

Abstract
RNA splicing is an indispensable step for expression of many eukaryotic genes. Combinations of 5′ and 3′ splice sites should be correctly selected in both constitutive and alternative splicing. Recent studies have revealed mechanisms of alternative splicing in some systems, in which specific regulators play vital roles in splice site selection. On the other hand, essential splicing factors such as SR proteins modulate splice site usage of general machinery. Specific regulators and splicing factors such as SR proteins have some common structural features. With these related components, a similar machinery of splice site selection is involved in constitutive and alternative splicing.
Keywords: Pre-mRNA, Splice site, Splicing, Alternative splicing, SR protein
IN higher eukaryotes, alternative splicing of messenger RNA precursors (pre-mRNAs) plays an important role in regulating gene expression; it can lead to the production of multiple mRNA species from a single gene during development and differentiation. The Drosophila sex-determination pathway is the most striking example, which involves a cascade of regulated alternative splicing events. Hence, one can easily imagine that studies on alternative splicing are indispensable for understanding developmental phenomena. Moreover, how certain combinations of 5′ and 3′ splice sites are correctly selected is a fundamental question for elucidation of the mechanism of constitutive as well as alternative pre-mRNA splicing. In addition to studies on the Drosophila genes, biochemical approaches, especially those using mammalian systems, have revealed some important aspects of the machinery involved in splice site selection, including the finding that many protein splicing factors have some common sequence motifs. One such motif is the RNA binding domain (RBD), which encompasses 80–90 amino acids, also referred to as the ribonucleoprotein consensus sequence (RNP-CS) or the RNA recognition motif (RRM) (Bandiziulis et al., 1989; Query et al., 1989; Rio, 1992; Birney et al., 1993; Mattaj, 1993). Another is the arginine-serine (RS)-rich sequence motif (for reviews, see Birney et al., 1993; Moore et al., 1993). Drosophila splicing regulators and splicing factors such as SR proteins contain the RBD and/or RS domains.
In this review, we first describe two specific examples of splicing regulation, both from Drosophila, which, because of genetics, is where the most is currently known about regulation. These examples show the negative control by the blockage of splice site usage and the positive control by the activation of suboptimal splice site. We then focus on the current idea, mainly based on the biochemical studies with mammalian systems, that a similar mechanism of splice site selection is involved in both constitutive and alternative splicing.
NEGATIVE REGULATION IN ALTERNATIVE SPLICING
Expression of one of the Drosophila somatic sex-determination genes, transformer (tra), is controlled by sex-specific splicing that is achieved by the alternative usage of two 3′ splice sites (Boggs et al., 1987). Choice of the upstream 3′ splice site leads to the inclusion of the second exon that contains a translational stop codon, generating nonfunctional mRNA in both female and male flies. The second exon is excluded only in females by the use of the downstream 3′ splice site, thereby producing mRNA that encodes functional Tra protein. Genetical studies revealed that Sex-lethal (Sxl), another member of the sex-determination genes, is necessary for the proper regulation of the sex-specific splicing of tra pre-mRNA (Nagoshi et al., 1988; Belote et al., 1989). The Sxl gene produces the functional protein that contains two copies of the RNA binding domain (RBD) (Bell et al., 1988) only in females. Using transgenic flies (Sosnowski et al., 1989) and the transfection system (Inoue et al., 1990), Sxl has been shown to repress the usage of the upstream 3′ splice site to induce the female-specific splicing of tra pre-mRNA. In the absence of functional Sxl gene product, the non-sex-specific splicing at the upstream 3′ splice site occurs exclusively in a default manner. Bacterially produced Sxl protein binds specifically to the uridine-rich sequence that lies in the pyrimidine cluster at the upstream 3′ splice site of tra pre-mRNA (Inoue et al., 1990). It is known that the pyrimidine cluster is indispensable for the splicing reaction and is recognized by some splicing factors, such as U2AF (for review, see Green, 1991). Therefore, Sxl protein functions as a negative regulator to compete out the splicing factors at the upstream 3′ splice site, resulting in the use of the downstream 3′ splice site of tra pre-mRNA in female flies (Fig. 1A) (Sosnowski et al., 1989; Inoue et al., 1990; Zamore et al., 1992; Valcarcel et al., 1993).
FIG. 1.
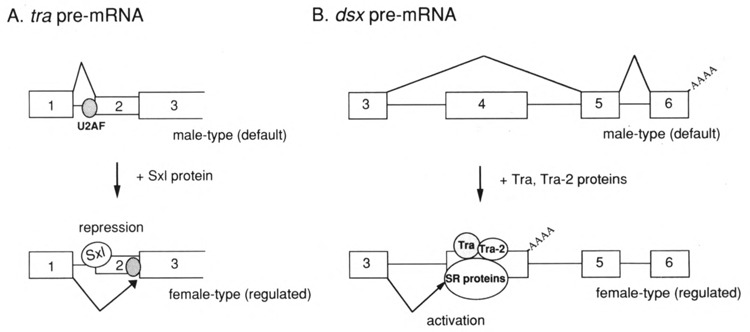
Mechanisms of alternative pre-mRNA splicing. (A) Sex-specific splicing of tra pre-mRNA is regulated by Sxl protein in Drosophila somatic cells. In male flies, splicing occurs at the upstream 3′ splice site; the first exon is spliced to the second exon (upper). In female flies, Sxl protein binds to the target sequence to prevent the binding of splicing factors such as U2AF at the upstream 3′ splice site, leading to the female-specific splicing at the downstream 3′ splice site (lower). (B) Sex-specific splicing of dsx pre-mRNA is controlled by Tra and Tra-2 proteins. In male flies, the third exon is spliced to the fifth exon in a default manner (upper). In females, Tra and Tra-2 bind specifically to the 13-nt repeat sequences in the fourth exon and recruit general splicing factors such as SR proteins, resulting in activation of the upstream 3′ splice site (lower). Polyadenylation reaction (shown as AAAA) occurs immediately downstream of the fourth exon in females. Boxes and the lines between boxes represent the exon and the intron sequences, respectively.
Expression of the Sxl gene is maintained by autoregulation of the sex-specific splicing (Bell et al., 1991). Male-specific Sxl mRNAs include the third exon, which contains the translational stop codon, generating nonfunctional proteins, whereas female-specific mRNAs exclude this exon and encode the functional proteins (Bell et al., 1988). The functional Sxl protein promotes the synthesis of its own female-specific mRNA by inhibiting the male-specific splicing, leading to constitutive production of Sxl protein only in female flies (Bell et al., 1991; Sakamoto et al., 1992). Because there exists the U-rich sequence at the male-specific 3′ splice site of Sxl pre-mRNA, a simple mechanism for the repression of male-specific splicing, as was the case with tra pre-mRNA, was proposed (Sosnowski et al., 1989). However, the mechanism seems to be more complicated than expected: multiple Sxl binding sequences located both upstream and downstream of the male-specific third exon are involved in the splicing regulation (Sakamoto et al., 1992; Horabin and Schedle, 1993), although the mechanism is not yet fully understood.
In addition to the cases of the sex-determination genes, other examples of negative regulation of alternative splicing are provided by Drosophila genes: the P-transposase gene (Laski et al., 1986) and the suppressor of white apricot locus [su(w a)] (Chou et al., 1987; Zachar et al., 1987). In the case of the P-transposase gene, the third intron containing the translational stop codon is removed in germ cells but not in somatic cells. Only the fully spliced mRNA encodes the functional transposase, and splicing of the third intron is inhibited in somatic cells. The intron is accurately spliced out in mammalian cell extracts, but the reaction is inhibited by the addition of Drosophila somatic cell extracts (Siebel and Rio, 1990). A multiprotein complex, which associates with the 5′ exon sequence, is involved in somatic inhibition (Siebel and Rio, 1990; Siebel et al., 1994).
POSITIVE REGULATION IN ALTERNATIVE SPLICING
In the Drosophila sex-determination cascade, expression of the doublesex (dsx) gene is regulated by two genes, tra and tra-2. In somatic cells, the tra-2 gene produces the same protein in both sexes (Amrein et al., 1988; Gorarski et al., 1989), whereas tra produces the functional protein only in females, as described above (Boggs et al., 1987). dsx pre-mRNA undergoes sex-specific RNA processing (splicing and polyadenylation reactions) (Burtis and Baker, 1989). In females, Tra and Tra-2 promote the female-specific processing of dsx pre-mRNA; the third exon is spliced to the female-specific fourth exon and the cleavage/polyadenylation reaction occurs immediately downstream of the fourth exon (Fig. 1B) (Burtis and Baker, 1989; Hoshijima et al., 1991; Hedley and Maniatis, 1991). .In contrast, in males, splicing between the third exon and the male-specific fifth exon occurs in a efault manner. Female-specific processing depen on activation of the female-specific splicing, be use neither the female-specific polyadenylation eaction nor the male-specific splicing is regulated Tra and Tra-2 proteins (Hoshijima et al., 1991; Ryner and Baker, 1991). The female-specific splicing cannot occur in the absence of the regulator proteins because the pyrimidine cluster at the 3′ splice site is not long enough to be recognized by splicing factors (Hoshijima et al., 1991). For activation of the female-specific splicing, six tandemly interspersed repeats of 13-nucleotide (nt) sequences in the female-specific fourth exon are necessary (Nagoshi and Baker 1990; Hoshijima et al., 1991; Inoue et al., 1992; Tian and Maniatis, 1992). Both Tra (Inoue et al., 1992) and Tra-2 (Hedley and Maniatis, 1991; Inoue et al., 1992) can bind directly to these 13-nt repeat sequences, although specific binding of Tra may require additional nuclear factors (Tian and Maniatis, 1992). It was suggested that Tra and Tra-2 are associated with each other on the repeat sequences (Inoue et al., 1992). Furthermore, in vitro studies have clearly showed that Tra and Tra-2 function by recruiting general splicing factors, including the SR proteins, to the repeat sequences (Tian and Maniatis, 1992, 1993). The yeast two-hybrid system and the far-western experiments demonstrated that both Tra and Tra-2 specifically interact with themselves, with each other, and with some members of the SR protein family (Wu and Maniatis, 1993; Amrein et al., 1994). Tra-2 contains the RNA binding domain (RBD) and the two arginine-serine (RS)-rich domains, whereas Tra possesses only the RS domains (Amrein et al., 1988; Gorarski et al., 1989). The RBD of Tra-2 protein is necessary but not sufficient for specific RNA binding in vitro. One of the RS domains that lies in the C-terminal region of Tra-2 is essential for specific RNA binding (Amrein et al., 1994). Furthermore, the RS domain has been shown to be required for protein-protein interactions (Amrein et al., 1994). Interestingly, Tra-2 functions as a negative regulator of alternative splicing of its own pre-mRNA in germ line cells (Mattox and Baker, 1991; Amrein et al., 1994).
SR PROTEINS AND SPLICE SITE SELECTION
It was previously reported that a HeLa cell S100 cytoplasmic extract is not able to support pre-mRNA splicing in vitro because it lacks an essential splicing factor, termed SF2 (Krainer et al., 1990, 1991). Cloning of SF2 cDNA revealed that it is identical with a mammalian splicing factor called ASF (alternative splicing factor), which had been identified on the basis of the activity to modulate the 5′ splice site selection of SV40 T/t antigen pre-mRNA (Ge and Manley, 1990; Ge et al., 1991). SC-35, another essential splicing factor that has similar properties to ASF/SF2, was identified by a monoclonal antibody raised against purified spliceosomes (Fu and Maniatis, 1990, 1992), and was subsequently shown to activate splicing upon addition to the HeLa cell S100 extract (Fu et al., 1992).
Subsequently, a group of proteins designated SR proteins has been identified in a variety of animal cells and tissues by the cross-reaction of the monoclonal antibody mAb104, which had been originally raised against Xenopus laevis oocyte nuclear proteins (Roth et al., 1991; Zahler et al., 1992). SR proteins include related polypeptides of approximately 20, 30, 40, 55, and 75 kDa, and addition of any of these SR proteins to the HeLa cell S100 extract is sufficient to activate splicing (Zahler et al., 1993a). Partial amino acid sequence analysis of the human 30 kDa protein revealed that it includes both ASF/SF2 and SC-35 polypeptides (Zahler et al., 1992). These data indicate a high degree of functional redundancy between the members of the SR protein family. In fact, the SR proteins have similar structural features, containing one or two RBDs and an RS domain (Zahler et al., 1993a).
The fact that ASF/SF2 was independently identified as an alternative splicing factor and a general splicing factor has raised an intriguing possibility that alternative splicing events can be regulated by the activity of some general splicing factor that is required for the basic splicing mechanism. Using pre-mRNAs with duplicated 5′ splice sites and a single 3′ splice site as in vitro splicing substrates, ASF/SF2 and SC-35 were shown to promote the utilization of the 5′ splice site proximal to the 3′ splice site (Fu et al., 1992). The mechanism by which ASF/SF2 and SF-35 exert their activities on 5′ splice site selection is not well understood. It has recently been suggested in the case of ASF/SF2 that the protein can specifically recognize 5′ splice sites and facilitates U1 snRNP binding to the sites via protein-protein interaction between ASF/SF2 and a U1 snRNP-specific protein, U1-70K (Zuo and Manley, 1994; Kohtz et al., 1994).
The activity of ASF/SF2 and SC-35 in promoting proximal 5′ splice site usage is antagonistically counteracted by the activity of the hnRNP Al protein that promotes the usage of distal 5′ splice site (Mayeda and Krainer, 1992). Moreover, each member of the SR protein family appears to have distinct activity on selecting 5′ splice sites of different pre-mRNAs (Fu, 1993; Zahler et al., 1993b). In addition, it has been suggested that the SR proteins and hnRNP A1 are differentially expressed in a variety of tissues (Zahler et al., 1993b; Mayeda et al., 1993). These results are intriguing in light of the fact that many pre-mRNAs are alternatively spliced in a tissue-specific manner. The relative amounts of SR proteins and hnRNP A1 protein expressed in each tissue might be one of the determinants of the alternative splicing pattern (Zahler et al., 1993b; Mayeda et al., 1993). ASF/SF2 and SC-35 also have the activity to promote the utilization of the proximal 3′ splice site in vitro, but this activity is not counteracted by hnRNP Al (Fu et al., 1992). Activity to promote distal 3′ splice site usage has also been identified in a HeLa nuclear extract, designated SF7 (Mayeda et al., 1993), but the molecular basis of this activity remains to be clarified.
EXON RECOGNITION
Although introns are essentially defined by the presence of a conserved 5′ splice site, a 3′ splice site, its preceding pyrimidine cluster, and a branch point, these elements are not likely to be sufficient to specify all introns. In several genes, specific exon sequences have been shown to function as cis elements for regulation of alternative splicing, as is the case with the 13-nt repeat sequences in the dsx female-specific exon (for review, see Green, 1991). In addition, exon sequences can be involved in general splice site selection. The role of exon sequence for selection of 3′ splice site was clearly demonstrated in the course of study on the splicing mechanism of the mouse immunoglobulin μ gene. The purine-rich sequence located within the last exon, M2, promotes splicing of distant upstream introns regardless of cell types (Watakabe et al., 1991). Furthermore, if the 13-nt repeat sequence region in dsx pre-mRNA is replaced by the purine-rich exon sequence, it can activate the usage of the female-specific 3′ splice site even in the absence of Tra and Tra-2 (Watakabe et al., 1993). Therefore, the purine-rich exon sequence of the immunoglobulin μ gene is thought to function as a general splicing enhancer. Similarly, purine-rich sequences from several other genes as well as some synthetic polypurine sequences have also been demonstrated to function as splicing enhancer in vitro (Tanaka et al., 1994). Because this finding is consistent with the exon recognition model, in which the definition of exon units is involved in specification of splice sites (Robberson et al., 1990; Talerico and Berget, 1990), such exonic splicing enhancer was designated exon recognition sequence (ERS) (Watakabe et al., 1993; Tanaka et al., 1994). Xu et al. (1993) also reported that the purine-rich sequence of the cardiac troponin T (cTNT) exon facilitates splicing of a heterologous intron in vivo, thus naming such sequence as exon splicing element (ESE). In the case of the human fibronectin EDA exon, there are both positive and negative modulator elements of splicing (Caputi et al., 1994).
The splicing enhancer sequences are thought to be most important when the splice sites to be selected are suboptimal or weak. Because such splice sites themselves cannot be well recognized by splicing machinery, additional information is necessary. The purine-rich exon sequences are likely to contribute such information (Watakabe et al., 1993). Presumably there exist variable purine-rich sequences in many exons, which modulate splice site selection. Similarly, it has been demonstrated that the downstream 5′ splice site sequence can stimulate the use of the weak upstream 3′ splice site across the exon (Robberson et al., 1990; Talerico and Berget, 1990; Kuo et al., 1991; Hoffman and Grabowski, 1992). The polyadenylation signal may play a similar role for 3′ terminal exons (Niwa and Berget, 1991). In addition, the 5′ cap structure stimulates splicing of 5′ proximal intron (Ohno et al., 1987; Inoue et al., 1989).
Some lines of evidence suggest that the interaction of U1 snRNP with the downstream 5′ splice site (Robberson et al., 1990; Talerico and Berget, 1990; Kuo et al., 1991; Hoffman and Grabowski, 1992) and with ERS (Watakabe et al., ,1993) facilitates spliceosome formation. Recently, it was demonstrated that the interaction of SR proteins with the splicing enhancer (designated SE) sequence in the human fibronectin ED1 exon stimulates U2 snRNP binding (Lavigueur et al., 1993). Moreover, ASF/SF2, one of the SR proteins, binds in a sequence-specific manner to the purine-rich sequence, named exonic splicing enhancer (ESE), in the last exon of bovine growth hormone pre-mRNA, thereby stimulating splicing of the upstream intron (Sun et al., 1993). The stimulation is counteracted by the addition of hnRNP A1. Interestingly, SC-35, another SR protein, neither binds to the sequence nor stimulates splicing of the intron (Sun et al., 1993). These findings indicate that SR proteins can function as specific positive factors via binding to the exon sequences.
CONCLUSION
Studies on the Drosophila sex-determination genes led to the finding that specific regulators play important roles in alternative splicing. These factors interact with specific regulatory sequences of pre-mRNA. In negative regulation, specific regulators prevent the association of splicing factors with pre-mRNA. On the other hand, the positive regulatory system in alternative splicing utilizes the general splicing machinery. Specific regulatory proteins such as Tra and Tra-2 recruit the essential splicing factors such as the SR proteins. The SR proteins are likely to play vital roles in splice site selection. They can interact with splice sites and the exon sequences (ERS/ESE), stimulating the usage of specific splice sites. Because SR proteins are differentially expressed in a variety of tissues and particular SR proteins have distinct functions in splice site selection, alternative splicing in many genes may be controlled directly by the SR proteins. In addition, other RNA binding proteins, such as hnRNP A1, may counteract or modify the effect of SR proteins.
ACKNOWLEDGEMENTS
We thank Tilmann Achsel, Kazuyuki Hoshijima, and Naoyuki Kataoka for helpful discussions and comments on the manuscript. The work presented from the authors’ laboratory was supported in part by grants-in-aid from the Ministry of Education, Science, and Culture of Japan, and by a grant from Senri Life Science Foundation to K. Inoue.
REFERENCES
- Amrein H., Gorman M., and Nöthiger R. (1988), Cell 55, 1025–1035. [DOI] [PubMed] [Google Scholar]
- Amrein H., Hedley M. L., and Maniatis T. (1994), Cell 76, 735–746. [DOI] [PubMed] [Google Scholar]
- Bandiziulis R. J., Swanson M. S., and Dreyfuss G. (1989), Genes Dev 3, 431–437. [DOI] [PubMed] [Google Scholar]
- Bell L. R., Maine E. M., Schedle P., and Cline T. W. (1988), Cell 55, 1037–1046. [DOI] [PubMed] [Google Scholar]
- Bell L. R., Horabin J. I., Schedle P., and Cline T. W. (1991), Cell 65, 229–239. [DOI] [PubMed] [Google Scholar]
- Belote J. M., McKeown M., Boggs R. T., Ohkawa R., and Sosnowski B. A. (1989), Dev Genet 10, 143–154. [DOI] [PubMed] [Google Scholar]
- Birney E., Kumar S., and Krainer A. (1993), Nucleic Acids Res 21, 5803–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggs R. T., Gregor P., Idriss S., Belote J. M., and McKeown M. (1987), Cell 50, 739–747. [DOI] [PubMed] [Google Scholar]
- Burtis K. C. and Baker B. S. (1989), Cell 56, 997–1010. [DOI] [PubMed] [Google Scholar]
- Caputi M., Casari G., Guenzi S., Tagliabue R., Sidoli A., Melo C. A., and Baralle F. E. (1994), Nucleic Acids Res 22, 1018–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T.-B., Zachar Z., and Bingham P. M. (1987), EMBO J 6, 4095–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X.-D. (1993), Nature 365, 82–85. [DOI] [PubMed] [Google Scholar]
- Fu X.-D. and Maniatis T. (1990), Nature 343, 437–441. [DOI] [PubMed] [Google Scholar]
- Fu X.-D. and Maniatis T. (1992), Science 256, 535–538. [DOI] [PubMed] [Google Scholar]
- Fu X.-D., Mayeda A., Maniatis T., and Krainer A. R. (1992), Proc Natl Acad Sci USA 89, 11224–11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H. and Manley J. L. (1990), Cell 62, 25–34. [DOI] [PubMed] [Google Scholar]
- Ge H., Zuo P., and Manley J. L. (1991), Cell 66, 373–382. [DOI] [PubMed] [Google Scholar]
- Goralski T. J., Edstrom J.-E., and Baker B. S. (1989), Cell 56, 1011–1018. [DOI] [PubMed] [Google Scholar]
- Green M. R. (1991), Annu Rev Cell Biol 20(7), 559–599. [DOI] [PubMed] [Google Scholar]
- Hedley M. L. and Maniatis T. (1991), Cell 65, 579–586. [DOI] [PubMed] [Google Scholar]
- Hoffman B. E. and Grabowski P. J. (1992), Genes Dev 6, 2554–2568. [DOI] [PubMed] [Google Scholar]
- Horabin J. I. and Schedle P. (1993), Mol Cell Biol 13, 7734–7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshijima K., Inoue K., Higuchi I., Sakamoto H., and Shimura Y. (1991), Science 252, 833–836. [DOI] [PubMed] [Google Scholar]
- Inoue K., Ohno M., Sakamoto H., and Shimura Y. (1989), Genes Dev 3, 1472–1479. [DOI] [PubMed] [Google Scholar]
- Inoue K., Hoshijima K., Sakamoto H., and Shimura Y. (1990), Nature 344, 461–463. [DOI] [PubMed] [Google Scholar]
- Inoue K., Hoshijima K., Higuchi I., Sakamoto H., and Shimura Y. (1992), Proc Natl Acad Sci USA 89, 8092–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohtz J. D., Jamison S. F., Will C. L., Zuo P., Lührmann R., Garcia-Blanco M. A., and Manley J. L. (1994), Nature 368, 119–124. [DOI] [PubMed] [Google Scholar]
- Krainer A. R., Conway G. C., and Kozak D. (1990), Genes Dev 4, 1158–1171. [DOI] [PubMed] [Google Scholar]
- Krainer A. R., Mayeda A., Kozak D., and Binns G. (1991), Cell 66, 383–394. [DOI] [PubMed] [Google Scholar]
- Kuo H.-C., Nasim F. H., and Grabowski P. J. (1991), Science 251, 1045–1050. [DOI] [PubMed] [Google Scholar]
- Laski F. A., Rio D. C., and Rubin G. M. (1986), Cell 44, 7–19. [DOI] [PubMed] [Google Scholar]
- Lavigueur A., Branche H. L., Kornblihtt A. R., and Chabot B. (1993), Genes Dev 7, 2405–2417. [DOI] [PubMed] [Google Scholar]
- Mattaj I. W. (1993), Cell 73, 837–840. [DOI] [PubMed] [Google Scholar]
- Mattox W. and Baker B. S. (1991), Genes Dev 5, 786–796. [DOI] [PubMed] [Google Scholar]
- Mayeda A. and Krainer A. R. (1992), Cell 68, 365–375. [DOI] [PubMed] [Google Scholar]
- Mayeda A., Helfman D. M., and Krainer A. R. (1993), Mol Cell Biol 13, 2993–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M. J., Query C. C., and Sharp P. A. (1993), in The RNA World (Gesteland R. F. and Atkins J. F., eds.), Cold Spring Harbor Laboratory Press, New York, pp. 303–357. [Google Scholar]
- Nagoshi R. N., McKeown M., Burtis K. K., Belote J. M., and Baker B. S. (1988), Cell 53, 229–236. [DOI] [PubMed] [Google Scholar]
- Nagoshi R. N. and Baker B. S. (1990), Genes Dev 4, 89–97. [DOI] [PubMed] [Google Scholar]
- Niwa M. and Berget S. M. (1991), Genes Dev 5, 2086–2095. [DOI] [PubMed] [Google Scholar]
- Ohno M., Sakamoto H., and Shimura Y. (1987), Proc Natl Acad Sci USA 84, 5187–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Query C. C., Bentley R. C., and Keene J. D. (1989), Cell 57, 89–101. [DOI] [PubMed] [Google Scholar]
- Rio D. C. (1992), Gene Expr 2, 1–5. [PMC free article] [PubMed] [Google Scholar]
- Robberson B. L., Cote G. J., and Berget S. M. (1990), Mol Cell Biol 10, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth M. B., Zahler A. M., and Stolk J. A. (1991), J Cell Biol 115, 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryner L. C. and Baker B. S. (1991), Genes Dev 5, 2071–2085. [DOI] [PubMed] [Google Scholar]
- Sakamoto H., Inoue K., Higuchi I., Ono Y., and Shimura Y. (1992), Nucleic Acids Res 20, 5533–5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebel C. W. and Rio D. C. (1990), Science 248, 1200–1208. [DOI] [PubMed] [Google Scholar]
- Siebel C. W., Kanaar R., and Rio D. C. (1994), Genes Dev 8, 1713–1725. [DOI] [PubMed] [Google Scholar]
- Sosnowski B. A., Belote J. M., and McKeown M. (1989), Cell 58, 449–459. [DOI] [PubMed] [Google Scholar]
- Sun Q., Mayeda A., Hampson R. K., Krainer A. R., and Rottman F. M. (1993), Genes Dev 7, 2598–2608. [DOI] [PubMed] [Google Scholar]
- Talerico M. and Berget S. M. (1990), Mol Cell Biol 10, 6299–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K., Watakabe A., and Shimura Y. (1994), Mol Cell Biol 14, 1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M. and Maniatis T. (1992), Science 256, 237–240. [DOI] [PubMed] [Google Scholar]
- Tian M. and Maniatis T. (1993), Cell 74, 105–114. [DOI] [PubMed] [Google Scholar]
- Valcarcel J., Singh R., Zamore P. D., and Green M. R. (1993), Nature 362, 171–175. [DOI] [PubMed] [Google Scholar]
- Watakabe A., Sakamoto H., and Shimura Y. (1991), Gene Expr 1, 175–184. [PMC free article] [PubMed] [Google Scholar]
- Watakabe A., Tanaka K., and Shimura Y. (1993), Genes Dev 7, 407–418. [DOI] [PubMed] [Google Scholar]
- Wu J. Y. and Maniatis T. (1993), Cell 75, 1061–1070. [DOI] [PubMed] [Google Scholar]
- Xu R., Teng J., and Cooper T. A. (1993), Mol Cell Biol 13, 3660–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachar Z., Chou T.-B., and Bingham P. M. (1987), EMBO J 6, 4105–4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore P. D., Patton J. G., and Green M. R. (1992), Nature 355, 609–614. [DOI] [PubMed] [Google Scholar]
- Zahler A. M., Lane W. S., Stolk J. A., and Roth M. B. (1992), Genes Dev 6, 837–847. [DOI] [PubMed] [Google Scholar]
- Zahler A. M., Neugebauer K. M., Stolk J. A., and Roth M. B. (1993a), Mol Cell Biol 13, 4023–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler A. M., Neugebauer K. M., Lane W. S., and Roth M. B. (1993b), Science 260, 219–222 [DOI] [PubMed] [Google Scholar]
- Zuo P. and Manley J. L. (1994), Proc Natl Acad Sci USA 91, 3373–3367. [Google Scholar]