

Abstract
The nuclear receptor superfamily of transcription factors, which includes the retinoic acid receptors and v-erb A, play important roles in the molecular control of hematopoiesis. To identify nuclear receptors expressed in hematopoietic cells, we screened a human bone marrow cDNA library using a degenerate oligonucleotide and isolated a 1.85-kb full-length cDNA encoding a new human member of this superfamily, the peroxisome proliferator activated receptor gamma (hPPARγ). Two different hPPARγ transcripts were expressed in hematopoietic cells: a 1.85-kb transcript, which corresponds to the full-length mRNA (PPARγ1), and a 0.65-kb transcript (PPARγ2), which cannot encode all of the nuclear receptor functional domains. Normal neutrophils and peripheral blood lymphocytes, as well as circulating leukemic cells from patients with AML, ALL, and CML, express only PPARγ2 on Northern blot analysis. In contrast, only the PPARγ1 transcript was detected in a variety of human leukemia cell lines and in cultured normal primary bone marrow stromal cells. Both transcripts were detected in various fetal and adult nonhematopoietic tissues. We mapped the location of the hPPARγ gene to human chromosome 3p25 by somatic cell hybridization and linkage analysis. PPARs have been shown to be activated by peroxisome proliferating agents, long-chain fatty acids and arachidonic acid. Human PPARγ, although homologous to the PPARγs of other species, has unique sequence and amino acid differences. Identification of hPPARγ will allow further understanding of its role in human cellular leukotriene, prostaglandin, and peroxide degradative or synthetic pathways, as well as its role in lipid metabolism and regulation of adipocyte differentiation.
Keywords: Human, Peroxisome proliferator activated receptor (PPAR), PPARγ, Hematopoiesis, cDNA, Nuclear receptor, Transcription factor, 3p25, Steroid-hormone receptor, Orphan receptor
THE nuclear hormone receptor superfamily of ligand modulated transcription factors can directly connect the cellular transcriptional response to extracellular signals such as retinoids and steroids, as well as long-chain fatty acids and arachidonic acid derivatives (Gottlicher et al., 1992). These receptors have been shown to regulate a number of major cellular processes including reproduction, development, homeostasis, differentiation, and oncogenesis (Forman and Samuels, 1990). Studies of the role of these receptors in hematopoiesis have focused primarily upon the retinoic acid receptor alpha (RARα) and v-erb A genes because of their important effects on hematopoietic cell differentiation (Desbois et al., 1991; Kakizuka et al., 1991).
The RARα gene on chromosome 17 is translocated to chromosome 15 and fused with the PML gene in virtually all cases of human acute promyelocytic leukemia (APL) (Rowley, 1988). This generates two fusion proteins, containing RARα and PML sequences, that have different transcriptional activating properties than the wild-type proteins (Kakizuka et al., 1991). Treatment of APL patients with the RARα ligand, all-trans retinoic acid, results in complete remissions in nearly all patients (Meng-er et al., 1988). V-erb A, an aberrant version of a thyroid hormone receptor, can block erythroid differentiation and induce malignant transformation, an ability that is correlated with the repression of retinoic acid receptor function (Schule et al., 1991; Sharif and Privalssky, 1991). .Both the RARs and v-erb A can interact with other transcription factors such as fos, jun (Desbois et al., 1991; Schule et al., 1991; Sharif and Privalsky, 1991), or other nuclear hormone receptors [e.g., retinoid × receptors (RXRs)] leading to heterodimer formation (Leid et al., 1992). RXRs can also heterodimerize with a number of different nuclear hormone receptors in vitro, including the peroxisome proliferator activated receptors (PPARs) (Kliewer et al., 1992).
Several different PPARs have been described: murine PPAR alpha (Issemann and Green, 1990) and gamma (Zhu et al., 1993), rat PPAR alpha (Gottlicher et al., 1992), human PPAR alpha (Sher et al., 1993), and Xenopus PPAR alpha, beta, and gamma (Dreyer et al., 1992), and we now report a human PPAR gamma. These are referred to as PPARα, β, and γ, and a fourth member of this family is represented by hNUC1, isolated from human osteosarcoma cells, which has some unique sequence characteristics (Schmidt et al., 1992). The α, β, and γ forms share significant amino acid identity, particularily in the DNA binding (C) and ligand binding (E) domains, but they are located on different chromosomes (Sher et al., 1993), have different A/B and D regions, have different expression patterns, and activate transcription differently in response to various ligands and PPAR response elements (Kliewer et al., 1994; Marcus et al., 1993; M. Greene, unpublished data).
Identification of the natural ligand(s) for the PPARs is not complete. Members of this family are known to be activated by the presence of peroxisome proliferator agents (Dreyer et al., 1992; Issemann and Green, 1990), fatty acids, and arachidonic acid (Banner et al., 1993; Gottlicher et al., 1992; Issemann et al., 1993). Serum activates the PPARs, and a study of HPLC fractionation of serum indicates that the activating compounds are arachidonic, oleic, linoleic, and palmitic acids (Banner et al., 1993). The peroxisome proliferator agents are a structurally diverse group of compounds and include fibrate hypolipidemic agents such as the pharmaceutical clofibrate and the synthetic compound WY-14643 (Auwerx, 1992; Beier et al., 1988; Dreyer et al., 1993; Issemann and Green, 1990), phthalate ester plasticizers, herbicides, and leukotriene D4 inhibitors (Zhu et al., 1993). These agents induce the proliferation of peroxisomes and peroxisomal enzymes, with subsequent hepatocellular carcinoma in rodents (Lock et al., 1989). Humans, primates, pigs, and dogs treated with clofibrate show distinct hypolipidemic effects, but they do not show dramatic peroxisomal proliferation in their hepatocytes, nor do they develop hepatocellular carcinoma (Auwerx, 1992; Blaauboer et al., 1990). Hepatic peroxisomal proliferation can also be elicited by high lipid diet or metabolic dysregulation to increase input of fatty acids to the liver (Auwerx, 1992; Kliewer et al., 1994). The peroxisome proliferating compounds have in common the fact that treatment with members of this diverse group of compounds results in intracellular accumulation of fatty acids (Auwerx, 1992). Activation of the PPARs by the addition of fatty acids, particularily arachidonic acid, at relatively high concentrations of approximately 150 mM, suggests that the natural ligand for the PPARs may be a long-chain fatty acid or arachidonic acid derivative, or that the natural ligand(s) are rapidly metabolized (Gottlicher et al., 1992). Activation studies in the presence of cyclooxygenase inhibitors suggest the natural ligand(s) are not products of this metabolic pathway (Gottlicher et al., 1993), and one of the most potent activators of PPARs is 5,8,11,14-eicostetraynoic acid (ETYA), a blocker of lipoxy-genases and cyclooxygenases, which fully activates xPPARα at a concentration of 1 μM, with an ED50 of 200 nM (Keller et al., 1993).
PPARs, as ligand-activated transcription factors in the steroid hormone receptor superfamily, modulate gene transcription in response to fatty acids, and interact with other members of the superfamily, including the retinoid receptor (Keller et al., 1993; Kliewer et al., 1992, 1994; Leid et al., 1992; Marcus et al., 1993) and thyroid hormone receptor subfamilies (Auwerx, 1992; Bogazzi et al., 1994; Qi et al., 1995), supporting the concept that there are important cross-signaling effects between the various steroid-related signaling pathways in the body. There is evidence that PPARs play important roles in gene regulation during development as well as providing a link between nutritional status and changes in gene expression in the adult organism (Auwerx, 1992; Blumberg et al., 1992; Beck et al., 1992).
To identify known or novel nuclear receptors expressed in the bone marrow compartment, a degenerate oligonucleotide was designed that could detect a variety of nuclear receptors, especially those in the thyroid-retinoid branch of the super-family (Blumberg et al., 1992; Evans, 1988; Forman and Samuels, 1990; Wahli and Martinez, 1991). A normal human bone marrow aspirate cDNA library was constructed and screened, and among the receptors obtained, two PPARγ cDNA clones (one full length) were isolated. The expression pattern of this gene was examined using RNA from a variety of normal and malignant hematopoietic cells and cell lines, and from adult and fetal nonhematopoietic tissues. Somatic cell hybrids and linkage analysis in multigeneration families were used to regionally map the chromosomal location of the hPPARγ gene.
MATERIALS AND METHODS
Generation and Screening of Human Bone Marrow cDNA Library
Oligo(dT)-primed first-strand cDNA was prepared from 2.6 mg poly(A) selected normal human bone marrow aspirate RNA (see below). The oligo(dT) primer (5′-ACTAGTGCGGCCGCCTAGGCCTCGAGTTTTTTTTTTTTTTT-3′) was designed to create an oriented cDNA library, using the method described by Blumberg et al. (1992). 5-Methyl dCTP was incorporated during first-strand synthesis to protect internal Xho I sites by hemimethylation interference. Residual 5-methyl dCTPs were removed, and second-strand synthesis was performed, incorporating the 3′ Xho I site. Internal EcoR I sites were then protected by EcoR I methylase treatment, the cDNA ends were made blunt-ended, and EcoR I linkers were added. After combined EcoR I and Xho I digestion, the cDNA was purified by Sepharose CL-4B chromatography and double-stranded cDNAs approximately 500 base pairs or greater in size were ligated to 1 ZAP II arms (Stratagene) and packaged in vitro with Gigapack II Gold (Strata-gene). This library contained 7.4 × 107 independent clones and was screened unamplified. The library was screened in duplicate with an HPLC-purified 32P-labeled synthetic 512-fold degenerate oligonucleotide (TGYGARGGNTGY-AARGGNTTYTT) under low-stringency conditions (1 M NaCl, 0.1 M Tris-HCl, pH 8.0, 6 mM EDTA, 125 units of heparin per ml, 0.05% sodium pyrophosphate, 100 mg/ml yeast RNA, 0.1% sodium dodecyl sulfate, at 46°C). Filters were washed at high stringency [two 15-min room temperature washes, one in 6 × SSC/0.05% sodium pyrophosphate, and one in 3 M tetramethylammonium chloride/0.05 M Tris-HCl (pH 8.0)/0.2 mM EDTA (3 M TMAC), then two 20-min washes at 58°C in 3 M TMAC, and one 15-min wash at room temperature in 6 × SSC/0.05% sodium pyrophosphate]. The degenerate oligonucleotide is a mixture of all possible DNA sequences encoding the highly conserved amino acid sequence CEGCKGFF, found in the first cysteine finger of the members of the thyroid-retinoid branch of the nuclear receptor superfamily (Blumberg et al., 1992; Evans, 1988; Wahli and Martinez, 1991). Positive plaques from the initial high-density screen of 1 × 106 clones were purified on secondary and tertiary screens and converted to plasmids by the automatic excision process (Stratagene Unizap) (Short et al., 1988). Purified plasmids were sequenced using a modified dideoxy chain termination method (Tabor and Richardson, 1989) and the screening oligonucleotide as the initial primer. Clones showing similarity to nuclear receptors were further characterized.
DNA sequences were analyzed using programs of Staden (1986), University of Wisconsin Genetics Computer Group (Devereaux et al., 1984), Feng and Doolittle (1987), DNASIS (Hitachi), GeneWorks release 2.2 (IntelliGenetics), and DNA Strider release 1.0 (Commissariat a l’Energie Atomique, France).
Preparation of RNA
Bone Marrow Aspirate RNA
Bone marrow (30 ml) was aspirated from the posterior iliac crest of a normal volunteer donor with a Jamshidi needle using standard sterile procedures. The aspirate was immediately placed on ice and allowed to clot. After the serum was removed, the remainder of the sample was transferred rapidly to 50-ml sterile tubes containing 1.5 volumes of 4 M guanidine isothiocyanate solution (4 M guanidine isothiocyanate, 0.5% N-lauroylsarcosine, 25 mM sodium citrate, 0.1 M 2-mercaptoethanol). Samples were immediately homogenized with a NaOH-cleaned, high-speed (Polytron) tissue homogenizer, and 360 μg total cellular RNA was prepared using a modified Chirgwin procedure (Chirgwin et al., 1979). Poly(A)+ RNA was selected according to a method by Cho (Blumberg et al., 1992) and its integrity was confirmed by gel electrophoresis.
Fractionated Peripheral Blood Cell RNA
Several leukapheresis packs were obtained from the American Red Cross to isolate monocytes, lymphocytes, and neutrophils. A Ficoll-Hypaque separation procedure (Kitano et al., 1991) was used to separate neutrophils from the mononuclear fraction, and the monocytes and lymphocytes were separated by overnight adherence to plastic. The monocytes were lysed in situ on the tissue culture dishes using the 4 M guanidine isothiocyanate solution following an overnight adherence step in Iscove’s modified Dulbecco’s media IMDM (Gibco) with 15% fetal bovine serum (Gemini) and 5% heat-inactivated human AB serum. Lymphocyte and monocyte total RNA was prepared and poly(A)+ RNA was then isolated using either the Fastrack kit (Invitrogen) or oligo(dT) columns (Pharmacia).
Leukemic Cell Line RNA
RNA (and DNA) was isolated from the following human hematopoietic cell lines: T-cell lines: CCRF-HSB-2, CCRF-CEM, MOLT-4, Jurkat; B-cell lines: Raji and NOR 25 (an EBV transformed normal adult peripheral blood B-cell line kindly provided by Nancy Perillo); myeloid leukemia cell lines: HL60, KG-1, K562, and THP-1. RNA was also prepared from the murine pre-B-cell lines 18.81, WEHI 231, and 70Z3 (kindly provided by O. Witte). These cell lines were cultured in IMDM with 10% fetal bovine serum, 100 U/ml penicillin G, 0.1 mg/ml streptomycin, and l-glutamine 0.4 mM. The NOR-25, THP-1, 18.81, WEHI 231, and 70Z3 media contained 5 × 10−5 M β-mercap-toethanol and the NOR-25 media also contained 0.01 M HEPES (Gibco). Poly(A)-selected RNA (20–40 mg) was prepared from 1 × 108 cells using the Fastrack kit.
Bone Marrow Stromal Cell RNA
Bone marrow stromal cells (kindly provided by J. Nolta and D. Kohn, M.D., L. A. Children’s Hospital) were obtained from a 200-μm wire mesh screen that was used to filter bone marrow obtained during a normal human donor bone marrow harvest. The cells were washed with saline and suspended in IMDM containing 15% horse serum, 15% fetal calf serum, 5 × 10−5 M β-mercaptoethanol, 10−3 M hydrocortisone, penicillin, and streptomycin. Cells were grown for a total of four passages before harvesting. Collagenase was used during the passaging of the cells to allow them to retain their ability to differentiate towards adipose as well as fibroblast cells (Nolta et al., 1992). Poly(A)-selected RNA was obtained from 1 × 108 cells using the Fastrack kit.
Patient Sample RNA
Patients seen at UCLA with active acute leukemia or chronic myelogenous leukemia provided informed consent (approved by the UCLA Human Subject Protection Committee) and donated 15–30 ml of peripheral blood. The mononuclear cell fraction was isolated by Ficoll-Hypaque density separation and total RNA was prepared as above.
Fetal RNA
This study reutilized existing Northern blots, previously prepared by KK, as part of an approved clinical study of the UCLA Clinical Genetics Laboratory (under the direction of B. Crandall). These blots were prepared as follows. Tissue samples from fetuses with no known genetic abnormality or chemical exposure were provided by elective surgical abortions performed between 11 and 24 weeks postconception, after obtaining the patient’s informed consent. Samples were obtained within minutes of pregnancy termination and were frozen immediately in liquid nitrogen and stored at −80°C. The conceptional age of abortuses was determined by measurement of fetal foot length. Prior to freezing, samples of fetal cortex were dissected away from the central diencephalon and the basal ganglia, and were freed of membrane and choroid plexus; the meninges were removed from spinal cords. Samples of placental tissue were obtained from the fetal aspect following removal of the amnion and chorion. Total RNA was prepared from frozen fetal cortex, cerebrum, pooled intact and partial spinal cords, kidney, lung, placenta, and liver tissue by homogenization in GITC using the methods of Chirgwin et al. (1979) or Chomczynski and Sacchi (1987); poly(A)+ RNA was isolated from total RNA by affinity chromatography (Aviv and Leder, 1972). Preparation of an additional Northern blot containing fetal liver RNA was as previously described (Kronquist et al., 1990).
Northern Blot Analysis
Formaldehyde agarose gels were loaded with either methyl mercury or heat-denatured total cellular or poly(A)-selected RNA. Gels were transferred to either Hybond (Amersham) or Nytran (Schleicher and Schuell) membranes using standard platform transfer techniques in 10 × or 20 × SSC. Prehybridizations and hybridizations were performed in 7% SDS, 0.5 M Na2PO4, pH 7.2, with 100 mg/ml salmon sperm DNA at high stringency (65°C) (Blumberg et al., 1992). A 1.6-kb PPARγ cDNA fragment was random prime labeled (Feinberg and Volgelstein, 1983) with a 32P dCTP, with specific activities from 0.4 to 1.0 × 109 cpm/mg (Pharmacia Oligolabeling kit) and was used as the probe for all Northern and Southern blot analyses (McBride et al., 1989) at concentrations of 3–5 × 106cpm/ml of hybridization solution. Exposures ranged from overnight to 10 days with Kodak XAR film, and 2 Dupont Quanta III screens, at −70°C.
Southern Blot Analysis
Genomic DNA was isolated from peripheral blood leukocytes of 10 normal volunteers. DNA from the normal volunteers was digested with restriction endonucleases EcoR I, Bam HI, Hind III, Xba I, Sac I, Taq I, Pvu II, Pst I, Bgl II, Msp I, EcoR V, and Kpn I. DNA fragments were fractionated by 0.7% or 0.8% agarose gel electrophoresis and transferred to positively charged nylon membranes in 0.5 N NaOH as described (McBride et al., 1989). A 0.8-kb Xho 1/EcoR I fragment containing the A, B, C, and D regions of hPPARγ was used as the probe for hybridization to normal volunteer DNA for Southern blot RFLP analyses. Probes were labeled as described above. The membranes were hybridized (at 42°C in 50% formamide) and washed (in 0.1 × SSC at 55–58°C) at high stringency, allowing less than 10% sequence divergence as described (McBride et al., 1989).
Somatic Cell Hybrids
The isolation and characterization of a panel of human-rodent somatic cell hybrids retaining subsets of human chromosomes have been described previously (McBride et al., 1982). Hybrid cell lines were characterized for the presence of all human chromosomes except Y by standard isoenzyme analyses, by Southern analysis with probes from previously localized genes, and, frequently, by cytogenetic analysis.
Linkage Analysis
Southern blots of Xba I digests of DNAs from 40 large, three-generation CEPH families (Dausset et al., 1990) were used for hybridization with a 0.8-kb A/B, C, and D region hPPARγ (PPARG) cDNA probe. RFLP typing was performed by standard Southern blot analysis under high-stringency conditions. All parental DNAs were initially examined, and all family members from informative matings (i.e., those with one or both heterozygous parents) were then examined. Two-point and multipoint linkage analysis of PPARG versus loci typed in the CEPH data base version 5 were performed using LINKAGE version 5.1 for microcomputers (Lathrop et al., 1984). An infrequent Taq I polymorphism (B1:B2 = 7.1:5.3 kb) detected in one CEPH parent was used to genotype CEPH family 1413 and the results with Xba I and Taq I were combined as a haplotype in this family.
GenBank Accession Number
The full-length nucleotide sequence for hPPARγ has been submitted to GenBank.
RESULTS
Isolation and Sequencing of the Human PPARγ cDNA
Two PPARγ clones were identified from the degenerate oligonucleotide screen of the normal human bone marrow cDNA library. A full-length PPARγ cDNA (clone 14) was isolated and sequenced from both ends (Fig. 1). This clone was 1844 base pairs (bp) long, and contained 5′ and 3′ flanking sequences, an open reading frame beginning at bp 173, and an in-frame stop codon at bp 1607. The 1434-bp ORF is predicted to encode a 478 amino acid nuclear receptor protein, with a predicted molecular weight of approximately 54.2 kDa. The beginning of the A/B region of the predicted PPARγ protein contains the amino acid sequence MVDT, as has been reported for other members of this family (Fig. 2A). Clone 14 contains all functional components characteristic of nuclear receptors, and shows similarities and conservative base changes with all other reported PPARs. Clone 14 shows the greatest similarity (96% overall amino acid identity) to murine PPARγ, and the next highest similarity (73% overall amino acid identity) to the Xenopus PPARγ cDNA (Fig. 2B) and is therefore identified as the human version of the PPARγ gene.
FIG. 1.

DNA sequence of the full-length human PPARγ cDNA (clone 14). The 1434-bp open reading frame is predicted to begin at bp 173 and end at bp 1607, producing a 478 aa nuclear receptor protein with a predicted mol. wt. of approximately 54 kDa.
FIG. 2A.

Comparison of the amino acid sequence of human PPARγ with its nearest relatives in the nuclear receptor superfamily, Xenopus PPARα, β, γ (Dreyer et al., 1992), murine PPARα (Issemann and Green, 1990), and human NUC1 (Schmidt et al., 1992), PPARα (Sher et al., 1993), and RARα (Giguere et al., 1987; Petkovich et al., 1987). The nuclear receptor superfamily structural regions are indicated by the shaded blocks as per Dreyer et al. (1992). The putative conserved tau 1 silencing domain (T1) (Forman and Samuels, 1990) is indicated. The location of a partially conserved activating motif (CAF) (Danielian et al., 1992) is within the last 20 amino acids of this family of receptors. Bold letters indicate amino acids identical to hPPARγ.
FIG. 2B.

Comparative amino acid identity between hPPARγ, other members of the PPAR family, and hRARα. The schematic amino acid alignment was prepared according to Fig. 2A. The percentage of amino acid identity among the various receptors and hPPARγ is indicated for each domain. The E domain is further subdivided to indicate the location of a highly conserved (tau 1 putative silencing) domain (Forman and Samuels, 1990). h = human, m = murine, x = Xenopus.
The second clone (clone 22) was incomplete, containing an artifactual splice at a Hind III site (bp 597) in the C region with loss of nine highly conserved C region amino acids. The next 1247 bp are identical to clone 14, confirming the accuracy of the DNA sequencing of clone 14.
Analysis of Hormone Receptor Functional Regions
The various regions of the PPARγ cDNA are identified, according to Dreyer et al. (1992) (Fig. 2B). Comparison (not shown) with the murine PPARγ shows 96% identity at the amino acid level and 71% identity at the nucleotide level (Zhu et al., 1993). Comparison with the other known members of the PPAR family (Fig. 2A,B) reveals other similarities at the amino acid level.
A/B Region
The 108–110 amino acid (aa) A/B region is approximately the same size as the murine PPARα (102 aa) (Issemann and Green, 1990) and the Xenopus PPARγ A/B region (112 aa) (Dreyer et al., 1992). It is 93% identical to the murine PPARγ receptor in this region, and 57% identical to the Xenopus PPARγ A/B region.
C Region
The highly conserved C region (containing the DNA binding and dimerization domains) has 100% identity with murine PPARγ and 97% identity with the Xenopus PPARγ receptor.
D Region
The D region, which contains the nuclear localization domain, is also referred to as the ligand 1 domain (Forman and Samuels, 1990) and is generally conserved in subfamilies binding the same ligand. The D region is 96% identical to the murine PPARγ D region and 70% identical to the Xenopus PPARγ D region.
E Region
The E regions (containing the ligand binding, dimerization, nuclear localization, and transactivation domains) of the murine and human PPARγ are 98% identical, whereas Xenopus and human PPARγs share 75% identity. Comparison of the Xenopus PPARγ with the human and murine PPARγ within the E region shows 7% identity in the first 49 aa after the D region, 90% identity in the 42 aa T1 transactivation silencing subdomain (Forman and Samuels, 1990), and 91% identity in the region extending from the ligand binding and dimerization domains to the carboxyl-terminus (148 aa). Although the murine and human PPARγ putative proteins are 100% identical in the first 49 aa after the D region, the Xenopus γ receptor is much more divergent in this region. In the remaining E region, beginning with the T1 domain, all three receptors share 90–91% identity.
Expression Pattern of PPARγ
Northern blot analysis revealed the presence of two different PPARγ mRNA transcripts in hematopoietic cells; two PPARγ transcripts of similar size are also present in the Xenopus system (Dreyer et al., 1992). The human 1.85-kb transcript is large enough to contain all the functional regions present in the 1428-bp open reading frame of the cDNA cloned from the bone marrow library (Figs. 1 and 2). The 0.65-kb transcript cannot contain all the functional regions of a nuclear receptor (e.g., the ligand binding domain is 678 bp, the DNA binding domain is 195 bp, and a short linker region would be 220–234 bp for a total of 1.1 kb). [The partial clone (clone 22) was not considered to represent the short 650–700-bp transcript based on length, because it contained 1010 bp identical to clone 14 full-length receptor open reading frame.]
The larger transcript (PPARγ1) is expressed in a variety of malignant hematopoietic cell lines, normal peripheral blood monocytes cultured overnight, and primary cultured stromal cells (Fig. 3). No PPARγ mRNA was detected in the Jurkat, MOLT4, CEM-CRFF, CCRF-HSB-2, Raji, K562, or KG-1 cell lines.
FIG. 3.
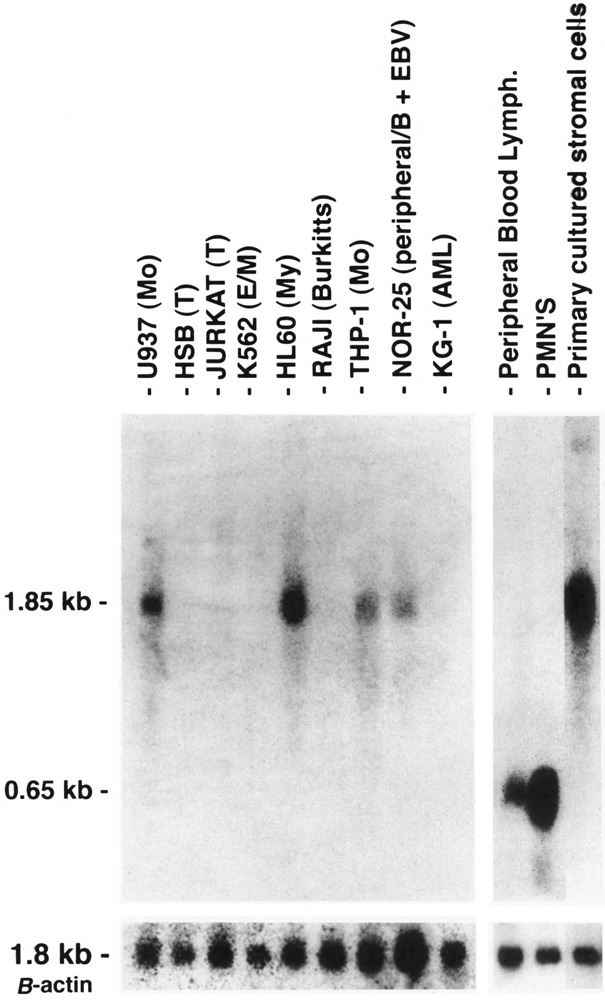
Northern blot analysis of hPPARγ mRNA expression in hematopoietic cell lines and primary peripheral blood cells as indicated. Mo = monocytic cell line, T = T-cell line, My = myeloid cell line, AML = acute myelogenous leukemia cell line, E/M = erythroid or myeloid cell line, B = B-cell line, PMN’s = polymorphonuclear leukocytes. Each lane contains 5 μg poly(A)-selected RNA. Probe shown is hPPARγ 1.6-kb (full-length) cDNA, hybridizing to hPPARγ transcripts either 1.8 kb or 0.65 kb in size. Control probe (β-actin, 1.85 kb) is shown below.
The shorter 0.65-kb transcript (PPARγ2) is the only form seen in freshly isolated peripheral blood neutrophils, lymphocytes cultured overnight (Fig. 3), and in the mononuclear fraction obtained from the peripheral blood of adult patients with acute myeloid leukemia (AML), chronic myeloid leukemia (CML), CML in blast crisis, and acute lymphocytic leukemia (ALL) (Fig. 4). Not all patient samples showed expression of PPARγ; mRNA was detected in the mononuclear fractions of 5/11 cases of the AML, 7/8 of the CML, 1/5 of the CML in blast crisis, 5/7 of the ALL, and weakly detected in one NK cell leukemia (Table 1). Blast counts varied somewhat in these samples. To exclude the possibility that some of the hybridization signal could have come from the few normal lymphocytes or monocytes remaining after a Ficoll-Hypaque separation, we prepared 20 mg total RNA from normal peripheral blood lymphocytes and found no signal under the same hybridization conditions (data not shown) [The lanes in Fig. 3 contain 5 mg poly(A)+ RNA.] Primary bone marrow stromal cells cultured for a total of four passages expressed only the 1.8-kb transcript (PPARγ1). In contrast to isolated hematopoietic cell populations, other human organ systems (colon, bladder, kidney, skeletal muscle, liver, spleen, placenta, and stomach) show expression of both transcripts, with the 1.85-kb mRNA being dominant in colon, muscle, placenta, and bladder (Fig. 5A,B). Both transcripts were also seen in fetal organs (16–19 gestational weeks); the 1.85-kb transcript was dominant in fetal kidney, and the 0.65-kb transcript was dominant in fetal liver and lung (Fig. 5B). Fetal liver specimens (19.2 and 16.4 weeks) contain significant amounts of hematopoietic cells, as the liver is the main source of hematopoesis at that time (Schwartz and Gill, 1983).
FIG. 4A.
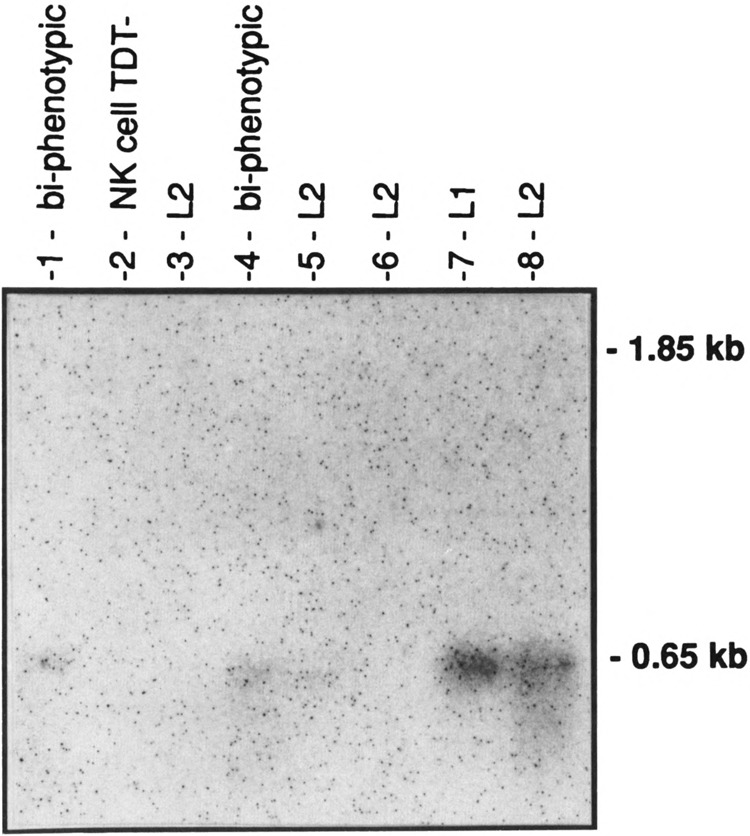
Northern blot analysis of hPPARγ in eight fresh acute lymphoid leukemia (ALL) cells, using the 1.6-kb (full-length) hPPARγ cDNA as probe. Each lane contains 10 μg total RNA isolated from the peripheral blood mononuclear cell fraction. Morphologic ALL sub-classification (L1-L2) is listed according to the French American British (FAB) classification (Sun, 1983). A Tdt (terminal deoxyribonucleotide transferase) negative, natural killer (NK) cell leukemia sample is included. The amount and integrity of the RNA in each lane of Figs. 4 and 5 were confirmed by ethidium bromide staining and by hybridizations with other probes (data not shown). Darker exposures failed to reveal hybridization in the 1.85-kb region for any of these blots (Figs. 4 and 5).
FIG. 4B.
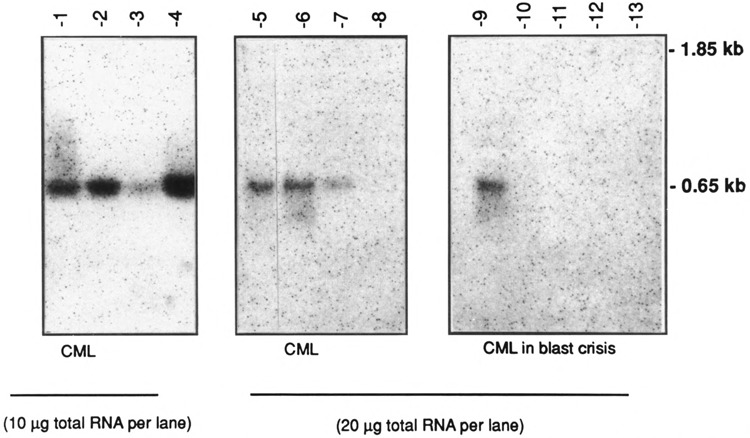
Northern blot analysis of hPPARγ RNA expression in the peripheral blood mononuclear fraction isolated from patients with chronic myelogenous leukemia (CML); 10 or 20 μg of total RNA was loaded per lane (as indicated).
FIG. 4C.
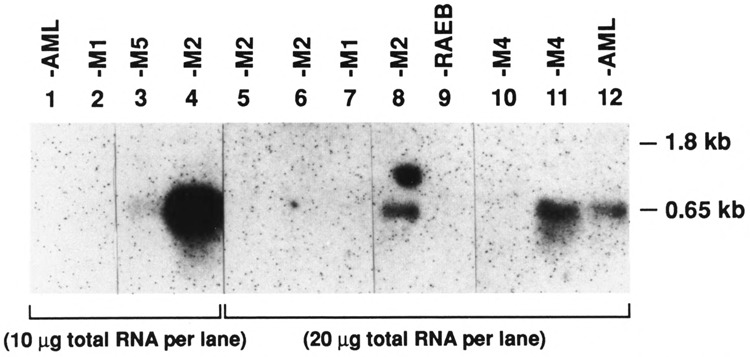
Northern blot analysis of hPPARγ RNA expression in peripheral blood mononuclear fractions from patients diagnosed with acute myeloid leukemias (listed according to the FAB classification). Lanes contain 10 or 20 μg total RNA as indicated. RAEB (lane 9) is from a patient with refractory anemia with excess blasts (Sun, 1983). Lane 8 has an artifact above the true signal.
TABLE 1.
SUMMARY OF HPPARγ AND RARα EXPRESSION IN HUMAN HEMATOPOIETIC CELLS
| Lineage | Name | PPARγ1(1.85 kb) | PPARγ2(0.65 kb) | RARα(3.6 and 2.4 kb) |
|---|---|---|---|---|
| Cultured Malignant Hematopoietic Cell Lines (Detectable Expression on 5 μg mRNA Northern Blots) | ||||
| Lymphoid | ||||
| T-Lymphoblastoid | HSB-2 | − | − | + + + |
| MOLT-4F | − | − | + + + | |
| CCRF-CEM | − | − | + + + | |
| T-cell | JURKAT | − | − | + + + |
| B-cell (Burkitts) | RAJI | − | − | + + + |
| B-cell (peripheral blood, EBV transformed) | NOR-25 | + + + | − | + + + |
| Myeloid | ||||
| Promyelocytic | HL60 | + + + | − | + + + |
| Myeloid-monocytic, (histiocytic, FC receptors, phagocytic, AML) | U937 | + + + | − | + + + |
| THP-1 | + + + | − | + + + | |
| KG-1 | − | − | + + + | |
| Erythroid | ||||
| Myeloid-erythroid heme + expression | K562 | − | − | + + + |
| Peripheral Blood, Bone Marrow Compartments (1-5 μg poly(A) or 10–20 μg total RNA Northern Blot Expression) | ||||
| Peripheral blood (fractionated, normal) | PMNs | − | + + + + | + + + + + + + |
| Lymphocytes (95% T) | − | + + + | + + + | |
| Monocytes (adherence purified, in culture overnight) | + | + + | + + + | |
| Total bone marrow aspirate stroma and hematopoietic cells | + + + + | + | + + + | |
| Cultured primary bone marrow stromal cells, fibroblastic and adipocytic differentiation | + + + | − | + + + | |
| Peripheral blood, leukemic patients | ||||
| AML | M1 | 0/2− | 0/2− | ND |
| M2 | 0/4− | 2/4 + | ND | |
| M3 | ND | ND | ND | |
| M4 | 0/2− | 1/2 + | ND | |
| M5 | 0/1− | 1/1 + | ND | |
| Unspecified | 0/2− | 1/2 + | ND | |
| CML | 0/8− | 7/8 + | ND | |
| CML in blast crisis | 0/5− | 1/5 + | ND | |
| ALL | L1 | 0/1− | 1/1 + | ND |
| L2 | 0/4− | 2/4 + | ND | |
| Biphenotypic | 0/2− | 2/2 + | ND | |
| NK cell, TDT− | 0/1− | ?1/1 + | ND | |
Relative strength of signal is approximately indicated on a + to + + + + subjective scale, with + + + + equal to a fourfold increase in signal strength relative to +. The PMN signal indicated as + + + + + + + + is at least eightfold stronger than a + signal. ?/1 + signal for the NK leukemia indicates a very faint signal. ND = not done. Adjustments are made to normalize differences in exposure times and amounts of RNA.
FIG. 5A.
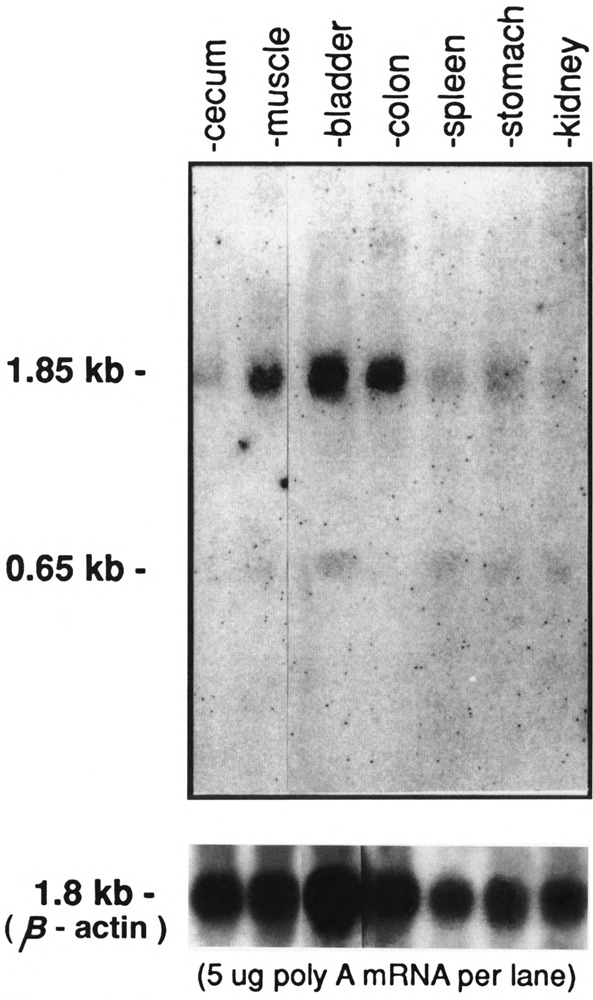
Northern blot analysis of hPPARγ mRNA expression in adult human organs. Each lane contain 5 μg poly(A)-selected RNA. The position of the two transcripts, 1.85 kb and 0.65 kb, is indicated. Hybridization using β actin cDNA is shown below.
FIG. 5B.
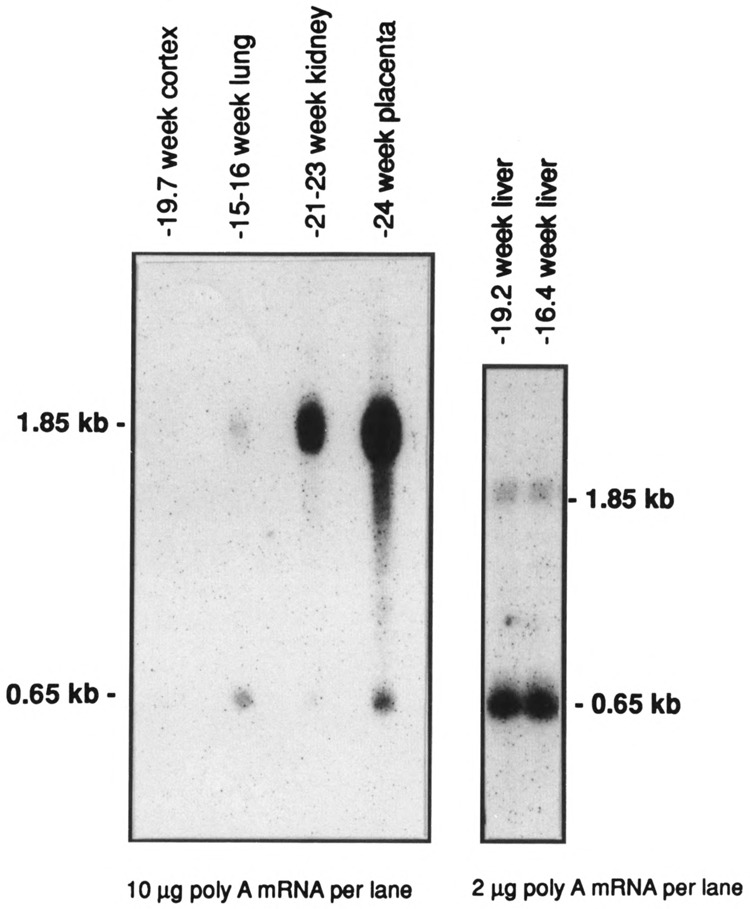
Northern blot analysis of hPPARγ expression in human fetal organs. Lanes contain either 10 μg or 2 μg of poly(A)-selected RNA as indicated. Cortex refers to the cerebral cortex.
Chromosomal Localization of the hPPARγ Gene (PPARG)
The gene was localized by Southern blot analysis of a panel of EcoR I-digested human-rodent somatic cell hybrid DNAs with a hPPARγ cDNA probe. The three hybridizing bands in human DNAs were readily distinguished from cross-hybridizing bands in rodent DNAs (not shown). All human bands cosegregated and the gene could be unambiguously assigned to human chromosome 3 (Table 2); it segregated discordantly (> 16%) with all other human chromosomes. The gene was further localized on the short arm of chromosome 3 by examination of two hybrids containing spontaneous breaks or deletions involving this chromosome. One hybrid contained a large deletion in the short arm with loss of distal 3p markers RAF1 (3p25), THRB (3p24.1-p22), and ACY1 (3p21), but retention of DNF15S2 (3p21.2-21.3) and CCHL1A2 (3p14.3) (Chin et al., 1991) as well as two other more centromeric 3p markers and all loci on 3q. The other hybrid contained a break in proximal 3q with retention of long arm markers and loss of all short arm markers. The hPPARγ gene was absent from both of these hybrids, indicating that it must be located in the region 3p21-p25.
TABLE 2.
SEGREGATION OF hPPARγ GENE IN SOMATIC CELL HYBRIDS, SHOWING hPPARγ SEGREGATES WITH HUMAN CHROMOSOME 3
| Human Chromosome | hPPARγ Gene/Chromosome | Discordancy (%) | |||
|---|---|---|---|---|---|
| +/+ | +/− | −/+ | −/− | ||
| 1 | 19 | 8 | 13 | 45 | 25 |
| 2 | 17 | 10 | 9 | 49 | 22 |
| 3 | 27 | 0 | 0 | 58 | 0 |
| 4 | 26 | 1 | 24 | 34 | 29 |
| 5 | 18 | 9 | 5 | 53 | 16 |
| 6 | 21 | 6 | 25 | 33 | 36 |
| 7 | 11 | 16 | 26 | 32 | 49 |
| 8 | 18 | 9 | 15 | 43 | 29 |
| 9 | 20 | 7 | 13 | 45 | 24 |
| 10 | 11 | 16 | 5 | 53 | 25 |
| 11 | 17 | 10 | 11 | 47 | 25 |
| 12 | 12 | 15 | 14 | 44 | 34 |
| 13 | 14 | 13 | 20 | 38 | 39 |
| 14 | 13 | 14 | 29 | 29 | 51 |
| 15 | 17 | 10 | 30 | 28 | 47 |
| 16 | 11 | 16 | 25 | 33 | 48 |
| 17 | 21 | 6 | 32 | 26 | 45 |
| 18 | 21 | 6 | 22 | 36 | 33 |
| 19 | 20 | 7 | 8 | 50 | 18 |
| 20 | 21 | 6 | 17 | 41 | 27 |
| 21 | 25 | 2 | 30 | 28 | 38 |
| 22 | 16 | 11 | 11 | 47 | 26 |
| X | 18 | 9 | 24 | 34 | 39 |
+/+ = number of somatic cell hybrids in which the hPPARγ gene was detected, and human chromosome z was present; +/− = hPPARγ gene detected, chromosome z not present; −/+ = hPPAR gene not detected, but chromosome z present; −/− neither hPPARγ gene nor chromosome z present. The human PPARG gene was detected as 1.35-, 3.4-, and 16.5-kb bands in EcoR I digests of human DNA and human-rodent somatic hybrid cell DNAs after Southern hybridization with a 0.8-kb cDNA probe. The human bands were all well resolved from cross-hybridizing 2.85-, 4.3-, and 15.3-kb, or 2.8- and 10.9-kb bands in EcoR I-digested Chinese hamster and mouse DNAs, respectively. The three human bands were either all present or all absent in any hybrid cell line. Detection of the human gene is correlated with the presence or absence of each human chromosome in the group of somatic cell hybrids. Discordancy represents presence of the gene in the absence of the chromosome (+/−), or absence of the gene despite the presence of the chromosome (−/+), and the sum of these numbers divided by the total number of hybrids examined (×100) represents percent discordancy. The 41 human–hamster hybrids consisted of 29 primary hybrids and 12 subclones; 12 were positive. The 44 human–mouse hybrids contained 16 primary clones and 28 subclones; 15 were positive.
The gene could be further localized to band 3p25 by genetic linkage analysis after identification of a high-frequency Xba I RFLP at the hPPARγ locus (PPARG) using the 0.8-kb hPPAR-γ cDNA probe; allelic bands of 4.0 kb (A2) and 9.0 kb (A1) were found in addition to constant bands of 3.0, 4.4, and 7.2 kb lengths. In 80 CEPH parents, the allele frequencies were: A1:A2 = 0.5: 0.5. The Xba I polymorphism was used for linkage analysis in the CEPH families. One or both parents was a heterozygote in 34 of the 40 total families, and both parents were heterozygotes in 18 of these families. All family members were examined in the informative families and the genotypes were entered into the CEPH data base and used for linkage analysis with other published markers from chromosome 3p (Table 3). Several errors were detected and corrected (see Table 3 footnotes).
TABLE 3.
TWO-POINT LOD SCORES FOR hPPARγ VERSUS OTHER LOCI ON CHROMOSOME 3p22-p26
| Locus† | Z at Ө* | Өmax | Zmax | Confidence Interval‡ | Physical Location | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0.05 | 0.1 | 0.2 | 0.3 | 0.4 | |||||
| D3S191 | − ∞ | 3.6 | 5.2 | 5.2 | 3.9 | 2.1 | 0.143 (0.092,0.196) |
5.5 (5.7) |
0.077–0.255 | pter-p25 |
| D3S18 | − ∞ | 19.5 | 20.6 | 18.1 | 13.0 | 6.5 | 0.096 (0.048,0.143) |
20.6 (21.3) |
0.055–0.139 | p26-p25 |
| RAF1 | − ∞ | 17.1 | 16.8 | 13.6 | 9.0 | 4.0 | 0.065 (0.056,0.071) |
17.2 (17.2) |
0.040–0.118 | P25 |
| D3S154 | − ∞ | 7.9 | 7.6 | 5.4 | 2.9 | 0.8 | 0.059 (0.119,0.001) |
8.0 (8.1) |
0.020–0.153 | p25 |
| D3S588 | − ∞ | 11.6 | 15.8 | 15.7 | 11.7 | 6.1 | 0.142 (0.136,0.146) |
16.5 (16.5) |
0.097–0.203 | p24 |
| THRB | − ∞ | 4.6 | 9.1 | 10.0 | 7.5 | 3.8 | 0.162 (0.116,0.213) |
10.3 (10.6) |
0.109–0.229 | p24.1-p22 |
The LOD scores (Z) at selected recombination fractions (Ө) are shown. The most likely recombination fraction (Өmax) and LOD scores (Z max) are also shown. [All values are computed assuming no sex difference in recombination frequencies except values in parentheses, which give sex-specific (male-female order), recombination fractions, and Z max]
Probe-enzyme combinations for these loci are: D3S191 = 38-96 with Msp I; D3S18 = L162 with Bam HI and Dra I enzymes combined as haplotype; RAF1 = = p628 with Taq I and Bgl I enzymes combined as a haplotype; D3S154 = LIB34-60 with Bg1 II;D3S588 = LIB12-69 with Hind III; THRB = pBH302 with Hind HIII.
Confidence intervals for recombination fractions over a 10-fold range of likelihood.
Technical comments: Several errors were detected and corrected. The genotypes for paternal grandparents 133311 and 133312 with L162/Dra I were deleted due to incompatibility. Genotypes for individuals 1204, 1208, 1703, and 1704 were omitted from the D3S18 haplotype because these individuals were recombinant between genotypes with Bam HI and Dra I. Individuals 6605 and 133208 were recombinant between RAF1/Bg1 II and RAF1/Taq I, and these individuals were deleted from the RAF1 haplotype. Finally, a phase error was detected in CEPH family 1420 between hPPARγ (PPARG) and both RAF1 and THRB. No errors could be found on reexamination of the autoradiographs for hPPARγ/Xba I in this family. This phase error was eliminated by the conservative change involving deletion of genotypes for paternal grandparents 142009 and 142010 for hPPARγ/Xba I.
Genotypes for all loci except PPARG were contributed to the CEPH data base by the laboratory of Dr. B. Zbar.
The results of two-point linkage analysis of PPARG with other published markers (Tory et al., 1992) in the 3p26–p22 region are shown in Table 3. Multipoint linkage analysis was used to order PPARG with respect to these other loci (Fig. 6). The results do not permit ordering of PPARG and RAF1 (odds of reversal only 1.4), but both loci are centromeric to genetic marker D3S18 (odds of reversal 2.3 × 105) and telomeric to D3S588 (odds of reversal 1.9 × 1027), and the thyroid hormone receptor β gene, THRB (odds of reversal 4.2 × 109), within band 3p25. Multipoint linkage analysis therefore establishes the location of hPPARγ within band 3p25.
FIG. 6.
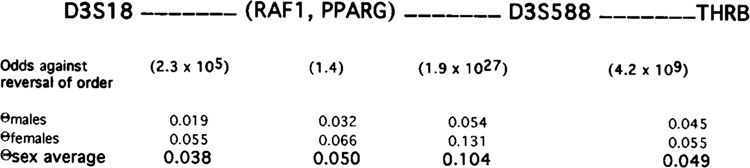
Multipoint linkage analysis of loci in the telomeric region of chromosome 3p. The result of the analysis of these values indicate that the most likely order of these loci is as shown. Note that based on all information available PPARG and RAF1 cannot be ordered. The values in parentheses indicate the odds against reversing the order of the adjacent loci. The most likely sex-specific recombination fraction (Q) between each locus is shown. Sex average values represent most likely recombinationfractions assuming no sex difference. PPARG = hPPARγ gene; RAF1 = RAF1 gene; THRB = thyroid hormone receptor, beta gene; D3S### = genetic marker, single copy DNA fragments, human chromosome 3.
DISCUSSION
We have isolated a new, functional member of the human nuclear receptor family of steroid hormone receptor transcription factors, hPPARγ. HPPARγ appears to be widely expressed in the human hematopoietic system, and in a variety of both human adult and fetal organs. The ratio of the two transcripts, 1.85 kb (hPPARγ1) and 0.65 kb (hPPARγ2), varies from organ to organ, and in different populations of hematopoietic cells. Two predominant transcripts are also seen in the Xenopus system (Dreyer et al., 1992) with the larger transcript (2.2 kb) seen in a variety of organs (e.g., liver, kidney, testes) and abdominal fat body, and the small transcript seen in Xenopus oocytes. In the human hematopoietic system, normal peripheral blood lymphocytes and neutro-phils, and circulating leukemic cells from patients with AML, ALL, CML, and CML in blast crisis, express only the shorter 0.65-kb PPARγ2 transcript. In contrast, human acute myeloid leukemia cell lines (e.g., HL60, U937, or THP-1) express only the larger 1.85-kb mRNA, and cultured stromal cells also only express the 1.85-kb transcript, raising the possibility that some aspect of culture conditions, such as rapid cell division, or the relative abundance of different growth factors or lipids may influence diffential expression of the two PPARγ transcripts. Another possibility is that metabolites produced following interactions between different cell types influence the expression of the short or the long transcript.
The likely explanation is that the transcript expressed reflects both cell type- and cell environment-specific requirements for PPAR function. Peripheral blood lymphocytes separated from monocytes by overnight adherence in culture media express the smaller transcript, whereas the monocytes express predominantly the larger transcript under identical culture conditions.
The 1.85-kb cDNA clone that we isolated is a full-length cDNA that corresponds to the 1.85-kb PPARγ1 mRNA and contains all functional nuclear receptor domains. Preliminary cotransfection studies with the hPPARγ cDNA and the acylcoA PPRE luciferase reporter plasmid (Kliewer et al., 1992) show activation of the reporter gene in the presence of the peroxisome proliferator WY 14643 (to be published separately). Although it is possible there is mRNA-specific rapid degradation of hPPARγ, we believe we have eliminated the possibility that the 0.65-kb message RNA reflects only general artifactual partial degradation of the RNA, because mRNA of this size is present on multiple human Northern blots, and hybridization of these blots with other cDNAs (c-fos, c-jun, RARα, and β-actin) showed no evidence for degradation of those mRNAs. In addition, a transcript of this size has been seen in the Xenopus system (Dreyer et al., 1992). Cloning and sequencing the short 0.65-kb mRNA will reveal whether it can bind DNA, or bind ligand, and whether it retains the heptad repeats or transactivating or silencing domains that permit functional protein-protein associations in transcriptional complexes. Additional studies will also reveal whether the short transcript is translated into protein.
The hPPARγ cDNA probe appears to detect a single copy gene (PPARG) located on human chromosome 3, although the presence of more than one gene copy at a single locus has not been rigorously excluded. A high-frequency, two-allele polymorphism (Xba I RFLP) has been identified at this locus, and it has been used to regionally localize the gene to band 3p25 in close proximity to RAF1 (Fig. 6). There is definite linkage between PPARG and all six of the loci in Table 3. PPARG is closely linked to RAF1, and the indicated recombination fraction between these two loci is probably an overestimate. No recombinants between these two loci were found in 68 informative meioses in 15 informative families, and a single recombinant was observed in each of three additional families. In contrast, at least four recombinants were found in a single family (CEPH 17). This suggests the presence of errors in family 17 but no correction was made because no error was found on CEPH 17 Xba I-digested DNA probed with the 0.8-kb hPPARγ cDNA. Despite this problem, PPARG and RAF1 could be ordered with respect to the other loci on chromosome 3.
Human PPARα was localized to a region slightly telomeric to 22q12-q13.1 (Sher et al., 1993), establishing these two human PPARs as separate genes on different chromosomes. The localization of hPPARγ to 3p25 has interesting implications. 3p deletions are commonly seen in a variety of carcinomas (Seizinger et al., 1991) whereas the 3p25-p21 deletion has been reported infrequently in patients with chronic lymphoproliferative disorders, including non-Hodgkins lymphoma (Mitelman et al., 1991). Whether loss or disruption of hPPARγ contributes to any human malignancy remains to be proven.
Studies demonstrate that PPARs heterodimerize with the 9-cis-retinoic acid receptor (RXR) and synergistically induce gene expression (Keller et al., 1993; Kliewer et al., 1992; Krey et al., 1993). These studies demonstrate a convergence between the retinoid-responsive pathways and the long-chain fatty acid/arachidonic acid-responsive pathways. The precise effects of PPARs in peroxide- and leukotriene-mediated aspects of host defense are unknown. Recent studies of the cellular response to PPAR activation have focused on fatty acid β oxidation and ω hydroxylation, and adipocyte differentiation (Chawla et al., 1994; Tontonoz et al., 1994). Target genes transcriptionally activated by PPARs include acyl-coA oxidase, the key enzyme regulating the fatty acid β oxidation pathway (Tugwood et al., 1992), peroxisome bifunctional enzyme (Zhu et al., 1993), the adipocyte P2 lipid binding protein gene (Tontonoz et al., 1994), mitochondrial HMG-CoA synthase (control site of the ketogenesis pathway) (Rodriquez et al., 1994), and CYP4A6 (cytochrome P-450 fatty acid ω hydroxylase), a key enzyme catalyzing the ω hydroxylation of arachidonic, lauric, and palmitic acids (Muerhoff et al., 1992). CYP4A6 is induced in rabbit liver in response to treatment with clofibric acid. In neutrophils, P-450 cytochrome(s) catalyze successive oxidations at the ω position to inactivate leukotriene B4, which is a potent stimulator of degranulation, chemotaxis, and adherence (Curnutte and Babior, 1990). Patients with peroxisome deficiency disorders do show impaired degradation of leukotrienes and excrete LTB4 in their urine (Mayatepek et al., 1993). The effect of peroxisome proliferator agents on LTB4 levels, or neutrophil ω oxidation, is not yet known.
Recently, the HIV-1 gene LTR modulatory region was identified as having a multiple nuclear receptor-responsive element activatable by PPAR/RXR heterodimers with increased expression in response to clofibric and 9-cis-retinoic acid (Ladias, 1994).
Specific hematopoietic gene promoters have not been tested for PPAR activation. Possible PPAR-responsive genes include many retinoid-responsive genes such as CD18, the leukocyte integrin β subunit, which has multiple combinations of imperfect PPAR/RAR/RXR type binding half-sites (TGACCT or AGGTCA) (Laudet et al., 1992; Umesono et al., 1991) and is retinoic acid inducible (Agura et al., 1992). The effects of PPAR upon myeloperoxidase, chloroacetate esterase, or catalase expression in myeloid cells also have not yet been examined.
Further study of the two forms of the PPARγ mRNA will be extremely useful for understanding their functions in hematopoietic cells as well as other organ systems.
ACKNOWLEDGEMENTS
This paper is dedicated to the memory of O. Wes McBride. We hope his many contributions to the Human Genome Project, as well as his personal help and fine work on this paper, will serve as reminders of his sorely missed presence, in many years to come.
We thank Eddy DeRobertis and Godfrey Getz for support; Walter Wahli, Janardan Reddy, Judith Gasson, Linda Gont, Gayle Baldwin, Wayne Grody, Dick Gatti, Linda Baum, Masayo Kornuc, Wilson Miller, and Maurice Wolin for helpful discussion and/or reagents; Timothy Beeker and Lareina Pedriquez for technical assistance; and Shari Hasford and Darlene Shannon for assistance in preparation of the manuscript. This research was supported by the UCLA Integrated Genetics Program (NRSA T32-GM 08243-03); University of Chicago Cancer Center Pilot Grant (G.L.G.); the University of Chicago Cardiovascular Pathophysiology Training Program (NRSA 5 T32 HL07237-17) (Godfrey Getz); by USPHS NIH grant ROl DK43025 (S.D.N.); by the Cigarette and Tobacco Surtax Fund of the State of California through the Tobacco-Related Disease Research Program of the State of California (KT45) (S.D.N.); by NCI CA 02897 (G.L.G.); and by USPHS NIH grant ROl HD27700-03 (Eddy DeRobertis).
REFERENCES
- Agura E. D., Howard M., and Collins S. J. (1992), Blood 79(3), 602-609. [PubMed] [Google Scholar]
- Auwerx J. (1992), Horm Res 38, 269–277. [DOI] [PubMed] [Google Scholar]
- Aviv H. and Leder P. (1972), Proc Natl Acad Sci USA 69, 1408–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banner C. D., Gottlicher M., Widmark E., Sjovall J., Rafter J. J., and Gustafsson J. A. (1993), J Lipid Res 34(9), 1583–1591. [PubMed] [Google Scholar]
- Beck F., Plummer S., Senior P. V., Byrne S., Green S., and Brammar W. J. (1992), Proc R Soc Lond B 247, 83–87. [DOI] [PubMed] [Google Scholar]
- Beier K., Volkl A., Hashimoto T., and Fahimi H. D. (1988), Eur J Cell Biol 46, 383–393. [PubMed] [Google Scholar]
- Blaauboer B. J., van Holsteijn C. W. M., Bleumink R., Mennes W. C., van Pelt F. N. A. M., Yap S. H., van Pelt J. F., van Iersel A. A. J., Timmerman A., and Schmid B. P. (1990), Biochem Pharmacol 40(3), 521–528. [DOI] [PubMed] [Google Scholar]
- Blumberg B., Mangelsdorf D. J., Dyck J. A., Bittner D. A., Evans R. M., and DeRobertis E. M. (1992), Proc Natl Acad Sci USA 89, 2321–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogazzi F., Hudson L. D., and Nikodem V. M. (1994), J Biol Chem 269(16), 11683–11686. [PubMed] [Google Scholar]
- Chawla A., Schwarz E. J., Dimaculangan D. D., and Lazar M. A. (1994), Endocrinology 135(2), 798–800. [DOI] [PubMed] [Google Scholar]
- Chin H., Kozak C. A., Kim H. L., Mock B., and McBride O. W. (1991), Genomics 11, 914–919. [DOI] [PubMed] [Google Scholar]
- Chirgwin J., Przybyla S., MacDonald R., and Rutter W. (1979), Biochemistry 18, 5294–5299. [DOI] [PubMed] [Google Scholar]
- Chomczynski P. and Sacchi N. (1987), Anal Biochem 162, 156–159. [DOI] [PubMed] [Google Scholar]
- Curnutte J. T. and Babior B. M. (1990), in Metabolism of Neutrophils (Williams W. J., ed.) McGraw-Hill, New York, pp. 777. [Google Scholar]
- Danielian P. S., White R., Lees J. A., and Parker M. G. (1992), EMBO J 11(3), 1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dausset J., Cann H., Cohen P., Lathrop M., Lalouel J.-M., and White R. (1990), Genomics 6, 575–577. [DOI] [PubMed] [Google Scholar]
- Desbois C., Aubert D., Legrand C., Pain B., and Samarut J. (1991), Cell 67, 731–740. [DOI] [PubMed] [Google Scholar]
- Devereaux J., Haeberli P., and Smithies O. (1984), Nucleic Acids Res 12(1), 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer C., Keller H., Mahfoudi A., Laudet V., Krey G., and Wahli W. (1993), Biol Cell 77, 67–76. [DOI] [PubMed] [Google Scholar]
- Dreyer C., Krey G., Keller H., Givel F., Helftenbein G., and Wahli W. (1992), Cell 68, 879–887. [DOI] [PubMed] [Google Scholar]
- Evans R. M. (1988), Science 240, 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A. P. and Vogelstein B. (1983), Anal Biochem 132, 6–13. [DOI] [PubMed] [Google Scholar]
- Feng D. F. and Doolittle R. F. (1987), J Mol Evol 25, 351–360. [DOI] [PubMed] [Google Scholar]
- Forman B. and Samuels H. (1990), Mol Endocrinol 4, 1293–1301. [DOI] [PubMed] [Google Scholar]
- Giguere V., Ong E. S., Segui P., and Evans R. M. (1987), Nature 330, 624–629. [DOI] [PubMed] [Google Scholar]
- Gottlicher M., Demoz A., Svensson D., Tollet P., Berge R. K., and Gustafsson J. A. (1993), Biochem. Pharmacol 46(12), 2177–2184. [DOI] [PubMed] [Google Scholar]
- Gottlicher M., Widmark E., Li Q., and Gustafsson J.-A. (1992), Proc Natl Acad Sci USA 89, 4653–4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issemann I. and Green S. (1990), Nature 347, 645–650. [DOI] [PubMed] [Google Scholar]
- Issemann I., Prince R. A., Tugwood J. D., and Green S. (1993), J Mol Endocrinol 11(1), 37–47. [DOI] [PubMed] [Google Scholar]
- Kakizuka A., Miller J. W. H., Umesono K., Warrell J. R. P., Frankel S. R., Murty V. V. V. S., Dmitrovsky E., and Evans R. M. (1991), Cell 66, 1–20 [DOI] [PubMed] [Google Scholar]
- Keller H., Dreyer C., Medin J., Mahfoudi A., Ozato K., and Wahli W. (1993), Proc Natl Acad Sci USA 90, 2160–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller H., Mahfoudi A., Dreyer C., Hihi A. K., Medin J., Ozato K., and Wahli W. (1993), Ann NY Acad Sci 684, 157–173. [DOI] [PubMed] [Google Scholar]
- Kitano K., Abboud C. N., Ryan D. H., Quan S. G., Baldwin G. C., and Golde D. W. (1991), Blood 77(8), 1625–1626. [PubMed] [Google Scholar]
- Kliewer S. A., Forman B. M., Blumberg B., Ong E. S., Borgmeyer U., Mangelsdorf D. J., Umesomo K., and Evans R. M. (1994), Proc Natl Acad Sci USA 91, 7355–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer S. A., Umesono K., Noonan D. J., Heymen R. A., and Evans R. M. (1992), Nature 358, 771–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey G., Keller H., Mahfoudi A., Medin J., Ozato K., Dreyer C., and Wahli W. (1993), J Steroid Biochem 47(1–6), 65–73. [DOI] [PubMed] [Google Scholar]
- Kronquist K. E., Dreazen E., Keener S. L., Nicholas T. W., and Crandall B. F. (1990), Prenat Diagn 10, 739–751. [DOI] [PubMed] [Google Scholar]
- Ladias J. A. A. (1994), J Biol Chem 269(8), 5944–5951. [PubMed] [Google Scholar]
- Lathrop G. M., Lalouel J. M., Julier C., and Ott J. (1984), Proc Natl Acad Sci USA 81, 3443–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudet V., Hanni C., Coll J., Catzeflis F., and Stehelin D. (1992), EMBO J 11(3), 1003–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leid M., Kastner P., Lyons R., Nakshatri H., Saunders M., Zacharewski T., Chen J.-Y., Staub A., Garnier J.-M., Mader S., and Chambon P. (1992), Cell 68, 377–395. [DOI] [PubMed] [Google Scholar]
- Lock E. A., Mitchell A. M., and Elcombe C. R. (1989), Annu Rev Pharmacol Toxicol 29, 145–163. [DOI] [PubMed] [Google Scholar]
- Marcus S. L., Miyata K. S., Zhang B., Subramani S., Rachubinski R. A., and Capone J. P. (1993), Proc Natl Acad Sci USA 90, 5723–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayatepek E., Lehmann W.-D., Fauler J., Tsikas D., Frolich J. C., Schutgens R. B. H., Wanders R. J. A., and Keppler D. (1993), J Clin Invest 91, 881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride O. W., Hieter P. A., Hollis G. F., Swan D., Otey M. C., and Leder P. (1982), J Exp Med 155, 1480–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride O. W., Pirtle I. L., and Pirtle R. M. (1989), Genomics 5, 561–573. [DOI] [PubMed] [Google Scholar]
- Meng-er H., Yu-chen Y., Shu-rong C., Jin-ren C., Jia-Xiang L., Lin Z., Long-jun G., and Zhen-yi W. (1988), Blood 72(2), 567–572.3165295 [Google Scholar]
- Mitelman F., Kaneko Y., and Trent J. (1991), Cytogenet Cell Genet 58, 1053–1079. [Google Scholar]
- Muerhoff A. S., Griffin K. J., and Johnson E. F. (1992), J Biol Chem 267(27), 19051–19053. [PubMed] [Google Scholar]
- Nolta J. A., Yu X. J., Bahner I., and Kohn D. B. (1992), J Clin Invest 90, 342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkovich M., Brand N. J., Krust A., and Chambon P. (1987), Nature 330(3), 444–450. [DOI] [PubMed] [Google Scholar]
- Qi J.-S., Desai-Jajnik V., Greene M. E., Raaka B. M., Samuels H. H. (1995), Mol Cell Biol 15, 1817–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriquez J. C., Gil-Gomez G., Hegardt F. G., and Haro D. (1994), J Biol Chem 269(29), 18767–18772. [PubMed] [Google Scholar]
- Rowley J. (1988), J Clin Oncol 6, 194. [DOI] [PubMed] [Google Scholar]
- Schmidt A., Endo N., Rutledge S. J., Vogel R., Shinar D., and Rodan G. A. (1992), Mol Endocrinol 6(4), 1634–1641. [DOI] [PubMed] [Google Scholar]
- Schule R., Rangarajan P., Yang N., Kliewer S., Ransone L., Bolado J., Verma I., and Evans R. (1991), Proc Natl Acad Sci USA 88, 6092–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz E. and Gill F. M. (1983), in Epochal Hematology: Hematology of the Newborn, Chapt. 5 (Williams W. J., ed.) McGraw-Hill, New York, p. 37. [Google Scholar]
- Seizinger B. R., Klinger H. P., Junien C., Nakamura Y., LeBeau M., Cavenee W., Emanuel B., Ponder B., Naylor S., Mitelman F., Louis D., Menon A., Newsham I., Decker J., Kaelbling M., Henry I., and Deimling A. v. (1991), Cytogenet Cell Genet 58, 1080–1096. [Google Scholar]
- Sher T., Yi H.-F., McBride O. W., and Gonzalez F. J. (1993), Biochemistry 32, 5598–5604. [DOI] [PubMed] [Google Scholar]
- Short J., Fernandez J., Sorge J., and Atuse W. (1988), Nucleic Acids Res 16, 7583–7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staden R. (1986), Nucleic Acids Res 14, 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N. C. J. (1983), in Hematology, an Atlas and Diagnostic Guide, W. B. Saunders, Philadelphia, PA, p. 150. [Google Scholar]
- Tabor S. and Richardson C. (1989), Proc Natl Acad Sci USA 86, 4076–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P., Hu E., Graves R. A., Budavari A. I., and Spiegelman B. M. (1994), Genes Dev 8, 1224–1234. [DOI] [PubMed] [Google Scholar]
- Tory K., Latif F., Modi W., Schmidt L., Wei M.-H., Li H., Coblerr P., Orcutt M. L., Delisio J., Geil L., Zbar B., and Lerman M. I. (1992), Genomics 13, 275–286. [DOI] [PubMed] [Google Scholar]
- Tugwood J. D., Issemann I., Anderson R. G., Bundell K. R., McPheat W. L., and Green S. (1992), EMBO J 11(2), 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesono K., Murakami K. K., Thompson C. C., and Evans R. M. (1991), Cell 65, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahli W. and Martinez E. (1991), FASEB J 5, 2243–2249. [DOI] [PubMed] [Google Scholar]
- Zhu Y., Alvares K., Huang Q., Rao M. S., and Reddy J. K. (1993), J Biol Chem 268(36), 26817–26820. [PubMed] [Google Scholar]