

Abstract
A pancreatic islet cell-specific enhancer element in the rat glucagon gene, Glu-G3, contains two domains, one of which, domain A, has been shown to be necessary for Glu-G3 activity. In the present study, the functions of the isolated domain A of Glu-G3 were investigated by using transient reporter fusion gene expression and DNA binding assays. A single copy of domain A was transcriptionally inactive in glucagonproducing islet cell lines, whereas it did confer activity when combined with domain B, suggesting that Glu-G3 may be a bipartite element. Multiple copies of domain A did function independently as transcriptional enhancer in phenotypically distinct islet cell lines but not in several nonislet cell lines. Sequences (PISCES, pancreatic islet cell-specific enhancer sequences), similar to that of domain A of Glu-G3 and present in cell-specific control elements of the rat insulin I (Ins-E1) and rat somatostatin genes (SMS-UE), are shown to be required for transcriptional activity of these elements. In addition, a protein was detected in islet cell lines that bound to the PISCES motifs within Glu-G3, Ins-E1, and SMS-UE. These results support the view that cell-specific control elements of the glucagon, insulin, and somatostatin genes share a functional regulatory sequence, PISCES, and provide direct evidence for the existence of an islet-specific, PISCES-binding transcription factor or closely related proteins being involved in the coordinate expression of islet hormone genes.
Keywords: Glucagon; Insulin; Somatostatin; Transcription, tissue-specific; DNA binding proteins
THE pancreatic islet hormones insulin, glucagon, and somatostatin are important regulators of blood glucose levels. Studies with both normal and transgenic mice have provided evidence suggesting that islet cells may arise from a common progenitor cell (Alpert et al., 1988). Insulin, glucagon, and somatostatin appear sequentially during development and are transiently coexpressed before terminal differentiation restricts expression to distinct islet cell types (Alpert et al., 1988). These observations raise the possibility that islet cells may share transcriptional regulatory proteins directing insulin, glucagon, and somatostatin gene transcription.
Glu-G3, Ins-E1, and SMS-UE are cis-acting transcriptional control elements in the 5′ flanking region of the rat glucagon (Philippe et al., 1988; Knepel et al., 1990, 1991), rat insulin I (Ohlsson and Edlund, 1986; Karlsson et al., 1987; Knepel et al., 1991), and rat somatostatin genes (Powers et al., 1989; Knepel et al., 1991; Vallejo et al., 1992a, 1992b), respectively, that are involved in the regulation of islet cell-specific expression of their genes. In vitro DNase I footprinting assays indicate that nuclear proteins or protein complexes binding to these control elements are not restricted to a particular islet cell type; they are present in all islet cell lines expressing the glucagon, insulin, or somatostatin genes, but not in several nonislet cell lines (Ohlsson and Edlund, 1986; Philippe et al., 1988; Powers et al., 1989; Ohlsson et al., 1991). Nuclear proteins in glucagon-producing InR1-G9 cells that bound to the glucagon G3 element and that, as indicated by mutational analysis, may represent the glucagon G3 transcription factor (Knepel et al., 1990), bound also to undefined sequences within the insulin E1 and somatostatin upstream elements (Knepel et al., 1991). By inspection, similarities are observed among the nucleotide sequences of these insulin, glucagon, and somatostatin enhancer elements (Knepel et al., 1991). These homologous 12-base pair sequences, which we now call PISCES (pancreatic islet cell-specific enhancer sequences), define a consensus motif (5′-TTTYACRCCTG/CA-3′) that may represent the binding site of a common islet-specific transcription factor or closely related proteins. However, the functional significance of the PISCES motif is not completely understood. For example, the PISCES motif in the rat insulin I gene has not yet been shown to mediate transcriptional activation. Furthermore, the glucagon G3, insulin E1, and somatostatin upstream elements are functionally not equivalent, exhibiting distinct transcriptional activities (Knepel et al., 1991). When placed in front of a truncated promoter, Glu-G3, but not SMS-UE, conferred transcriptional activation in a glucagon-producing islet cell line (InR1-G9), whereas in a somatostatin-expressing islet cell line (RIN 1027-B2), SMS-UE, but not Glu-G3, stimulated transcription. Ins-E1 showed strong activity in the insulin-producing islet cell line HIT, but was only weakly active in InR1-G9 cells (Knepel et al., 1991). Therefore, despite their partial sequence similarity, Glu-G3, Ins-E1, and SMS-UE are functionally distinct islet cell-specific transcriptional regulatory elements.
Besides Glu-G3, two additional control elements (G1 and G2) have been characterized in the rat glucagon gene 5′ flanking region that act together and confer, at least in part, pancreatic A-cell-specific expression to the glucagon gene (Philippe et al., 1988). The structural organization of the glucagon G3 enhancer-like element has been studied in greater detail (Knepel et al., 1990). Using an electrophoretic mobility shift assay and nuclear extracts from islet cell lines, it was shown that the sequence of the Glu-G3 element comprises two distinct protein binding domains: a more upstream domain A (5′-CGCCTGA-3′), and a more downstream domain B (5′-GATTGAAGGGTGTA-3′). These binding motifs are recognized by distinct nuclear proteins. However, mutating domain B within Glu-G3 did not affect the transcriptional activity of the Glu-G3 element detected in a transient expression assay in vivo or in a cell-free transcription assay in vitro. However, transcriptional activity was abolished after mutating Glu-G3 domain A. Based on these results, it was concluded that, although it contains two distinct protein binding domains (A and B), the Glu-G3 element forms a functional enhancer-like element by a single copy of only one motif (domain A) (Knepel et al., 1990). Noteworthy, the PISCES motif within Glu-G3 (5′-TTTCACGCCTGA-3′, from −263 to −252, defined by homology with sequences in Ins-E1 and SMS-UE) contains the bases of domain A of Glu-G3 (5′-CGCCTGA-3′, from −258 to −252, characterized by mutational analysis) (Knepel et al., 1990, 1991).
In the present study, the properties of the isolated domain A of Glu-G3 were investigated by using transient reporter fusion gene expression and DNA binding assays. The results obtained provide direct evidence for the existence of a pancreatic islet-specific PISCES binding protein that may be critical for the distinct cell-specific transcriptional activities of the three enhancer elements of the islet hormone-producing genes.
MATERIALS AND METHODS
Plasmid Constructions
The plasmid −136GluLuc was prepared by subcloning a BamHI-HincII fragment of −136GluCAT (Philippe et al., 1988), containing the rat glucagon gene promoter from −136 to +58, into the BamHI-SmaI sites of poLuc (Brasier et al., 1989). The plasmid −60GluLuc was prepared by subcloning the BamHI-HincII fragment of −60GluCAT (Philippe et al., 1988), containing the rat glucagon gene promoter from −60 to +58, into the BamHI-SmaI sites of pXP2 (Nordeen, 1988). One, two, or four copies, as indicated, of oligodeoxynucleotides with 5′-GATC overhangs were added in the forward orientation to the BamHI site of the respective plasmid. All constructs were confirmed by sequencing. The plasmids pT81Luc (Nordeen, 1988) and −65SM-SLuc (Schwaninger et al., 1993a) have been described previously.
Cell Culture and DNA Transfection
InR1-G9 (Takaki et al., 1986), αTC2 (Powers et al., 1990), HIT-T15 (Santerre et al., 1981), RIN 1027-B2 (Philippe et al., 1987), NG108 (Fishman and Spector, 1981), HepG2, HeLa S3, and JEG-3 cell lines were cultured as described (Fishman and Spector, 1981; Philippe et al., 1988; Schwaninger et al., 1993b). Islet cell lines were trypsinized and transfected in suspension by the DEAE-dextran method with 2 μg of indicator plasmid per 6-cm dish. Nonislet cell lines were transfected by the calcium phosphate technique with 10 μg of indicator plasmid per 6-cm dish. RSV-CAT or RSV-Luc (0.4 or 2 μg per 6-cm dish for islet or nonislet cell lines, respectively) was added as a second reporter to check for transfection efficiency. Cells were harvested 48 h after transfection. Transfection, cell extract preparation, and reporter enzyme assays were performed as described (Philippe et al., 1988; Schwaninger et al., 1993b).
Electrophoretic Mobility Shift Assay
Nuclear extracts were prepared by the method of Dignam et al. (1983) with the modification that after the chromatin extraction step an ammonium sulfate (0.3 g/ml) precipitation was included before dialysis. Oligodeoxynucleotides with 5′-GATC overhangs were labeled by a fill-in reaction using [α-32P]dCTP and Klenow enzyme. Using 15 μg of protein of nuclear extracts, the electrophoretic mobility shift assay was performed as described (Knepel et al., 1990).
RESULTS
A Single Copy of Domain A of Glu-G3 Is Not Transcriptionally Active
The Glu-G3 element extends from −264 to −238 as defined by DNase I footprinting assay (Philippe et al., 1988; Knepel et al., 1990). It has been shown previously that a synthetic Glu-G3 oligodeoxynucleotide (from −274 to −234) stimulates basal activity when linked to 136 base pairs of the rat glucagon promoter and transiently transfected into the glucagon-producing islet cell line, InR1-G9 (Knepel et al., 1990, 1991). This activity depends on domain A of Glu-G3, as indicated by a mutational analysis (Knepel et al., 1990). To investigate whether domain A of Glu-G3 is sufficient to confer transcriptional activation, oligodeoxynucleotides containing either the isolated domain A (G3A, from −262 to − 247) or the entire Glu-G3 sequence (wild-type or mutant) were inserted upstream from the truncated rat glucagon gene promoter (from −136 to +58) linked to the coding region of the luciferase reporter gene (plasmid −136GluLuc) (Fig. 1). The G3A oligodeoxynucleotide also includes, extending the sequence of domain A of Glu-G3, the Glu-G3 PISCES motif (except for a single thymine at the 5′ end) (Fig. 1). Consistent with previous findings, Glu-G3 wild-type sequence increased transcription about 18-fold or 9-fold after transfection into the glucagon-producing islet cell lines InR1-G9 (Fig. 1A) or αTC2 (Fig. 1B), respectively. In contrast, the isolated domain A of Glu-G3 (G3A, from −262 to −247) did not stimulate transcription (Fig. 1A,B). Also inactive or only very weakly active were the Glu-G3 mutants mB1, mB2, and mB3 (Fig. 1A,B). In these mutants, 7–10 bases within Glu-G3 were mutated or deleted; however, the bases within domain A of Glu-G3 and the PISCES motif remained unaltered (Fig. 1A,B). These results demonstrate that a single copy of the isolated domain A of Glu-G3 does not confer transcriptional activation. Thus, the domain A is necessary for Glu-G3 activity but it is not sufficient.
FIG. 1.
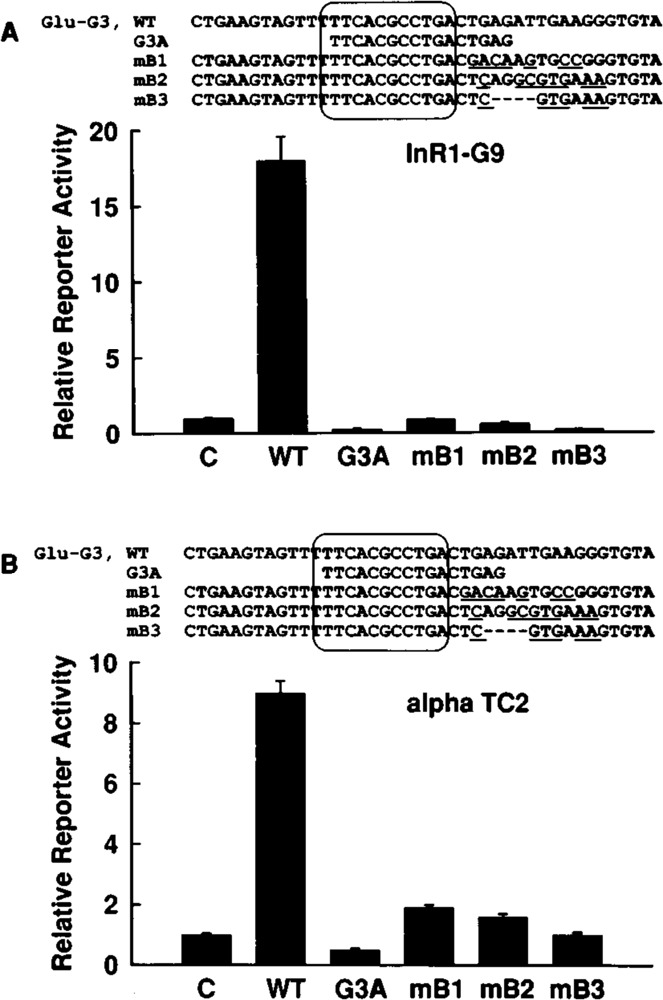
A single copy of domain A of Glu-G3 is not sufficient for transcriptional activity. The Glu-G3 oligodeoxynucleotide with wild-type sequence (WT) or the indicated oligodeoxynucleotides containing deletions or mutations were placed in front of the glucagon promoter (from −136 to +58) fused to the luciferase reporter gene (plasmid −136GluLuc). The constructs were transfected into the glucagon-expressing InR1-G9 (A) or αTC2 (B) cell lines. All oligodeoxynucleotides contain domain A of Glu-G3 (5′-CGCCTGA-3′). The mutated bases are underlined. Internal base deletions are indicated. The PISCES motif is enclosed by a solid line. Glu-G3 contains the bases from −274 to −234 of the rat glucagon gene, relative to the transcription start site. The sequences of the oligodeoxynucleotides used are presented at the top of the figure panels (except for the linker bases with 5′-GATC overhang). Reporter enzyme activity is expressed relative to the mean value in each experiment of the activity observed in the controls (C) with the promoter alone (− 136GluLuc). Values are means ± SE of three independent experiments, each done in duplicate.
G3A Has Latent Transcriptional Activity
In addition to the oligodeoxynucleotides containing Glu-G3 or only domain A of Glu-G3 (G3A), the oligodeoxynucleotides shown in Fig. 2 containing domain B of Glu-G3 (G3B, from −247 to −234) or a mutant of the domain A of Glu-G3 (G3Amut) were inserted either separately or in combination upstream of the truncated glucagon promoter (from −136 to +58) linked to the luciferase reporter gene. In G3Amut, domain A of Glu-G3 is mutated within the Glu-G3 PISCES motif in a way that, when introduced into full-length Glu-G3, destroys Glu-G3 activity (mutant 6) (Knepel et al., 1990). These constructs were transfected into the glucagon-producing islet cell lines InR1-G9 and αTC2. Like domain A of Glu-G3, a single copy of domain B of Glu-G3 did not have transcriptional activity (Fig. 2). However, the combination of domain A with domain B led to increased transcription, the activity of one of which (G3B + G3A in αTC2 cells) was even higher than that of Glu-G3 native sequence (Fig. 2). In InR1-G9 cells, G3A + G3B and G3B + G3A combinations (the oligodeoxynucleotide inserted 5′ to the other one is mentioned first) increased transcription 4- and 12-fold, respectively (Fig. 2A). In αTC2 cells, G3A + G3B and G3B + G3A combinations elevated transcriptional activity 4- and 34-fold, respectively (Fig. 2B). The activity observed was completely abolished by the mutations in G3A (Fig. 2). By far, the strongest activity was observed when two copies of G3A were combined, stimulating transcription 58-fold in InR1-G9 cells (Fig. 2A) and 128-fold in αTC2 cells (Fig. 2B). These data demonstrate that neither the domain A nor the domain B has significant enhancer activity alone, but the two can function as enhancers in combination. G3A can also function independently when two copies are combined. Thus, the domain A of Glu-G3 has strong latent transcriptional activity that becomes apparent when synergizing with itself or other elements in glucagon-producing islet cell lines. Because the synergism between two copies of domain A of Glu-G3 resulted in transcriptional activity that was higher than that resulting from the synergism between one copy of G3A and one copy of G3B, the latent transcriptional activity of domain A of Glu-G3 may be stronger than that of domain B.
FIG. 2.
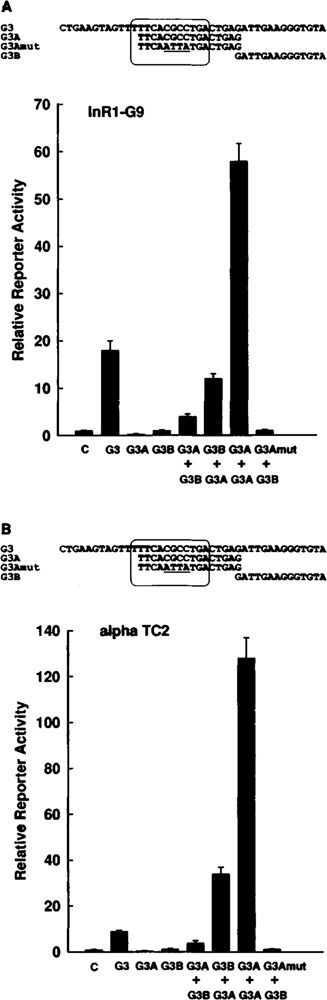
G3A can function as a transcriptional enhancer when combined with G3B or with a second copy of G3A. The indicated oligodeoxynucleotides were inserted either separately or in combination (the oligodeoxynucleotides inserted 5′ to the other one is mentioned first) in front of the truncated glucagon promoter (from −136 to +58) fused to the luciferase reporter gene (plasmid − 136 GluLuc). The constructs were transfected into the glucagon-expressing islet cell lines InR1-G9 (A) or αTC2 (B). The sequences of the oligodeoxynucleotides used are presented at the top of the figure panels (except for the linker bases with 5′ -GATC overhang). The mutated bases are underlined. The PISCES motif is enclosed by a solid line. Reporter enzyme activity is expressed relative to the mean value in each experiment of the activity observed in the controls (C) with the promoter alone (−136GluLuc). Values are means ± SE of three independent experiments, each done in duplicate.
G3A Confers Pancreatic Islet-Specific Activity
The above results show that two copies of G3A have independent transcriptional activity in glucagon-producing islet cell lines. To study the cell specificity of this activity, four copies of G3A or G3Amut were placed in front of the truncated non-cell-specific thymidine kinase promoter (−81 to +52) of herpes simplex virus linked to the luciferase reporter gene (pT81Luc). These constructs were transfected into phenotypically distinct islet cell lines as well as into nonislet cell lines. As shown in Fig. 3A, G3A showed transcriptional activity in all islet cell lines tested. G3A stimulated transcription about 62-fold and 166-fold in the glucagon-producing islet cell lines αTC2 and InR1-G9, respectively, about 62-fold in the insulin-producing islet cell line HIT, and about 3.5-fold in the somatostatin-producing islet cell line RIN 1027-B2 (Fig. 3A). However, G3Amut was inactive; it did not enhance transcriptional activity beyond the level reached by the promoter alone in the islet cell lines tested (Fig. 3A). In the nonislet cell lines tested, G3A was inactive (HepG2, hepatoma; HeLa, cervical carcinoma) or had weak activity that was not mutationally sensitive (JEG, choriocarcinoma; NG108, glioma × neuroblastoma) (Fig. 3B). Similar results were obtained using constructs with the truncated rat glucagon promoter (−136 to +58) in place of the viral thymidine kinase promoter (not shown). These results demonstrate that the transcriptional activity of G3A is islet specific. Because the G3A sequence contains a PISCES motif and because G3A activity depends on the PISCES motif, as indicated by the mutant, these results suggest that the PISCES motif as found in Glu-G3 has islet-specific transcriptional activity.
FIG. 3.
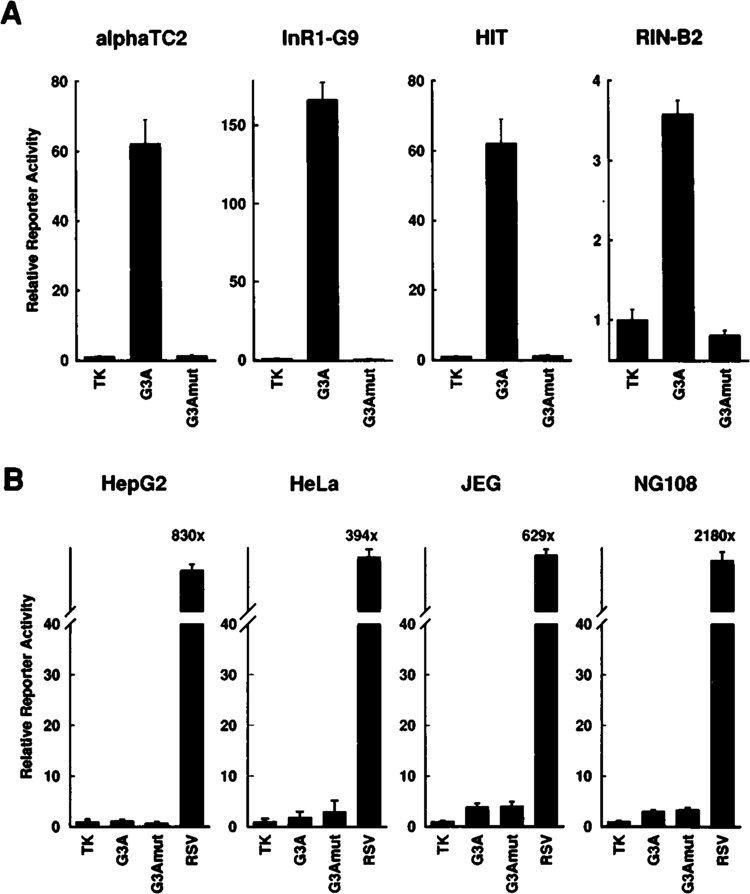
G3A confers pancreatic islet-specific transcriptional activity. Four copies of the oligodeoxynucleotides G3A or the mutant G3Amut were inserted upstream of the truncated thymidine kinase promoter (−81 to +52) of herpes simplex virus linked to the luciferase reporter gene (plasmid pT81Luc). The constructs were transfected into phenotypically distinct islet cell lines (A) as well as into several nonislet cell lines (B). As a positive control, the plasmid pRSVLuc (RSV) was transfected into the nonislet cell lines. Reporter enzyme activity is expressed relative to the mean value in each experiment of the activity observed in the respective controls with the promoter alone (TK, thymidine kinase). Values are means ± SE of three independent experiments, each done in duplicate.
Pancreatic Islet-Specific Distribution of Specifically G3A Binding Nuclear Proteins
Protein binding was studied by electrophoretic mobility shift assay using the oligodeoxynucleotides G3A and G3Amut as probes. When labeled G3A was incubated with nuclear extracts prepared from the glucagon-producing αTC2 cell line, a major retarded band was detected representing a protein-DNA complex (Fig. 4, indicated by an arrow). This protein binding was sequence specific, because this complex was not formed with labeled G3Amut (Fig. 4). A complex of similar mobility and sequence specificity was found with labeled G3A and nuclear proteins extracted from the insulin-producing HIT cells and the somatostatin-producing RIN 1027-B2 cells (Fig. 4). These complexes (indicated by an arrow) comigrated when also the binding reactions with αTC2 nuclear extracts were run on the same gel as the binding reactions with HIT and RIN 1027-B2 extracts (not shown). RIN 1027-B2 cell extracts produced additional faster migrating complexes, binding to G3A but not to G3Amut (Fig. 4), as has been reported previously for InR1-G9 cell extracts (Knepel et al., 1990). In nuclear extracts from nonislet cell lines (HeLa, HepG2), only weak protein binding to G3A was detected and it was not mutation sensitive (Fig. 4). Thus, by this assay a G3A binding protein or protein complex of similar mobility is found in islet cell lines but not in nonislet cell lines, the binding of which is abolished by the same mutation that also destroys G3A transcriptional activity (Fig. 3). Labeled G3Amut binds one or more proteins that form slower migrating complexes (Fig. 4); in view of the lack of transcriptional activity of G3Amut (Fig. 3), the functional significance of these proteins remains unclear.
FIG. 4.
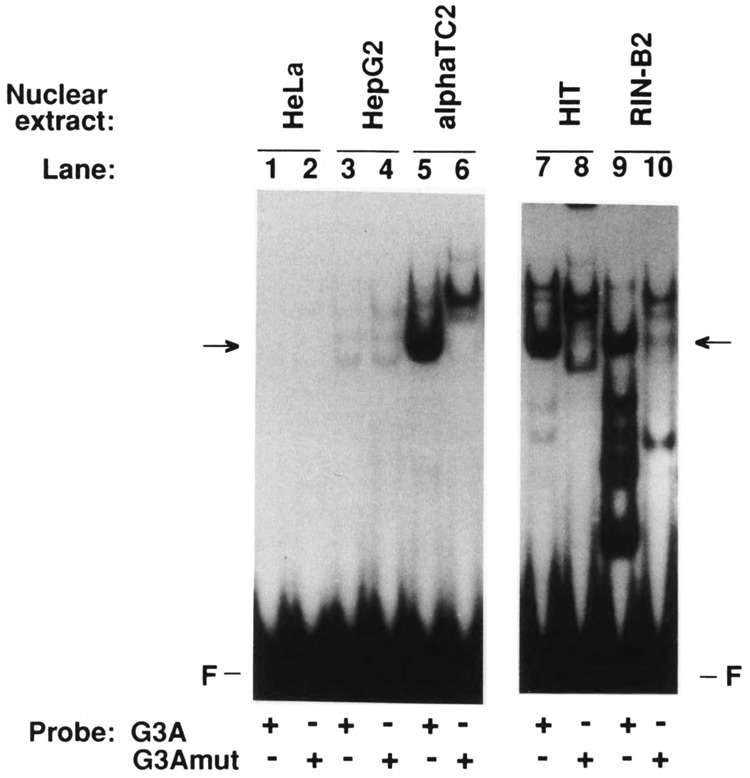
Pancreatic islet-specific distribution of G3A binding nuclear proteins as revealed by the electrophoretic mobility shift assay. Labeled G3A or G3Amut were incubated with nuclear extracts from the cell lines indicated and the reaction mixture was resolved by electrophoresis on nondenaturing polyacrylamide gels. F, free probe. The arrows indicate complexes of similar mobility formed with labeled G3A, but not with labeled G3Amut, and nuclear proteins in phenotypically distinct islet cell lines.
The PISCES Motifs Within the Enhancer-Like Elements Insulin E1 and Somatostatin-UE Are Necessary for Transcriptional Activity, as in Glucagon G3
It has been shown previously (Knepel et al., 1990) that 4- and 3-base mutations (designated mutants 6 and 5, respectively) of the sequence 5′-CGCCTGA-3′ within Glu-G3 (called domain A and located within the PISCES motif; see Fig. 5) result in a total loss of enhancer-like Glu-G3 activity. To study the functional significance of the PISCES motif within Ins-E1 and SMS-UE, the PISCES motifs within these elements were mutated in the same way as the PISCES motif has been mutated within Glu-G3. As shown in Fig. 5, the homologous bases within the PISCES motifs were converted into the same mutant bases (ATTA or GAC). Wild-type and mutant oligodeoxynucleotides were placed in front of a truncated promoter linked to a reporter gene and the resulting plasmids were transfected into preferentially glucagon-producing (InR1-G9, αTC2), insulin-producing (HIT), or somatostatin-producing (RIN 1027-B2) cell lines, respectively. As shown in Fig. 5, wild-type Glu-G3, Ins-E1, and SMS-UE enhanced transcription 6.3-, 7.7-, and 11.3-fold, respectively. Both types of mutations within the PISCES motifs virtually abolished the enhancer activity of Glu-G3, Ins-E1, and SMS-UE (Fig. 5).
FIG. 5.

The PISCES motif within the cell-specific enhancer-like elements Glu-G3, Ins-E1, and SMS-UE of the rat glucagon, rat insulin I, and rat somatostatin genes, respectively, is required for their transcriptional activity. The sequences of the oligodeoxynucleotides used are shown (except for the linker bases with 5′-GATC overhang). The PISCES motif within each element is indicated by the solid line. Two sets of mutants (m5, m6) were created with each element by converting homologous bases within the PISCES motif into the same mutant bases as indicated. WT, wild-type sequence. The numbers given in parentheses correspond to the 5′ and 3′ ends of the DNA sequence, relative to the transcription start site of the gene. These oligodeoxynucleotides were inserted in front of a minimal promoter linked to a reporter gene (−136GluCAT for Glu-G3, −65SMSLuc for SMS-UE, −60GluLuc for Ins-E1). The constructs were transfected into the cell lines indicated. Reporter enzyme activity is expressed relative to the mean value in each experiment of the activity observed in the respective controls (C) with the promoter alone. Values are means ± SE of three independent experiments, each done in duplicate.
Similar results as obtained in InR1-G9 cells were also obtained in αTC2 cells (not shown). These results demonstrate that the PISCES motifs of these cell-specific enhancer-like elements are critical for function.
Binding Specificity of the Islet-Specific Nuclear Protein Complex Formed on G3A: PISCES-Dependent Binding to Ins-E1 and SMS-UE as Well as to Glu-G3
The binding specificity of islet nuclear protein complexes formed on labeled G3A was studied by competition experiments in electrophoretic mobility shift assays using nuclear proteins extracted from phenotypically distinct islet cell lines. As shown in Fig. 6A, G3A, but not G3Amut, did compete for binding of the nuclear protein complex formed on labeled G3A in αTC2 nuclear extracts (marked by an arrow), confirming the sequence specificity of binding of this complex that was observed using labeled G3Amut (Fig. 4). Wild-type Glu-G3, Ins-E1, and SMS-UE were able to compete for binding, whereas the respective mutants 5 and 6 were weak competitors or did not compete at all (Fig. 6A). Similar competition experiments were performed using HIT (Fig. 6B) and RIN 1027-B2 nuclear extracts (Fig. 6C). The complexes of similar mobility (indicated by an arrow) were also competed by wild-type Glu-G3, Ins-E1 and SMS-UE, whereas the respective mutants 5 and 6 had strongly reduced ability or were unable to compete (Fig. 6B,C). These competition experiments demonstrate that the protein complexes of similar mobility formed with G3A and nuclear extracts from phenotypically distinct islet cell lines are able to bind to the PISCES motifs in the cell-specific enhancer-like elements of the three hormone-coding genes. The variability in the ability of some of the Ins-E1 and SMS-UE mutants to compete for G3A binding may suggest that identical mutant bases within a somewhat distinct sequence context may be less detrimental to binding. The observed decrease in binding is, however, sufficient to virtually abolish function (Fig. 5).
FIG. 6.
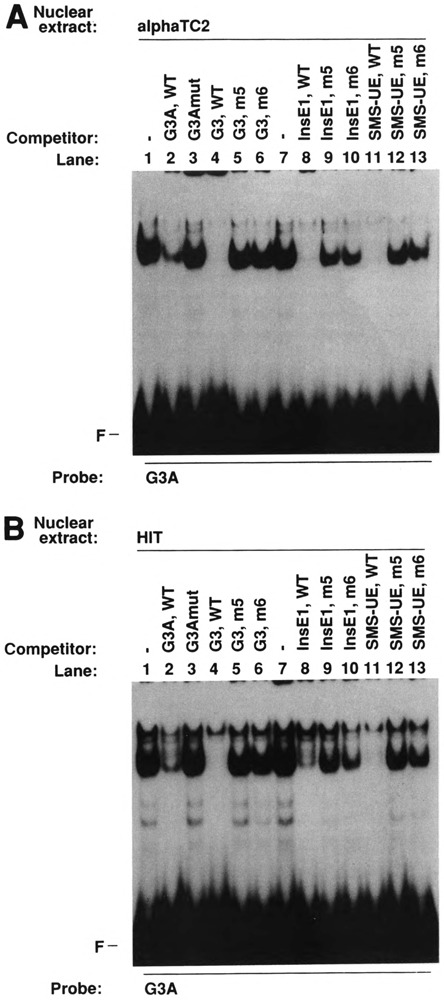
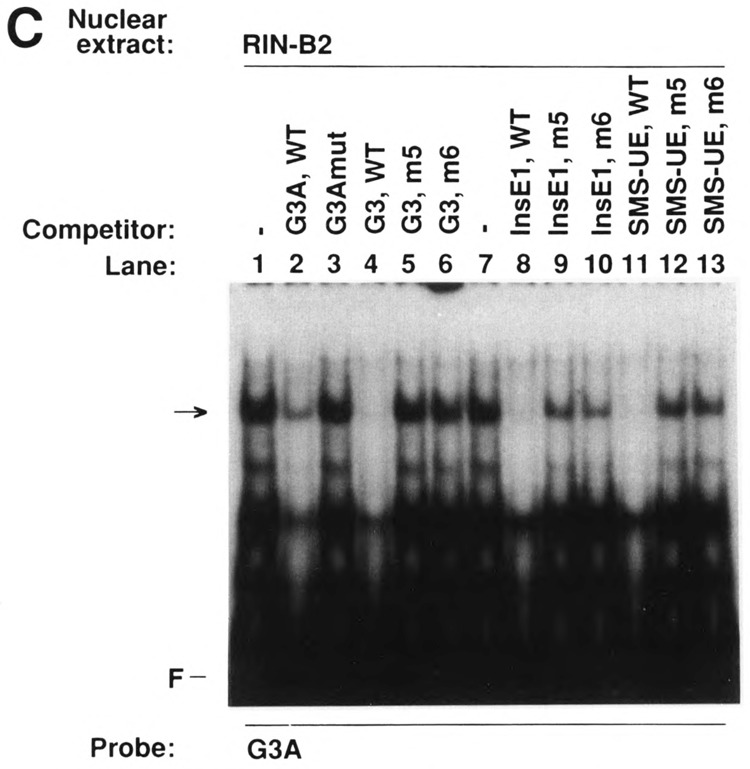
Binding specificity of islet nuclear protein complexes formed on G3A: PISCES-dependent binding to Glu-G3, Ins-E1, and SMS-UE as revealed by competition experiments in an electrophoretic mobility shift assay. Labeled G3A was incubated with nuclear extracts from glucagon-producing αTC2 (A), insulin-producing HIT (B), and somatostatin-producing RIN 1027-B2 cells (C). Competitors were added at a 50-fold molar excess as indicated. The sequences of the oligodeoxynucleotides are shown in Figs. 2 and 5. WT, wild-type sequence; F, free probe. The arrows indicate the complexes of similar mobility formed with G3A and nuclear extracts from phenotypically distinct islet cell lines.
DISCUSSION
Evidence for a Bipartite Nature of Glu-G3
The domain A of Glu-G3 is necessary for islet cell-specific transcriptional activity of Glu-G3 as was demonstrated by mutating base sequences within domain A (Knepel et al., 1990). In a previous study, 3- or 4-base mutations within domain B did not decrease transcriptional activity (Knepel et al., 1990). However, in the present study, more extensive 7- to 10-base mutations outside domain A eliminated Glu-G3 activity. Furthermore, the isolated domain A of Glu-G3 was not transcriptionally active. Therefore, base sequences within domain A of Glu-G3, although necessary, are not sufficient for Glu-G3 activity. Thus, this study suggests that Glu-G3 may be a bipartite element with at least two functionally interdependent domains. Consistent with this assumption, G3A was shown to have latent transcriptional activity. G3A and G3B did function in combination, but not separately, as a transcriptional enhancer. When G3A was placed 5′ to G3B, transcriptional activity was less than that conferred by wild-type Glu-G3, possibly due to an altered spacing because seven additional bases were inserted in between by linker bases. However, when placed 3′ to G3B, G3A conferred strong transcriptional activity exceeding Glu-G3 wild-type activity in αTC2 cells. Similar to the suggested bipartite nature of Glu-G3, numerous viral and cellular enhancer elements are composed of multiple sequence motifs that act together in a synergistic fashion to give a specific enhancer effect. The virtually ubiquitous activity of the SV40 enhancer is based on the presence of multiple binding sites for different transcription factors, each with a distinct and often broad cellular distribution (Maniatis et al., 1987). In the tyrosine aminotransferase gene, synergy between constitutive, liver-specific elements and hormone-responsive elements determines the tissue specificity of the hormonal control (Nitsch et al., 1993). The islet cell-specific activity of the FF minienhancer in the 5′ flanking region of the rat insulin I gene depends on the synergistic interaction of at least two sequence elements, the Far element and the FLAT element (German et al., 1992a, 1992b). Interestingly, FF minienhancer activity could not be reconstituted by combining oligodeoxynucleotides containing only the Far and only the FLAT elements (German et al., 1992a), indicating that the two elements can only function when properly juxtaposed. Furthermore, multiple copies of either the Far element or the FLAT element by themselves could not enhance transcription (German et al., 1992a). In contrast, this study shows that multiple copies of G3A exhibit strong transcriptional activity, demonstrating that the domain A of Glu-G3 can function independently as transcriptional enhancer. Although G3A and the G3A binding protein or protein complex may form one module, the structure and function of the second module remains to be defined. Noteworthy, results obtained using the in vitro DNase I footprinting assay suggest that protein binding to domain B may depend on protein binding to domain A, because Glu-G3 mutant 6, which is substituted within domain A, failed to compete the protection of the entire Glu-G3 sequence, including domain B (Knepel et al., 1990).
PISCES-Dependent Transcription and Protein Binding
The PISCES motif is a partial sequence homology between Glu-G3, Ins-E1, and SMS-UE, which was found just by sequence comparison (Knepel et al., 1991), and its significance was unclear. The present study strongly suggests that the PISCES motif is a regulatory sequence with islet-specific activity as is indicated by the mutational analysis of Ins-E1, Glu-G3, and SMS-UE as well as by G3A activity. Transcriptional activity of multiple copies of domain A of Glu-G3 was not restricted to glucagon-producing islet cell lines but was also found in insulin- or somatostatin-producing islet cell lines but not in several nonislet cell lines. This islet-specific activity of domain A of Glu-G3 may be conferred by the binding of an islet-specific transcription factor, because a domain A–binding protein complex with the same cellular distribution and mutation sensitivity as domain A activity was detected by the electrophoretic mobility shift assay. Domain A of Glu-G3 contains and depends on the Glu-G3 PISCES motif. A regulatory role for the PISCES motif within Ins-E1 has not been demonstrated previously, whereas the PISCES motifs within Glu-G3 and SMS-UE comprise the bases required for transcriptional activation by Glu-G3 (Knepel et al., 1990) and SMS-UE (Vallejo et al., 1992a). The present study confirms and extends these observations. Identical 3- or 4-base mutations within the PISCES motifs of Ins-E1, Glu-G3, and SMS-UE were found to abolish transcriptional activity of these enhancer elements, suggesting that Ins-E1, Glu-G3, and SMS-UE share a functional regulatory sequence, PISCES. Furthermore, the islet-specific G3A binding protein was demonstrated to specifically bind to the PISCES motifs in all three enhancer elements of the islet hormone genes. Thus, by cellular distribution and binding specificity, this protein or protein complex stands as a likely candidate for representing a common islet-specific transcription factor or related factors directing insulin, glucagon, and somatostatin gene transcription through the PISCES motif. This factor(s) appears to bind similarly to the PISCES motifs found in Glu-G3, Ins-E1, and SMS-UE, although the DNA sequences vary from each other, raising the possibility that the PISCES binding protein is recognizing other factors that are bound to DNA in this region.
The characterization of PISCES-dependent transcription and protein binding by the present study provides direct evidence for the existence of an islet-specific PISCES binding transcription factor or closely related factors being involved in the coordinate expression of the insulin, glucagon, and somatostatin genes in distinct islet cell types. This PISCES binding transcription factor could be involved in the transcriptional activation of additional genes expressed in pancreatic islets. The proposed role of the PISCES binding transcription factor is reminiscent of that of cell-specific transcription factors in other tissues. The liver-enriched transcription factor C/EBPα activates the transcription of several liver-specific genes (Umek et al., 1991). Myogenin and additional closely related proteins are expressed exclusively in skeletal muscle and stimulate transcription from the E box DNA motif found in the regulatory regions of most known muscle-specific genes (Hughes, 1992). Whereas growth hormone and prolactin are expressed in distinct anterior pituitary cell types, both the growth hormone and prolactin genes are activated by the pituitary-specific POU domain transcription factor Pit-1, which is ultimately present in somatotrophs, lactotrophs, and thyrotrophs (Voss and Rosenfeld, 1992). There is evidence to suggest that these cell-specific transcription factors may also act as developmental regulators. C/EBPα is expressed late during development and may participate in cell growth arrest and terminal differentiation (Umek et al., 1991), whereas myogenin and Pit-1 are expressed early during development and are involved in the commitment or maintenance of cell lineages (Hughes, 1992; Voss and Rosenfeld, 1992). These examples raise the possibility that the PISCES binding transcription factor could have a role not only in regulating islet hormone gene transcription but also in specifying endocrine cell lineage in the developing pancreatic islets.
Despite their similarities, Ins-E1, Glu-G3, and SMS-UE are functionally distinct islet cell-specific transcriptional enhancer elements (Knepel et al., 1991). If, as the present study suggests, these control elements share a functional PISCES motif binding identical or related factors, we speculate that the PISCES motif combines, in each case, with different adjacent regulatory sequences within the control elements to yield unique cell-specific activities. Consistent with this explanation, evidence for a modular organization has been presented for SMS-UE (Vallejo et al., 1992a) and Glu-G3 (this study). A mutational analysis of the rat insulin I gene 5′ flanking region demonstrated a threefold reduction in activity when 8 bases within the El region and adjacent to the PISCES motif were mutated (Karlsson et al., 1987), suggesting that Ins-E1 may also contain regulatory sequences in addition to the PISCES motif. The nuclear proteins that recognize sequence motifs adjacent to or overlapping the PISCES motif have not been characterized for Glu-G3, whereas these nuclear proteins may include βTF-1 for Ins-E1 (Kruse et al., 1993) and CREB, STF-1, βTF-1 or an α-CBF-like transcription factor for the SMS-UE (Vallejo et al., 1992b; Kruse et al., 1993; Leonard et al., 1993).
In addition to the band comigrating with complexes in αTC2 and HIT nuclear extracts, RIN 1027-B2 cell extracts produced three faster migrating complexes of similar binding specificity as resolved by native gel electrophoresis using G3A as probe. The relationship between the bands of different mobility is not obvious. Because the transcriptional activity of G3A in RIN 1027-B2 cells was less than in αTC2 and HIT cells, it is one possibility that these proteins modulate transcriptional activity.
In a subset of patients with non-insulin-dependent diabetes mellitus an 8-base pair repeat was found from −322 to −315 in the 5′ flanking region of the insulin gene (Olansky et al., 1992) disrupting a sequence that is highly homologous to the PISCES motif. The transcriptional activity of the mutant human insulin promoter was found to be reduced (Olansky et al., 1992), suggesting that variants of the PISCES motif could contribute to the diabetes phenotype.
The molecular structure of the PISCES binding transcription factor is unknown. In a recent study, the trancription factor CREB was found to bind to a region of SMS-UE containing the PISCES motif (Vallejo et al., 1992b). However, several lines of evidence suggest that CREB is not related to the PISCES binding transcription factor as characterized by electrophoretic mobility shift assay in the present study (PISCES-TF): i) CREB is ubiquitously expressed, whereas the PISCES-TF is islet specific; ii) various CREB binding sites (CREs) do not compete for PISCES-TF binding; and iii) a specific anti-CREB antiserum did not inhibit or “supershift” PISCES-TF binding (manuscript in preparation). The β- and δ-cell-specific protein STF-1/IDX-1 and a novel β-cell-specific factor not yet cloned, βTF-1, were reported to bind to sites partly overlapping the PISCES motifs in SMS-UE (Leonard et al., 1993; Miller et al., 1994) and Ins-E1 (Kruse et al., 1993), respectively. The isolation of cDNAs encoding PISCES binding proteins may provide tools that will be helpful to identify the PISCES binding transcription factor(s).
ACKNOWLEDGEMENTS
We thank D. Scholz and I. Cierny for expert technical assistance, A. Neubauer for critical reading of the manuscript, and H. Kuhlenkamp for typing the manuscript. This work was supported by Grant Kn220/5-1 and SFB 402/A3 from the Deutsche Forschungsgemeinschaft, Bonn, Germany.
REFERENCES
- Alpert S., Hanahan D., and Teitelman G. (1988), Cell 53, 295–308. [DOI] [PubMed] [Google Scholar]
- Brasier A. R., Tate J. E., and Habener J. F. (1989), Biotechniques 7, 1116–1122. [PubMed] [Google Scholar]
- Dignam J. D., Lebovitz R. M., and Roeder R. G. (1983), Nucleic Acids Res 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman M. C. and Spector I. (1981), Proc Natl Acad Sci USA 78, 5245–5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German M. S., Moss L. G., Wang J., and Rutter W. J. (1992a), Mol Cell Biol 12, 1777–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German M. S., Wang J., Chadwick R. B., and Rutter W. J. (1992b), Genes Dev 6, 2165–2176. [DOI] [PubMed] [Google Scholar]
- Hughes S. M. (1992), Nature 360, 536–537. [DOI] [PubMed] [Google Scholar]
- Karlsson O., Edlund T., Moss J. B., and Rutter W. (1987), Proc Natl Acad Sci USA 84, 8819–8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepel W., Jepeal L., and Habener J. F. (1990), J Biol Chem 265, 8725–8735. [PubMed] [Google Scholar]
- Knepel W., Vallejo M., Chafitz J. A., and Habener J. F. (1991), Mol Endocrinol 5, 1457–1466. [DOI] [PubMed] [Google Scholar]
- Kruse F., Rose S. D., Swift G. H., Hammer R. E., and MacDonald R. J. (1993), Genes Dev 7, 774–786. [DOI] [PubMed] [Google Scholar]
- Leonard J., Peers B., Johnson T., Ferreri K., Lee S., and Montminy M. R. (1993), Mol Endocrinol 7, 1275–1283. [DOI] [PubMed] [Google Scholar]
- Maniatis T., Goodbourn S., and Fischer J. A. (1987), Science 236, 1237–1245. [DOI] [PubMed] [Google Scholar]
- Miller C. P., McGehee R. E. Jr., and Habener J. F. (1994), EMBO J 13, 1145–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsch D., Boshart M., and Schütz G. (1993), Proc Natl Acad Sci USA 90, 5479–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordeen S. K. (1988), Biotechniques 6, 454–457. [PubMed] [Google Scholar]
- Ohlsson H. and Edlund T. (1986), Cell 45, 35–44. [DOI] [PubMed] [Google Scholar]
- Ohlsson H., Thor S., and Edlund T. (1991), Mol Endocrinol 5, 897–904. [DOI] [PubMed] [Google Scholar]
- Olansky L., Welling C., Giddings S., Adler S., Bourey R., Dowse G., Serjeantson S., Zimmet P., and Permutt M. A. (1992), J Clin Invest 89, 1596–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe J., Chick W. L., and Habener J. F. (1987), J Clin Invest 79, 351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe J., Drucker D. J., Knepel W., Jepeal L., Misulovin Z., and Habener J. F. (1988), Mol Cell Biol 8, 4877–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers A. C., Tedeschi F., Wright K. E., Chan J. S., and Habener J. F. (1989), J Biol Chem 264, 10048–10056. [PubMed] [Google Scholar]
- Powers A. C., Efrat S., Mojsov S., Spector D., Habener J. F., and Hanahan D. (1990), Diabetes 39, 406–414. [DOI] [PubMed] [Google Scholar]
- Santerre R. F., Cook R. A., Crisel R. M. D., Sharp J. D., Schmidt R. J., Williams D. C., and Wilson C. P. (1981), Proc Natl Acad Sci USA 7, 4339–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaninger M., Blume R., Oetjen E., Lux G., and Knepel W. (1993a), J Biol Chem 268, 23111–23115. [PubMed] [Google Scholar]
- Schwaninger M., Lux G., Blume R., Oetjen E., Hidaka H., and Knepel W. (1993b), J Biol Chem 268, 5168–5177. [PubMed] [Google Scholar]
- Takaki R., Ono J., Nakamura M., Yokogawa Y., Kumae S., Hiraoka T., Yamaguchi K., Hamaguchi K., and Uchida S. (1986), In Vitro Cell Dev Biol 22, 120–126. [DOI] [PubMed] [Google Scholar]
- Umek R. M., Friedman A. D., and McKnight S. L. (1991), Science 251, 288–292. [DOI] [PubMed] [Google Scholar]
- Vallejo M., Miller C. P., and Habener J. F. (1992a), J Biol Chem 267, 12868–12875. [PubMed] [Google Scholar]
- Vallejo M., Penchuk L., and Habener J. F. (1992b), J Biol Chem 267, 12876–12884. [PubMed] [Google Scholar]
- Voss J. W. and Rosenfeld M. G. (1992), Cell 70, 527–530. [DOI] [PubMed] [Google Scholar]