

Abstract
The response of the cellular RNA processing machinery to herpes simplex virus type 1 (HSV-1) infection was studied at the ultrastructural level in HeLa cells and compared to the distributions of RNA polymerase II molecules and viral RNA. Immunogold labeling of RNA polymerase II molecules revealed that viral genome transcription was restricted to filaments in an intranuclear, virus-induced region. This region also contained viral RNAs as revealed by in situ hybridization of two biotinylated viral DNA probes: a probe encompassing a limited portion of the viral genome (the F fragment) and a probe for the total genome. In addition, the latter probe revealed large amounts of viral RNA within the clusters of interchromatin granules, intranuclear structures of normal cells that became enlarged during HSV-1 infection. Components of spliceosomes were localized by in situ hybridization with biotinylated U1 and U2 DNA probes. The large viral region contained only traces of U1 and U2 RNAs, probably because of the low frequency of splices of viral transcripts. The clusters of interchromatin granules, however, accumulated U1 and U2 RNAs with the same frequency as in noninfected cells. Poly(A) RNA was detected by in situ hybridization of a biotinylated poly(dT) probe. Some was present over the filaments of the virus-induced region but most was accumulated in the clusters of interchromatin granules. Our data suggest, therefore, that the clusters of interchromatin granules, in addition to their involvement in spliceosome component assembly, might also be a transient storage site for some families of viral mRNA, possibly a sorting site that regulates their migration.
Keywords: Biotinylated probes, HeLa cells, HSV-1 infection, In situ hybridization, Interchromatin granules, Poly(A) RNA, RNA polymerase II, Spliceosome, Transcription, U1 RNA, U2 RNA, Ultrastructure, Viral RNA
A major goal of cell biology is the identification of intranuclear domains (or structures) involved in messenger RNA (mRNA) synthesis. Previous electron microscope studies clearly established that decondensed chromatin is involved in the synthesis of the heterogeneous nuclear RNA (hn-RNA) molecules (Croston and Kadonaga, 1993), the morphological support of these growing molecules being the well-known perichromatin fibrils, and that processing of premessenger RNA (pre-mRNA) occurs in the interchromatin space (Bachellerie et al., 1975; Nash et al., 1975; Fakan et al., 1976; Fakan, 1978, 1994; Fakan and Puvion, 1980; Puvion and Moyne, 1978, 1981; Puvion and Viron, 1981; Gallinaro et al., 1983). It must be emphasized, however, that the early part of the splicing events does occur in association with the perichromatin fibrils, as demonstrated by the detection at their level of small amounts of the various components of spliceosomes, including small nuclear ribonucleoprotein (snRNP) antigens (Fakan et al., 1984; Puvion et al., 1984) and U1 and U2 small RNAs (Visa et al., 1993a; Puvion-Dutilleul et al., 1994).
Large amounts of spliceosome components accumulate within three well-delineated structures, that is, the clusters of interchromatin granules, the so-called interchromatin granule-associated zones (Visa et al., 1993a), and the coiled bodies, all structures that are enclosed within the interchromatin space (Lamond et al., 1990; Carmo-Fonseca et al., 1991a, 1991b; Spector, 1993; Visa et al., 1993a, 1993b). As suggested by the authors, coiled bodies and interchromatin granule-associated zones might be involved in the maturation of individual snRNP particles whereas the clusters of interchromatin granules might be involved in the assembly of functional spliceosomes, although an additional role of these three structures in snRNP recycling cannot be excluded.
The size of the clusters of interchromatin granules is highly variable depending on the physiological state of the cell. It is well established that the intranuclear development of DNA viruses, such as herpes simplex virus type 1 (HSV-1) (Dupuy-Coin et al., 1978; Schwartz and Roizman, 1969; Scotto et al., 1979; Puvion-Dutilleul and Laithier, 1987) and adenovirus (Martinez-Palomo, 1968; Martin et al., 1987; Puvion-Dutilleul, 1991), induces a marked enlargement of the clusters of interchromatin granules. In adenovirus infection in which the multiple splicing events in late primary viral transcripts require the host splicing machinery, we detect spliceosome components mainly in a virus-induced intranuclear domain that has the appearance of a fibrillogranular network and that is the active transcription site for viral genomes, as well as over the clusters of interchromatin granules (Puvion-Dutilleul et al., 1994). Therefore, in adenovirus development, which is characterized by a high frequency of splices, the formation of new compartments or structures containing snRNP components is not observed. We demonstrate, furthermore, that it is the relocalization of clusters of interchromatin granules and active replication sites that is responsible for the progressive transformation of the speckled distribution of snRNPs into the peripheral shell observed at the optical level (Jimenez-Garcia and Spector, 1993).
In HSV-1 infection, a series of biochemical studies clearly established that viral mRNA, like cellular and adenovirus mRNA molecules, are capped at the 5′ terminus and polyadenylated at the 3′ end (Bachenheimer and Roizman, 1972; Silverstein et al., 1973, 1976; Bartkoski and Roizman, 1976, 1978; Moss et al., 1977; Stringer et al., 1977). An important difference resides in the degree of splicing of the HSV-1 transcripts (Roizman and Sears, 1990; Smiley et al., 1991). That is, only a few HSV-1 mRNAs are spliced during their biogenesis and they need the cellular splicing machinery (Watson et al., 1981; Frink et al., 1981, 1983). This low frequency of splices in viral mRNA and the progressive shutoff of the synthesis of cellular mRNA during HSV-1 infection (Nishioka and Silverstein, 1978) diminish the use of the cellular splicing machinery.
Studies of the intranuclear rearrangement of cellular spliceosome components in herpes infection are instructive in this system in which splicing events become minimal. Recent optical studies revealed that the distribution of antigens of snRNPs shifted from a diffuse speckled pattern to a punctate distribution in which coiled bodies were no longer detectable (Phelan et al., 1993). The HSV-induced punctuate distribution was considered to be the result of the intense labeling of the virus-enlarged clusters of interchromatin granules in which snRNP antigens accumulate (Martin et al., 1987).
In addition to the low frequency of splices, HSV-1 transcripts differ from cell and adenovirus transcripts by the shorter length of the intranuclear poly(A) chains (about 30 adenosine residues instead of 150–200 adenosines) (Silverstein et al., 1976; Philipson et al., 1971). Our previous electron microscope detection of poly(A) RNA in the clusters of interchromatin granules of noninfected cells (Visa et al., 1993b) and adenovirus-infected cells (Puvion-Dutilleul et al., 1994) demonstrates that, in addition to the involvement of these structures in some cellular and viral splicing events, they might be a sorting site for the mature mRNAs, thereby regulating their transport to the cytoplasm, an assumption that is reinforced by the detection of large amounts of adenovirus RNAs in the clusters of interchromatin granules (Puvion-Dutilleul et al., 1992).
HSV-1-infected cells are a good model system to ascertain the role of the clusters of interchromatin granules in pre- and/or postsplicing events in a system in which mRNA synthesis is intense but rarely requires splicing. To do this, we used infected HeLa cells to undertake at ultrastructural level, first, the immunogold localization of the cellular RNA polymerase II molecules involved in HSV-1 genome transcription (Rice et al., 1994) and, second, in situ hybridization and immuno-gold detection of the hybrids to detect U1 snRNA, U2 snRNA, and poly(A)+ RNA. In addition, we reinvestigated the precise distribution of HSV-1 RNA by in situ hybridization of two different biotinylated specific DNA probes. We report evidence that the growing HSV-1 transcripts were exclusively localized within the clear virus-induced regions of the infected nuclei whereas clusters of interchromatin granules, which accumulated viral RNA and poly(A)+ RNA, might be transient accumulation sites for mature HSV-1 RNA and, therefore, might be a sorting site of viral mRNA, allowing the regulation of their migration to the cytoplasm.
MATERIALS AND METHODS
Cells, Virus, and Infection
HeLa cells (5 × 105 cells per 5-cm plastic culture dish) were grown at 37°C, in the presence of 5% CO2, in Eagle’s minimum essential medium supplemented with 5% calf serum. Twenty-four hours later, the cells were near confluence. Some cultures were fixed (see below) whereas others were infected at 10 plaque-forming units (pfu) per cell with KOS strain of HSV-1. After a 30-min adsorption at 37°C, 5 ml fresh culture medium was added. Infected cultures were incubated for 17 h before fixation.
Fixation and Embedding
Both noninfected and infected cell cultures were fixed for 1 h at 4°C with either 4% formaldehyde (Merck, Darmstadt, Germany) in 0.1 M phosphate buffer, pH 7.3, or 1.6% glutaraldehyde (Taab Lab. Equip. Ltd, Reading, UK) in the same buffer. During fixation, the cells were scrapped from the plastic substratum and centrifuged. The resulting pellets were dehydrated in increasing concentrations of methanol and embedded in Lowicryl K4M (Chemische Werke Lowi, Waldkraiburg, Germany) at low temperature according to Roth (1989). Polymerization was carried out for 5 days at −30°C under long wavelength UV light (Philips fluorescent tubes TL 6W). Ultrathin sections were collected on formvar-carbon-coated gold grids (200 mesh) and stored until use.
Antibody Against RNA Polymerase II and Immunogold Detection
A monoclonal antibody (7C2) specific to RNA polymerase II was produced using a polypeptide (YSPTSPS)3 that corresponds to a threefold repetition of the consensus heptapeptide that is repeated 52 times at the carboxy-terminal domain (CTD) of the largest subunit (hRPB1 or hRPB220) of mammalian RNA polymerase II (Corden et al., 1985; Wintzerith et al., 1992). The peptide, which was synthesized with an additional Cys residue at the C-terminus, was coupled to ovalbumin (Har-low and Lane, 1988) and injected into high-responding Biozzi mice, as described previously (Brou et al., 1993). Hybridoma culture supernatants were screened for nuclear staining of COS-7 cells. Positive hybridomas were further tested by ELISA with the synthetic peptide, as well as by Western blot analysis, using partially purified RNA polymerase II fractions (Kim and Dahmus, 1988). Specific hybridomas were subcloned twice on soft agar. The selected clone 7C2 produced IgG1(κ) antibodies into the supernatant of the hybridoma culture, which was used at a 1/10 dilution in PBS.
Grids bearing sections of formaldehyde- and glutaraldehyde-fixed HeLa cells with or without 17-h HSV-1 infection were floated for 1 h at room temperature, at the surface of 10 μl drops of 7C2 monoclonal antibody. After washing in PBS, the grids were floated for 30 min, at room temperature, on 10 μl drops of goat anti-mouse IgG conjugated to gold particles, 10 nm in diameter (Biocell Res. Lab., Cardiff, UK) diluted 1/25 in PBS. After washing in PBS, grids were rinsed in distilled water, air-dried, and stained for 10 min with 5% aqueous uranyle acetate. Four different assays were made that gave similar results. In addition, some grids were incubated for longer periods of incubation (4 h, 8 h, overnight) in the presence of the primary antibody.
Biotinylated Probes
Two commercial viral DNA probes biotinylated by nick-translation were used. Probe 1, which was purchased from Enzo Biochem, Inc. (New York, NY, lot No. 7DBA3), was described previously (Puvion-Dutilleul and Puvion, 1989, 1991). It was a mixture of two clones of HSV (1 and 2) DNA sequences at the concentration of 60 μg/ml and consisted of double-stranded DNA with fragment sizes ranging between 200 and 2000 base pairs in nondenaturing conditions. The insert size of HSV-1 DNA cloned in pBR322 is 8.0 kb and corresponds to the fragment F of the genomic map of HSV-1 as determined by restriction enzymes (Locker and Frenkel, 1979; Pellet et al., 1985). The fragment includes the UL48 gene coding for the α trans-inducing factor (αTif) (Post et al., 1981; Batterson and Roizman, 1983; Campbell et al., 1984; Pellet et al., 1985). The αTif protein, which is located in the tegument of mature viruses, is required for the induction of immediate-early genes (Everett, 1987; Roizman et al., 1988). Probe 2, which was purchased from Kreatech Biotechnology B.V. (Amsterdam, The Netherlands, lot 90HV01) at the concentration of 10 μg/ml, is a total genomic DNA probe (153 kb) with a fragment size ranging between 200 and 600 bases in denaturing conditions.
The poly(dT) probe (54 μg/ml) was a synthetic polynucleotide with an average length of 195 nt (Pharmacia P-L Biochemicals Inc., Milwaukee, WI). It was biotinylated with biotin-16-dUTP (Bethesda Research Lab.) in the presence of terminal deoxynucleotidyl transferase (Cook et al., 1988; Kumar et al., 1988) and added to 200 μg/ml carrier tRNA. As previously reported (Visa et al., 1993b), a tail of 2–3 biotin-16-dUTP per poly(dT) molecule was obtained.
The U1 and U2 probes (60 μg/ml) were cloned in vectors pSP64 and were biotinylated by nick-translation of whole plasmid DNA using biotinylated dATP (biotin 14-dATP, Bethesda Research Lab., Bethesda, MD). The U1 DNA probe, which was kindly provided by J. E. Dahlberg, corresponded to a 260 bp BglII-Ddel fragment of human DNA encompassing the entire U1 RNA coding region (Lund and Dahlberg, 1984). The U2 DNA probe, which was kindly provided by T. Pederson, corresponded to the entire human U2 RNA coding sequence plus 12 3′ flanking nucleotides (Kleinschmidt and Pederson, 1987). The two probes containing 200 μg/ml Escherichia coli DNA have been described in our previous work (Visa et al., 1993a).
Hybridization Solutions
The hybridization solutions for the DNA probes (U1 DNA, U2 DNA, and viral DNA) each contained 50% deionized formamide, 2 × SSC (1 × SSC = 0.15 M sodium chloride, 0.015 M sodium citrate), 10% dextran sulfate, 400 μg/ml of either Escherichia coli DNA (U1 and U2 DNA probes) or salmon sperm DNA (viral probes 1 and 2), and biotinylated probe. The biotinylated probe was at the final concentration of 10 μl/ml except for the viral probe 2, the final concentration of which was about 2 μ/ml. To denature double-stranded DNA, each hybridization solution was plunged into boiling water for 4 min and immediately chilled in melting ice and used about 10 min later.
The hybridization solution for the poly(dT) probe, which consisted of formamide, SSC, dextran, and biotinylated probe (final concentration 10 μg/ml) as above was not added with either Escherichia coli DNA or salmon sperm DNA. Due to the absence of double-stranded DNA in the poly(dT)-containing hybridization solution, the latter was used for in situ hybridization without a previous heat denaturation step.
Electron Microscope In Situ Hybridization
The protocols used for the detection of defined RNA sequences have been extensively described in technical reviews (Puvion-Dutilleul, 1993a, 1993b). Briefly, grids bearing sections of formaldehyde-fixed infected and noninfected HeLa cells were floated on the surface of 1–2 μl drops of hybridization solution distributed at the surface of a parafilm sheet for various conditions of duration and temperatures depending on the probe used. For the detection of U1 RNA, U2 RNA, and poly(A) RNA the grids were incubated for 90 min, whereas for the detection of viral RNA, incubations were 90 min and 210 min. Hybridizations of poly(dT) probe and viral probes were performed at 37°C whereas U1 DNA and U2 DNA probes were used at 64°C. These experimental conditions gave the best hybridization signals in our previous works (Puvion-Dutilleul and Puvion, 1991; Visa et al., 1993a, 1993b). After hybridization, the grids were rinsed at the surface of PBS drops. Then they were incubated for 30 min at room temperature in the presence of 10 μl drops of goat anti-biotin antibody conjugated to gold particles, 10 nm in diameter (Biocell Research Lab.) diluted 1/25 in PBS. Then they were sequentially rinsed in PBS and a jet of distilled water and air-dried. Finally, the grids were stained with 5% aqueous uranyl acetate for 10 min.
To increase the specificity of the labeling and/ or the accessibility of the nucleic acid sequences to the probes, some grids were submitted to enzymatic digestions performed at 37°C prior to hybridization. For protease digestion, grids were floated for 15 min at the surfaces of 10 μl drops of 0.2 mg/ml protease (protease from Streptomyces grissus, type VI, Sigma, St. Louis, MO). For RNase and DNase digestion, grids were floated for 1 h on the surfaces of 10 μl drops of 1 mg/ml RNase A (from bovine pancreas, BRL Biochemicals, Ltd., Poole, UK) and 1 mg/ml DNase 1 (Worthington Biochemical Corp., Freehold, NJ), respectively, both in 10 mM Tris-HCl buffer, pH 7.3. Detections of U1 RNA, U2 RNA, and poly(A) RNA were performed on nondigested grids and on protease-digested grids. For the detection of HSV-1 RNA, hybridizations always were performed on DNase-treated grids to eliminate hybridization of the viral probes with the single-stranded portions known to be present along the viral genomes. In parallel, additional protease treatments of sections prior to hybridizations were performed to increase the hybridization signal over viral RNA. To verify that each probe was detecting RNA exclusively, some grids were digested with RNase with or without previous protease or protease-DNase treatment. Four different assays were made with each biotinylated probe, which gave identical results.
Quantitative Analysis
Micrographs of randomly selected nuclei originating from grids processed in parallel were used for quantitative evaluations of gold particle density following either in situ hybridization of infected and noninfected cells with biotinylated probes or immunogold labeling with 7C2 monoclonal antibody. Twenty-five individual measurements were made for each quantitative evaluation. The areas of the various nuclear regions were measured using a Hewlett-Packard 9845B desktop computer interfaced with a 9111A graphic tablet. The number of gold particles within the measured areas was manually counted and the density of labeling (number of gold particles per square micron) was calculated.
RESULTS
General Morphology of Noninfected and Infected Nuclei
In the absence of HSV-1 infection, condensed chromatin was rare in HeLa cells. It appeared as narrow layers at the border of the nuclei and around the nucleoli and as small clumps scattered in the interchromatin spaces (Fig. 1a,c). The inter-chromatin spaces contained small pleomorphic clusters of interchromatin granules, elongated fibrillar masses, the so-called interchromatin granules-associated zones (Visa et al., 1993a), and a few coiled bodies (Fig. 1b).
FIG. 1.
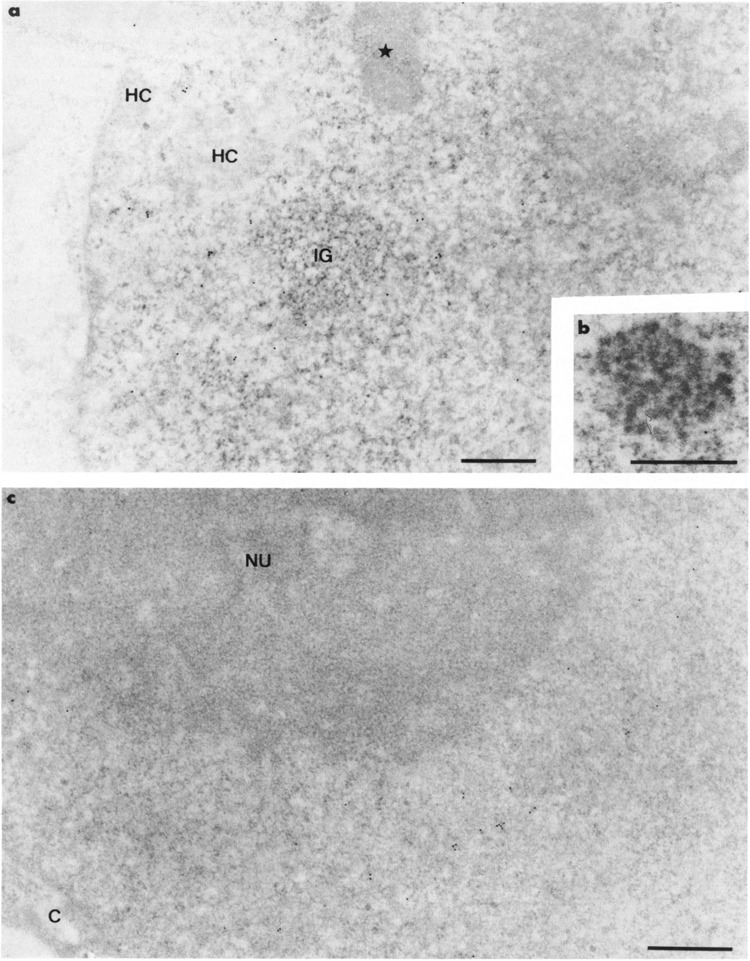
Intranuclear distribution of RNA polymerase II molecules in noninfected HeLa cells using 7C2 monoclonal antibody. Glutaraldehyde fixation and Lowicryl K4M embedding. Uranyl acetate staining. In (a), gold particles are present in the interchroma-tin space excluding the cluster of interchromatin granules (IG) and its associated zone (star). The condensed chromatin masses (HC) also are devoid of labeling. The coiled body in (b) and the nucleolus (NU) in (c) are entirely devoid of labeling. C: cytoplasm. Bars represent 0.5 μm.
Changes induced by the intranuclear development of HSV-1 in HeLa cells were similar to those previously described for HSV-1-infected rabbit fibroblasts (Puvion-Dutilleul et al., 1985a, 1985b; Puvion-Dutilleul, 1988). Seventeen hours after infection, almost all nuclei showed typical, large, clear regions that filled the nuclei and pushed the host chromatin and nucleoli to the nuclear peripheries (Fig. 2a). When progeny viruses were rare, host chromatin was moderately compacted. It became highly compacted in cells containing numerous progeny viruses (Puvion-Dutilleul and Besse, 1994). The virus-induced regions contained viral DNA, viral capsids, and dense bodies (Puvion-Dutilleul, 1988; Puvion-Dutilleul and Puvion, 1989) (Fig. 2b). These viral regions were locally separated from the adjacent marginated host chromatin by spherical clusters of interchromatin granules (Fig. 2a). Neither coiled bodies nor interchromatin granule-associated zones were observed.
FIG. 2.
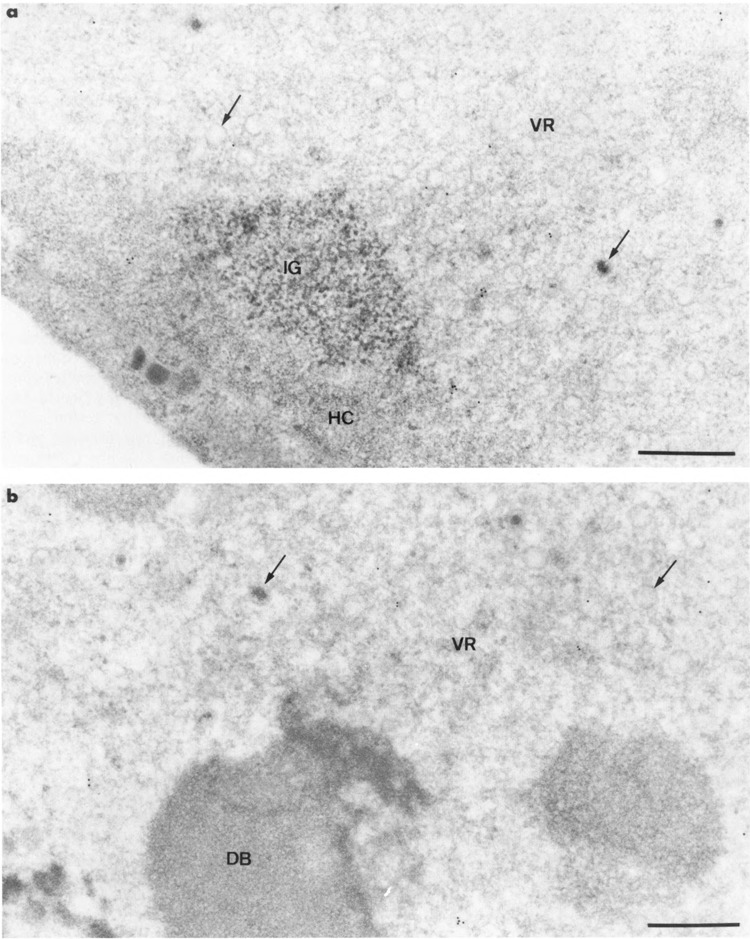
Intranuclear distribution of RNA polymerase II molecules in HSV-1-infected HeLa cells using 7C2 monoclonal antibody. Formaldehyde fixation and Lowicryl K4M embedding. Uranyl acetate staining. Gold particles are present over the filaments of the large virus-induced region (VR) in (a) and (b) and absent over the viral capsids (arrows). The cluster of interchromatin granules (IG) and the marginated host chromatin (HC) in (a) and the intranuclear dense body (DB) in (b) are not labeled. Bars represent 0.5 μm.
Intranuclear Distribution of Host RNA Polymerase II
A monoclonal antibody (7C2) directed against the CTD of mammalian RNA polymerase II was prepared, as described in Materials and Methods. The specificity of this antibody was demonstrated by immunoblot analysis of the polypeptides from a HeLa cell extract (Fig. 3). Although a polypeptide with the size of the polymerase largest subunit (about 220 kDa) was most readily detectable (lane 1), additional minor bands became visible only with higher amounts of extract (compare lanes 1–4). Interestingly, both phosphorylated and un-phosphorylated forms of this subunit (Payne et al., 1989; Lu et al., 1991; Corden, 1990) could be detected with this antibody using higher resolution electrophoresis conditions (not shown). In addition, in vitro transcription was not impaired by the antibody. Altogether, these observations indicated that both transcribing and nontranscribing polymerase molecules could be visualized with the 7C2 antibody.
FIG. 3.

Specificity of the 7C2 antibody. Increasing amounts of whole-cell extract (Manley et al., 1983) prepared from non-infected HeLa cells (10, 20, 40, and 80 μg in lanes 1–4, respectively) were loaded on a 10% polyacrylamide SDS gel (Laemmli, 1970). After electrophoresis, the proteins were transferred to a nitrocellulose filter, incubated with the 7C2 antibody, and protein/antibody complexes were revealed by the ECL Western blotting detection system (Amersham), as previously described (Chatton et al., 1994).
With this in mind, the 7C2 antibody was applied on sections of cells before and after HSV-1 infection to localize the transcription sites of cellular and viral mRNA. We observed that the intensity of the labeling was similar whatever the fixative used (formaldehyde or glutaraldehyde) and whatever the duration of the incubation with 7C2 antibody (1 h to overnight). Only the 1-h incubation period, however, gave clean grids (i.e., entirely devoid of protein aggregates and background). Therefore, the results presented below were from 1-h incubation.
In noninfected HeLa cells, gold particles were restricted to the interchromatin space (Fig. 1a). The coiled bodies (Fig. 1b), the clusters of interchromatin granules, and their associated masses were entirely devoid of labeling (Fig. 1a). No labeling occurred over the areas of condensed chromatin, whatever their location in the nuclei (perinuclear, perinucleolar, and intranucleoplasmic) or over nucleoli and cytoplasms (Fig. 1a,c).
In infected cell cultures, changes in the distribution of the labeling occurred that were independent of the fixative (formaldehyde or glutaraldehyde) used. The quantitative estimations presented below were from formaldehyde-fixed HeLa cells infected with HSV-1. Nuclei of cells with a few or no progeny viruses showed a gold labeling of the filaments of the large virus-induced region (5 ± 2 gold particles per μm2) without any labeling of the enclosed dense bodies and viral capsids, and of the surrounding clusters of inter-chromatin granules (Fig. 2a,b). A few gold particles were present over areas of the marginated host chromatin (about 1 gold particle per μm2). Similar intranuclear distribution of labeling occurred, but at a somewhat lower level, in cells containing innumerable progeny viruses.
Localization of HSV-1 RNA
We previously demonstrated that the specific detection of HSV-1 RNA molecules in rabbit fibroblasts by in situ hybridization requires a protease-DNase pretreatment of Lowicryl sections prior to hybridization of the probe (Puvion-Dutilleul and Puvion, 1991). The protease digestion increases the accessibility of the probe to the viral RNA present at the surface of the sections, whereas the DNase digestion suppresses the possible hybridization of the probe with the homologous single-stranded portions of the HSV-1 genomes (Puvion-Dutilleul et al., 1989).
We studied the distribution of viral RNA in formaldehyde-fixed HeLa cells infected for 17 h with HSV-1 by using the same probe restricted to the 8-kb-long F fragment of the genomic map as determined by restriction enzymes that we designated probe 1. In addition, to detect much more viral RNA families, we used a biotinylated genomic HSV-1 probe. This larger probe was designated probe 2.
Double protease-DNase digestion of sections of 17-h-infected HeLa cells totally dissolved the viral and cellular chromatins and, especially, eliminated the electron opacity of marginated host chromatin and encapsidated viral genomes. Following the use of probe 1, which mainly recognizes the RNA originated from the UL 48 gene (see Materials and Methods), the distribution of labeling in infected HeLa cells was similar to that previously obtained with infected rabbit fibroblasts (Puvion-Dutilleul and Puvion, 1991). Indeed, whatever the duration of the hybridization step (90 min or 210 min), gold particles always were rare and mainly restricted to the ribosome-containing cytoplasmic areas and the intranuclear viral regions excluding their enclosed dense bodies. Clusters of interchromatin granules were unlabeled except for a few that contained 1–4 gold particles (not shown). Progeny viruses, host chromatin, nucleoli, and mitochondria were entirely devoid of labeling.
Following the use of probe 2, which does recognize all viral RNA molecules that are present at the surface of the sections, labelings of the ribosome-rich cytoplasmic areas and intranuclear viral regions of 17-h-infected HeLa cells were more intense, even in cells containing numerous progeny viruses. In addition, about two-thirds of the clusters of interchromatin granules clearly were labeled (Fig. 4). A few gold particles were observed over some nucleoli, mainly in association with the enzyme-bleached intranucleolar chromatin. Virus-induced intranuclear dense bodies, host chromatin, and progeny viruses were entirely devoid of labeling. The intensity and distribution of the labeling were similar for 90 min and 210 min of incubation of sections in the presence of probe 2.
FIG. 4.
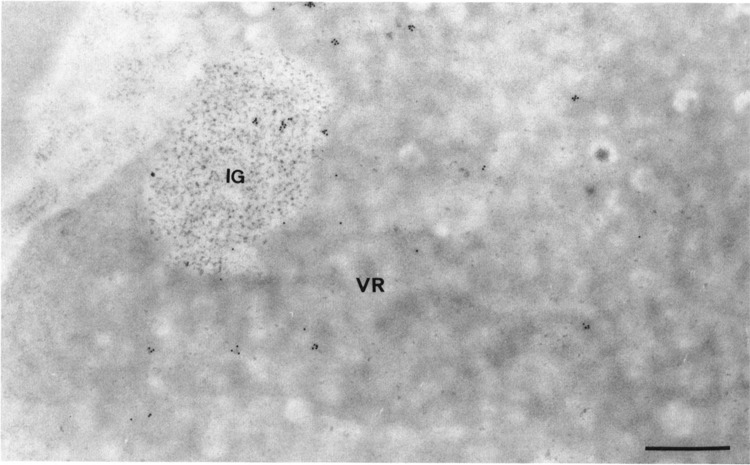
Intranuclear distribution of HSV-1 RNA in a HSV-1-infected HeLa cell by in situ hybridization of probe 2, a genomic probe. Formaldehyde fixation and Lowicryl K4M embedding. Protease-DNase pretreatment of sections prior to hybridization. Uranyl acetate staining. Gold particles are present over both the filaments of the virus-induced region (VR) and the cluster of interchromatin granules (IG). Bar represents 0.5 μm.
In the absence of protease pretreatment of sections, we always observed a markedly reduced rate of labeling over cytoplasm, viral region, and clusters of interchromatin granules (not shown).
A protease-RNase digestion of sections of formaldehyde-fixed infected cells completely abolished the labeling of ribosome-containing cytoplasmic areas and clusters of interchromatin granules following the use of probes 1 and 2. A very occasional labeling still occurred, however, that was associated with some filaments of the viral region and some viral nucleoids, due to the specific binding of the probes to the homologous single-stranded portions of the HSV-1 genomes as previously described (Puvion-Dutilleul and Puvion, 1991). No labeling was observed when hybridizations with probes 1 and 2 were performed on protease-DNase-digested sections of formaldehyde-fixed noninfected HeLa cells.
Localization of Poly(A)+ RNA by In Situ Hybridization
The biotinylated poly(dT) probe was applied on sections of formaldehyde-fixed HeLa cells. Comparable results were obtained with or without previous protease digestion.
In the nuclei of 17-h-infected cells, we found a more intense labeling following protease pretreatment of sections than in nondigested sections (Fig. 5a,b, Table 1). Gold particles accumulate over the clusters of interchromatin granules of cells with or without progeny viruses (19 ± 7 gold particles per μm2 for digested sections instead of 10 ± 4 per μm2 for nondigested sections), whereas they were scattered over the virus-induced region (3 ± 1 gold particles per μm2 for digested sections instead of about 1 gold particle per μm2 for nondigested sections). Occasionally, a few gold particles were observed within the marginated host chromatin and at the peripheries of the strands of intranucleolar chromatin (not shown). The nucleoli, viral capsids, and virus-induced dense bodies were unlabeled.
FIG. 5.

Intranuclear distribution of poly(A) RNA in HSV-1-infected cells by in situ hybridization of poly(dT) probe. Formaldehyde fixation and Lowicryl K4M embedding. Uranyl acetate staining. Comparison of the intensity of the labeling without (a) and with (b) protease digestion of section prior to hybridization. In the presence of the proteins of the section in (a), only a few gold particles are detectable over the viral region (VR) and the cluster of interchromatin granules (IG). Following the removal of the proteins of the section in (b), labeling is more intense, especially over the cluster of interchromatin granules (IG). The RNA-containing areas of the nucleolus, both fibrillar threads (F) and large, round masses of granules (G), are not labeled. Bars represent 0.5 μm.
TABLE 1.
DENSITY OF LABELING IN NUCLEI OF NONINFECTED CELLS AND HSV-1-INFECTED CELLS AFTER ULTRASTRUCTURAL IN SITU HYBRIDIZATION WITH THE POLY(dT) PROBE
| Infected Cells Protease Pretreatment | Noninfected Cells Protease Pretreatment | |||
| −* | +† | −* | +† | |
| Clusters of interchromatin granules | 10 ± 4 | 19 ± 7 | 7 ± 5 | 8 ± 5 |
| Virus-induced regions‡ | 1 | 3 ± 1 | ||
| Nucleoplasm§ | 2 ± 1 | 2 ± 1 | ||
In situ hybridization was performed on Lowicryl sections of formaldehyde-fixed infected and noninfected cells with biotinylated poly(dT) probe to detect poly(A) RNA. The gold particle densities are expressed as number of particles per square micron. Number of individual measurements for each nuclear area is 25.
Hybridizations performed on nondigested sections.
Hybridizations performed on protease-digested sections.
Density of labeling over the whole intranuclear virus-induced region of infected cells excluding the virus-induced dense bodies.
Density of labeling over the extranucleolar regions of noninfected nuclei excluding the clusters of interchromatin granules and the masses of condensed chromatin.
In the nuclei of noninfected cells, gold particles were present over the nucleoplasms and the clusters of interchromatin granules (Table 1). Almost all nucleoli were devoid of gold particles, although a few (less than 5%) contained up to 5 highly dispersed gold particles. The interchromatin granules-associated zones, coiled bodies, and condensed chromatin were unlabeled, as previously described (Puvion-Dutilleul et al., 1994; Visa et al., 1993b). In contrast to infected nuclei, a protease pretreatment of sections of noninfected nuclei did not significantly modify the intensity of the labeling (Table 1).
In the cytoplasms of infected and noninfected cells, gold particles were scattered over the ribosome-rich regions and the mitochondria without changes in the intensity of the labelings in the presence or absence of protease pretreatments (not shown).
An additional treatment of sections with RNase A (high concentration of RNase A and low ionic strength) prior to hybridization resulted in a total absence of labeling over sections of formaldehyde-fixed infected and noninfected HeLa cells (Fig. 6).
FIG. 6.

Control: hybridization with poly(dT) probe following protease-RNase pretreatment of section. HSV-1-infected HeLa cell. Formaldehyde fixation and Lowicryl K4M embedding. Uranyl acetate staining. The viral region (VR) and the cluster of interchroma-tin granules (IG) are entirely devoid of labeling. HC: host chromatin; C: cytoplasm. Bar represents 0.5 μm.
Localization of U1 and U2 snRNA by In Situ Hybridization
When hybridizations with the U1 DNA probe were performed on sections of formaldehyde-fixed HeLa cells infected for 17 h with HSV-1, labeling was intense over each cluster of interchromatin granules, even in cells containing innumerable progeny viruses (Fig. 7a). In addition, gold particles were present over the filaments of the viral region, whereas the viral capsids, virus-induced dense bodies, and nucleoli were entirely devoid of labeling (Fig. 7b). A few gold particles occasionally were seen over some areas of the marginated host chromatin and over cytoplasm.
FIG. 7.

Localization of U1 RNA in HSV-1-infected HeLa cells by in situ hybridization of an U1 DNA probe. Formaldehyde fixation and Lowicryl K4M embedding. Uranyl acetate staining. (a,b) Nondigested sections. Gold particles are numerous in the nucleus over the clusters of interchromatin granules (IG) and the virus-induced regions (VR). In (a), a few gold particles also are present in the cytoplasm (C) over its ribosome-rich areas whereas the viruses are devoid of labeling. In (b), the virus-induced intranuclear dense body (DB) is not labeled, (c) Protease pretreatment of section prior to hybridization. Elimination of the proteins of the section does not modify the intensity and the distribution of labeling. Gold particles are still associated with the intranuclear virus-induced region (VR) and the cluster of interchromatin granules (IG). Bars represent 0.5 μm.
When the hybridization solution containing the biotinylated human U1 DNA was applied on the surface of the sections of formaldehyde-fixed noninfected HeLa cells, as previously described (Puvion-Dutilleul et al., 1994; Visa et al., 1993a), gold particles accumulated over each cluster of interchromatin granules and its associated zone whereas the gold labeling of the coiled bodies was poor. Gold particles also were scattered over the interchromatin space and the ribosome-rich cytoplasmic areas.
Following the use of the biotinylated human U2 DNA probe, the interchromatin space of noninfected cells, the viral intranuclear regions of infected cells, and the clusters of interchromatin granules of both cells were labeled (Fig. 8a,b). In noninfected cells, the coiled bodies were intensely labeled whereas the interchromatin granules-associated zones were devoid of labeling, as previously described (Puvion-Dutilleul et al., 1994; Visa et al., 1993a) (Fig. 8a). Only a few gold particles were observed over the cytoplasms of infected and noninfected cells.
FIG. 8.

Localization of U2 RNA by in situ hybridization of an U2 DNA probe. Formaldehyde fixation and Lowicryl K4M embedding. Uranyl acetate staining, (a) Noninfected HeLa cell. Gold particles are scattered over the interchromatin space and the clusters of interchromatin granules (IG). They are more numerous over the coiled body (CB) and totally absent over the interchromatin granule-associated zone (star), (b) HSV-1-infected HeLa cell. Gold particles are present over the cluster of interchromatin granules (IG) and, although at a lower extent, over the filaments of the viral region (VR). Arrow: viral capsid. Bars represent 0.5 μm.
A protease pretreatment of sections prior to hybridization with U1 and U2 DNA probes did not modify the intensity and distribution of the labeling (Fig. 7c). By contrast, a RNase digestion of sections with or without protease digestion entirely suppressed the labeling of infected and noninfected cells (not shown).
DISCUSSION
This study performed at the electron microscope level using refined in situ hybridization and immunocytological techniques reveals the functional compartmentalization of the nuclei of HSV-1-infected cells, which is related to the successive steps of viral mRNA synthesis. It also demonstrates the involvement of the clusters of interchromatin granules in the posttranscriptional events of the viral RNA transcripts prior to their migration to the cytoplasm.
The immunogold detection on Lowicryl sections of the RNA polymerase II molecules with 7C2 monoclonal antibody allows the precise localization of the DNA actively engaged in the synthesis of RNA polymerase II transcripts. Indeed, a correlation of immunogold data with those of autoradiography following incorporation of tritiated uridine by noninfected and infected HeLa cells (Fakan et al., 1976; Puvion-Dutilleul et al., 1985b) clearly indicates that the antibody mainly revealed those RNA polymerase II molecules that were actively engaged in transcription. In noninfected cells, only the transcription sites of cellular nonnucleolar DNA are revealed. It is clear that the distribution of the RNA polymerase II molecules is superimposed upon that of decondensed chromatin, which, in HeLa cells, is abundant and dispersed in the interchromatin space. The data confirm previous autoradiographic and in situ hybridization data, which demonstrated that condensed chromatin is not engaged in transcription (Croston and Kadonaga, 1993) and that clusters of interchromatin granules, their associated zones, and coiled bodies, three structures involved in pre- and/or postsplicing events, are not sites of synthesis of pre-mRNA(Visa et al., 1993a, 1993b).
In HSV-1-infected cells, both cellular and viral transcription sites were simultaneously detected by the anti-RNA polymerase II antibody because the synthesis of both viral mRNA and most of cellular mRNA is initiated by the cellular RNA polymerase II, although the expression of most cellular genes is repressed (Smiley et al., 1991). Recent biochemical studies demonstrated a virus-induced aberrant phosphorylation of the large subunits of the RNA polymerase II molecules that might explain the preferential recruitment of the molecules by the viral genomes instead of by the cellular genes (Rice et al., 1994). In situ hybridization data, which clearly established a strict segregation of cellular DNA and nonencapsidated HSV-1 genomes into two concentric nuclear compartments (Puvion-Dutilleul and Besse, 1994), allow us to identify with certainty the kinds of DNA present within the labeled areas, which are therefore engaged in transcription. Gold particles that were scattered over the filaments of the intranuclear viral regions are related to viral transcription, whereas the gold particles that occasionally were observed over the surrounding host chromatin compartment represent the sites of host pre-mRNA synthesis. The study of the distribution of cellular RNA polymerase II, therefore, provides information about the concentration of viral transcriptional activity within the virus-specific region, and the persistence at a reduced rate of the activity of marginated host DNA throughout the infectious cycle. It must be emphasized that the present in situ hybridization detection of a few poly(A)+ RNA molecules over marginated host chromatin, as well as other previous approaches, including biochemical studies (Wagner and Roizman, 1969; Jacquemont and Roizman, 1975), electron microscope autoradiography performed on infected cultures exposed for a brief tritiated uridine pulse before fixation (Puvion-Dutilleul et al., 1985b), and direct visualization of transcription complexes in infected cultures accomplished through the Miller procedure (Puvion-Dutilleul, et al., 1982), also indicate that the synthesis of host mRNA continues at a reduced rate throughout HSV-1 infection. The effects of HSV infection on cellular gene expression, however, are complex (Smiley et al., 1991). Indeed, although alteration of translation of host mRNA occurs during HSV infection, as revealed by biochemical studies that clearly demonstrate the degradation of most of the host mRNA and the subsequent shutoff of host-protein synthesis (Sydiskis and Roizman, 1967; Fenwick, 1984; Kwong and Frenkel, 1987; Kwong et al., 1988), some cellular transcripts are stabilized. However, the latter do not appear to direct host-protein synthesis (Mayman and Nishioka, 1985; Kemp and Latchman, 1988).
Transcribing HSV-1 genomes segregate within the intranuclear viral region of infected nuclei, a region that is also their replicating site and the accumulation site for viral proteins (Puvion-Dutilleul, 1988). On the other hand, only traces of U1 and U2 snRNAs are detected within this virus-induced region. The low level of detection of these spliceosome components in a region that contains viral transcripts is consistent with the low frequency of splices of herpes transcripts. At present, the transcripts of only four genes are known to be spliced (Watson et al., 1981; Frink et al., 1981, 1983). Conversely, in normal (Visa et al., 1993a) and adenovirus-infected cells (Puvion-Dutilleul et al., 1994), in which many cellular and viral genes contain introns, significant amounts of U1 and U2 snRNAs were detected in those nuclear regions that contain actively transcribed DNA.
Large amounts of spliceosome components always accumulate within clusters of interchromatin granules, as demonstrated herein and in previous electron microscope in situ hybridizations in normal (Visa et al., 1993a, 1993b) and adenovirus-infected cells (Puvion-Dutilleul et al., 1994). The clusters of interchromatin granules also contain some spliceosome proteins. Indeed, ultrastructural studies performed on normal (Fakan et al., 1984; Puvion et al., 1984; Carmo-Fonseca et al., 1991a, 1991b; Visa et al., 1993a) and herpes-infected cells (Martin et al., 1987) revealed an intense labeling of the interchromatin granule clusters with Y12 antibody, which recognizes a 28 kDa polypeptide found in the U1, U2, U4, and U6 snRNPs (Lerner et al., 1981) as well as with anti-RNP antibody specific for the 70 kDa polypeptide associated with the U1 snRNP complex (Billings et al., 1982). Electron microscopy also revealed that the clusters of interchromatin granules are one component of the so-called speckled staining pattern observed in optical studies in normal and HSV-l-infected cells (Gale and McCarty, 1986; Carmo-Fonseca et al., 1989, 1991a, 1991b; Bachmann et al., 1989). The low level of splices in herpes-infected cells, therefore, does not alter the intranuclear distribution of the spliceosome components. Interestingly, however, it has been reported that HSV-1 infection alters the composition of the speckles that become devoid of a cellular protein, the La protein. This protein of 50 kDa molecular weight, which has been reported to be either homogeneously distributed in the nucleus (Nyman et al., 1986) or concentrated in certain nuclear domains (Carmo-Fonseca et al., 1989) depending on the antibody used, is considered to be a transcription/termination factor of RNA polymerase III (Gottlieb and Steitz, 1987). It associates with U1 and U6 snRNPs (Madore et al., 1984; Kunkel et al., 1986) and also binds to small RNA tRNA, 5S RNA, and Ro RNA (Rinke and Steitz, 1982; Hendrick et al., 1981). Curiously, HSV-1 infection of CV-1 cells alters the cellular shuttling of the La protein before and after the initiation of the replication of viral genomes by inducing the migration of the La protein from the intranuclear speckle to, first, cytoplasm and, later, distinct intranuclear patches that might be the sites of synthesis and/or processing of early viral mRNA (Bachmann et al., 1989). Changes in the distribution of cellular La protein, which is associated in normal cells with various small RNA excluding U RNA, may be responsible for the HSV-l-induced alterations of cellular metabolism, including a strong inhibition of cellular RNA splicing (Sandri-Goldin and Mendoza, 1992) and cellular protein synthesis (Fenwick et al., 1979).
As expected, because HSV-1 transcripts are polyadenylated (Silverstein et al., 1976; McLauchlan and Simpson, 1989), in situ hybridization of specific probes reveals the presence of both viral RNA and poly(A)+ RNA within the intranuclear, virus-induced region that is the exclusive site of transcription of viral genomes. It is a general phenomenon that poly(A)+ RNA is associated with growing RNA fibrils. Indeed, poly(A)+ RNA is associated with the perichromatin fibrils of normal nuclei (Visa et al., 1993b), and the easily identifiable compartment devoted to the transcription of viral genomes in adenovirus-infected nuclei (Puvion-Dutilleul et al., 1994). In HSV-infected nuclei, however, the polyadenylation process is more complex. In this case, HSV-infected cells contain poly(A) chains of highly variable lengths (Silverstein et al., 1976). The length of the poly(A) chain is especially short in the infected nuclei in which 30 adenoside (A30) is the most frequent species. In extracts of total RNAs, however, poly(A) chains of 150 adenosine (A150) are abundant, although shorter poly(A) chains of only 30, 50, and 80 adenosine (A30, A50, A80) are also present. The data strongly suggest that polyadenylation of HSV-1 RNA occurs in two steps, the second being concomitant with processing and transport to the cytoplasm. The short length of intranuclear poly(A) chains in HSV-1 infected nuclei might explain why protease digestion of a section is required prior to hybridization to obtain a clear hybridization signal.
An important finding is the detection of significant amounts of HSV-1 RNA in the clusters of interchromatin granules of infected nuclei, even in cells containing innumerable progeny viruses. The data suggest that the clusters of interchromatin granules might be transient storage sites of mature viral mRNA prior to their migration into the cytoplasm, a role already proposed in adenovirus-infected cells in which clusters of interchromatin granules also contain viral RNA and poly(A)+ RNA (Puvion-Dutilleul et al., 1994). The clusters of interchromatin granules might contribute to the regulation of the migration of herpes and adenovirus mRNAs into the cytoplasm. In the present work, however, only traces of viral RNA were observed following the use of probe 1, which is restricted to the 8 kb portion of the viral genome containing the gene UL48. The total absence of labeling when viral probes 1 and 2 were applied on sections of noninfected cells clearly indicates that the hybridization signals obtained with the two viral probes on infected cells are specific for HSV-1 RNA and strongly suggests that the occasional slight labeling of clusters of interchromatin granules with probe 1 may signal a minimal level of RNA products of gene UL48 in these structures at 17 h postinfection. Further studies are needed, however, to confirm the role of clusters of interchromatin granules in the regulation of the transport of HSV-1 mRNA and reveal whether this role concerns each HSV-1 mRNA family or only defined families. On the other hand, it cannot be excluded that the viral RNAs that were trapped within the clusters of interchromatin granules represent a stable population of viral RNAs that never leave the nucleus. Further studies to distinguish between these interpretations will require the isolation and biochemical characterization of clusters of interchromatin granules from HSV-1-infected cells.
A role for the nucleolus in maturation, transport, and/or degradation of mRNA has become the topic of the day since the demonstration of myc mRNA in nucleoli (Bond and Wold, 1993). We must emphasize, however, that our data do not prove with certainty that viral mRNA is present within the nucleoli of HSV-1-infected cells because nucleolar labeling occurred only sporadically and minimally, even after hybridization of the viral genomic probe. Furthermore, more convincing data are needed about the eventual presence of poly(A)+ RNA within the nucleoli of HSV-l-infected and noninfected cells. We must emphasize that neither viral RNA nor poly(A)+ RNA were detected within the dense bodies, those enigmatic HSV-1-induced structures that contain some viral (Puvion-Dutilleul and Pichard, 1986; Puvion-Dutilleul and Cebrian, 1988) and nucleolar proteins (Puvion-Dutilleul et al., 1985b; Lopez-Iglesias et al., 1988). Therefore, a possible role for nucleoli and dense bodies, which are nucleolus-related structures, in some steps of HSV-1 mRNA synthesis also remains to be ascertained.
In conclusion, the present study, which is a further demonstration of the involvement of clusters of interchromatin granules in the assembly of mature spliceosomes, also suggests a possible involvement of the clusters of interchromatin granules in some steps of viral gene expression.
ACKNOWLEDGEMENTS
The authors are very indebted to Profs. C. Kedinger (LGME, Strasbourg, France) and E. H. Leduc (Brown University, Providence, RI) for critical reading of the manuscript. They thank Drs. J. E. Dahlberg (University of Wisconsin, Madison, WI) and T. Pederson (Worcester Foundation for Experimental Biology, Shrewsbury, MS) for the gift of plasmids used in this study. They also thank Dr. Y. Lutz (LGME, Strasbourg, France) for producing monoclonal antibodies against RNA polymerase II, Dr. J. P. Bachellerie (Centre de Recherche et de Génétique du CNRS, Toulouse) for his gift of biotinylated probes, and P. Sheldrick for his gift of infected cultures. This work was funded by the Centre National de la Recherche Scientifique (CNRS) and by special grants from the Association pour la Recherche sur le Cancer (Villejuif/France) and the Fondation pour la Recherche Médicale. S. Besse is a recipient of a fellowship from the Comité Départemental du Val-de-Marne de la Ligue Nationale contre le Cancer. F. Puvion-Dutilleul is a member of the Institute National de la Santé et de la Recherche Médicale (INSERM).
REFERENCES
- Bachellerie J. P., Puvion E., and Zalta J. P. (1975), Eur J Biochem 58, 327–337. [DOI] [PubMed] [Google Scholar]
- Bachenheimer S. L. and Roizman B. (1972), J Virol 10, 875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann M., Falke D., Schröder H. C., and Müller W. E. (1989), J Gen Virol 70, 881–891. [DOI] [PubMed] [Google Scholar]
- Bartkoski M. J. Jr. and Roizman B. (1976), J Virol 20, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkoski M. J. Jr. and Roizman B. (1978), Virology 85, 146–156. [DOI] [PubMed] [Google Scholar]
- Batterson W. and Roizman B. (1983), J Virol 46, 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings P. B., Allen R. W., Jensen F. C., and Hoch S. C. (1982), J Immunol 128, 1176–1180. [PubMed] [Google Scholar]
- Bond V. C. and Wold B. (1993), Mol Cell Biol 13, 3221–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brou C., Chaudhary S., Davidson I., Lutz Y., Wu J., Egly J.-M., Tora L., and Chambon P. (1993), EMBO J 12, 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell M. E. M., Palfreyman J. W., and Preston C. M. (1984), J Mol Biol 180, 1–19. [DOI] [PubMed] [Google Scholar]
- Carmo-Fonseca M., Pfeifer K., Schröder H. C., Vaz M. F., Fonseca J. E., Müller W. E. G., and Bachmann M. (1989), Exp Cell Res 185, 73–85. [DOI] [PubMed] [Google Scholar]
- Carmo-Fonseca M., Pepperkok R., Sproat B., Ansorge W., Swanson M., and Lamond A. I. (1991a), EMBO J 10, 1863–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo-Fonseca M., Tollervey D., Pepperkok R., Barabino S., Merdes A., Brunner C., Zamore P., Green M., Hurt E., and Lamond A. I. (1991b), EMBO J 10, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatton B., Bocco J. L., Goetz J., Gaire M., Lutz Y., and Kedinger C. (1994), Oncogene 9, 375–385. [PubMed] [Google Scholar]
- Cook A. F., Vuokolo E., and Brakel C. L. (1988), Nucleic Acids Res 16, 4077–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corden J. L. (1990), Trends Biochem Sci 15, 383. [DOI] [PubMed] [Google Scholar]
- Corden J. L., Cadena D. L., Ahearn J. M. Jr., and Dahmus M. E. (1985), Proc Natl Acad Sci USA 82, 7934–7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croston G. E. and Kadonaga J. T. (1993), Curr Opin Cell Biol 7, 417–423. [DOI] [PubMed] [Google Scholar]
- Dupuy-Coin A. M., Arnoult J., and Bouteille M. (1978), J. Ultrastruct Res 65, 60–72. [DOI] [PubMed] [Google Scholar]
- Everett R. D. (1987), Curr Opin Cell Biol 7, 589–604. [Google Scholar]
- Fakan S. (1978), in The Cell Nucleus, vol. 5 (Busch H., ed.), Academic Press, New York, pp. 3–54. [Google Scholar]
- Fakan S. (1994), Trends Cell Biol 4, 8–90. [DOI] [PubMed] [Google Scholar]
- Fakan S., Leser G., and Martin T. E. (1984), J Cell Biol 98, 358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakan S. and Puvion E. (1980), in International Review of Cytology, vol. 65 (Bourne G. H. and Danielli J. F., eds.), Academic Press, Orlando, pp. 255–299. [DOI] [PubMed] [Google Scholar]
- Fakan S., Puvion E., and Spohr G. (1976), Exp Cell Res 99, 155–164. [DOI] [PubMed] [Google Scholar]
- Fenwick M. L. (1984), in Comprehensive Virology, vol. 19 (Fraenkel-Conrat H. and Wagner R. R., eds.), Plenum Press, United Kingdom, pp. 359–390. [Google Scholar]
- Fenwick M., Morse L. S., and Roizman B. (1979), J Virol 29, 825–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frink R. J., Anderson K. P., and Wagner E. K. (1981), J Virol 39, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frink R. J., Eisenberg R., Cohen G., and Wagner E. K. (1983), J Virol 45, 634–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale A. and McCarty M. D. (1986), Adv Rheumatol 70, 237–261. [Google Scholar]
- Gallinaro H., Puvion E., Kister L., and Jacob M. (1983), EMBO J 2, 953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb E. and Steitz J. A. (1987), Mol Biol Rep 12, 243. [Google Scholar]
- Harlow E. and Lane D. (1988), Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- Hendrick J. P., Wolin S., Rinke J., Lerner M., and Steitz J. (1981), Mol Cell Biol 12, 1138–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont B. and Roizman B. (1975), J Gen Virol 29, 155–165. [DOI] [PubMed] [Google Scholar]
- Jimenez-Garcia L. F. and Spector D. L. (1993), Cell 73, 47–59. [DOI] [PubMed] [Google Scholar]
- Kemp L. M. and Latchman D. S. (1988), Eur J Biochem 174, 443–449. [DOI] [PubMed] [Google Scholar]
- Kim W.-Y. and Dahmus M. E. (1988), J Biol Chem 263, 18880–18885. [PubMed] [Google Scholar]
- Kleinschmidt A. and Pederson T. (1987), Mol Cell Biol 7, 3131–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Tchen P., Roullet F., and Cohen J. (1988), Anal Biochem 169, 376–382. [DOI] [PubMed] [Google Scholar]
- Kunkel G. R., Maser R. L., Calvet J. P., and Pederson T. (1986), Proc Natl Acad Sci USA 83, 8575–8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong A. D. and Frenkel N. (1987), Proc Natl Acad Sci USA 84, 1926–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong A. D., Kruper J. A., and Frenkel N. (1988), J Virol 62, 912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U. K. (1970), Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Lamond A. I., Barabino S., Blencowe B. J. (1990), in Nucleic Acids and Molecular Biology, vol. 4 (Eckstein F. and Lilley D., eds.), Springer-Verlag, Berlin, pp. 243–257. [Google Scholar]
- Lerner E. A., Lerner M. R., Janeway C. A., and Steitz J. (1981), Proc Natl Acad Sci USA 78, 2737–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locker H. and Frenkel N. (1979), J Virol 32, 429–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Iglesias C., Puvion-Dutilleul F., Cebrian J., and Christensen M. E. (1988), Eur J Cell Biol 46, 259–269. [PubMed] [Google Scholar]
- Lu H., Florees O., Weinmann R., and Reinberg D. (1991), Proc Natl Acad Sci USA 88, 10004–10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E. and Dahlberg J. E. (1984), J Biol Chem 259, 2013–2021. [PubMed] [Google Scholar]
- Madore S. J., Weiden E. D., and Pederson T. (1984), J Biol Chem 259, 1929–1933. [PubMed] [Google Scholar]
- Manley J. L., Fire A., Samuels M., and Sharp P. A. (1983), Methods Enzymol 101, 568–592. [DOI] [PubMed] [Google Scholar]
- Martin T. E., Barghusen S. C., Leser G. P., and Spear P. (1987), J Cell Biol 105, 2069–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Palomo A. (1968), Pathol Microbiol 31, 147–164. [DOI] [PubMed] [Google Scholar]
- Mayman B. and Nishioka Y. (1985), J Virol 53, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLauchlan J., Simpson S., and Clements J. B. (1989), Cell 59, 1093–1105. [DOI] [PubMed] [Google Scholar]
- Moss B., Gershowitz A., Stringer J. R., Holland L. E., and Wagner E. K. (1977), J Virol 23, 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash R. E., Puvion E., and Bernhard W. (1975), J Ultrastruct Res 53, 395–405. [DOI] [PubMed] [Google Scholar]
- Nishioka Y. and Silverstein S. (1978), J Virol 27, 619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyman U., Hallman H., Hadlaczky G., Pettersson I., Sharp G., and Ringertz N. R. (1986), J Cell Biol 102, 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne J. M., Laybourn P. J., and Dahmus M. E. (1989), J Biol Chem 264, 19621–19629. [PubMed] [Google Scholar]
- Pellet P. E., McKnight J. L. C., Jenkins F. J., and Roizman B. (1985), Proc Natl Acad Sci USA 82, 5870–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan A., Carmo-Fonseca M., McLauchlan J., Lamond A. I., and Clements J. C. (1993), Proc Natl Acad Sci USA 90, 9056–9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipson L., Wall R., Glickman G., and Darnell J. E. (1971), Proc Natl Acad Sci USA 68, 2806–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post L. E., Mackem S., and Roizman B. (1981), Cell 24, 555–565. [DOI] [PubMed] [Google Scholar]
- Puvion E. and Moyne G. (1978), Exp Cell Res 115, 79–88. [DOI] [PubMed] [Google Scholar]
- Puvion E. and Moyne G. (1981), in The Cell Nucleus, vol. 8 (Bush H., ed.), Academic Press, New York, pp. 59–115. [Google Scholar]
- Puvion E. and Viron A. (1981), J Ultrastruct Res 74, 351–360. [DOI] [PubMed] [Google Scholar]
- Puvion E., Viron A., Assens C., Leduc E. H., and Jeanteur Ph. (1984), J Ultrastruct Res 87, 180–189. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul F. (1988), in Electron Microscopic Review, vol. 1 (Harris J. R., ed.), Pergamon Press, plc, New York, pp. 279–339. [Google Scholar]
- Puvion-Dutilleul F. (1991), J Histochem Cytochem 39, 669–680. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul F. (1993a), in Hybridization Techniques for Electron Microscopy (Morel G., ed.), CRC Press, Boca Raton, pp. 269–299. [Google Scholar]
- Puvion-Dutilleul F. (1993b), Eur J Dermatol 3, 415–424. [Google Scholar]
- Puvion-Dutilleul F., Bachellerie J. P., Visa N., and Puvion E. (1994), J Cell Sci 13, 1457–1468. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul F. and Besse S. (1994), Chromosoma 103, 104–110. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul F. and Cébrian J. (1988), J Ultrastruct Mol Struct Res 98, 229–242. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul F. and Laithier M. (1987), Biol Cell 61, 129–139. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul F., Laithier M., and Sheldrick P. (1985a), J Gen Virol 66, 15–30. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul F., Pédron J., Laithier M., and Sheldrick P. (1982), Biol Cell 44, 249–260. [Google Scholar]
- Puvion-Dutilleul F. and Pichard E. (1986), Biol Cell 58, 15–22. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul F., Pichard E., Laithier M., and Puvion E. (1989), Eur J Cell Biol 50, 187–200. [PubMed] [Google Scholar]
- Puvion-Dutilleul F., Pichard E., Sheldrick P., Amalric F., and Puvion E. (1985b), Eur J Cell Biol 39, 458–468. [PubMed] [Google Scholar]
- Puvion-Dutilleul F. and Puvion E. (1989), Eur J Cell Biol 49, 99–109. [PubMed] [Google Scholar]
- Puvion-Dutilleul F. and Puvion E. (1991), J Electron Microsc Technol 18, 336–353. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul F., Roussev R., and Puvion E. (1992), J Struct Biol 108, 209–220. [DOI] [PubMed] [Google Scholar]
- Rice S. A., Long M. C., Lam V., and Spencer C. A. (1994), J Virol 68, 988–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinke J. and Steitz J. A. (1982), Cell 29, 149–159. [DOI] [PubMed] [Google Scholar]
- Roizman B., Kristie T., McKnight J. L. C., Michael N., Mavromara-Nazos P., and Spector D. (1988), Biochimie 70, 1031–1043. [DOI] [PubMed] [Google Scholar]
- Roizman B. and Sears A. E. (1990), in Virology, 2nd ed. (Fields B. N. and Knipe D. M., eds.), Raven Press, New York, pp. 1795–1841. [Google Scholar]
- Roth J. (1989), in Methods in Cell Biology (Tartakoff A. M., ed.), Academic Press, New York, pp. 513–551. [Google Scholar]
- Sandri-Goldin R. M. and Mendoza G. E. (1992), Genes Dev 6, 848–863. [DOI] [PubMed] [Google Scholar]
- Schwartz J. and Roizman B. (1969), J Virol 4, 879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotto J. M., Sauron B., Dupuy-Coin A. M., and Gautier M. (1979), J Submicrosc Cytol 11, 229–241. [Google Scholar]
- Silverstein S., Bachenheimer S. L., Frenkel N., and Roizman B. (1973), Proc Natl Acad Sci USA 70, 2101–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein S., Millette R., Jones P., and Roizman B. (1976), J Virol 18, 977–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley J. R., Panning B., and Smibert C. A. (1991), in Herpesvirus Transcription and Its Regulation (Wagner E. K., ed.), CRC Press, Boca Raton, pp. 151–179. [Google Scholar]
- Stringer J., Holland L., Swanstrom R., Pivo K., and Wagner E. (1977), J Virol 21, 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector D. L. (1993), Curr Opin Cell Biol 5, 442–447. [DOI] [PubMed] [Google Scholar]
- Sydiskis R. J. and Roizman B. (1967), Virology 32, 678–686. [DOI] [PubMed] [Google Scholar]
- Visa N., Puvion-Dutilleul F., Bachellerie J. P., and Puvion E. (1993a), Eur J Cell Biol 60, 308–321. [PubMed] [Google Scholar]
- Visa N., Puvion-Dutilleul F., Harper F., Bachellerie J. P., and Puvion E. (1993b), Exp Cell Res 208, 19–34. [DOI] [PubMed] [Google Scholar]
- Wagner E. K. and Roizman B. (1969), J Virol 4, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson R. J., Sullivan M., and Van de Woude G. F. (1981), J Virol 37, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wintzerith M., Acker J., Vicaire S., Vigneron M., and Kedinger C. (1992), Nucleic Acids Res 20, 910. [DOI] [PMC free article] [PubMed] [Google Scholar]