

Abstract
The human complement system is represents the main effector arm of innate immunity and its ambivalent function in cancer has been subject of ongoing dispute. Glioma stem-like cells (GSC) residing in specific niches within glioblastomas (GBM) are capable of self-renewal and tumor proliferation. Recent data are indicative of the influence of the complement system on the maintenance of these cells. It appears that the role of the complement system in glial tumorigenesis, particularly its influence on GSC niches and GSC maintenance, is significant and warrants further exploration for therapeutic interventions.
Introduction
Traditionally, the human complement system is regarded to be a main effector arm of the innate immunity and comprises around thirty soluble and membrane-associated proteins. Innate immunity forms the first line of defense against invading micro-organisms, and is ancient compared to the adaptive immune system. Analogues of components of the mammalian alternative complement pathway have been identified in deuterostomes/protostomes over 1000 million years ago, whereas the first molecules from the jawed-invertebrate specific adaptive immune system emerged at least 400 million years later [63]. After breaching the host’s environmental barriers, invading microbes are detected by the pattern recognition molecules (PRM) of the classical (C1q) and lectin (MBL, ficolins) complement pathways (CP, LP) [75, 96]. (Fig. 1). Further, activation of the complement cascade through the AP is achieved through insufficient inhibition of spontaneous hydrolysis of C3 (C3-H20) by the microbe (AP). All three activation pathways converge at the level of C3 which, after formation of the C3 and C5 convertase complexes, continues with the formation of the terminal complement complex (TCC) either as the pore-like membrane attack complex (MAC) or as cell-activating sC5b-9 [51]. MAC assembly in the cell membrane causes prompt colloid osmotic lysis [56, 59].
Fig. 1.

Schematic and simplified representation of the complement system. Complement regulatory proteins, both fluid-phase and membrane- bound are coloured light grey. C1INH: C1-inhibitor; CD46: Membrane Cofactor Protein; CD55: Complement decay-accelerating factor; FI: Complement factor I; CR1: Complement receptor type 1; FH: Complement factor H; C4BP: C4bbinding protein
Over a century after the initial discovery of the complement system by Buchner et al. it was realized that the actions of complement are not constrained to an effector mechanism of the innate immunity, but are also involved in directing the adaptive immune response, angiogenesis, tissue regeneration, fat metabolism and development of the central nervous system (CNS) [75]. The complement system acts as intricate immune surveillance system that is able to discriminate between healthy host tissue, cellular debris, apoptotic cells and foreign intruders (Fig. 1) and contributes to a large variety of inflammatory-, immune-, ischemic-, age-related pathologic processes of the CNS [75]. A wide repertoire of specific complement inhibitors has been developed against a variety of diseases of which eculizumab, an antibody against C5 and C1 (C1-INH) became FDA approved for the treatment of paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome and hereditary angiogoedema [1]. In recent years it became clear that many factors of the complement system are expressed in the brain [60, 93]. The production of the complement proteins in CNS is constrained to microglia, oligodendrocytes, astrocytes and, to a lesser extent, ependymal cells [93]. The complement system appears to be a key player in CNS homeostasis as complement effector mechanisms have been identified in neurogenesis and regulating synaptic pruning [54]. Various pathological conditions cause an imbalance between complement activation and inhibition.
Activation of the complement system contributes to a wide variety of CNS diseases including Alzheimer disease, CNS inflammation, traumatic brain injury and tumors [6, 55]. The cross-link between inflammation and cancer is generally accepted as chronic and insidious inflammation is recognized to play decisive roles at different stages of tumor development, including initiation, promotion, malignant conversion, invasion, and metastasis [28, 52]. The actions of the immune system against tumors in progress can be referred to as the cancer immunoediting process, which is composed of three distinct phases: elimination, equilibrium and escape. During the elimination phase and the equilibrium phase the immunological response is able to prevent tumor progression. In contrast, during the escape phase acquired adaptations of malignant cells and the host immune system response allow for expansion of the tumor cell population [94]. By acting as an intrinsic effector mechanism and by forming a functional bridge between the innate and the adaptive immune system, the complement system is an integral component of the antitumor immune response [94]. Complement activation following recognition of damage-associated molecular patterns (DAMPs) expressed by tumor cells, or improper regulation, allow for a potent anti-tumor response [37]. The potent antitumor response by complement has been utilized for antibody-based cancer immunotherapies by eliciting complement-dependent cytotoxicity, exemplified by the use of rituximab and ofatumumab in the treatment of B cell lymphomas and chronic lympocytic leukaemia, respectively [89]. However, the complement system also shows another face. Recent pre-clinical cancer models showed that the activated complement system contributes to a tumor facilitating micro-environment [1]. This adverse capacity seems to be a consequence of imbalanced, rather than physiological, complement activation [74]. Various studies have reported significant reductions of orthotopic tumor growth following complement system inhibition within the cascade [74]. Further, diverse complement effectors are implicated in other cancer-related phenomena as sustained proliferative signaling, angiogenesis and invasion and metastasis [33, 74].
Contributory to treatment resistance of glial neoplasms is the presence of glioma stem-like cells (GSCs) [85]. GSCs reside in specific anatomical niches within the tumor and propagate glioma repopulation by converting into either a differentiated tumor cell, or a new cancer stem cell [46]. The maintenance of GSCs requires specific intrinsic factors within the cells and various paracrine cues from adjacent cells [46]. The complement system represents an as yet unidentified effector in GSC maintenance, and unraveling its interplay will reveal new targets for therapeutic intervention.
Complement and GSC maintenance: Intrinsic regulation
Factors that are involved in GSC maintenance comprise of metabolic, genetic and epigenetic regulatory mechanisms [90]. Although the mechanisms underlying GSC plasticity are largely unknown, several intrinsic regulatory mechanisms are known to be involved in reprogramming differentiated GBM cells into stem-like cells. Among these are Sex Determining Region Y -Box 2 (SOX-2) [88], signal transducer and activator of transcription 3 (STAT-3), octamer-binding transcription factor 4 (OCT-4) and mammalian target of rapamycin (mTOR) signaling [23, 82]. The GSCs maintain their multipotent state through autocrine stimulation of the C3a- and C5a-receptors on the plasma membrane by secretion of alternative pathway C3-convertase components (C3, factor D and factor B) and subsequent extracellular cleavage of C3, as observed in resting T-cells (Fig. 2) [87]. The C3 and C5 convertases (Fig. 1) are responsible for the release of their respective bioactive fragments C3a and C3b, and C5a and C5b. The anaphylotoxins C3a and C5a signal through the G protein coupled receptors C3aR and C5aR (CD88) respectively. Interaction of several downstream signal transduction pathways followed by C3aR and C5aR activation with recognized GSC regulatory mechanisms effectors may therefore aid to GSC maintenance. Figure 2 presents a schematic overview of the interaction of autocrine derived complement with GSC regulatory mechanisms. C3a-C3aR interaction activates STAT-3 and causes an increase of Wnt2b and SOX-2 expression in a serine protease MAPK dependent fashion, as was shown in a model of retinal regeneration [36]. C5aR1 activation contributes to the maintenance of pluripotency of OCT-4 positive human induced pluripotent stem cells (hPSC) through extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) activation [35]. In vitro administration of recombinant C5a to C5aR expressing gastric cancer cells promotes the activation of PI3K/Akt and downregulates p21 activation [11]. Inhibition of p21 is a key mechanism of GSC self-renewal and prevention of differentiation [99].
Fig. 2.
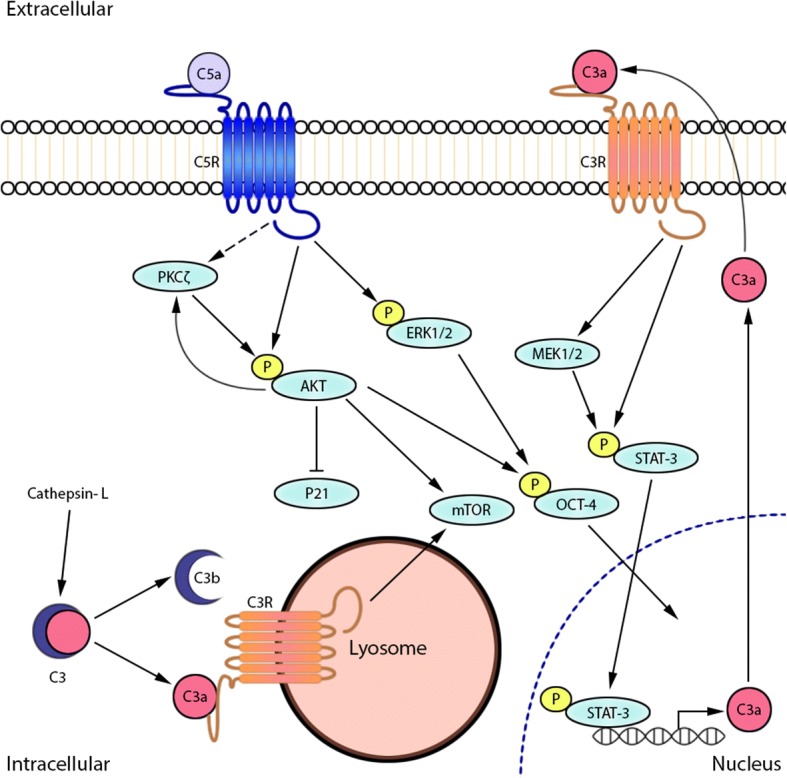
Proposed interaction of complement C3a and C5a with GSC regulatory mechanisms. C5a-C5aR interaction activates PI3K/Akt/mTOR signaling and PKCζ but suppresses p21 with subsequent OCT-4 activation. Intracellular activation of C3a by cathepsin-L may occur, thereby sustaining basal mTOR activation. Either intracellular or extracellular derived C3a phosphorylates STAT-3 and causes an increase of SOX-2 expression
Further, complement C3 activation is not limited to the extracellular space because intracellular C3 activation is an ubiquitous event within human cells [50]. Resting T-lymphocytes contain intracellular pools of C3 that are activated by cysteine protease cathepsin-L. This ‘tonic’ intracellular C3a generation engages C3aR present on lysosomes and sustains the basal mammalian target of rapamycin (mTOR) activation required for T-cell survival [50]. Activation of the PI3K/Akt/mTOR signaling cascade is involved in GSC phenotype maintenance [18]. In addition, GBM tissue shows the profound presence of cathepsin L, which has been shown to negatively affect apoptosis and promotes invasion [104]. Intracellular C3a activation by cathepsin-L may provide for an as yet unidentified additional intrinsic GSC effector mechanism. Noteworthy is the critical role of autocrine derived C5a-C5aR1 signalling in affecting the fate of Neural Progenitor Cells (NPCs). C5a was found actively secreted within neural rosettes [16]. Atypical protein kinase C zeta (PKCζ) dependent C5a-C5aR1 signaling regulates the apicobasal polarity of the NPC, thereby maintaining symmetrical self-renewing cell division [16]. PKCζ inhibition reverses C5a-C5aR induced p42/44 phosphorylation (ERK) and attenuates the mitotic activity of NPCs [16]. PKCζ is overexpressed in GBM cells and its downregulation inhibits in vitro migration and invasion of GBM cells. Another mechanism through which the GSC could generate C5 in order to self-activate C5aR is by expressing cell membrane-bound serine protease, as was found in different cancer cell types [62].
Complement and GSC maintenance
The neuropil constitutes the extracellular matrix of the brain in which endothelial and perivascular cells, microglia, tumor-associated macrophages and non-neoplastic astrocytes are present and networks of cytokines and growth factors are active [32]. Together with neoplastic cells the microenvironment coalesces into the tumor mass. GSC are enriched in areas of hypoxia, in perivascular niches and at the invasive edge of the tumor [46]. A graphical representation of the various cells involved is provided in Fig. 2. The actions of the complement factors within the various glioma niches are summarized in Table 1.
Table 1.
Complement protein in the GBM tumor
| Complement proteins | Niche | Mechanism | Proposed effect |
|---|---|---|---|
| C1q | GSC | Wnt activation | Stimulate GSC differentiation |
| Perivascular | Priming SDF-1 gradient | GSC migration | |
| Perivascular | gC1qR interaction | Potentiate tumor cell invasiveness, chemoattractant | |
| Perivascular | cC1qR interaction (IL-8, MCP-1 and IL-6 secretion) | GSC migration | |
| Microenvironment | GAM-M2 induction | Immunosuppression | |
| Invasive | gC1qR (Bradykinin induction) | Tumor cell invasiveness | |
| MBL | Microenvironment | GAM-M2 induction | Immunosuppression |
| C3 | Microenvironment | MDSC recruitment | Immunosuppression |
| C3b | Microenvironment | Treg induction (ligand for CD46) | Immunosuppression |
| Microenvironment | GAM-M2 induction | Immunosuppression | |
| C3a/C3aR | Hypoxic | STAT-3 activation | GSC maintenance |
| GSC | mTOR activation | GSC maintenance | |
| Hypoxic | NOX-4 activation, (STAT-3, HIF-α) | GSC maintenance | |
| GSC | SOX-2 activation | GSC maintenance | |
| Microenvironment | Chemotaxis immune cells | ||
| CD46 | Hypoxic | Jagged-1-Notch disruption | GSC maintenance |
| Perivascular | |||
| C5a/C5aR | Invasive nice | PKCζ activation | Potentiate tumor cell invasiveness |
| GSC | PI3K/Akt/mTOR | GSC maintenance | |
| GSC | P21 inhibition | GSC maintenance | |
| GSC | OCT-4 expression | GSC maintenance | |
| Perivascular | MMP-9, MT1-MMP activation | Increase tumor cell invasiveness | |
| Perivascular | MCP-1 secretion | Increase tumor cell invasiveness | |
| Perivascular | TGF-β | GSC differentation into vascular pericytes | |
| Perivascular | NO secretion (iNOS/eNOS induction) | GSC maintenance | |
| Perivascular | VEGF expression | Vascular tube formation | |
| Microenvironment | Chemotaxis immune cells | ||
| Microenvironment | Treg induction (High concentration) | Immunosuppression | |
| Microenvironment | GAM-M1 activation (balanced C activation) | Anti-tumor response | |
| Microenvironment | MDSC recruitment (ROS) | Immunosuppression | |
| C5b-C9 | Perivascular | bFGF release | GSC dedifferentiation |
Hypoxic niche
The inadequate neo-angiogenesis in GBM results in hypoxic areas within the tumor. The neoplastic cells respond to low oxygen levels by the expression of members of the hypoxia inducible factor (HIF) family of transcriptional factors [49]. HIFs are upregulated in GSC and its forced expression induces a stem-cell like phenotype in glioma cells [49]. Transcriptional targets of HIFs include angiogenic genes like Vascular Endothelial Growth Factor (VEGF) as well as stem cell markers [49]. Areas of hypoxia optimally accommodate complement activation as provide for damage-associated molecular patterns (DAMPs) that are recognized by C1q. Hypoxic conditions induce (HIF-dependent) down-regulation of complement regulatory genes CD55, CD46 and factor H and upregulate C3, C3a and C3aR and enhance C3a-C3aR engagement [27, 66]. The constituents of the complement system have been identified to interact with HIF associated signaling pathways and may therefore act as an additional effector mechanism in HIF dependent GSC survival, self-renewal and tumor growth. Firstly, the complement system contributes to facilitate HIF transcription through STAT-3 activation that is critical for the transcription of HIF-1α in GSCs and tumor-associated myeloid cells [69]. The production of reactive oxygen species, as a result of overexpression of nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX-4), was identified as the molecular mechanism underlying hypoxia-induced STAT-3 activation in GBM cells [103]. In a model of renal ischemia/reperfusion injury, oxidative stress induces an increased expression of NOX-4 in tubular cells and NOX-2 infiltrating monocytes and myeloid dendritic cells [84]. This effect is dramatically reduced after the administration of the complement 1-inhibitor (C1-INH). In vitro administration of C3a to cultured proximal tubular cells induces NOX-4 expression regardless of hypoxic conditions [84].
Secondly, through C3aR and C5aR interaction on the GSC, complement may provide for additional signal transduction pathways for PI3K- or mitogen-activated protein kinase (MAPK)/ERK1/2-dependent HIF-1α protein translation [68, 69]. HIF-1α and the components of the complement cascade converge at the level of the Notch signaling pathway. Notch activation restricts glioma cell differentiation and stimulates astrocytes into a neural stem-like cell state [69]. HIF-1/2α driven GSC maintenance requires Notch signaling [69]. In resting T-cells, CD46 sequesters the Notch ligand Jagged-1, thereby preventing the interaction between Jagged-1 and Notch that activates T-cells [48]. Hypoxia-mediated downregulation of the expression of CD46 or CD46-C3b interaction following complement activation may allow for Notch-Jagged-1 interaction. A direct contribution of CD46 downregulation in maintaining the undifferentiated state of the GSC remains to be elucidated. C3a inhibits SDF-1α induced neuronal differentiation of NPCs through ERK1/2 phosphorylation regulation [83]. SDF-1α is a HIF-1α target gene in GBM cells [22]. Importantly, SDF-1α induces recruitment of bone-marrow derived CD45+ myeloid cells, endothelial and pericyte progenitor cells to GBM [22].
Lastly, HIF-1α modulated, Wnt/ β-catenin activation has been identified to stimulate GSC differentiation and therefore promotes a less-aggressive, neuronal tumor phenotype. Subsequent β-catenin mediated Notch inhibition further allows for GSC differentiation [71]. The role of Wnt activation in regulating the GSC state remains controversial as many reports claim that Wnt activation promotes GSC maintenance and expansion [42]. C1q is an activator of canonical Wnt signalling through binding with the Fz-receptor and subsequent induction of C1s dependent cleavage of low-density lipoprotein receptor-related protein 6 (LRP6) [61]. Intriguingly, C1q-induced activation of Wnt signalling attenuates the proliferation of muscle stem cells [61].
Perivascular niche
As a result of vigorous and abnormal angiogenesis, local regions of hypoxia develop with subsequent complement activation, facilitating complement mediated GSC regulation. The actions of the complement system extend into the perivascular niche, where GSC survival and angiogenesis are mediated. In the adult mammalian brain, neuronal stem cells reside in two germinal niches of which the subventricular zone is highly vascularized. Adult stem cells lie in close proximity to the vasculature, where communication occurs through direct cell-cell interactions and soluble secreted cues [81]. In contrast to germinal niches, where the rate of stem cell proliferation is low, the perivascular niche within GBM contributes to the generation of the GSCs and tumor growth [9]. Besides endothelial cells (EC) and GSCs, major cell types that have been recognized to reside in the perivascular niche include pericytes, immune cells, fibroblasts and astrocytes, all of which provide an additional biological source of complement proteins [9, 93]. The complement system establishes an active interplay with the perivascular niche constituents and GSCs and fulfills an active role in the migration of GSC and promotes angiogenesis.
Several components of the (activated) complement system are powerful chemoattractants attracking blood borne cells and GSCs to the perivascular niche [75]. The complement system potentiates the migration of GSC towards the perivascular niche just like it does to mesenchymal stem cells (MSCs), NPCs and hematopoietic stem/progenitor cells (hPSC). In GBM, GSCs move towards the tumor vasculature and engage in cell-cell contact [9]. The interaction of Stromal Cell-Derived Factor 1 (SDF1) with its receptor C-X-C Motif Chemokine Receptor 4 (CXCR4) is operative to guide the tumor cells to the peri-endothelial space [72]. C1q primes chemotactic SDF-1-dependent migration of human umbilical cord blood derived- Mesenchymal Stem Cells (MSC), in part by upregulating the expression of CXCR4 and through interaction with its globular heads binding receptor (gC1qR), which is ubiquitously expressed [70]. The activated complement system potentiates the SDF1-CXCR4 chemotaxis, independent of C3aR, as has been observed in hematopoietic stem/progenitor cells (HSPCs) [38]. C3a modulates concentration-dependent SDF1-CXCR4 induced migration of NPCs [83]. C5a attracts MSCs in a C5aR dependent fashion and its degradation product C5adesArg causes an increased secretion of MMP-9 and MT1-MMP, creating a highly proteolytic microenvironment in favor of cell-migration [79]. In addition, IL-8 secreted by ECs stimulates GSC migration and maintains its stemness properties, in part by upregulating the expression of its cognate receptors CXCR1 and CXCR2 [39]. The interaction of C1q with its receptor present on the EC (cC1qR) initiates the release of IL-8, macrophage chemoattractant protein-1 (MCP-1) and IL-6, that contribute to the homing of GSCs and MSCs [92, 97]. IL-8 mediated GSC homing and maintenance is amplified by C5a as observed in whole blood cells and in human dermal microvascular endothelial cells (HVEC-d) [97]. Lastly, C5a contributes to GSC migration by increasing the expression of MCP-1/CCL2, as observed in HVEC-d in a dose dependent matter [102].
Complement modulated Perivascular niche-GSC interaction
GSCs form intimate contacts along the length of the endothelial tube, essential for their survival and inducing secretion of soluble factors by ECs that keep the GSCs in an undifferentiated state [80]. Several pathways constitute EC orchestrated GSC regulation, including the transforming growth factor-β (TGF-β) pathway [80]. EC-derived TGF-β induces the differentiation of GSC into pericytes that contribute to vascular formation in GBM [12]. Crosstalk of TGF-β signaling and complement activation is observed in various cells. C5a has been shown to upregulate TGF-β transcript expression, and vice-versa TGF-β upregulates the expression of C5aR [29]. Further, TGF-β and C5a signaling converge downstream at the level of SMAD independent pathways including, PI3K/AKT/mTOR and MAPK/ERK1/2 signaling pathways [77, 105]. A second pathway through which the GSC phenotype is maintained is the nitric oxide (NO) signaling [10]. The biological source of NO is EC-derived by the expression of endothelial NO synthase (eNOS) or alternatively, through the expression of inducible NOS (iNOS) by the GSC [10]. Activation of eNOS requires phosphorylation of AKT, suggestive of a contributory role for the activated complement system [21]. The complement system mediates the expression of iNOS and NO levels as shown in models of gastrointestinal ischemia by inhibition of C3 and C5 [57]. Lastly, EC derived basic Fibroblast Growth Factor (bFGF) induces the reversion of differentiated GBM cells [25]. However, the underlying mechanism resulting in functional expression of bFGF remains poorly defined, given that hypoxia does not induce bFGF expression in human vascular smooth muscles cells [7]. Interestingly, small amounts of C5b-9 (membrane attack complex) releases bFGF from human umbilical vein endothelial cells (HVEC) [4]. Active and inactive forms of C1s are found to form aggregates with bFGF, thereby reducing its activity [76].
The complement system influences GSC mediated angiogenesis
Indications for the close interaction of GSC and endothelial cells emerged from the finding that xenotransplanted tumors derived from GSCs were characterized by widespread angiogenesis that was not encountered in their non-GSC counterparts [3]. GSCs secrete VEGF and treatment with bevacizumab blocks the pro-angiogenic effects of VEGF by hampering microvascular endothelial cell migration and vascular tube formation and inhibiting the growth and vascularity of GSC derived xenotransplants [3]. Bioactive complement products have been identified as important effectors in pathological neovascularization in age-related macular degeneration (ARMD), diabetic retinopathy, and retinopathy of prematurity [100]. Interaction of C5a with its receptor C5aR1 induces VEGF expression in a dose-dependent matter in retinal pigmented epithelium (RPE) in-vivo and in-vitro [15]. The induction of oxidative stress in RPE cells reduces the surface expression of DAF, CD55 and CD59 and impairs complement regulation at the level of factor H, resulting in complement activation and complement-dependent VEGF expression [91]. Consequently, inhibiting the AP using a recombinant factor H reduces the expression of VEGF and subsequent angiogenesis in a mouse model of choroidal neovascularization [91]. Conversely, the inhibition of VEGF causes a decrease of the complement inhibitory proteins (CIPs) factor H, CD46 and CD59 in human RPE-cells and glomerular endothelial cells through VEGFR2/PKC-α/CREB signaling [44]. These observations imply that VEGF protects neo-angiogenesis by local inhibition of the complement system. It remains undetermined whether complement activation directly contributes to VEGF expression or VEGF suppresses complement activation through CIP induction. In a mouse model of ovarian cancer, C3 and C5aR were shown to be closely involved in neo-angiogenesis [63]. Tumors derived from partial C3, C5aR and complete C5aR knock out mice displayed decreased microvascular density compared to their WT-littermates [63]. Additional in vivo assays showed significant impairment of angiogenesis for complete C3 and C5aR knock-out mice. Interestingly, direct functional effect of C5a comparable to VEGF-A on tube formation of endothelial cells was also observed. This effect was found to be reversible using the C5aR inhibitor PMX-53. PMX-53 also significantly impaired VEGF165 mediated HMEC tube formation [63]. In addition to C3 and C5aR, microvascular density was significant decreased in tumors in C1q deficient mice bearing a syngeneic B16 melanoma compared to their WT-littermates [8].
Complement and immune cell crosstalk in the perivascular niche
Activation of the complement system by means of C3a and C5a plays an important role in the inflammatory process by recruiting immune cells such as mast cells, monocytes, macrophages, neutrophils, MDSCs and adaptive immune cells including T cells [75]. The BBB consists of highly specialized endothelial cells that communicate with pericytes and astrocytes to protect the CNS from the chemical variations in the bloodstream, and establishes a strictly controlled interface for immune cell trafficking. In GBM the BBB’s integrity is disrupted due to the abnormal tumor microvasculature, resulting in an increased vascular permeability and consequently, an increase in immune cell infiltration including monocyte-derived cells, microglia and T-lymphocytes [19, 24]. C5a/C5aR neutralization alleviates the BBB breakdown in models of traumatic brain injury and systemic lupus erythematodus and it is likely that the activated complement system also affects the BBB in GBM, with possible consequences for the passage of immune cells [40].
Lymphocytic infiltration and PD-1
In glioma, tumor infiltrating lymphocytes (TILs) consisting of CD4+ and CD8+ cells are present [65]. Glioma TILs show a predominant regulatory T-cell population (CD4 + CD25 + Foxp3+) [65]. Regulatory T cells (Tregs) are believed to be the primary regulators of immunosuppression in the glioma microenvironment [65]. The proportions of CD3+ and CD8+ over Foxp3+ cells reportedly correlate with the clinical course of GBM patients [78]. The activated complement system by means of CD46 may account for an increased proportion of Tregs. The C3 cleavage fragment, C3b, is a natural ligand for CD46 on T cells. Stimulation of naïve CD4+ T cells with anti-CD46 monocolonal antibodies (mAb) or C3b dimers in the presence of IL-2 induces a differentiation towards a IL-10 producing type 1 regulatory T cell (Tr1) [45]. However, CD4+ Foxp3+ regulatory T cells present in GBM are predominantly thymus derived (tTregs) rather than peripheral induced IL-10 producing regulatory T-cells [95]. In the presence of CD46 stimulation, cell contact-mediated tTreg function is impaired [47]. Instead, tTregs differentiate to IL-10 secreting Tr1 cells [47]. In several human cancers a potent immunosuppressive subpopulation of IL-10 producing Tregs has been identified and these Tregs suppress CD8+ T-cell effector functions which is associated with poor survival [64].
In models of melanoma and non-small cell lung cancer combined with genetic ablation or mAb blocking of programmed death 1/programmed death ligand 1 (PD-1/PD-L1) and C3aR appears to be more effective in restraining tumor growth than only blocking PD-1 therapy alone [2]. In glioma, the expression of PD-L1 is correlated with glioma grade and has been identified as a negative prognostic factor. Recently, therapeutic blockade of PD-1 in the GL-261 murine glioma model induced an impressive prolonged survival, with TILs showing a shift towards CD8+ T cells [20]. The dual role of complement activation in the tumor micro-environment was illustrated by tumor progression in tumor-bearing mice with either high- or low C5a-producing syngeneic lymphoma cells [30]. High C5a producing tumors showed a significant increased tumor progression associated with an overall decrease CD4+ and CD8+ T cells in the tumor [30]. Further, it was shown that in vitro polarization of CD4+ cells is observed to be C5a concentration dependent. A low C5a concentration promotes Th1 cell differentiation while high concentrations (> 500 ng/ml) promotes Treg induction [30]. Taken together, imbalanced complement activation may be associated with an immunosuppressive micro-environment and is therefore contributory to tumor progression.
Glioma associated microglia and macrophages (GAMs)
Glioma associated microglia and macrophages (GAMs) are considered to be the most prominent glioma-infiltrating immune cells, constituting up to 30% of all immune cells within the tumor microenvironment. GAMs are recruited into the tumor microenvironment through various glioma derived factors that contribute to polarization from a tumor-suppressive to a tumor-promoting phenotype. Activated complement factors may contribute to the constant recruitment of GAMs to the tumor micro-environment, either directly, or through interactions with glioma factors, and influence their polarization. Several factors have been recognized to attract GAMs to the tumor environment, among which are MCP-1 and SDF-1 [31]. C1q and C5a have been shown to potentiate the actions of these chemokines [67, 70]. Upon lipopolysaccharide (LPS), IFNγ, or C3a and C5a induced activation (M1) GAMs produce inflammatory mediators, phagocytose tumor cells, present antigens to immune cells and induce a T-cell response [34]. In contrast, GAMs activated through factors such as CSF-1, IL-10, TGF-β and the complement opsonins C1q and C3b acquire a pro-tumorigenic phenotype (M2) which include promoting and facilitating immunosuppression, glioma proliferation and invasiveness [98]. Unbalanced complement activation induces Il-10 and TGF-β expression, thereby promoting a pro-tumorigenic phenotype [29, 47]. Within the scope of autoimmunity the complement opsonins C1q and MBL regulate macrophage polarization towards a M2 macrophage phenotype [5, 26].
Myeloid-derived suppressor cells (MDSC)
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of cells containing myeloid cells in various differentiation stages. MDSCs are found to be significantly increased in the peripheral blood of glioblastoma patients [73]. MDSCs are believed to elicit distinct immunosuppressive actions within the glioma micro-environment including generation of oxidative stress through the production of ROS and thereby inducing T-cell inhibition [24]. Intriguingly, in a TC-1 syngeneic model of cervical cancer in mice, pharmacological inhibition of C5aR resulted in an increase in CD8+ T-cell infiltration along with deceleration of tumor growth comparable to the effects of paclitaxel [53]. C5a mediated suppression of the antitumor CD8+ T-cell response is associated with an increase of MDSC in the tumor microenvironment and a subsequent increase in ROS production [53]. In a syngeneic mouse model for lung cancer, MDSCs appeared to be reduced in a subpopulation analysis of splenocytes after C5aR blockade [14]. Concordantly, in a murine model of ovarian cancer C3 silencing increases the number infiltrating CD8+ T-cells infiltrating the tumor by 10-fold and reduced the number of MDSCs by 80% [13]. However, the observed reduction in tumor growth was found to be independent on the number of CD8+ T-cells [13].
Invasive niche
High-grade gliomas show aggressive invasiveness in two compartments: the perivascular space and the brain parenchyma [17]. The bradykinin and the SDF-1/CXCR4 axis act as a chemoattractant that guide glioma cells towards blood vessels [58]. The bradykinin-forming cascade and the classical complement pathway share many elements, including cross-activation, shared binding proteins and control mechanisms [43]. C1 inhibitor (C1 INH) inhibits the bradykinin-forming cascade at several levels: its local absence at the glioma invasive edge may further initiate the classic complement cascade by activating C1r [43]. Vice-versa, the presence of the ubiquitously expressed receptor for the globular head of C1q (gC1qR) allows for bradykinin production on the endothelial cell surface [43]. The activated complement system further stimulates glioma cell migration through facilitating local degradation of extracellular matrix proteins. In C5aR expressing colon and bile duc cancer cells C5a enhances cell invasiveness by increasing the expression of several matrix metalloproteinases (MMPs), including MMP-1 and MMP-9 in C5aR expressing colon and bile duct cancer cells [62]. Soluble C5b-9, which is as a pro-inflammatory mediator, induces MMP-2 expression by microglia upon activation [101]. The molecular regulation of MMP-2 and MMP-9 is poorly characterized. The activated complement system signaling may provide an alternative mechanism for the NF-κB mediated overexpression of MMP-2 and MMP-9 in glioma cells by means of C5a-C5aR [86]. C1q may activate the canonical Wnt/β-catenin signaling pathway, which increases MMP-2 and MMP-9 expression by glioma cells [41].
Conclusion remarks and future directions
A wide variety of molecular pathways that are characteristic of the aggressive nature of GBM are known to be induced or modulated by the activated complement system (Table 1 and Fig. 3). The unbalanced activation in the tumor microenvironment may suppress the antitumor inflammatory response. There is increasing data that the complement system could serve as an unidentified intrinsic and extrinsic regulatory mechanism for GSC maintenance, thereby supporting treatment resistance. The activated complement system may contribute to evade the immune response, activate invasion and induce angiogenesis. Because several tumor promoting pathways are activated by complement the elucidation of the role of complement activation is necessary to discover new targets for therapy in malignant glioma.
Fig. 3.

Graphical summary of the potential actions of the complement system in the glial tumor microenvironment
Abbreviations
- AP
Alternative Pathway
- BBB
Blood Brain Barrier
- bFGF
basic Fibroblast Growth Factor
- C1-INH
C1-inhibitor
- C3aR
C3a Receptor
- C5aR1
C5a Receptor-1
- C5b-9
Membrane Attack Complex
- cC1qR
collagen C1q Receptor (Calreticulin)
- CNS
Central Nervous System
- CREB
Cellular Transcription Factor
- CSF 1
Colony Stimulating Factor 1
- CXCR1
C-X-C Motif Chemokine Receptor 1
- CXCR2
C-X-C Motif Chemokine Receptor 2
- CXCR4
C-X-C Motif Chemokine Receptor 4
- DAMP
Damage-Associated Molecular Pattern
- EC
Endothelial Cells
- eNOS
endothelial Nitric Oxide synthase
- ERK1/2
Extracellular-signal regulated kinases
- Fz
Frizzled
- GBM
Glioblastoma Multiforme
- gC1qR
Globular C1q Receptor
- GSC
Glioma Stem-like Cell
- HIF
Hypoxia Inducible Factor
- HMEC
Human Mammary Epithelial Cells
- hPSC
human induced Pluripotent Stem Cells
- HVEC
Human Umbilical Vein Endothelial Cells
- HVEC-d
Human Dermal Microvascular Endothelial Cells
- IFNγ
Interferon-γ
- LPS
lipopolysaccharide
- LRP6
Low-density lipoprotein receptor-related protein 6
- MAPK
Mitogen-Activated Protein Kinase
- MBL
Mannose Binding Lectin
- MCP-1
Macrophage Chemoattractant Protein-1
- MMP-9
Matrix metallopeptidase 9
- MSC
mesenchymal stem cells
- MSDC
Myeloid-Derived Suppressor Cell
- MT1-MMP
Membrane-Type 1 Matrix MetalloProteinase
- mTOR
mammalian target of rapamycin
- NO
Nitric Oxide
- NOX-2
Nicotinamide adenine dinucleotide phosphate OXidase 2
- NOX-4
Nicotinamide adenine dinucleotide phosphate OXidase 4
- NPC
Neural Progenitor Cell
- OCT-4
Octamer-Binding Transcription Factor-4
- PD-1
Programmed Cell Death protein 1
- PD-L1
Programmed Cell Death ligand 1
- PI3K
Phosphatidylinositol 3-kinase
- PKC-α
Protein kinase C- α
- PKCζ
Protein Kinase C zeta
- ROS
Reactive Oxygen Species
- SDF-1α
Stromal Cell-Derived Factor 1α
- SOX-2
Sex Determining Region Y -Box 2
- STAT-3
Signal Transducer and Activator of Transcription-3
- TGF-β
Transforming Growth Factor-β
- TIL
Tumor Infiltrating Lymphocytes
- VEGF
Vascular Endothelial Growth Factor
- VEGFR2
Vascular Endothelial Growth Factor Receptor 2
- Wnt
Wingless-related integration site
Authors' contributions
TB conceived of the presented idea. TB wrote the manuscript with support from JK, LA, RW, DM and PH. PS and JK supervised the project. All authors read and approved the final manuscript.
Competing interest
The authors whose names are listed above certify that they have no affiliations with or involvement in any organization or entity with any fi nancial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Afshar-Kharghan V. The role of the complement system in cancer. J Clin Invest. 2017;127:780–789. doi: 10.1172/JCI90962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ajona D, Ortiz-Espinosa S, Moreno H, Lozano T, Pajares MJ, Agorreta J, Bertolo C, Lasarte JJ, Vicent S, Hoehlig K, et al. A combined PD-1/C5a blockade synergistically protects against lung Cancer growth and metastasis. Cancer discovery. 2017;7:694–703. doi: 10.1158/2159-8290.CD-16-1184. [DOI] [PubMed] [Google Scholar]
- 3.Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, Shi Q, McLendon RE, Bigner DD, Rich JN. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 4.Benzaquen LR, Nicholson-Weller A, Halperin JA. Terminal complement proteins C5b-9 release basic fibroblast growth factor and platelet-derived growth factor from endothelial cells. J Exp Med. 1994;179:985–992. doi: 10.1084/jem.179.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohlson SS, O'Conner SD, Hulsebus HJ, Ho MM, Fraser DA. Complement, c1q, and c1q-related molecules regulate macrophage polarization. Front Immunol. 2014;5:402. doi: 10.3389/fimmu.2014.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouwens TA, Trouw LA, Veerhuis R, Dirven CM, Lamfers ML, Al-Khawaja H. Complement activation in glioblastoma multiforme pathophysiology: evidence from serum levels and presence of complement activation products in tumor tissue. J Neuroimmunol. 2015;278:271–276. doi: 10.1016/j.jneuroim.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 7.Brogi E, Wu T, Namiki A, Isner JM (1994) Indirect angiogenic cytokines upregulate VEGF and bFGF gene expression in vascular smooth muscle cells, whereas hypoxia upregulates VEGF expression only. Circulation 90:649–652 [DOI] [PubMed]
- 8.Bulla R, Tripodo C, Rami D, Ling GS, Agostinis C, Guarnotta C, Zorzet S, Durigutto P, Botto M, Tedesco F. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat Commun. 2016;7:10346. doi: 10.1038/ncomms10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M et al (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11:69–82 [DOI] [PubMed]
- 10.Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, Holland EC. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell. 2010;6:141–152. doi: 10.1016/j.stem.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Li GQ, Zhang L, Tang M, Cao X, Xu GL, Wu YZ. Complement C5a/C5aR pathway potentiates the pathogenesis of Gastric Cancer by down-regulating p21 expression. Cancer Lett. 2018;412:30–36. doi: 10.1016/j.canlet.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, et al. Glioblastoma Stem Cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153:139–152. doi: 10.1016/j.cell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho MS, Vasquez HG, Rupaimoole R, Pradeep S, Wu S, Zand B, Han HD, Rodriguez-Aguayo C, Bottsford-Miller J, Huang J, et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014;6:1085–1095. doi: 10.1016/j.celrep.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, Rouzaut A, Pajares MJ, Montuenga LM, Pio R. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. 2012;189:4674–4683. doi: 10.4049/jimmunol.1201654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cortright DN, Meade R, Waters SM, Chenard BL, Krause JE. C5a, but not C3a, increases VEGF secretion in ARPE-19 human retinal pigment epithelial cells. Curr Eye Res. 2009;34:57–61. doi: 10.1080/02713680802546658. [DOI] [PubMed] [Google Scholar]
- 16.Coulthard LG, Hawksworth OA, Li R, Balachandran A, Lee JD, Sepehrband F, Kurniawan N, Jeanes A, Simmons DG, Wolvetang E, et al. Complement C5aR1 Signaling Promotes Polarization and Proliferation of Embryonic Neural Progenitor Cells through PKCzeta. J Neurosci. 2017;37:5395–5407. doi: 10.1523/JNEUROSCI.0525-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuddapah VA, Robel S, Watkins S, Sontheimer H. A neurocentric perspective on glioma invasion. Nat Rev Neurosci. 2014;15:455–465. doi: 10.1038/nrn3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniele S, Costa B, Zappelli E, Da Pozzo E, Sestito S, Nesi G, Campiglia P, Marinelli L, Novellino E, Rapposelli S, et al. Combined inhibition of AKT/mTOR and MDM2 enhances glioblastoma Multiforme cell apoptosis and differentiation of cancer stem cells. Scientific reports. 2015;5:9956. doi: 10.1038/srep09956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies DC. Blood-brain barrier breakdown in septic encephalopathy and brain tumours. J Anat. 2002;200:639–646. doi: 10.1046/j.1469-7580.2002.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dejaegher J, Verschuere T, Vercalsteren E, Boon L, Cremer J, Sciot R, Van Gool SW, De Vleeschouwer S (2017) Characterization of PD-1 upregulation on tumor-infiltrating lymphocytes in human and murine gliomas and preclinical therapeutic blockade. Int J Cancer 141: 1891–1900 Doi 10.1002/ijc.30877 [DOI] [PubMed]
- 21.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 22.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du Z, Jia D, Liu S, Wang F, Li G, Zhang Y, Cao X, Ling EA, Hao A. Oct4 is expressed in human gliomas and promotes colony formation in glioma cells. Glia. 2009;57:724–733. doi: 10.1002/glia.20800. [DOI] [PubMed] [Google Scholar]
- 24.Eder K, Kalman B. The dynamics of interactions among immune and glioblastoma cells. NeuroMolecular Med. 2015;17:335–352. doi: 10.1007/s12017-015-8362-x. [DOI] [PubMed] [Google Scholar]
- 25.Fessler E, Borovski T, Medema JP. Endothelial cells induce cancer stem cell features in differentiated glioblastoma cells via bFGF. Mol Cancer. 2015;14:157. doi: 10.1186/s12943-015-0420-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fraser DA, Bohlson SS, Jasinskiene N, Rawal N, Palmarini G, Ruiz S, Rochford R, Tenner AJ. C1q and MBL, components of the innate immune system, influence monocyte cytokine expression. J leukoc biol. 2006;80:107–116. doi: 10.1189/jlb.1105683. [DOI] [PubMed] [Google Scholar]
- 27.Greijer AE, van der Groep P, Kemming D, Shvarts A, Semenza GL, Meijer GA, van de Wiel MA, Belien JA, van Diest PJ, van der Wall E. Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1) J Pathol. 2005;206:291–304. doi: 10.1002/path.1778. [DOI] [PubMed] [Google Scholar]
- 28.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu H, Mickler EA, Cummings OW, Sandusky GE, Weber DJ, Gracon A, Woodruff T, Wilkes DS, Vittal R. Crosstalk between TGF-beta1 and complement activation augments epithelial injury in pulmonary fibrosis. FASEB J. 2014;28:4223–4234. doi: 10.1096/fj.13-247650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gunn L, Ding C, Liu M, Ma Y, Qi C, Cai Y, Hu X, Aggarwal D, Zhang HG, Yan J. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J Immunol. 2012;189:2985–2994. doi: 10.4049/jimmunol.1200846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19:20–27. doi: 10.1038/nn.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 33.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 34.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 35.Hawksworth OA, Coulthard LG, Taylor SM, Wolvetang EJ, Woodruff TM. Brief report: complement C5a promotes human embryonic stem cell pluripotency in the absence of FGF2. Stem cells. 2014;32:3278–3284. doi: 10.1002/stem.1801. [DOI] [PubMed] [Google Scholar]
- 36.Haynes T, Luz-Madrigal A, Reis ES, Echeverri Ruiz NP, Grajales-Esquivel E, Tzekou A, Tsonis PA, Lambris JD, Del Rio-Tsonis K (2013) Complement anaphylatoxin C3a is a potent inducer of embryonic chick retina regeneration. Nat Commun 4: 2312 Doi 10.1038/ncomms3312 [DOI] [PMC free article] [PubMed]
- 37.Hernandez C, Huebener P, Schwabe RF. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene. 2016;35:5931–5941. doi: 10.1038/onc.2016.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Honczarenko M, Ratajczak MZ, Nicholson-Weller A, Silberstein LE. Complement C3a enhances CXCL12 (SDF-1)-mediated chemotaxis of bone marrow hematopoietic cells independently of C3a receptor. J Immunol. 2005;175:3698–3706. doi: 10.4049/jimmunol.175.6.3698. [DOI] [PubMed] [Google Scholar]
- 39.Infanger DW, Cho Y, Lopez BS, Mohanan S, Liu SC, Gursel D, Boockvar JA, Fischbach C. Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res. 2013;73:7079–7089. doi: 10.1158/0008-5472.CAN-13-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacob A, Alexander JJ. Complement and blood-brain barrier integrity. Mol Immunol. 2014;61:149–152. doi: 10.1016/j.molimm.2014.06.039. [DOI] [PubMed] [Google Scholar]
- 41.Kahlert UD, Maciaczyk D, Doostkam S, Orr BA, Simons B, Bogiel T, Reithmeier T, Prinz M, Schubert J, Niedermann G, et al. Activation of canonical WNT/beta-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012;325:42–53. doi: 10.1016/j.canlet.2012.05.024. [DOI] [PubMed] [Google Scholar]
- 42.Kalani MY, Cheshier SH, Cord BJ, Bababeygy SR, Vogel H, Weissman IL, Palmer TD, Nusse R. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc Natl Acad Sci U S A. 2008;105:16970–16975. doi: 10.1073/pnas.0808616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaplan AP, Ghebrehiwet B. The plasma bradykinin-forming pathways and its interrelationships with complement. Mol Immunol. 2010;47:2161–2169. doi: 10.1016/j.molimm.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 44.Keir LS, Firth R, Aponik L, Feitelberg D, Sakimoto S, Aguilar E, Welsh GI, Richards A, Usui Y, Satchell SC, et al. VEGF regulates local inhibitory complement proteins in the eye and kidney. J Clin Invest. 2017;127:199–214. doi: 10.1172/JCI86418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kemper C, Chan AC, Green JM, Brett KA, Murphy KM, Atkinson JP. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature. 2003;421:388–392. doi: 10.1038/nature01315. [DOI] [PubMed] [Google Scholar]
- 46.Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell Stem Cell. 2011;8:482–485. doi: 10.1016/j.stem.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le Buanec H, Gougeon ML, Mathian A, Lebon P, Dupont JM, Peltre G, Hemon P, Schmid M, Bizzini B, Kunding T, et al. IFN-alpha and CD46 stimulation are associated with active lupus and skew natural T regulatory cell differentiation to type 1 regulatory T (Tr1) Cell. Proc Natl Acad Sci U S A. 2011;108:18995–19000. doi: 10.1073/pnas.1113301108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le Friec G, Sheppard D, Whiteman P, Karsten CM, Shamoun SA, Laing A, Bugeon L, Dallman MJ, Melchionna T, Chillakuri C, et al. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat Immunol. 2012;13:1213–1221. doi: 10.1038/ni.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, RE ML, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, Subias M, Pickering MC, Drouet C, Meri S, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector Differentiation. Immunity. 2013;39:1143–1157. doi: 10.1016/j.immuni.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mamidi S, Hone S, Kirschfink M. The complement system in cancer: ambivalence between tumour destruction and promotion. Immunobiology. 2017;222:45–54. doi: 10.1016/j.imbio.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 52.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 53.Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, Coukos G, Lambris JD. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9:1225–1235. doi: 10.1038/ni.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mastellos DC. Complement emerges as a masterful regulator of CNS homeostasis, neural synaptic plasticity and cognitive function. Exp Neurol. 2014;261:469–474. doi: 10.1016/j.expneurol.2014.06.019. [DOI] [PubMed] [Google Scholar]
- 55.McGeer PL, Lee M, McGeer EG. A review of human diseases caused or exacerbated by aberrant complement activation. Neurobiol Aging. 2017;52:12–22. doi: 10.1016/j.neurobiolaging.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 56.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Montalto MC, Hart ML, Jordan JE, Wada K, Stahl GL. Role for complement in mediating intestinal nitric oxide synthase-2 and superoxide dismutase expression. Am J Physiol Gastrointest Liver Physiol. 2003;285:G197–G206. doi: 10.1152/ajpgi.00029.2003. [DOI] [PubMed] [Google Scholar]
- 58.Montana V, Sontheimer H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J Neurosci. 2011;31:4858–4867. doi: 10.1523/JNEUROSCI.3825-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morgan BP, Boyd C, Bubeck D (2017) Molecular cell biology of complement membrane attack. Seminars in cell & developmental biology: Doi. 10.1016/j.semcdb.2017.06.009 [DOI] [PubMed]
- 60.Morgan BP, Gasque P. Expression of complement in the brain: role in health and disease. Immunol Today. 1996;17:461–466. doi: 10.1016/0167-5699(96)20028-F. [DOI] [PubMed] [Google Scholar]
- 61.Naito AT, Sumida T, Nomura S, Liu ML, Higo T, Nakagawa A, Okada K, Sakai T, Hashimoto A, Hara Y, et al. Complement C1q activates canonical Wnt signaling and promotes aging-related phenotypes. Cell. 2012;149:1298–1313. doi: 10.1016/j.cell.2012.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nitta H, Murakami Y, Wada Y, Eto M, Baba H, Imamura T. Cancer cells release anaphylatoxin C5a from C5 by serine protease to enhance invasiveness. Oncol Rep. 2014;32:1715–1719. doi: 10.3892/or.2014.3341. [DOI] [PubMed] [Google Scholar]
- 63.Nunez-Cruz S, Gimotty PA, Guerra MW, Connolly DC, Wu YQ, DeAngelis RA, Lambris JD, Coukos G, Scholler N. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia. 2012;14:994–1004. doi: 10.1593/neo.121262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O'Garra A, Barrat FJ, Castro AG, Vicari A, Hawrylowicz C. Strategies for use of IL-10 or its antagonists in human disease. Immunol Rev. 2008;223:114–131. doi: 10.1111/j.1600-065X.2008.00635.x. [DOI] [PubMed] [Google Scholar]
- 65.Ooi YC, Tran P, Ung N, Thill K, Trang A, Fong BM, Nagasawa DT, Lim M, Yang I. The role of regulatory T-cells in glioma immunology. Clin Neurol Neurosurg. 2014;119:125–132. doi: 10.1016/j.clineuro.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 66.Pedersen ED, Froyland E, Kvissel AK, Pharo AM, Skalhegg BS, Rootwelt T, Mollnes TE. Expression of complement regulators and receptors on human NT2-N neurons--effect of hypoxia and reoxygenation. Mol Immunol. 2007;44:2459–2468. doi: 10.1016/j.molimm.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 67.Peerschke EI, Ghebrehiwet B. cC1qR/CR and gC1qR/p33: observations in cancer. Mol Immunol. 2014;61:100–109. doi: 10.1016/j.molimm.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 68.Perianayagam MC, Balakrishnan VS, King AJ, Pereira BJ, Jaber BL. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int. 2002;61:456–463. doi: 10.1046/j.1523-1755.2002.00139.x. [DOI] [PubMed] [Google Scholar]
- 69.Qiang L, Wu T, Zhang HW, Lu N, Hu R, Wang YJ, Zhao L, Chen FH, Wang XT, You QD, et al. HIF-1alpha is critical for hypoxia-mediated maintenance of glioblastoma Stem Cells by activating Notch signaling pathway. Cell Death Differ. 2012;19:284–294. doi: 10.1038/cdd.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qiu Y, Marquez-Curtis LA, Janowska-Wieczorek A. Mesenchymal stromal cells derived from umbilical cord blood migrate in response to complement C1q. Cytotherapy. 2012;14:285–295. doi: 10.3109/14653249.2011.651532. [DOI] [PubMed] [Google Scholar]
- 71.Rampazzo E, Persano L, Pistollato F, Moro E, Frasson C, Porazzi P, Della Puppa A, Bresolin S, Battilana G, Indraccolo S, et al. Wnt activation promotes neuronal differentiation of glioblastoma. Cell Death Dis. 2013;4:e500. doi: 10.1038/cddis.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rao S, Sengupta R, Choe EJ, Woerner BM, Jackson E, Sun T, Leonard J, Piwnica-Worms D, Rubin JB. CXCL12 mediates trophic interactions between endothelial and tumor cells in glioblastoma. PLoS One. 2012;7:e33005. doi: 10.1371/journal.pone.0033005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, Garcia J, Vogelbaum MA, Finke J. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro-Oncology. 2011;13:591–599. doi: 10.1093/neuonc/nor042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD (2017) Complement in cancer: untangling an intricate relationship. Nature reviews Immunology: Doi. 10.1038/nri.2017.97 [DOI] [PMC free article] [PubMed]
- 75.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sakiyama H, Kaji K, Nakagawa K, Nagino K. Inhibition of bFGF activity by complement C1s: covalent binding of C1s with bFGF. Cell Biochem Funct. 1998;16:159–163. doi: 10.1002/(SICI)1099-0844(199809)16:3<159::AID-CBF779>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 77.Sarma JV, Ward PA. New developments in C5a receptor signaling. Cell health and cytoskeleton. 2012;4:73–82. doi: 10.2147/CHC.S27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sayour EJ, McLendon P, McLendon R, De Leon G, Reynolds R, Kresak J, Sampson JH, Mitchell DA. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer immunology, immunotherapy : CII. 2015;64:419–427. doi: 10.1007/s00262-014-1651-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schraufstatter IU, Discipio RG, Zhao M, Khaldoyanidi SK (2009) C3a and C5a are chemotactic factors for human mesenchymal stem cells, which cause prolonged ERK1/2 phosphorylation. Journal of immunology (Baltimore, Md : 1950) 182: 3827–3836 [DOI] [PubMed]
- 80.Sharma A, Shiras A. Cancer stem cell-vascular endothelial cell interactions in glioblastoma. Biochem Biophys Res Commun. 2016;473:688–692. doi: 10.1016/j.bbrc.2015.12.022. [DOI] [PubMed] [Google Scholar]
- 81.Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- 82.Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma Stem Cells. Stem cells (Dayton, Ohio) 2009;27:2383–2392. doi: 10.1002/stem.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shinjyo N, Stahlberg A, Dragunow M, Pekny M, Pekna M. Complement-derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells. Stem cells (Dayton, Ohio) 2009;27:2824–2832. doi: 10.1002/stem.225. [DOI] [PubMed] [Google Scholar]
- 84.Simone S, Rascio F, Castellano G, Divella C, Chieti A, Ditonno P, Battaglia M, Crovace A, Staffieri F, Oortwijn B, et al. Complement-dependent NADPH oxidase enzyme activation in renal ischemia/reperfusion injury. Free Radic Biol Med. 2014;74:263–273. doi: 10.1016/j.freeradbiomed.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 85.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 86.Speidl WS, Kastl SP, Hutter R, Katsaros KM, Kaun C, Bauriedel G, Maurer G, Huber K, Badimon JJ, Wojta J. The complement component C5a is present in human coronary lesions in vivo and induces the expression of MMP-1 and MMP-9 in human macrophages in vitro. FASEB j. 2011;25:35–44. doi: 10.1096/fj.10-156083. [DOI] [PubMed] [Google Scholar]
- 87.Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, Shapiro VS, Dubyak GR, Heeger PS, Medof ME. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like Cell. Cell. 2014;157:580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Taylor RP, Lindorfer MA. Cytotoxic mechanisms of immunotherapy: harnessing complement in the action of anti-tumor monoclonal antibodies. Semin Immunol. 2016;28:309–316. doi: 10.1016/j.smim.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 90.Thomas TM, Yu JS. Metabolic regulation of glioma stem-like cells in the tumor micro-environment. Cancer Lett. 2017;408:174–181. doi: 10.1016/j.canlet.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thurman JM, Renner B, Kunchithapautham K, Ferreira VP, Pangburn MK, Ablonczy Z, Tomlinson S, Holers VM, Rohrer B. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J Biol Chem. 2009;284:16939–16947. doi: 10.1074/jbc.M808166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.van den Berg RH, Faber-Krol MC, Sim RB, Daha MR. The first subcomponent of complement, C1q, triggers the production of IL-8, IL-6, and monocyte chemoattractant peptide-1 by human umbilical vein endothelial Cell. J Immunol. 1998;161:6924–6930. [PubMed] [Google Scholar]
- 93.Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Mol Immunol. 2011;48:1592–1603. doi: 10.1016/j.molimm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 95.Wainwright DA, Sengupta S, Han Y, Lesniak MS. Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro-Oncology. 2011;13:1308–1323. doi: 10.1093/neuonc/nor134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 97.Wang L, Han G, Wang R, Chen G, Xu R, Xiao H, Li X, Geng S, Li Y, Li X, et al. Regulation of IL-8 production by complement-activated product, C5a, in vitro and in vivo during sepsis. Clin Immunol. 2010;137:157–165. doi: 10.1016/j.clim.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 98.Wurdinger T, Deumelandt K, van der Vliet HJ, Wesseling P, de Gruijl TD (2014) Mechanisms of intimate and long-distance cross-talk between glioma and myeloid cells: how to break a vicious cycle. Biochim Biophys Acta 1846: 560–575 Doi 10.1016/j.bbcan.2014.10.003 [DOI] [PubMed]
- 99.Yan K, Wu Q, Yan DH, Lee CH, Rahim N, Tritschler I, DeVecchio J, Kalady MF, Hjelmeland AB, Rich JN. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev. 2014;28:1085–1100. doi: 10.1101/gad.235515.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yanai R, Thanos A, Connor KM. Complement involvement in neovascular ocular diseases. Adv Exp Med Biol. 2012;946:161–183. doi: 10.1007/978-1-4614-0106-3_10. [DOI] [PubMed] [Google Scholar]
- 101.Yang C, Yang L, Liu Y. Soluble complement complex C5b-9 promotes microglia activation. J Neuroimmunol. 2014;267:16–19. doi: 10.1016/j.jneuroim.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 102.Yang YH, Tsai IJ, Chang CJ, Chuang YH, Hsu HY, Chiang BL. The interaction between circulating complement proteins and cutaneous microvascular endothelial cells in the development of childhood Henoch-Schonlein purpura. PLoS One. 2015;10:e0120411. doi: 10.1371/journal.pone.0120411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yu MO, Park KJ, Park DH, Chung YG, Chi SG, Kang SH (2015) Reactive oxygen species production has a critical role in hypoxia-induced Stat3 activation and angiogenesis in human glioblastoma. J Neuro-Oncol [DOI] [PubMed]
- 104.Zajc I, Hreljac I, Lah T. Cathepsin L affects apoptosis of glioblastoma cells: a potential implication in the design of cancer therapeutics. Anticancer Res. 2006;26:3357–3364. [PubMed] [Google Scholar]
- 105.Zhang YE (2009) Non-Smad pathways in TGF-beta signaling. Cell Res 19:128–139. 10.1038/cr.2008.328 [DOI] [PMC free article] [PubMed]