essential for transcription activation of the G1/S transition.
FIG. 5.
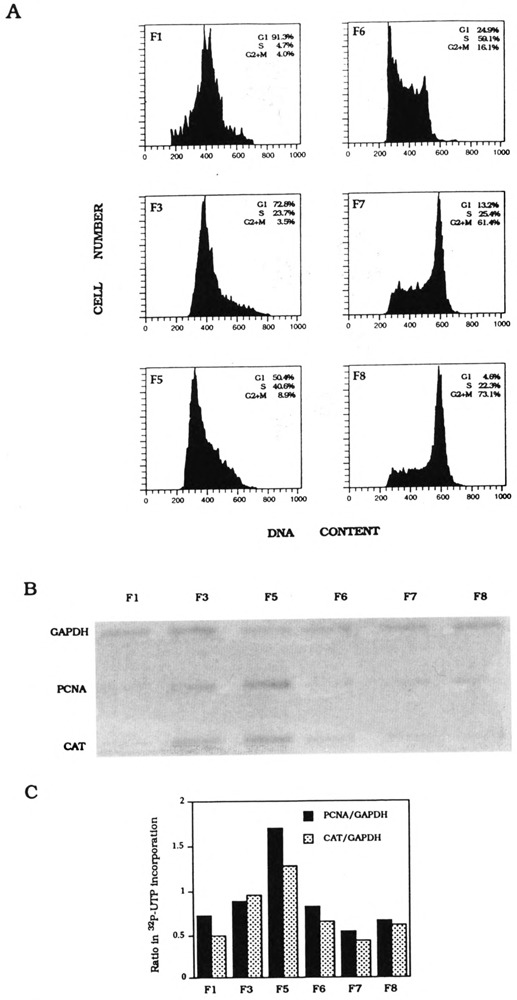
K562 PCNA-HH-CAT stable transfectant cell cycle synchronization and transcription analysis. (A) Flow cytometry analysis of DNA contents of counterflow centrifugal elutriated cells (F1–F8) as determined by FACScan. Percentage of G1 S-phase, and G2 × M cells in each synchronized cell fraction is shown in each inset. (B) Hybridization of nascent nuclear transcripts to PCNA, CAT, and GAPDH DNA probes. (C) Quantitation of nascent PCNA and CAT transcripts normalized with nascent GAPDH transcripts, respectively.
FIG. 6.
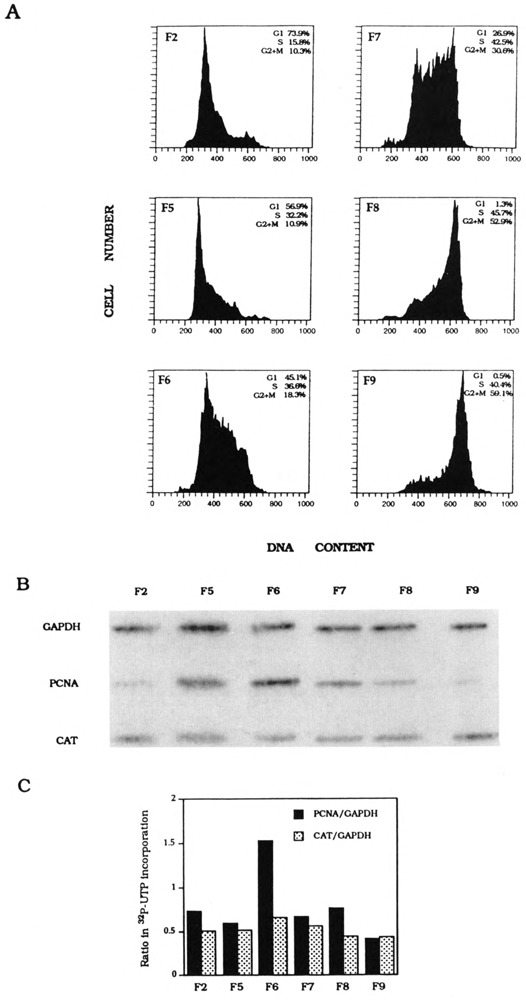
K562 PCNA-HN-CAT stable transfectant cell cycle synchronization and transcription analysis. (A) Flow cytometry analysis of DNA contents of counterflow centrifugal elutriated cells (F1–F9) as determined by FACScan. Percentage of G1 S-phase, and G2 × M cells in each synchronized cell fraction is shown in each inset. (B) Hybridization of nascent nuclear transcripts to PCNA, CAT, and GAPDH DNA probes. (C) Quantitation of nascent PCNA and CAT transcripts normalized with nascent GAPDH transcripts, respectively.
PCNA Gene First Intron Contained Conserved E2F Sites
When the three PCNA intron 1 sequences were compared, we detected a conserved motif TTTTC-GCGCCAAA in human, TTTTCCCGCCAAA in rat, and TTTCCGCGCCAAA in mouse (Fig. 7), which resembles the E2F sites of adenovirus E2 and mouse dihydrofolate reductase (DHFR) genes, TTTTCGCGCTTAAA and ATTTCGCGCCAAA, respectively. Furthermore, 18 bp downstream of this intron site, there is yet another conserved sequence (GCGGGAAAA in all three genes) related to the E2F site in the human thymidine kinase promoter (Kim and Lee, 1992). E2F was previously identified as an early adenovirus gene transcription factor. It is now shown to be involved in cell cycle control as a functional target of the tumor suppressor protein pRb that is encoded by the retinoblastoma susceptibility gene (Nevins, 1992; Johnson et al., 1993). The disruption of E2F-pRb interaction is apparently effected by pRb phosphorylation at the G1/S transition (reviewed in Cobrinik et al., 1992; Hamel et al., 1992), and/or cyclin D-pRb complex formation (Dowdy et al., 1993; Ewen et al., 1993).
FIG. 7.

Conserved first intron sequences in the PCNA genes. Alignment of human, mouse, and rat PCNA intron sequences with the GCG pileup program and displayed with PrettyBox. Shown are human intron sequence from base 75 to 600 (Travali et al., 1989), mouse intron sequence from base 71 to 581 (Yamaguchi et al., 1991), and rat intron sequence from base 66 to 579 (Liu, 1992). The positions of E2F sites are indicated. The 35-bp negative regulatory element (Alder et al., 1992) is between base positions 48 and 82 in the human sequence. The 3′ junctions of the deletion constructs PCNA-EHΔ60/133-CAT and PCNA-EHΔ60/378-CAT are indicated.
Activation of PCNA Intron Reporter Genes by E2F-1/DP-1 Heterodimer
Recent cloning of the E2F-1 cDNA has facilitated analysis of E2F-mediated cell cycle gene regulation (Helin et al., 1992; Kaelin et al., 1992; Shan et al., 1992). Subsequently, another cDNA, named DP-1 for DRTF-polypeptide-1, which encodes a second E2F site binding protein, was isolated (Girling et al., 1993; Helin et al., 1993b). E2F-1 and DP-1 can heterodimerize in vitro as well as in vivo, leading to cooperative transactivation of the E2F site (Bandara et al., 1993; Helin et al., 1993b; Krek et al., 1993). The PCNA intron E2F sites can be recognized by E2F-1 recombinant protein in vitro (our unpublished observation). To test whether this E2F-1/DP-1 heterodimer can regulate PCNA gene expression, we transfected E2F-1 and DP-1 expression plasmids, together with one of the following reporter genes: PCNA-HH-CAT, PCNA-HN-CAT, and DHFR-CAT (Fig. 8B). Mouse DHFR promoter contains an E2F binding site and can be activated by E2F-1 expression (Slansky et al., 1993). After β-gal normalization, both PCNA-HH-CAT and DHFR-CAT activities were stimulated either four- or seven-fold by E2F-1 and DP-1 expression, respectively. In contrast, the PCNA-HN-CAT gene was not activated by E2F-1 and DP-1 expression.
FIG. 8.
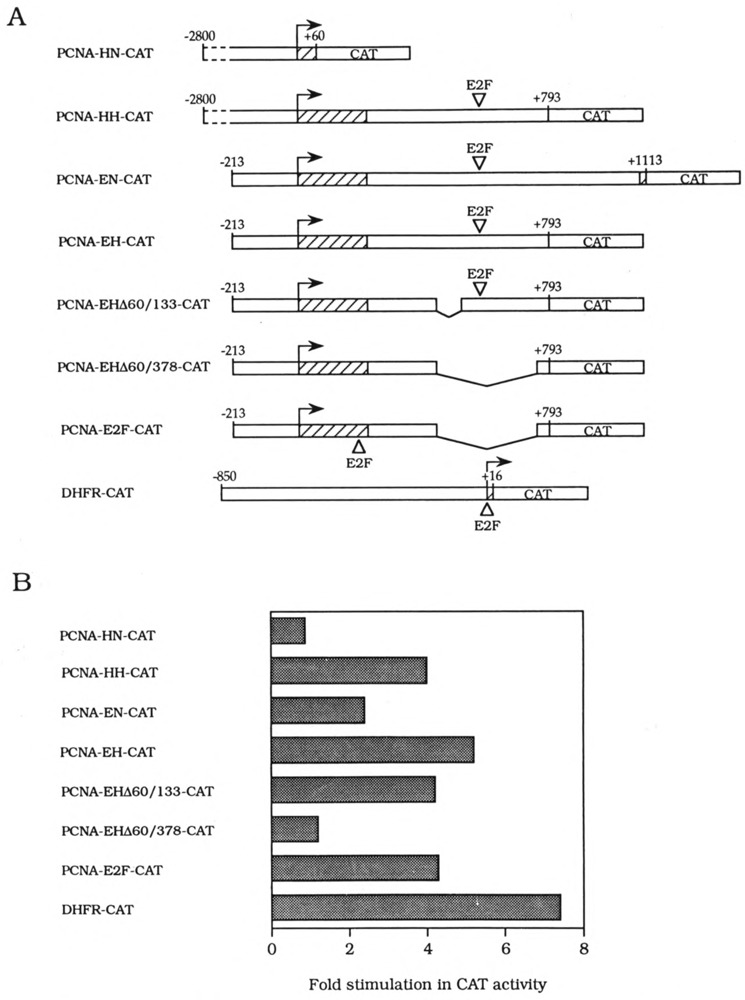
Regulation of PCNA promoter activities by CMV E2F-1 and DP-1 expression plasmids. (A) The structure of PCNA reporter genes is shown with the position of E2F site as indicated. (B) The ability of human E2F-1 and DP-1, transfected into human Saos-2 cells, to activate transcription of PCNA-HN-CAT, PCNA-HH-CAT, PCNA-EN-CAT, PCNA-EH-CAT, PCHA-EHΔ60/133-CAT, PCNA-EHΔ60/378-CAT, PCNA-E2F-CAT, or DHFR(−850/+16)pCAT (Schmidt et al., 1990) reporter genes was determined. CAT activities, derived from duplicate transfections, were normalized to the β-galactosidase activities produced by an internal control plasmid, pβJ3. Shown is the stimulation of promoter-driven CAT activity in response to CMV E2F-1 and CMV DP-1 (Helin et al., 1993b) divided by the activity obtained with the CMV vector alone, Activation of PCNA-HH-CAT, PCNA-EN-CAT, PCNA-EH-CAT, PCNA-EHΔ60/133-CAT, and PCNA-E2F-CAT by E2F-1 and DP-1 was observed reproducibly in at least three independent transfections.
The PCNA-HH-CAT gene contains over 1500 bp of 5′ flanking region for which the nucleotide sequence has not been reported. To better define cis-acting element(s) required for activation of the PCNA-HH-CAT gene, first we deleted over 2 kb of 5′ flanking sequences to remove this unknown region (Fig. 8A). Cotransfection of this PCNA-EH-CAT gene containing 413 bp intron sequences or a similar PCNA-EN-CAT construct, which contains an additional 295 bp of intron sequences (Fig. 8A), with E2F-1 and DP-1 expression plasmids resulted in activation of both reporter genes (Fig. 8B). Next we made internal deletions of the PCNA first intron: PCNA-EHΔ60/133-CAT is missing a known negative region in this intron (Alder et al., 1992), and PCNA-EHΔ60/378-CAT lacks both this negative region and E2F binding sites (Fig. 8A). As expected, PCNA-EHΔ60/378-CAT is not inducible by E2F-1 and DP-1, whereas PCNA-EHΔ60/133-CAT still is (Fig. 8B). Finally, we reinserted the E2 and DHFR-like E2F site (TTTTCGCGCCAAA) back into the PCNA-EHΔ60/378-CAT plasmid downstream of the transcription initiation site (Fig. 8A). This E2F binding site, originally derived from the first intron, is able to confer inducibility by E2F-1 and DP-1 to this PCNA-E2F-CAT gene (Fig. 8B). We conclude that E2F-1- and DP-1-responsive sequences in the PCNA intron are likely the E2F binding sites.
Cyclin D1 promoter (Herber et al., 1994; Philipp et al., 1994) contains putative E2F binding sites (TTTCGGGCA and TTTGGCGCC) but expression of E2F-1 and DP-1 stimulated the cyclin D1-CAT reporter gene less than twofold (data not shown). Therefore, although cyclin D1 and PCNA can associate with each other (Xiong et al., 1992b), they are independently regulated at the transcriptional level.
The DNA topoisomerase I promoter also contains two putative E2F binding sites (TTTGCCCCG and CGGCGGAAAA), but this reporter gene was inhibited by E2F-1 and DP-1 (data not shown). Further work will be required to determine whether these sites are recognized by the E2F complex in vivo, or if their flanking sequences contain antagonistic motif(s).
DISCUSSION
Characterization of Cell Cycle-Regulated Promoters
We have examined the promoters of three cell cycle regulatory proteins. The human cyclin D1 promoter activity is restricted to G1; the human DNA topoisomerase I promoter is active throughout S-phase; the human PCNA promoter is transiently activated at the G1/S transition. Specific cell cycle-regulated elements of these three promoters have not been defined in our present study, but we have mapped the location of such elements within a region of 1 kb for human cyclin D1 and DNA topoisomerase I promoters, and within a 733-bp region of the PCNA first exon and intron. Further mapping of cis-acting elements by promoter deletions, combined with transcription analysis in transfected nuclei, after cell cycle synchronization, should yield definitive information about cell cycle control elements for each promoter.
In previous studies, activation of cell cycle-regulated promoters was only demonstrated in G0-arrested cells stimulated with serum. These promoters included the human PCNA promoter (Chang et al., 1990; Ottavio et al., 1990), human DNA polymerase α promoter (Pearson et al., 1991), the human cdc2 promoter (Dalton, 1992), the mouse DHFR promoter (Slansky et al., 1993), the mouse B-myb promoter (Lam and Watson, 1993), and the human cyclin D1 promoter (Herber et al., 1994). We made the following modification to the analysis of cell cycle-regulated promoters in cell cycle-synchronized cells. First, we choose to use human K562 erythroleukemia cells instead of HeLa cells. Both HeLa and K562 grow in suspension cultures, but the latter is the cell line of choice for cell cycle synchronization by counterflow centrifugal elutriation (Kauffman et al., 1990). HeLa cells also express endogenous HPV-18 E7 protein, which can disrupt interaction between pRb protein and transcription factors (Pagano et al., 1992), including E2Fs (Flemington et al., 1993; Hagemeier et al., 1993; Helin et al., 1993a; Lees et al., 1993), ATF-2 (Kim et al., 1992), MYC (Rustgi et al., 1991), and Sp-1 (Udvadia et al., 1993). Secondly, we use nuclear transcription reactions to analyze reporter gene expression. Our results thus give a more direct measurement of transcription rates than RNA analyses or reporter enzyme assays.
Regulation of the PCNA Gene by E2F
The E2F binding site complex consists of five polypeptides (Huber et al., 1993), four of which have been cloned: E2F-1 (Helin et al., 1992; Kaelin et al., 1992; Shan et al., 1992), DP-1 (Girling et al., 1993; Helin et al., 1993b), E2F-2 and E2F-3 (Ivey-Hoyle et al., 1993; Lees et al., 1993). The E2F-1 and DP-1 expression plasmids (Helin et al., 1992; Kaelin et al., 1992; Helin et al., 1993b) can activate the E2F site-containing PCNA, DHFR, DNA polymerase α, and cdc2 promoters (our unpublished observation). In contrast, cyclin D1 and DNA topoisomerase I promoters are not activated by E2F-1 and DP-1. The identification of transcriptional regulators for the latter promoters will be facilitated by further characterization of cis-acting elements important for cell cycle activation.
The PCNA E2F site, unlike other E2F sites (Nevins, 1992), is localized in the first intron. This intron contains about 500 bp of evolutionarily conserved DNA sequences. Basal activity of the PCNA-EH-CAT gene is about sevenfold lower than the PCNA-EHΔ60/133-CAT gene, perhaps due to the negative regulatory region (Alder et al., 1992). The latter region and E2F site are within 100 bp of each other, thus their binding proteins may interact, and possibly may contribute to the transient activation of this gene.
The use of a common transcription factor represents the simplest mechanism for coordinating S-phase gene expression. This mechanism is used by yeast for coordinating the transcription of many S-phase genes. DNA topoisomerase I is activated during S-phase, but unlike the PCNA promoter, it is not E2F inducible. At least one of the two E2F site-related sequences (TTTGCCCCG) can bind E2F, but transactivation may be inhibited by either trans-acting or cis-acting mechanisms. At least two additional E2F-l-related cDNAs have been cloned (Lees et al., 1993), and whether distinct subsets of E2F sites can be activated by the expression of individual E2F/DRTF polypeptide or specific combinations thereof is being tested. Another possibility is that transcription of the DNA topoisomerase I gene during S-phase is regulated by other cell cycle transcription factor(s) for which binding site(s) reside within the 1-kb 5′ flanking region (Heiland et al., 1993).
In summary, we carried out cell cycle transcription analysis of three promoters. Only one of them, the PCNA intron-containing promoter, was shown to be transiently activated at G1/S and also E2F inducible. The above results, taken together, identified the human PCNA gene as a bona fide cell cycle- and E2F-regulated gene. Many promoters (e.g., c-myc) also contain an E2F site, and presumably can be activated by E2F expression. However, c-myc is an early G1 gene and E2F is thought to be associated with pRb during most of G1, and this complex acts as a transcriptional repressor (Flemington et al., 1993; Hagemeier et al., 1993; Helin et al., 1993a). Therefore, the presence of E2F site(s) may not infer cell cycle regulation by E2F. The latter process, the coordinate activation of S-phase genes, by E2F and its associated proteins, may be controlled by new E2F synthesis (Slansky et al., 1993; Shan et al., 1994), heterodimerization between E2F-1 and DP-1 or related polypeptide(s) (Bandara et al., 1993; Helin et al., 1993b; Krek et al., 1993), phosphorylation of pRb and inhibition of pRb-E2F interaction, and/or disruption of pl07-E2F interaction by unknown mechanisms (Schwarz et al., 1993; Zamanian and La Thangue, 1993; Zhu et al., 1993). A better understanding of E2F regulatory mechanisms will be facilitated by the identification of other E2F-regulated promoters as we have shown here for the PCNA gene.
ACKNOWLEDGEMENTS
We thank Andrew Arnold for cyclin Dl/PRAD1 cDNA, Renato Baserga for the human PCNA gene and cDNA, David Beach and Yue Xiong for the human cyclin D1 genomic clone, Kristian Helin and Ed Harlow for CMV E2F-1 and CMV DP-1, Bill Kaelin for CMV RBAP-1, Rowena Girling and Nic La Thangue for SV DP-1, Chi-Hon Lee for DNA topoisomerase I-CAT, Gary Merrill for DHFR-CAT, and Rick Westerman for PrettyBox. We also thank Heinz-Peter Nasheuer and Teresa S.-F. Wang for helping with counterflow centrifugal elutriation of K562 cells, Chiou-Fen Chuang and Mei-Ling Liu for technical assistance. The work in the laboratory of S.Y.N. is supported by Academic Sinica and National Science Council (ROC).
REFERENCES
- Alder H., Yoshinouchi M., Prystowsky M. B., Appasamy P., and Baserga R. (1992), Nucleic Acids Res 20, 1769–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews B. J. and Hershowitz I. (1990), J Biol Chem 265, 14057–14060. [PubMed] [Google Scholar]
- Baldin V., Lukas J., Marcote M. J., Pagano M., and Draetta G. (1993), Genes Dev 7, 812–821. [DOI] [PubMed] [Google Scholar]
- Bandara L. R., Buck V. M., Zamanian M., Johnson L. H., and La Thangue N. B. (1993), EMBO J 12, 4317–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baserga R. (1991), J Cell Sci 98, 433–436. [DOI] [PubMed] [Google Scholar]
- Bravo R., Frank R., Blundell P. A., and Macdonald-Bravo H. (1977), Nature 326, 515–517. [DOI] [PubMed] [Google Scholar]
- Chang C.-D., Ottavio L., Travali S., Lipson K. E., and Baserga R. (1990), Mol Cell Biol 10, 3289–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charollais R.-H., Alder H., Ferber A., Koniecki J., Sell C., and Baserga R. (1992), Gene Exp 2, 285–296. [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P. and Sacchi N. (1987), Anal Biochem 162, 156–159. [DOI] [PubMed] [Google Scholar]
- Cobrinik D., Dowdy S. F., Hinds P. W., Mittnacht S., and Weinberg R. A. (1992), Trends Biochem Sci 17, 312–315. [DOI] [PubMed] [Google Scholar]
- Dalton S. (1992), EMBO J 11, 1797–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdy S. F., Hinds P. W., Louie K., Reed S. I., Arnold A., and Weinberg R. A. (1993), Cell 73, 499–511. [DOI] [PubMed] [Google Scholar]
- Ewen M. E., Sluss H. K., Sherr C. J., Matsushime H., Kato J-y., and Livingston D. A. (1993), Cell 73, 487–497. [DOI] [PubMed] [Google Scholar]
- Flemington E. K., Speck S. H., and Kaelin W. G. Jr. (1993), Proc Natl Acad Sci USA 90, 6914–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girling R., Partridge J. F., Banara L. R., Burden N., Totty N. F., Hsuan J. J., and La Thangue N. B. (1993), Nature 362, 83–87; 365, 468. [DOI] [PubMed] [Google Scholar]
- Greenberg M. E. and Ziff E. B. (1984), Nature 311, 433–437. [DOI] [PubMed] [Google Scholar]
- Hagemeier C., Cook A., and Kouzarides T. (1993), Nucleic Acids Res 21, 4998–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel P. A., Gallie B. L., and Phillips R. A. (1992), Trends Genet 8, 180–185. [DOI] [PubMed] [Google Scholar]
- Heiland S., Knippers R., and Kunze N. (1993), Eur J Biochem 217, 813–822. [DOI] [PubMed] [Google Scholar]
- Helin K., Lees J. A., Vidal M., Dyson N., Harlow E., and Fattaey A. (1992), Cell 70, 337–350. [DOI] [PubMed] [Google Scholar]
- Helin K. and Harlow E. (1993), Trends Cell Biol 3, 43–46. [DOI] [PubMed] [Google Scholar]
- Helin K., Harlow E., and Fattaey A. (1993a), Mol Cell Biol 13, 6501–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K., Wu C.-L., Fattaey A. R., Lees J. A., Dynlacht B. D., Ngwu C., and Harlow E. (1993b), Genes Dev 7, 1850–1861. [DOI] [PubMed] [Google Scholar]
- Herber B., Truss M., Beato M., and Miller R. (1994), Oncogene 9, 1295–1304. [PubMed] [Google Scholar]
- Hochhauser D., Stanway C. A., Harris A. L., and Hickson I. D. (1992), J Biol Chem 267, 18961–18965. [PubMed] [Google Scholar]
- Huber H. E., Edwards G., Goodhart P. J., Patrick D. R., Huang P. S., Ivey-Hoyle M., Barnett S. F., Oliff A., and Heimbrook D. C. (1993), Proc Natl Acad Sci USA 90, 3525–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba T., Matsushime H., Valentine M., Roussel M. F., Sherr C. J., and Look A. T. (1992), Genomics 13, 565–574. [DOI] [PubMed] [Google Scholar]
- Ivey-Hoyle M., Conroy R., Huber H. E., Goodhart P. J., Oliff A., and Heimbrook D. C. (1993), Mol Cell Biol 13, 7802–7812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D. G., Schwarz J. K., Cress W. D., and Nevins J. R. (1993), Nature 365, 349–352. [DOI] [PubMed] [Google Scholar]
- Johnston L. H. (1992), Trends Cell Biol 2, 353–357. [DOI] [PubMed] [Google Scholar]
- Kaelin W. G. Jr., Krek W., Sellers W. R., De-Caprio J. A., Ajchenbaum F., Fuchs C. S., Chittendan T., Li Y., Farnham P. J., Blanar M. A., Livingston D. M., and Flemington E. K. (1992), Cell 70, 351–364. [DOI] [PubMed] [Google Scholar]
- Kauffman M. G., Noga S. J., Kelly T. J., and Donnenberg A. D. (1990), Anal Biochem 191, 41–46. [DOI] [PubMed] [Google Scholar]
- Kim S. J., Wagner S., Liu F., O’Reilly M. A., Robbins P. D., and Green M. R. (1992), Nature 358, 331–334. [DOI] [PubMed] [Google Scholar]
- Kim Y. K. and Lee A. S. (1992), J Biol Chem 267, 2723–2727. [PubMed] [Google Scholar]
- Krek W., Livingston D. M., and Shirodkar S. (1993), Science 262, 1557–1560. [DOI] [PubMed] [Google Scholar]
- Kunze N., Yang G., Dolberg M., Sundarp R., Knippers R., and Richter A. (1991), J Biol Chem 266, 9610–9616. [PubMed] [Google Scholar]
- Labrie C., Morris G. F., and Mathews M. B. (1993), Mol Cell Biol 13, 1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam E. W.-F. and Watson R. J. (1993), EMBO J 12, 2705–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Thangue N. B. (1994) Trends Biochem Sci 19, 108–114. [DOI] [PubMed] [Google Scholar]
- Lees J. A., Saito M., Vidal M., Valentine M., Look T., Harlow E., Dyson N., and Helin K. (1993), Mol Cell Biol 13, 7813–7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.-H., Ma J.-T., Yueh A. Y., Lees-Miller S. P., Anderson C. W., and Ng S.-Y. (1993), J Biol Chem 268, 21147–21154. [PubMed] [Google Scholar]
- Liu S.-H., Lee H.-H., Chen J.-J., Chuang C.-F., and Ng S.-Y. (1994), Cell Growth Differen 5, 447–455. [PubMed] [Google Scholar]
- Liu Y.-C. (1992), GenBank X67329.
- Margolskee R. F., Kavathas P., and Berg P. (1988), Mol Cell Biol 8, 2837–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney J. D. and Heintz N. (1991), Trends Biochem Sci 16, 430–435. [DOI] [PubMed] [Google Scholar]
- Motokura T. and Arnold A. (1993), Curr Opin Genet Dev 3, 5–10. [DOI] [PubMed] [Google Scholar]
- Nevins J. R. (1992), Science 258, 424–429. [DOI] [PubMed] [Google Scholar]
- Ng S.-Y., Gunning P., Liu S.-H., Leavitt J., and Kedes L. (1989), Nucleic Acids Res 17, 601–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottavio L., Chang C.-D., Rizzo M.-G., Travali S., Casadevall C., and Baserga R. (1990), Mol Cell Biol 10, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M., Dürst M., Joswig S., Draetta G., and Jansen-Dürr P. (1992), Oncogene 7, 1681–1686. [PubMed] [Google Scholar]
- Pardee A. B. and Keyomarsi K. (1992), Curr Opin Cell Biol 4, 186–191. [DOI] [PubMed] [Google Scholar]
- Pearson B. E., Nasheuer H.-P., and Wang T. S.-F. (1991), Mol Cell Biol 11, 2081–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipp A., Schneider A., Vasrik I., Finke K., Xiong Y., Beach D., Alitalo, and Eilers M. (1994), Mol Cell Biol 14, 4032–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prelich G. and Stillman B. (1988), Cell 53, 117–126. [DOI] [PubMed] [Google Scholar]
- Quelle D. E., Ashman R. A., Shurtleff S. A., Kato J.-y., Bar-Sagi D., Roussel M. F., and Sherr C. J. (1993), Genes Dev 7, 1559–1571. [DOI] [PubMed] [Google Scholar]
- Rustgi A. K., Dyson N., and Bernards R. (1991), Nature 352, 541–544. [DOI] [PubMed] [Google Scholar]
- Salier J.-L. and Kurachi K. (1989), Biotechniques 7, 30–31. [PubMed] [Google Scholar]
- Schmidt E. E., Owen R. A., and Merrill G. F. (1990), J Biol Chem 265, 17397–17400. [PubMed] [Google Scholar]
- Schwarz J. K., Devoto S. H., Smith E. J., Chellappan S. P., Jakoi L., and Nevins J. R. (1993), EMBO J 12, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B., Zhu X., Chen P.-L., Durfee T., Yang Y., Sharp D., and Lee W.-H. (1992), Mol Cell Biol 12, 5260–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B., Chang C.-Y., Jones D., and Lee W.-H. (1994), Mol Cell Biol 14, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr C. J. (1991), Trends Genet 7, 398–402. [DOI] [PubMed] [Google Scholar]
- Sherr C. J. (1993), Cell 73, 1059–1065. [DOI] [PubMed] [Google Scholar]
- Slansky J. E., Li Y., Kaelin W. G. Jr., and Farnham P. G. (1993), Mol Cell Biol 13, 1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spandidos D. A. (1987), Gene Anal Technol 4, 50–56. [DOI] [PubMed] [Google Scholar]
- Travali S., Ku D.-H., Rizzo M. G., Ottavio L., Baserga R., and Calabretta B. (1989), J Biol Chem 264, 7466–7472. [PubMed] [Google Scholar]
- Udvadia A. J., Rogers K. T., Higgins P. D. R., Murata Y., Martin K. H., Humphrey P. A., and Horowitz J. M. (1993), Proc Natl Acad Sci USA 90, 3265–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindelov L. L. and Christensen I. J. (1990), Cytometry 11, 753–770. [DOI] [PubMed] [Google Scholar]
- Waga S. and Stillman B. (1994), Nature 369, 207–212. [DOI] [PubMed] [Google Scholar]
- Wang J. C. (1991), J Biol Chem 266, 6659–6662. [PubMed] [Google Scholar]
- Xiong Y., Menninger J., Beach D., and Ward D. C. (1992a), Genomics 13, 575–584. [DOI] [PubMed] [Google Scholar]
- Xiong Y., Zhang H., and Beach D. (1992b), Cell 71, 505–514. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M., Hayashi Y., Hirose F., Matsuoka S., Moriuchi T., Shiroishi T., Moriwaki K., and Matsukage A. (1991), Nucleic Acids Res 19, 2403–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian M. and La Thangue N. B. (1993), Mol Biol Cell 4, 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Xiong Y., and Beach D. (1993), Mol Biol Cell 4, 897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L., van den Heuvel S., Helin K., Fattaey A., Ewen M., Livingston D., Dyson N., and Harlow E. (1993), Genes Dev 7, 1111–1125. [DOI] [PubMed] [Google Scholar]