

Abstract
The bacteriophage T4 Alt gene product is a component of the phage head and enters the host cell in the process of infection together with the phage DNA. It immediately ADP-ribosylates host RNA polymerase, presumably at only one of the two α-subunits. Transcription from T4 “early” promoters, therefore, might be catalyzed, at least in part, by an altered RNA polymerase. The T4 alt gene was cloned into the expression vector pBluescript. E. coli cells, transformed with this recombinant vector, overexpressed the 76 kDa Alt gene product, which was purified to homogeneity. The purified enzyme not only ADP-ribosylates the α-subunit of RNA polymerase, but also subunits β and β′, as well as the σ70-factor. The recombinant enzyme behaved like the native enzyme isolated from mature phage particles. The effect of the ribosylation reaction on the transcription activity of host RNA polymerase was investigated in vivo. It results in a modulation of T4 “early” promoter strengths, presumably, in a number of cases, leading to an overexpression of T4 “early” genes. The degree of overexpression, in some cases, should reach 50%, and seems to be well dosed for each promoter, controlling an individual transcription unit.
Keywords: ADP-ribosyltransferase; Expression vector; Promoters, prokaryotic; T-phages; Transcription regulation; RNA polymerase, bacterial
E. coli RNA polymerase core enzyme is engaged in all transcription in a T4-infected cell: T4-directed RNA synthesis remains sensitive to rifampicin (Di-Mauro et al., 1969; Haselkorn et al., 1969; Zillig et al., 1970). However, up to 13 phage-coded proteins stepwise modify the RNA polymerase or associate with it, leading to the recognition of three different classes of promoters: early, middle, and late (Mosig and Hall, 1994; Wilkens and Rüger, 1994; Stitt and Hinton, 1994; Williams et al., 1994).
The first modification observed in this sequence of events is the ADP-ribosylation of the α-subunits. Initially this reaction is catalyzed by the product of T4 alt gene. This gene was sequenced and codes for a 76 kDa protein (Koch and Rüger, 1994). Originally, the Alt protein had been identified as a component of the phage head (Paulson et al., 1976; Steven et al., 1976). The protein is processed during phage head maturation and enters the host cell together with the infecting DNA. It was reported to ADP-ribosylate about half of the α-subunits of host RNA polymerase (Rohrer et al., 1975). The ADP-ribosyl residue is transferred preferentially to amino acid Arg265 of this subunit (Horvitz, 1974; Goff, 1974; Ovchinnikov et al., 1977); however, other residues might also be ADP-ribosylated. There is conflicting data on the stability of the ADP-ribose moieties linked to the α-subunits; some reports suggest that the ADP-ribosylation is reversible (Horvitz et al., 1974) whereas others find it stable (Goff and Setzer, 1980). The native, processed Alt gene product ribosylates other RNA polymerase subunits, as well as proteins such as lysozyme and bovine serum albumin, and it performs an autoribosylation reaction (Rohrer et al., 1975). It therefore seems that the native Alt protein acts as a multitarget enzyme.
Later in the infection cycle ADP-ribosylation of both α-subunits is brought to completion by a second, newly synthesized ADP-ribosyltransferase, the 26 kDa Mod gene product. In contrast to Alt, this enzyme reacts more specifically towards RNA polymerase (Skorko et al., 1977). The biological effects of the ADP-ribosylation on gene expression are not precisely known. A double mutant (alt −, mod −) does not show any significant defects in phage burst or latent period (Goff and Setzer, 1980). It has been presumed that ADP-ribosylation of host RNA polymerase might be involved in the shutdown of bacterial RNA synthesis in the T4-infected cell (Mailhammer et al., 1975; Goldfarb and Palm, 1981; Goldfarb and Malik, 1984; Drivdahl and Kutter, 1990). Because host RNA polymerase seems to be the preferential target for alteration and modification, the consequences of these two reactions on T4-specific transcription regulation remain to be clarified.
To address this question, we cloned the T4 alt gene into the expression vector pBKS and purified the recombinant ADP-ribosyltransferase. We show that the overexpressed enzyme behaves as the described native and processed counterpart in ADP-ribosylating not only the α-subunits of E. coli RNA polymerase but also β, β′, and σ 70, as well as some other proteins. We also constructed a plasmid vector pTKRI, expressing gpAlt constitutively. This construct is compatible to plasmid vector pKWIII, suitable for estimating the strengths of T4 “early” promoters in vivo (Wilkens and Rüger, 1994). The vectors cotransformed into E. coli result in a two-plasmid system allowing ADP-ribosylation to be monitored on host cell-directed transcription in vivo. The results demonstrate that ADP-ribosylation of E. coli RNA polymerase modulates T4 promoter strengths. With eight out of 21 T4 “early” promoters, the transcriptional activity increased up to twofold, 12 promoter activities remained unchanged, and one promoter showed a decreased transcription activity compared to controls of unribosylated, native RNA polymerase.
MATERIALS AND METHODS
Materials
DNA-dependent RNA polymerase, alkaline phosphatase, and unlabeled nucleoside triphos-phates were purchased from Boehringer (Mannheim, Germany). [32P]UTP was obtained from Amersham Buchler. [Adenylate-32P]NAD was from NEN Dupont. Acrylamid, N,N′-bisacrylamid, and antibiotics were from Serva (Heidelberg, Germany). T4 DNA ligase was purchased from Stratagene (La Jolla, CA). All restriction enzymes came from Gibco-BRL (Gaithersburg, MD). Agarose was obtained from FMC (Rock-land, ME). Chromatography resins were purchased from Pharmacia-LKB (Uppsala, Sweden). Enzymatic DNA manipulations were performed according to Ausubel et al. (1993).
Bacteria and Plasmids
T4 dC-DNA was prepared by growing T4 mutant alc536 [42−(amC87)-56− (amE51)-denB−-(amS19)-alc− (unf39)] in E. coli B834 [gal−(U56), (met−, gal−(U), su−)]. For the cloning experiments we used plasmid vector pBluescript KS (pBKS) from Stratagene. Plasmid vector pT7/T3 was obtained from Pharmacia-LKB. E. coli K38 (HfrCλ) harboring plasmid pGP1-2 was a gift from Dr. S. Tabor (Harvard). In vivo strengths of T4 “early” promoters were determined, taking advantage of vector pKWIII (Wilkens and Rüger, 1994).
Overexpression of the T4 ADP-Ribosyltransferase
Overexpression of the alt gene was performed using the T7 RNA polymerase/promoter system of Tabor and Richardson (1985). E. coli K38 cells harboring plasmids pGP1-2 and pBKS:T4alt (see Results section) were grown at 30°C in standard broth [1% (w/v) Bacto tryptone, 0.5% (w/v) yeast extract, 0.5% (w/v) NaCl, and 40 μg/ml of kanamycin and of ampicillin] to a titer of 5 × 108 cells/ml. Overexpression of the alt gene was triggered by thermoinduction of the gene for T7 RNA polymerase for 30 min at 42°C. Incubation of the induced culture was continued for 2 h at 37°C, cells were harvested by centrifugation, and overexpression of the alt gene was monitored on 10% SDS-polyacrylamide gels (Laemmli, 1970).
Purification of the T4 ADP-Ribosyltransferase (gpAlt)
Ten grams of cells overexpressing gpAlt were resuspended in 30 ml of lysis buffer (50 mM Tris-HCl, pH 7.5, 2 mM EDTA, 1 mM DTT, 0.1 M NaCl, 20 μm phenylmethylsulfonyl fluorid). Cells were opened by sonic oscillation (0°C for 8 × 15 s) and centrifuged to remove cellular debris (10,000 × g for 20 min). The supernatant contained 673 mg of protein in a total volume of 27 ml. Streptomycin sulfate solution (10%, w/w) was added with stirring to the supernatant to a final concentration of 2.5%. After 20 min on ice the precipitate was collected by centrifugation (27,000 × g for 30 min at 4°C). The supernatant was adjusted to 30% saturation by adding crystalline ammonium sulfate. The precipitate again was removed by centrifugation. The ammonium sulfate concentration of the supernatant was increased to 40% with slow stirring and the precipitate containing the Alt protein was collected as above. The sediment was dissolved in 1 ml of TGED-100 (20 mM Tris-HCl, pH 8.0, 5% glycerol, 0.1 mM EDTA, 0.1 mM DTT, and 0.1 M NaCl) and dialyzed against the same buffer.
After dialysis the preparation (1.5 ml) was applied to a 70-ml Blue Sepharose column equilibrated with TGED-100. First, the column was washed with 90 ml of TGED-100, then developed with a linear gradient from 0.1 to 1.0 M NaCl in TGED over a time of 60 min. The flow rate was adjusted to 1 ml/min and 0.5-ml fractions were collected. The elution and all further steps were monitored on 7.5% SDS-polyacrylamide gels. Fractions containing the peak of the Alt protein were pooled and precipitated (80% saturation with ammonium sulfate). The precipitate was collected by centrifugation as above, resuspended in 150 μl 20 mM Tris-HCl, pH 8.0, 50 mM NaCl, and loaded onto a Superose 6 HR 10/30 column, equilibrated with the same buffer. Peak fractions containing gpAlt were pooled and dialyzed against TGED-100 containing 50% glycerol (v/v), and were stored at −20°C.
Purification of the Native Alt Gene Product and Protein Sequencing
The purification of the native T4 Alt gene product from phage particles had originally been described by Rohrer et al. (1975). These authors disintegrated phages by osmotic shock. This method results in large volumes, difficult to concentrate without hydrophobic reaggregation of the capsid proteins. To determine the NH2-terminal amino acid sequence, we therefore isolated Alt from preparative 7.5% SDS-polyacrylamide gels (13 × 10 × 0.2 cm). The Alt protein was identified by comparison of the band patterns obtained with T4 wild-type and T4 alt− phages (kindly provided by J. Hageman, Colorado). The corresponding band was cut out and, taking advantage of a volatile salt solution (10 mM NH4-carbonate), the protein was electroeluted from the gel slice. After lyophilization, the sample was applied to a 470A gas-phase protein sequencer and a 120A on-line phenylthiohydantoin (PTH) amino acid analyzer (Applied Biosystems).
ADP-Ribosyltransferase Assay
ADP-ribosyltransferase activity was determined essentially according to Rohrer et al. (1975). Incubation mixtures contained in a final volume of 0.1 ml 35 μg RNA polymerase (holoenzyme), 1 μCi [32P]NAD (specific activity: 800 Ci/mmol) in transferase buffer [0.05 M Tris-acetate, pH 7.5, 0.01 M Mg(OAc)2, 0.022 M NH4Cl, 1 mM EDTA, 0.01 M 2-mercaptoethanol, 10% glycerol], and 2.5–3 μg of the recombinant transferase. The reaction mixture was incubated for 30 min at 15°C and the reaction was stopped by precipitation with 5% trichloroacetic acid. The precipitate was collected on nitrocellulose filters, washed, and subjected to scintillation counting.
The activity of the purified transferase was also assayed by radioactive labeling of the proteins in vitro, followed by gel electrophoresis and autora-diography. The labeling reaction was performed in a final volume of 100 μl of transferase buffer (see above) containing, in addition, 40 μg RNA polymerase (holoenzyme), 1 μCi [adenylate-32P] NAD, and 10 μg of the recombinant transferase. After incubation for 30 min at 15°C the proteins were precipitated with 10% trichloroacetic acid. The precipitate was centrifuged, the sediment was washed with 70% acetone/water (v/v), dried, and dissolved in electrophoresis buffer. Samples were incubated at 95 °C for 5 min and separated on a 10% SDS-polyacrylamide gel (Laemmli, 1970). The gel was stained with Coomassie brilliant blue, dried, and autoradiographed on Fuji-RX films.
Construction of the pTKRI Plasmid
Plasmid pTKRI, expressing the Alt gene product constitutively, was constructed from three components (see Fig. 4). First, a 3-kb DNA fragment harboring the T4 alt gene was obtained from plasmid pBKS : T4alt by digestion with KpnI and BamHI. Second, plasmid pGP1-2 (Tabor and Richardson, 1985) was cleaved with PstI, phenolized, and treated with mungbean nuclease to generate blunt ends. Redigestion with BamHI yielded a 3-kb DNA fragment, harboring the gene for kanamycin resistance and the p15A origin. As a third component promoter Plac was taken as a 0.4-kb PvuII-KpnI fragment from plasmid pT7/T3 (Pharmacia LKB). The three DNA fragments were isolated each from preparative agarose gels, pooled, and ligated. On the grounds of their unique cleavage sites, there is only one possibility to form a circularly closed plasmid.
FIG. 4.
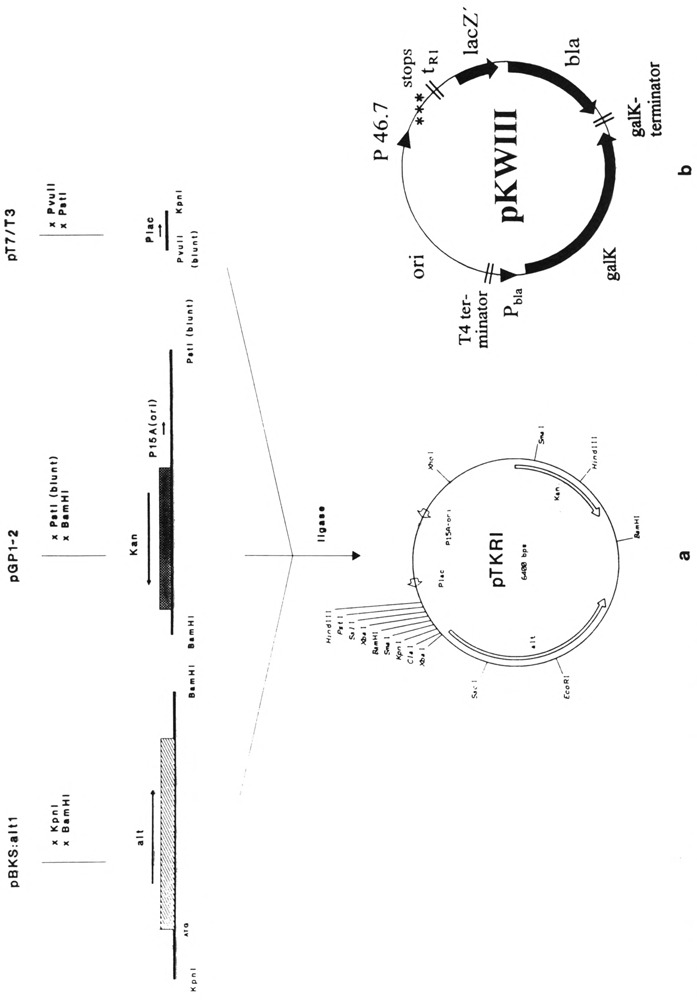
(a) Construction of vector pTKRI. Only elements and restriction sites important for the construction or for vector function are given. For explanation see text, (b) Vector pKWIII. Downstream from the promoter cloning site the following elements were inserted: stop codons in all three reading frames, the Rho-dependent terminator tR1 of phage λ, lacZ′ and the reading frame of β-lactamase (bla) as a reporting unit. Expression of β-lactamase in E. coli, transformed with pKWIII, depends on the strengths of the inserted T4 promoters. The 6-phospho-β-galactosidase (galK) reading frame inserted in opposite orientation serves as internal standard for bacterial transcription activity. Expression of galK is initiated at promoter Pbla.
In Vitro Determination of T4 “Early” Promoter Strengths
E. coli CMK603 cells (5 ml) harboring either pKWIII, or pKWIII and pTKRI, were grown at 30°C up to 1.0–1.2 at OD590 and harvested by centrifugation for 10 min at 10,000 × g and 4°C. For the preparation of crude extracts, cells were resuspended in 1 ml of Tris-HCl buffer (10 mM, pH 7.0), disrupted by sonic oscillation, and cellular debris was removed by a second centrifuga-tion. Aliquots (50 μl) of the cell extracts were pipetted into 950 μl of a phosphate buffer (10 mM Na2HPO4, 4.4 mM KH2PO4, pH 7.0) containing ampicillin in a final concentration of 0.25 mg/ml. The β-lactamase activity in the cell extracts (depending on the cloned T4 promoters) was monitored by the hydrolysis of ampicillin on a Philips PU8735 UV/Vis spectrophotometer at OD233. The 6-phospho-β-galactosidase activity (under control of an E. coli promoter Pbla) was determined according to the method of Hengstenberg and Morse (1968). Rates of hydrolysis of o-nitro-phenyl-β-galactoside-6-phosphate (0.5 mM) were monitored by increasing OD412. The ratios of corresponding β-lactamase and 6-phospho-β-galactosidase activities were taken as a measure for the relative promoter strengths (Wilkens and Rüger, 1994).
RESULTS
Cloning, Overexpression, and Purification of the T4 ADP-Ribosyltransferase (gpAlt)
Starting material for the cloning of the alt gene was restriction fragment XhoI-10, situated between 120.089 kb and 126.000 kb on the genomic map of bacteriophage T4 (Kutter et al., 1992). For an overview of this region and the fragments cloned see Fig. 1. To recover the 5.9-kb DNA fragment, 80 μg T4 dC-DNA was digested with XhoI and loaded onto 0.8% preparative agarose gels. Fragment XhoI-10 was cut out, electroeluted, and redigested with SphI. The resulting two restriction fragments (1.3 and 4.6 kb in size) were phenolized and treated with mungbean nuclease to render all cleavage sites blunt ended. The two fragments then were incubated with endonuclease ClaI, cleaving the 4.6-kb fragment into two sub-fragments, 1.6 and 3 kb, respectively. The three fragments were separated in a second electrophoresis and the 3-kb ClaI-blunt fragment was recovered. This 3-kb DNA fragment then was cloned into the ClaI-SmaI sites of vector pBKS, resulting in the recombinant plasmid pBKS : alt1. The nucleotide sequence of the insert has been determined (Koch, 1993; Koch and Rüger, 1994). The sequence data showed that the insert DNA harbors the alt gene and the C-terminal part of gene 30.
FIG. 1.
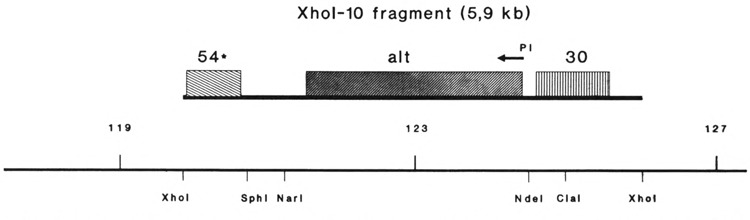
The T4 genomic region harboring the alt gene is shown together with the flanking genes and the map positions (Kutter et al., 1992). Restriction sites important to the cloning procedure are indicated.
To clone the alt gene downstream from the T7 promoter without flanking regions and in proper orientation for transcription, the recombinant plasmid pBKS : alt1 was linearized by digestion with NdeI, phenolized, and treated with mungbean nuclease. This digestion separates the alt gene from the flanking gene 30 portion. To gain a restriction terminus that rendered the alt fragment clonable, it was redigested with NarI. The remaining 2.2-kb alt gene fragment was separated from the rest of the DNA by gel electrophoresis and cloned into the ClaI-SmaI sites of vector pBKS, resulting in the recombinant plasmid pBKS: T4alt.
For the overexpression of the alt gene the T7 RNA polymerase-based system of Tabor and Richardson (1985) was used. The plasmid pBKS : T4alt was transformed into E. coli K38/pGP1-2 cells and grown as described in Materials and Methods. Upon thermoinduction of T7 RNA polymerase, the expression of the cloned alt gene was achieved. The synthesis of the gpAlt (76 kDa) was monitored on a Coomassie-stained SDS-polyacrylamide gel (Fig. 2).
FIG. 2.

Purification of T4 ADP-ribosyltransferase. Proteins were separated on a 10% SDS-polyacrylamide gel and stained with Coomassie brilliant blue. The left lane shows BSA (68 kDa) as a molecular weight marker. Lanes 1 and 2: E. coli K38/pGP1-2 containig plasmid pBKS : T4alt, uninduced and induced, respectively (overexpression of protein was performed as described in Materials and Methods); lane 3: crude cell extract, 75 μg of protein loaded onto gel; lane 4: ammonium sulfate fractionation, 75 μg of protein; lane 5: pooled fractions after Blue Sepharose affinity column, 15 μg of protein; lanes 6 and 7: pooled fractions after Superose 6 HR column, 3 and 10 μg of the purified ADP-ribosyltransferase, respectively. The position of the gpAlt is indicated. The radioactivity incorporated by the reaction of the ribosyltransferase was measured with aliquots of protein taken from the fractions shown on the gel. The counts incorporated were corrected to 10 μg of total protein in the test and are as follows: lane 3: 780 cpm; lane 4: 2220 cpm; lane 5: 3060 cpm, and lane 7: 8420 cpm.
Essentially, T4 ADP-ribosyltransferase was purified by affinity chromatography on Blue Sepharose. A detailed description of the purification of the enzyme is given in Materials and Methods, and steps of the procedure are presented in Fig. 2. The starting material for the purification of the ADP-ribosyltransferase was 10 g of cells overexpressing the protein. Cells were opened by sonic oscillation and centrifuged. The resulting supernatant was first prepurified by a streptomycin sulfate treatment and then fractionated in two steps with ammonium sulfate. The Alt protein precipitates by increasing the sulfate concentration from 30% to 40%. After centrifugation, resuspension of the pellet, and dialysis, 185 mg of protein was recovered and chromatographed on Blue Sepharose. The dextran blue component of this resin is known to behave as a nucleotide analogue and, therefore, is suited to adsorb proteins with NAD binding domains (Thompson et al., 1975). The T4 gpAlt shows a high affinity to the Blue Sepharose resin. The protein eluted from the column only at 0.9 M NaCl. Electrophoresis of the pooled peak fractions showed that the enzyme preparation at this step contains only minor contaminations with other proteins (Fig. 2). Further purification of the enzyme was achieved by gel sieve chromatography on Superose 6. The final concentration of the enzyme preparation was 12 mg/ml.
NH2-Terminal Amino Acid Sequencing of gpAlt
The recombinant protein and the native, processed Alt protein, isolated from phage particles on preparative SDS-polyacrylamide gels (see Materials and Methods), were submitted to NH2-terminal sequencing. The comparison showed the identity of both amino acid sequences; however, the native Alt lacked the first six amino acids (M E L I T E). Earlier work revealed that the Alt protein together with most of the T4 prohead proteins is processed during phage head maturation. The amino acid sequence of the Alt protein around the determined cleavage site coincides with the processing site recognized by the T4 prohead proteinase gp21 (Isobe et al., 1976; Black et al., 1994)
Determination of the ADP-Ribosyltransferase Activity of the Purified Enzyme
The ADP-ribosyltransferase activity of the purified gpAlt was determined as described above with RNA polymerase holoenzyme as an acceptor protein. ADP-ribosylation reaction products were analyzed by SDS-polyacrylamide gel electrophoresis and subsequent autoradiography (Fig. 3). The results obtained demonstrate that the recombinant gpAlt, when isolated from the overexpressing cells, showed the same specificity towards E. coli RNA polymerase as reported for the “altering” ADP-ribosyltransferase purified from the disrupted phage particles (Rohrer et al., 1975). It can be seen in Fig. 3 that the recombinant ADP-ribosyltransferase labels all polymerase subunits. Furthermore, the recombinant transferase is auto-ADP-ribosylated, even in the presence of RNA polymerase. All labeled bands of the RNA polymerase, as well as the transferase band, migrate in the gel with velocities corresponding to those of the unlabeled proteins. In the absence of the recombinant ADP-ribosyltransferase no label was incorporated.
FIG. 3.
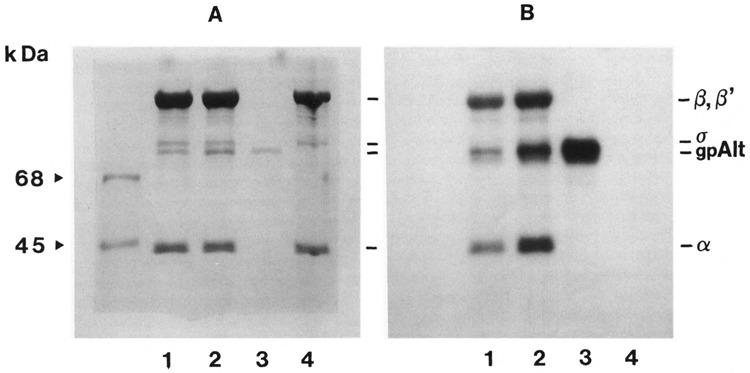
Electrophoresis and autoradiography of E. coli RNA polymerase and T4 ADP-ribosyltransferase labeled with [32P]NAD in vitro. RNA polymerase holoenzyme (40 μg of protein) was incubated with two concentrations of purified transferase at 15°C for 30 min and subsequently submitted to electrophoresis as described in Materials and Methods. (A) Coomassie-stained gel; (B) autoradio-graph. The left lane shows the marker proteins BSA (68 kDa) and ovalbumin (45 kDa). Lanes 1: RNA polymerase incubated with 7.5 μg of transferase; lanes 2: RNA polymerase incubated with 10.5 μg of transferase; lanes 3: transferase without RNA polymerase (10 μg of protein); lanes 4: RNA polymerase holoenzyme without transferase. The positions of the RNA polymerase subunits β, β′, σ, and α, as well as the position of gpAlt, are indicated.
The experiments have shown that the gene product Alt overexpressed in vivo is not completely ADP-ribosylated. It can be seen in Fig. 2 that the Alt product is considerably ADP-ribosylated in vitro, despite the presence of NAD in the overexpressing cell. This rises the question as to whether the availability of intracellular NAD or its cellular concentration play a role in the degree and the specificity of the ribosylation reaction. Because radioactive label attached to an RNA polymerase subunit cannot be chased by the addition of large amounts of cold NAD (not shown), the Alt-catalyzed ADP-ribosylation does not seem to be reversible.
Effect of ADP-Ribosylation of E. coli RNA Polymerase on T4 “Early” Promoter Strengths
To express the alt gene in cells that at the same time carry T4 promoters on a test plasmid and to study the transcription initiated at this promoter in the presence of the T4 alt gene product, we constructed vector pTKRI (see Materials and Methods and Fig. 4). Because pTKRI with its p15A origin is a low copy plasmid and the Lac promoter is relatively weak, the expression of gpAlt in pTKRI-harboring cells is moderate. Nevertheless, the synthesis of the Alt protein can be followed on polyacrylamide gels after a concentration on Blue Sepharose (not shown). On the other hand, the gpAlt expression in these cells should be sufficient to simulate the T4 infection phase in E. coli, because only 40 gpAlt molecules are injected together with the T4 DNA (Horvitz, 1974). The compatibility of plasmids pTKRI and pKWIII (Wilkens and Rüger, 1994) allows investigating the effect of ADP-ribosylation on transcription initiated at a bacterial promoter (Pbla) and at different T4 “early” promoters in the identical in vivo test system.
Plasmid pKWIII harbors two promoters: an E. coli promoter upstream from the 6-phospho-β-galactosidase (Pbla) and mutually exchangeable T4 promoters in front of the β-lactamase gene. The expression of 6-phospho-β-galactosidase under the control of a bacterial promoter remained constant, also in the presence of ADP-ribosylated RNA polymerase. However, the expression of β-lactamase, under the control of some of the T4 “early” promoters tested, increased up to 50%. This, and the observation that E. coli cells expressing gpAlt grow regularly, inferred that in our system the recombinant Alt protein did not shut down transcription initiated at bacterial promoters.
The T4 “early” promoters that were tested had originally been isolated by Liebig and Rüger (1989) using vector M13 HDL 17. We recloned 21 of these well-defined promoter sequences into pKWIII and tested the correct insertion of the cloned promoters by sequencing. The strengths of these promoters were monitored in the presence of gpAlt (Table 1) with the two-plasmid system described above. The promoters PA1 and PA2 from bacteriophage T7, and PH207 from bacteriophage T5 served as controls. As shown in Table 1, eight out of the 21 T4 “early” promoters (P3.6, P12.8, P131.7, P148.6, P8.1, P54.4, P161.1, and P128.6) showed an approximately twofold increase of their transcription activity in the presence of gpAlt. Promoter P164.5 was the only one that the strength decreased. With the other T4, and the T5/T7 promoters, no change in the promoter strengths was observed. The strengths of these control promoters, measured with the aid of pKWIII, were equal to the strengths determined by other authors in other test systems (Knaus and Bujard, 1988, 1990; Deuschle et al., 1986).
TABLE 1.
COMPARISON OF IN VIVO PROMOTER ACTIVITIES DETERMINED IN THE PRESENCE AND THE ABSENCE OF gpAlt WITH THE AID OF TWO-PLASMID VECTOR SYSTEM pKWIII AND pTKRI
| Promoter Tested | Without gpAlt | With gpAlt | ||||
|---|---|---|---|---|---|---|
| β-Lact (ΔE/min · ml) | 6-p-Gal (ΔE/min · ml) (E. coli Prom.) | Ratio | β-Lact (ΔE/min · ml) | 6-p-Gal (ΔE/min · ml) (E. coli Prom.) | Ratio | |
| Control promoters | ||||||
| PA1(T7) | 3.15 | 0.14 | 22.50 | 2.26 | 0.10 | 22.60 |
| PA2(T7) | 1.82 | 0.18 | 10.11 | 1.75 | 0.19 | 9.21 |
| P207(T5) | 3.41 | 0.14 | 24.36 | 1.90 | 0.08 | 23.75 |
| T4 “early” promoters | ||||||
| P3.6 | 1.67 | 0.16 | 10.44 | 2.43 | 0.11 | 22.09 |
| P128.2 | 0.60 | 0.07 | 8.57 | 0.55 | 0.07 | 7.86 |
| P12.8 | 1.11 | 0.10 | 11.10 | 3.33 | 0.13 | 25.62 |
| P65.6 | 0.89 | 0.15 | 5.93 | 1.34 | 0.19 | 7.05 |
| P131.7 | 1.60 | 0.15 | 10.66 | 4.30 | 0.19 | 22.63 |
| P148.6 | 2.09 | 0.23 | 9.09 | 3.91 | 0.24 | 16.29 |
| P50.0 | 2.42 | 0.11 | 22.00 | 3.64 | 0.15 | 24.27 |
| P1.4–3.9 | 0.71 | 0.15 | 4.73 | 0.59 | 0.11 | 5.36 |
| P57.9 | 2.56 | 0.14 | 18.29 | 1.86 | 0.11 | 16.91 |
| P8.1 | 2.27 | 0.11 | 20.64 | 7.08 | 0.14 | 50.57 |
| P54.4 | 2.11 | 0.22 | 9.59 | 5.13 | 0.21 | 24.43 |
| P161.1 | 0.96 | 0.06 | 16.00 | 3.42 | 0.14 | 24.43 |
| P164.5 | 1.67 | 0.06 | 27.83 | 0.70 | 0.10 | 7.00 |
| P73.0 | 1.91 | 0.14 | 13.64 | 1.95 | 0.13 | 15.00 |
| P41.0 | 4.88 | 0.23 | 21.22 | 3.69 | 0.15 | 24.60 |
| P11.5 | 1.63 | 0.16 | 10.19 | 1.93 | 0.19 | 10.15 |
| P128.6 | 1.75 | 0.15 | 11.66 | 3.67 | 0.12 | 30.58 |
| P46.7 | 0.72 | 0.12 | 6.00 | 1.12 | 0.15 | 7.46 |
| P40.4 | 0.78 | 0.13 | 6.00 | 0.97 | 0.16 | 6.06 |
| P5.9–7.3 | 0.67 | 0.14 | 4.79 | 0.95 | 0.16 | 5.93 |
| P35.3 | 0.81 | 0.19 | 4.26 | 1.26 | 0.13 | 4.39 |
The values given are averaged from three independent tests each. The ratio of β-lact to 6-p-gal reflects the strengths of promoters tested. For details see text.
DISCUSSION
The cloning of the ADP-ribosyltransferase gene and its overexpression in E. coli is a first step for a fast and efficient purification of the enzyme that allows the consequences of the ADP-ribosylation in vivo and in vitro to be investigated. As had been reported recently (Koch and Rüger, 1994), the introduction of two stop codons into the reading frame of the alt gene led to the overexpression of a truncated 30 kDa NH2-terminus of its product. The truncated polypeptide still binds to Blue Sepharose and can be competed off by NAD, suggesting that the NAD binding domain of the enzyme lies in the NH2-terminus. Moreover, secondary structure predictions performed according to Chou and Fasman (1978) and Gamier et al. (1978) indicated a 150 amino acid NH2-terminal portion of the polypeptide, which could represent a nucleotide binding cleft (Rossman et al., 1974). The ability of gpAlt to bind to this resin allows purification and concentration of the enzyme by affinity chromatography, essentially in one step. Minor contaminants can easily be removed on a gel sieve.
The purified, recombinant enzyme shows transferase activity with radioactively labeled NAD as a substrate. The data presented above clearly indicate that this protein ADP-ribosylates not only the α-subunits of E. coli RNA polymerase but also subunits β, β′, and σ 70. In addition, it performs an autoribosylation reaction. By these criteria the recombinant Alt protein is a multitarget enzyme and behaves like the native gpAlt, previously described by Rohrer et al. (1975).
The comparison of the NH2-termini of the purified recombinant alt gene product on the one hand, and of the SDS gel-purified protein from mature phage particles on the other, revealed that the latter protein lacks the first six amino acids. It is highly probable that this difference is due to protein processing, known to play an important role in the morphogenesis of virus particles (Hellen and Wimmer, 1992). The morphogenesis of the T4 phage head is initiated by the assembly of a prohead structure consisting of unprocessed proteins. This structure then is converted by the T4 prohead proteinase gp21 to the mature capsid, which also contains 40 copies of the processed Alt protein. A putative processing of gpAlt has been described by Horvitz (1974). He reported that a 79-kDa precursor peptide of gpAlt was processed to a protein of about 69 kDa comigrating with the BSA marker protein. The difference in the molecular masses of the native and the recombinant proteins on SDS-polyacrylamide gels had also been observed in our experiments. The loss of the first six amino acids reduces the molecular mass of gpAlt by only 1 kDa. Digesting the native and the recombinant proteins with hydroxylamine (Bornstein and Balian, 1977) results in two protein fragments each, 53 and 24 kDa for the native, and 52 and 24 kDa for the recombinant enzyme (Raudonikiene and Rüger, unpublished results). This suggested that the divergent velocities of migration, observed with the two forms of the Alt protein, must be due to conformational differences rather than to a more extended loss of molecular mass. It should be noted that hydroxylamine treatment removes ADP-ribosyl residues and that the electrical charge added to the RNA polymerase subunits by the transfer of these residues does not result in a noticeable band shift (compare A and B in Fig. 3). The nature of this structural difference remains to be elucidated.
It had not been known whether the processing step would be required to activate the Alt protein or if it is incidental to it. Horvitz suggested (1974) that the processing of gpAlt was necessary to activate the ADP-ribosyltransferase. Our experiments prove that processing is not linked with the activity of the Alt protein and, therefore, this step seems to be more important for correct assembly of the T4 phage head. This conclusion is supported by the observation that only processed gpAlt is incorporated into phage heads. T4 mutants failing to process gpAlt result in petit phage, and phage with overlength DNA (Wu et al., 1991; Black et al., 1994).
Because T4 gpAlt is a component of the phage head and is injected together with the viral DNA, transcription initiated at T4 “early” promoters is probably mediated by the altered host RNA polymerase. To investigate the in vivo function of ADP-ribosylation, we compared T4 “early” promoter strengths in E. coli cells in the presence and the absence of gpAlt. In contrast to the earlier assumption that expression of gpAlt stops E. coli transcription (Seifert et al., 1969; Geiduschek and Kassavetis, 1988; Drivdahl and Kutter, 1990), we did not observe an influence of the gpAlt overexpression on the viability of the cells and the expression of gpAlt was not markedly reduced. On the other hand, experiments performed with a two-plasmid system, where one of the plasmids expresses the Alt gene product constitutively and the other carries different T4 “early” promoters, indicate that ADP-ribosylation modulates T4 promoter strengths. The ADP-ribosylated enzyme doubles its transcriptional activity on a sizeable number of T4 “early” promoters. Promoter P164.5 was the only one among candidates tested that responded negatively to the ADP-ribosylation. This promoter is one of the two transcription initiation sites located in the T4 D region, which was reported to show low transcriptional activity under the influence of a RNA polymerase with mod-modified α-subunits in vitro (Goldfarb and Palm, 1981).
Severinov et al. (1994) recently showed that transcription by RNA polymerase assembled with a mod-ribosylated α-subunit and unribosylated β, β′, and σ70 subunits, purified from overexpressing cells did not stimulate transcription from the rrnB P1 promoter with the UP element, and these authors did not observe a stimulation of transcription from CAP-regulated promoters. These results suggest that transcription initiated at some E. coli promoters may be downmodulated, but not entirely stopped. Hence, ADP-ribosylation of RNA polymerase cannot be the only event responsible for the shutdown of host cell transcription observed 1 min after infection (Drivdahl and Kutter, 1983; Drivdahl and Kutter, 1990). Own experiments performed in vitro with ADP-ribosylated RNA polymerase and with the unribosylated enzyme did not reveal any detectable differences in the overall transcriptional activity on E. coli chromosomal DNA (not shown). This also agrees with the observation, that T4 alt and mod mutants in vivo shut off host transcription normally (Goff and Setzer, 1980).
A factor of about 2, by which ADP-ribosylation of RNA polymerase stimulates transcription from T4 “early” promoters, at first sight does not seem to be impressive. Nevertheless, it means that at least twice as many transcripts are initiated within a given time interval at these phage promoters than at E. coli promoters (only few bacterial promoters reach the strengths of phage promoters). Consequently, T4-encoded proteins translated from these messengers should come up about twice as fast as bacterial proteins, possibly designed to interfere with the infecting DNA. In accelerating transcription from phage promoters and possibly decelerating transcription from bacterial promoters, ADP-ribosylation of RNA polymerase would help to moderately overexpress T4 “early” gene products. This strategy could be useful in securing the supremacy of the T4 genetical program, and in economizing the limited metabolic resources of an infected host cell through to viral DNA replication and coat protein synthesis. We would not be surprised if ADP-ribosylation catalyzed by the Mod protein turned out to serve a similar strategy at the onset of middle mode transcription with its different factor requirements. These functions would be a rationale for the existence of two different genes coding for two different ADP-ribosylating activities acting at different times in the infection cycle. The recombinant Alt enzyme will be a valuable tool to further investigate the effects of ADP-ribosylation on RNA polymerase and on several other E. coli proteins.
ACKNOWLEDGEMENTS
The authors thank J. Hageman (University of Colorado at Boulder) for sending the T4 alt− mutant strain, and S. Tabor (Harvard University, Cambridge) for the gift of the T7 plasmids. Our thanks are also due to H. Meyer (Institut für Physiologische Chemie, Ruhr-University, Bochum sequencing the NH2-terminus of the T4-encoded ADP-ribosyltransferases and to R. Allard for reading the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft, and by stipends of the Wilhelm and Günter Esser Found (T.K.) and the Alexander von Humboldt Foundation (A.R.). Some of the results are part of the thesis of T. Koch (1993), Faculty for Biology, I Universität Bochum, Germany.
REFERENCES
- Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., and Struhl K. (1993), in Current Protocols in Molecular Biology, Greene Publishing and Wiley-Interscience, New York. [Google Scholar]
- Black L. W., Showe M. K., and Steven A. C. (1994), in Molecular Biology of Bacteriophage T4 (Karam J., Black L., Carlson K., Eiserling F., Hall D., Kreuzer K., Kutter E., and Mosig G., eds.), American Society for Microbiology, Washington, DC, pp. 218–258. [Google Scholar]
- Bornstein P. and Balian G. (1977), Methods Enzymol 47, 132–145. [DOI] [PubMed] [Google Scholar]
- Chou P. Y. and Fasman G. D. (1978), Adv Enzymol 47, 45–148. [DOI] [PubMed] [Google Scholar]
- Deuschle U., Kammerer W., Gentz R., and Bujard H. (1986), EMBO J 5, 2987–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro E., Snyder L., Marino D., Lamberti A., Coppo A., and Tochini-Valentini G. P. (1969), Nature 222, 533–537. [DOI] [PubMed] [Google Scholar]
- Drivdahl R. M. and Kutter E. (1983), Fed Proc 42, 1881. [Google Scholar]
- Drivdahl R. M. and Kutter E. (1990), J Bacteriol 172, 2716–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamier J., Osguthorpe D. J., and Robson B. (1978), J Mol Biol 120, 97–120. [DOI] [PubMed] [Google Scholar]
- Geiduschek E. P. and Kassavetis G. A. (1988), in The Bacteriophages (Calendar R., ed.), Plenum Press, New York, pp. 93–116. [Google Scholar]
- Goff C. G. (1974), J Biol Chem 249, 6181–6190. [PubMed] [Google Scholar]
- Goff C. G. and Setzer J. (1980), J Virol 33, 547–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb A. and Malik S. (1984), J Mol Biol 177, 87–105. [DOI] [PubMed] [Google Scholar]
- Goldfarb A. and Palm P. (1981), Nucleic Acids Res 9, 4863–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haselkorn R., Vogel M., and Brown R. D. (1969), Nature 285, 634–641. [DOI] [PubMed] [Google Scholar]
- Hellen C. U. T. and Wimmer E. (1992), Experientia 48, 201–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengstenberg W. and Morse M. L. (1968), Carbohydr Res 7, 180–183. [Google Scholar]
- Horvitz H. R. (1974), J Mol Biol 90, 739–750. [DOI] [PubMed] [Google Scholar]
- Isobe T., Black L. W., and Tsugita A. (1976), Proc Natl Acad Sci USA 73, 4205–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus R. and Bujard H. (1988), EMBO J 7, 2919–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus R. and Bujard H. (1990), in Nucleic Acids and Molecular Biology, Vol. 4 (Eckstein F. and Lilley D. M. J., eds.), Springer Verlag, Berlin-Heidelberg, pp. 110–122. [Google Scholar]
- Koch T. (1993), Die ADP–ribosyltransferase (gpAlt) des Bakteriophagen T4: Sequenzierung, Reinigung und biochemische Charakterisierung. Thesis, Fakultät für Biologie, Ruhr-Universität Bochum, Bochum, Germany. [Google Scholar]
- Koch T. and Rüger W. (1994), Virology 203, 294–298. [DOI] [PubMed] [Google Scholar]
- Kutter E., Guttmann B., Mosig G., and Rüger W. (1992), in Genetic Maps, Sixth Edition (O’Brian S. J., ed.), Laboratory of Viral Carcinogenesis, Natl. Cancer Institute, Cold Spring Harbour Laboratory Press, pp. 1.1–1.26. [Google Scholar]
- Laemmli U. K. (1970), Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Liebig H. D. and Rüger W. (1989), J Mol Biol 208, 517–536. [DOI] [PubMed] [Google Scholar]
- Mailhammer R., Yang H. L., Reiness G., and Zubay G. (1975), Proc Natl Acad Sci USA 72, 4928–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosig G. and Hall D. H. (1994) in Molecular Biology of Bacteriophage T4 (Karam J., Black L., Carlson K., Eiserling F., Hall D., Kreuzer K., Kutter E., and Mosig G., eds.), American Society for Microbiology, Washington, DC, pp. 127–208. [Google Scholar]
- Ovchinnikov Y. A., Lipkin V. M., Modyanov N. N., Chertow O. Y., and Smirnow Y. V. (1977), FEBS Lett 76, 108–111. [DOI] [PubMed] [Google Scholar]
- Paulson J. R., Lazaroff S., and Laemmli U. K. (1976), J Mol Biol 103, 99–109. [DOI] [PubMed] [Google Scholar]
- Rohrer H., Zillig W., and Mailhammer R. (1975), Eur J Biochem 60, 227–238. [DOI] [PubMed] [Google Scholar]
- Rossman M. G., Moras D., and Olson K. W. (1974), Nature 250, 194–199. [DOI] [PubMed] [Google Scholar]
- Seifert W., Quasba P., Walter G., Palm P., Schachner M., and Zillig W. (1969), Eur J Biochem 9, 319–324. [DOI] [PubMed] [Google Scholar]
- Severinov K., Ross W., Tang H., Goldfarb A., Ebright R. H., and Gourse R. L. (1994), Abstract of the 1994 Gargnano Meeting, Italy. [Google Scholar]
- Skorko R., Zillig W., Rohrer H., Fujiki H., and Mailhammer R. (1977), Eur J Biochem 79, 55–66. [DOI] [PubMed] [Google Scholar]
- Steven A. C., Couture E., Aebi U., and Showe M. K. (1976), J Mol Biol 106, 187–221. [DOI] [PubMed] [Google Scholar]
- Stitt B. and Hinton D. (1994), in Molecular Biology of Bacteriophage T4 (Karam J., Black L., Carlson K., Eiserling F., Hall D., Kreuzer K., Kutter E., and Mosig G., eds.), American Society for Microbiology, Washington, DC, pp. 142–160. [Google Scholar]
- Tabor S. and Richardson C. C. (1985), Proc Natl Acad Sci USA 82, 1074–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S. T., Cass K. H., and Stellwagen E. (1975), Proc Natl Acad Sci USA 72, 669–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkens K. and Rüger W. (1994), in Molecular Biology of Bacteriophage T4 (Karam J., Black L., Carlson K., Eiserling F., Hall D., Kreuzer K., Kutter E., and Mosig G., eds.), American Society for Microbiology, Washington, DC, pp. 132–141. [Google Scholar]
- Williams K. P., Kassavetis G. A., Herendeen D. R., and Geiduschek E. P. (1994), in Molecular Biology of Bacteriophage T4 (Karam J., Black L., Carlson K., Eiserling F., Hall D., Kreuzer K., Kutter E., and Mosig G., eds.), American Society for Microbiology, Washington, DC, pp. 161–175. [Google Scholar]
- Wu D. G., Wu C. H., and Black L. W. (1991), J Mol Biol 218, 705–721. [DOI] [PubMed] [Google Scholar]
- Zillig W., Fuchs E., Palm P., Rabussay D., and Zechel K. (1970), in RNA Polymerase and Transcription (Silvestri L., ed.), North Holland Publishing Co., London, Amsterdam, pp. 167–189. [Google Scholar]