

Abstract
Flow cytometry provides highly sensitive multi-parameter analysis of cells and particles, but has been largely limited to the use of a single focused sample stream. This limits the analytical rate to ~50K particles/s and the volumetric rate to ~250 μl/min. Despite the analytical prowess of flow cytometry, there are applications where these rates are insufficient, such as rare cell analysis in high cellular backgrounds (e.g. circulating tumor cells and fetal cells in maternal blood), detection of cells/particles in large dilute samples (e.g. water quality, urine analysis), or high throughput screening applications. Here we report a highly parallel acoustic flow cytometer that uses an acoustic standing wave to focus particles into 16 parallel analysis points across a 2.3-mm wide optical flow cell. A line focused laser and wide-field collection optics are used to excite and collect the fluorescence emission of these parallel streams onto a high-speed camera for analysis. With this instrument format and fluorescent microsphere standards, we obtain analysis rates of 100K/s and flow rates of 10 mL/min, while maintaining optical performance comparable to that of a commercial flow cytometer. The results with our initial prototype instrument demonstrate that the integration of key parallelizable components, including the line focused laser, particle focusing using multi-node acoustic standing waves, and a spatially arrayed detector, can increase analytical and volumetric throughputs by orders of magnitude in a compact, simple and cost effective platform. Such instruments will be of great value to applications in need of high throughput yet sensitive flow cytometry analysis.
FOR TOC ONLY
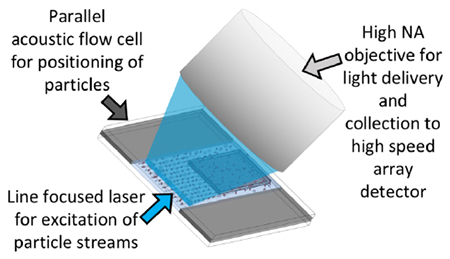
A typical flow cytometer sensitively measures up to twenty optical parameters from individual cells on a cell-by-cell basis at analytical rates as high as ~50,000 cells/s and volumetric rates of up to 250 μl/min.1,2 This analytical power makes it the technology of choice for many applications including cellular phenotyping (e.g. CD4+ T-cell counts), cell cycle analysis, and apoptosis measurements.1,2 Additionally, flow cytometry is valuable for rare cell detection,3–6 high throughput screening for pharmaceutical lead compounds,7–9 analysis of environmental samples for algae, plankton, other microbes,10–12 and as a method of process monitoring for food safety.13 However, use of flow cytometry for high throughput applications is limited by the analytical and volumetric rates. Increasing these rates would reduce the need for cell enrichment strategies for rare cell applications, improve the rate of high throughput screening, and increase the likelihood of detection of microbes or algae of interest in both food safety and environmental applications.
Analytical and volumetric rate limitations in conventional flow cytometers are largely due to the analytical paradigm that uses a tightly focused laser to interrogate a single focused stream of particles or cells.2 In a conventional flow cytometer the laser spot is focused to an elliptical spot (~10 μm high by 50 μm wide) that interrogates a sample stream that is focused by a high velocity sheath fluid to a core diameter of only a few microns.2,14 This approach allows a cell or particle to occupy most of the interrogation volume (~ 1 pL) as it passes through the focused spot of the laser. This enables a wash-free sensitive measurement, which is a critical analytical advantage for flow cytometry compared to other techniques. The high analytical rate of a flow cytometer is achieved in part by flowing sample at linear velocities up to 10 m/s. The onset of turbulence precludes higher linear velocities.14
These limitations on linear velocity and the width of the sample stream restrict the volumetric throughput of a hydrodynamic focusing flow cytometer. However, the volumetric throughput of a cytometer can be increased by means other than increasing the overall velocity of the system if tight particle focusing can be achieved using a different approach. One such approach uses acoustic standing waves to focus particles in a cylindrical capillary upstream of the analysis point. Acoustic focusing enables sample delivery rates of about 1 ml/min, while maintaining the precision measurements typical of a flow cytometer.15–17 Much like a conventional flow cytometer, this method analyzes the particles in a single stream, therefore the analytical rate of both these methods is limited by coincident events occurring due to the stochastic arrival of cells in the interrogation volume.14,16,17
The limitations of single sample stream flow cytometers have led to the development of parallel stream flow cytometers to increase volumetric and analytical event rates. Most parallel flow cytometer systems rely on hydrodynamics or tight microfluidic channels to create parallel focused streams of particles for analysis. Modestly parallel systems have been constructed using simple multiplexing of conventional fluidics and optics to create systems of up to 4 flow streams with overall analysis rates of ~250,000 particles/s.18 Alternatively, microfluidic approaches have led to hydrodynamically focused flow cytometers with 24 parallel sample streams that have sample delivery rates of up 830 μl/min and analytical rates of 240,000 cells per second.19 Though these systems, which are commercially available, do show improved throughput compared to a conventional flow cytometer they are very large, relatively expensive, and significantly more complex than standard flow cytometers.19 Additional work has demonstrated parallel analysis through the use of up to four inexpensive flow cytometers operating in parallel to increase the sampling rate of high-throughput screening (HTS) flow cytometry systems.20 This technique shows promise, but it demonstrates the need to decrease the cost per analysis point/cytometer (currently ~$50K) to maximize the effectiveness of this approach.20
As much of the bulk and complexity of parallel cytometer systems is due to the hydrodynamic focusing, inertial focusing in parallel microfluidic channels may also offer a path to highly parallel flow cells.21 However, this approach requires channels only slightly wider than the particles, which requires care to prevent clogging and limited volumetric throughput.
By contrast, multinode acoustic standing waves offer a highly parallel and high throughput method to tightly focus flowing particles for analysis or separations without the use of microfluidic channels.2,22 At a resonant frequency condition, an integer number of half-wavelengths are excited across the focusing plane of the channel. The superposition of the reflecting waves within the focusing chamber creates an acoustic standing wave. The magnitude of the primary acoustic force on a particle within an acoustic standing wave system depends upon the pressure of the standing wave, the applied frequency, the position within the wave, the volume of the particle and the relative properties of the media and particle described by an acoustic contrast factor. Prior results using such standing waves demonstrate the ability to create highly parallel acoustic flow cells that support up to 37 discrete focused streams in a single channel23 and up to 300 focused streams in a multichannel microfabricated flow cell.24 Additionally, the wide aspect ratio and correspondingly large cross-sectional area of these flow cells reduces clogging and allows for sheathless, precise, and highly parallel focusing of particles at low linear velocities, but high volumetric throughputs (25 mL/min).24
Based on these considerations, we present here a fundamentally new methodology for parallelizing high throughput flow cytometry analysis. Integrating a highly parallel acoustic focusing flow cell, a line focused excitation laser, wide field collection optics, and a spatially arrayed detector creates a relatively simple, high performance instrumentation platform with both high volumetric delivery rates and high analytical rates. We discuss the scalability of this approach and the value of this work for flow cytometry applications requiring high volumetric sample delivery rates or high analytical rates.
MATERIALS AND METHODS
1. Multinode Acoustic Flow Cell
Device Design.
The acoustic flow cell used for this study is fabricated in house. A 200 μm thick silicon wafer is etched completely through across an area of ~2,300 μm width by 5 cm length and anodically bonded with glass on both sides to create a 2,300 μm × 200 μm × 5 cm transparent channel (W × H × L) (Figure 1A, 1B). The transparent flow chamber minimizes excess heat and optical scatter that would otherwise occur due to absorption and scattering from an opaque silicon background.
Figure 1. System description.
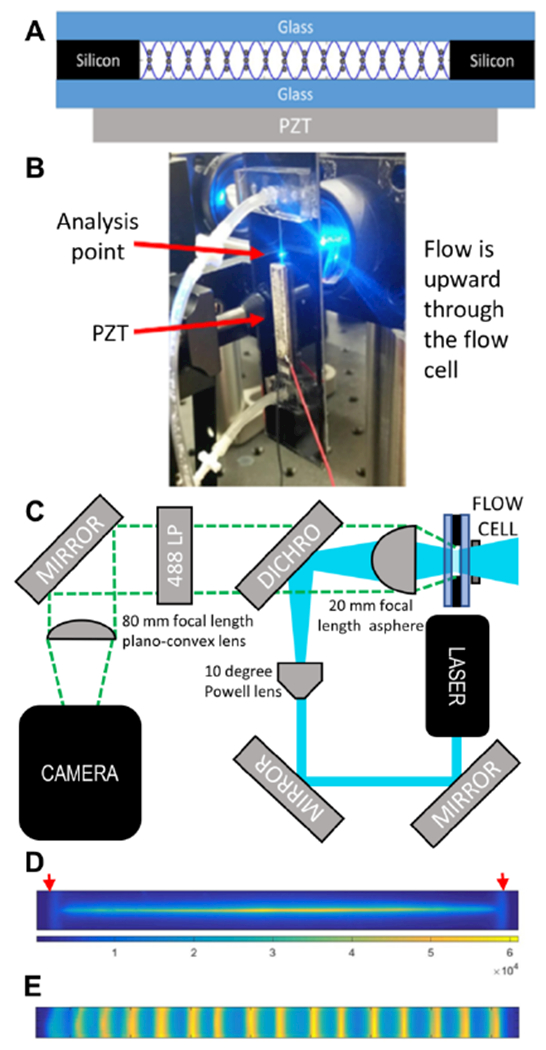
(A) Cross-sectional schematic of the acoustic focusing flow cell. The standing acoustic pressure wave (sine wave) focuses particles to the 16 pressure nodes across the width of the flow cell. (B) Image of the mounted flow cell. (C) Schematic of optics. (D) Laser excitation profile. The red arrows show the positions of the walls, that were 2.3 mm apart. (E) Stacked image of acoustically focused fluorescent particles.
Photolithography and Deep Reactive-Ion Etching (DRIE).
Using a 10 cm silicon substrate wafer, AZ 4620 positive photoresist is spin coated to form a uniform 6-10 μm layer. The spin-coated wafer is developed and washed before etching. A 90-minute Bosch process is used to etch through the device. Once the device is processed through the DRIE, it is left in acetone for 1-2 hours (or as needed) to dissolve the remaining photoresist, rinsed with DI water and dried with N2. The etched silicon is then piranha-cleaned prior to the anodic bonding process.
Anodic-Bonding.
The cleaned silicon device is placed on an aluminum plate on top of a ceramic hot plate. A piranha cleaned glass slide is placed flush on top of the silicon device. The silicon wafer is connected to the (−) output and the glass slide is connected to the (+) output of a DC power supply. The hot plate is set to 500°C. After temperature equilibration, the voltage is increased from 100V to 800V over ~4 minutes. Successful bonding is indicated by a color change of the glass to silicon contact area. Once bonding is complete, the procedure is repeated on other side of the device. Each side takes 3-6 hours to bond, resulting in a sandwiched silicon etched device between two borosilicate glass slides (Figure 1A). Punctured polydimethylsiloxane (PDMS) seals the fluidic connections made via polyether ether ketone (PEEK) tubing.
Transducer Selection and Attachment.
A 4.48 MHz PZT (30 × 5 mm, APC International, Mackeyville, PA) is superglued under the channel and driven at 5.08 MHz to create 16 pressure nodes (Figure 1A). This agrees with the predicted number of nodes (n) using a half-wavelength resonance condition n = 2Lf/c, where c is the speed of sound (~1490 m/s), L is the width of the flow cell (2.3 mm), and f is the drive frequency.
2. Optical Setup
The flow cell is placed in front of the aspheric lens that serves as both the collection lens and focusing lens in an epifluorescent configuration (Figure 1B). The full optical schematic is shown in Figure 1C. Excitation is provided by a beam from 150 mW 488 nm laser (Mini-WhisperIT, Pavilion Integration Corporation, San Jose CA) steered by two mirrors through a 10 degree Powell lens (Laserline Optics Canada Inc, Canada) onto a reflective 488 nm dichroic beamsplitter (Semrock, Rochester, NY) to create a line-shaped excitation beam that is focused into the acoustic flow cell using a 25 mm diameter aspheric lens with a 20 mm focal length (AL2520-A, ThorLabs, Inc. Newton, NJ) (Figure 1C). The analysis position is just above the PZT (Figure 1B). The emitted light is collected back into the aspheric lens, passed through the beamsplitter, filtered with a 488 nm long-pass filter (488nm Edge Basic, Semrock, Rochester, NY), and imaged onto an sCMOS sensor (Hamamatsu Orca flash 4.0 v2) using a 25 mm diameter plano-convex lens with an 80-mm focal length. At the sample illumination point, the line-shaped excitation beam, detected via emission of Fluorescein dye solution in the flow cell, (Figure 1D) has dimensions of ~2.3 mm width and ~10-20 μm height, matching both the width of the flow cell and the active area (8 × 2048 pixels) of the camera’s field of view when the camera is operated at it maximum acquisition rate of 25,655 frames per second. To visually display the streams in flow, 75K frames were taken over 3 seconds of acoustically focused fluorescent particles flowing through active region of the camera. The frames were summed and averaged into a single 8 × 2048 aggregate frame. This aggregate frame has been stretched 20× vertically to more easily visualize focused positions of particles in the flow cell (Figure 1E).
3. Analysis of Frames Data
Two data collection methods are used for the high-speed camera data from the Hamamatsu Orca Flash 4.0 v2 (operated at 25,655 frames/s or ~39 μs exposure). The full uncompressed raw data from the camera (6.7 Gb/s) is stored either on the hard drive for post processing in MATLAB or analyzed in real-time using a custom data acquisition package (Kytos Data Acquisition system, built by DarklingX, Los Alamos, NM). The stored raw data files allow for detailed analysis and a means of testing image analysis algorithms, while the Kytos acquisition system provides substantial real-time information compression of the data to generate standard flow cytometry data sets.
Figure 2 (panels A & B), shows stored image data acquired by the sCMOS detector after post-experiment processing. The raw stored data consists of a simple series of frames that are 8 pixels in width by 2048 pixels in length. During collection, the long 2048 axis of the frame is oriented to be perpendicular (⊥) to the direction of flow (Figure 2). In panel A, the long axis of the collected frame is presented along the vertical axis and labeled as perpendicular (⊥) pixels. Only the pixels that spanned locations where particles flowed are shown, which consisted of 1600 pixels of the overall length of the original 2048 pixel frame. The horizontal axis of panel A consists of a series of 180 frames that each show the pixels 2 through 7 (left to right) of the 8 pixel wide frames. As these pixel rows are oriented parallel to the flow of the system, they are termed parallel (∥) pixels. Only pixels 2-7 across the width of each frame are shown and were used for analysis, with pixel positions 1 and 8 of each row row rendered black to make individual frames more distinguishable and to eliminate any possible edge effects during analysis. Each bright streak corresponds to a particle traversing the excitation beam. Figure 2B shows frames 71 through 88, where the perpendicular pixels (⊥) 300 – 400 of the frame are plotted on the vertical axis and parallel pixels (2-7) for each respective frame were plotted on the horizontal axis. In this figure panel, the particle enters the detection window from on the right side of a frame (frame 71), traverses through the frame (frames 71-87), and exits the left side of the frame (frame 87). In the real-time acquisition mode such frame regions are used to calculate optical parameter data for each passing particle (Figure 2C). To accomplish this, the user first defines regions of interest (typically 50 – 100 perpendicular pixels) corresponding to the position of each of focused streams. For the majority of the data presented here we used 16 acoustically focused streams. Thus, we defined 16 discrete detection regions, each corresponding to a focused stream. Each such region is termed a stream window and is monitored individually in each frame. Event triggering in each stream window occurs by comparison with a user-defined threshold level in one of two possible modes. In a peak intensity mode, the largest single pixel value, averaged with its eight nearest neighbors, must exceed the user-defined threshold. In an integrated mode, the integral of all pixels in the region of interest must exceed the user-defined threshold. Around each event trigger, several frames of the stream window are analyzed to provide signal parameters in real time. Several measurement parameters are collected. The parameters used primarily for this work were the peak intensity parameter, which is the single brightest pixel from a particle’s traverse through the laser beam and the peak integrated intensity parameter, which is defined as the maximum integrated pixel value of a single frame within a particle’s time of flight through the laser beam. Additional parameters extracted include the column or X position of the brightest pixel of an event (BPX), the time of flight (number of frames) through the laser, the frame number, and the event number. Beyond these values, the integrated fluorescent intensity for each event is stored as a waveform across several frames (Figure 2C). The frames included are all frames in which signals are above the chosen threshold value described above, as well as several pre-event frames extracted from buffered data and several post-event frames. While not used here, waveforms fit to a Gaussian can be used to eliminate irregular events such as doublets and the fit parameters (Amplitude, R2, and standard deviation) used to further quantify the fluorescent parameters. Singlet waveforms have a high R2 and outliers (doublets) can be excluded based on R2 values.
Figure 2. Analysis of camera data.
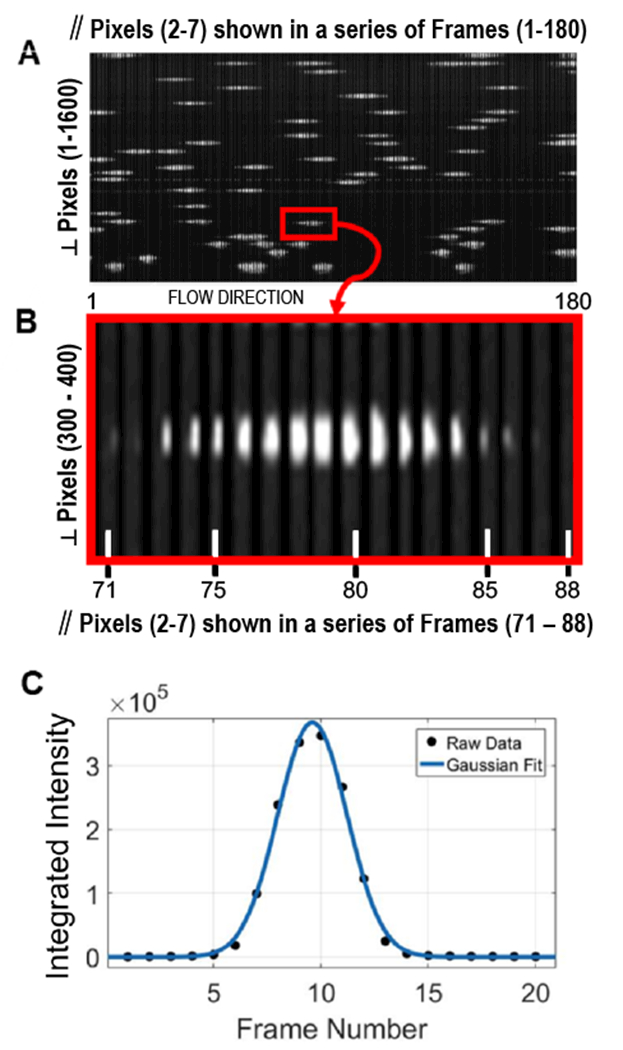
(A) A series of 180 frames displayed in sequential order to create a pseudo image of fluorescent intensity of particles passing through the system. As described in the text each 6 × 1600 pixel frame shown is derived from the original 8 × 2048 pixel frame. The red box is expanded in panel (B), where a region of interest 6 pixels wide (∥ pixels 2-7) and 100 pixels high (⊥ pixels 300 to 400) is displayed sequentially for frames 71 to 88. Each of the 18 frames consists of a 6 by 100-pixel image. A small width spacing (completely black in color) is introduced between each frame to make them individually distinguishable. (C) Waveform and Gaussian fit of the integrated fluorescence intensity derived from a series of frames capturing a single particle passing through the laser. The details of this analysis are provided in the text.
Notably, our approach does not require precise flow control or optical tracking approaches used in image cytometry approaches.25,26 Rather, we are simply using the sCMOS detector as an approximate 1D array that simplifies direct collection of data across a wide field of view.
4. FCS Data Analysis
After acquisition, data are stored in flow cytometry format and analyzed using a commercial software package (FCS Express, De Novo Software, Glendale, CA).
RESULTS
1. Acoustic Focusing ON/OFF
Like all flow cytometers, the performance of our system depends upon having a consistent interaction between the particle and the interrogating laser. Thus, the use of acoustic focusing within the flow cell greatly improves the system’s performance, leading to very predictable and consistent particle/laser interactions across all of the focused streams. Figure 3A shows the variation observed in the positioning of the particles across one stream of a multinode parallel acoustic flow cell with the acoustic focusing field OFF and ON. It is clear that focusing provides tight lateral positioning within a given sample stream, as the X position of the brightest pixel (BPX) is much more tightly distributed when the acoustic field is on. Similar performance is seen across all streams in a parallel flow cell (Supporting Information Figure S-1). For data analysis purposes, the data acquisition system defines a range of pixels as a stream, e.g. from 1020 to 1090 for stream 10 in Figure 3A. The large number of events on the edge of the window results from any partial event at the edge of window being recorded with a BPX value of the edge pixel value, which biases the histogram data as any event in the unfocused data that just clips the window is recorded as the last pixel of the window. For the data in figure 3, 16 streams were defined and each event is assigned both a BPX value, based on where its brightest pixel occurred on the sensor, and a stream number based on which stream window it appeared in (Supporting Information Figure S-1). As each event within a stream window is identified with a stream number, it is simple to correlate events based on the stream and for events in each stream to be analyzed separately. Our results show that a similar number of particles are found in each of the 16 focused streams (Supporting Information Figure S-1).
Figure 3. Acoustic focusing increases system performance.
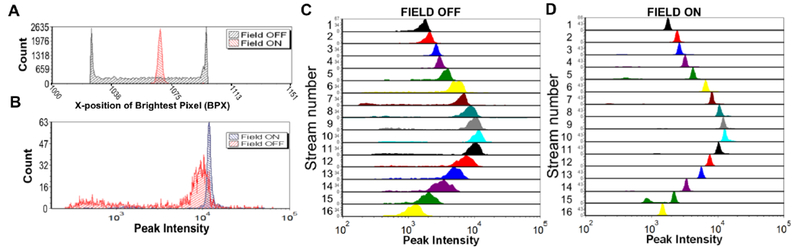
(A) Histogram of the position of particles across stream 10 of a flow cell supporting 16 acoustic focusing nodes. When the field is on (red) the focusing is clear as particles appear in a tight Gaussian distribution, whereas when the focusing is off (black) the particles are broadly distributed across the collection window (B) Histogram of the fluorescence peak intensity for a single focusing stream (stream 10), with the focusing field on (blue) and off (red). (C & D) Stacked histogram of the peak intensity of the particles across all 16 focusing regions with the acoustic focusing field off (C) and on (D).
Using standard flow cytometry gating approaches on the stream parameter, we can analyze the fluorescence peak intensity of events from a given stream in focused and unfocused states (Figure 3B). There is a large increase in system performance in terms of both fluorescence precision (unfocused 76% CV, focused 6% CV) and optical collection (mean intensity is brighter for focused particles) when the acoustic field tightly focuses the particles. Within a given stream, the particle positions clearly differ in focused and unfocused scenarios (Figure 3A). As such, much of the broadening of the optical collection of the unfocused beads population might be due to the variation across the 1D width of the laser excitation profile.
The non-uniform laser excitation (Figure 1D) and the difficulty of efficiently collecting light over a wide field of view creates variation in the fluorescent intensity profile across the 16 focusing regions. Nonetheless, it can be seen that unfocused particles have significantly lower precision (Figure 3C) as compared to focused particles (Figure 3D), regardless of stream position. Though there is variation in optical excitation and collection efficiency, the benefit of focusing is clear. In future work, a per-channel gain could be applied to normalize across streams. On an optimized system where the laser profile and the Powell lens are fully matched, it is expected that ~5% variation along the focused laser line could be achieved.
2. Evaluation of Optical Sensitivity and Resolution
Eight peak ultra-rainbow calibration beads (3μm diameter, RCP-30-5A Spherotech, Lake Forest, IL) are used as a calibration standard to characterize the performance of the parallel cytometer. Figure 4A shows the optical performance across 17 focusing nodes. While full resolution of the calibration beads is seen in the center of the channel where both the laser excitation and optical collection efficiency are highest, only the brightest 4-5 populations are seen at the edges where the laser excitation is lowest and the optical collection is least efficient. Looking at the highest performing stream (stream 10), seven fluorescent peaks are clearly visible with good population separation and CV’s comparable to those acquired on a commercial cytometer (~3-6%) (Figure 4B). An observed eighth population might correspond to blank beads, but further work is needed to confirm this conclusion. Similar performance was seen with 6 μm Rainbow calibration beads (Supporting Information Figure S-2).
Figure 4. Eight peak rainbow beads.
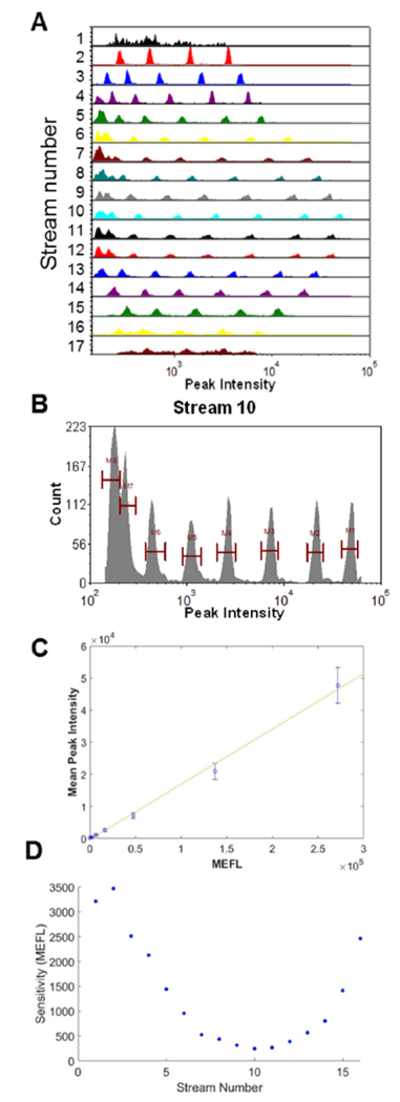
(A) Histogram of the peak intensity for all 17 streams. (B) Histogram of peak intensity for a stream 10 (C) Mean peak intensity of each bead population in stream 10 plotted vs. the estimated mean equivalent number of Fluorescein molecules per bead. Error bars are the standard deviation of each bead population. (D) Current maximum sensitivity to a threshold that is two standard deviations above the background noise of the system.
In this study, we also find that increasing the applied laser power increases the sensitivity across the entire channel width (see Supporting Information Figure S-3). Our current laser power per area (150 mW over 20 μm × 2000 μm) is ~5X less than that used by a typical flow cytometer (20 mW over 20μm × 50 μm). This observation suggests that we are far from any power saturation effects, so fluorescence sensitivity should increase with higher applied laser power. Thus, full resolution of all bead populations in all streams may be possible with a higher power laser well matched to the Powell lens.
Using the microsphere standards, we see that our system has a linear response when plotted as estimated Molecules of Equivalent Fluorescein (MEFL) vs fluorescence peak intensity. We note that these commercial microspheres have a broad fluorescence emission spectra. As our optical collection system collects light from any wavelengths >488 (through our long pass dichroic beam splitter), the use of Mean Equivalent Fluoresceins (MEFL) is simply used as an approximation of linearity and sensitivity. Thus, the sensitivity values given here are not directly comparable to a standard fluorescein channel on typical commercial cytometers, which typically have filter sets that select for tighter spectral bands. Therefore, our estimates of sensitivity in terms of absolute MEFL are potentially optimistic. Nonetheless, the standard curve of peak intensity vs. estimated MEFL value of each microsphere (as provided by the manufacturer) shows high linearity that demonstrates the quantitative potential of the system (Figure 4C). With regards to sensitivity and resolution, at the center of our system we can clearly resolve the dimmest fluorescently tagged bead, giving us an approximated sensitivity of ~800 MEFL or better. An interpolation of the sensitivity down to a level limited by the background signal plus system noise (background level plus 2 times the standard deviation of the background) suggests the approximate sensitivity for the highest performing stream is a few hundred MEFL (~250 MEFL), while at the system edges the sensitivity is a few thousand MEFL (~3000 MEFL) (Figure 4D). While our sensitivity numbers may be optimistic, they demonstrate the potential of this optical collection paradigm. Future work to optimize such systems with regards to laser power, optical collection efficiency, and signal processing algorithms are likely to be able to achieve sensitivity similar to a conventional flow cytometer.
3. Performance vs. Flow Rate
The performance of our parallel system depends on a consistent interaction between the interrogating laser and the particles of interest. For smaller particle sizes and higher flow rates with larger linear velocities, it becomes increasingly difficult for the acoustic field to tightly focus the particles. Additionally, as the linear velocity through the excitation region increases, the transit time decreases, so the absolute number of photons collected and the sensitivity is reduced. Therefore, we investigated the system performance with varying bead sizes and flow rates.
Three sizes of rainbow calibration beads are analyzed for volumetric flow rates of 250 μl/min up to 10 mL/min. As expected, with a constant camera frame rate (~25 k/s), the peak intensity and the system sensitivity decrease with increasing linear velocity (Figure 5). In addition, the acoustic focusing width of the particles across each of the 16 focusing nodes increases at the highest flow rates and linear velocities (Supporting Information Figure S-4). With this increase in focusing width, the laser/particle interactions broaden and we see increased CVs of the fluorescent parameters of each bead population.
Figure 5. Standard calibration beads vs. flow rate (Stream 10).
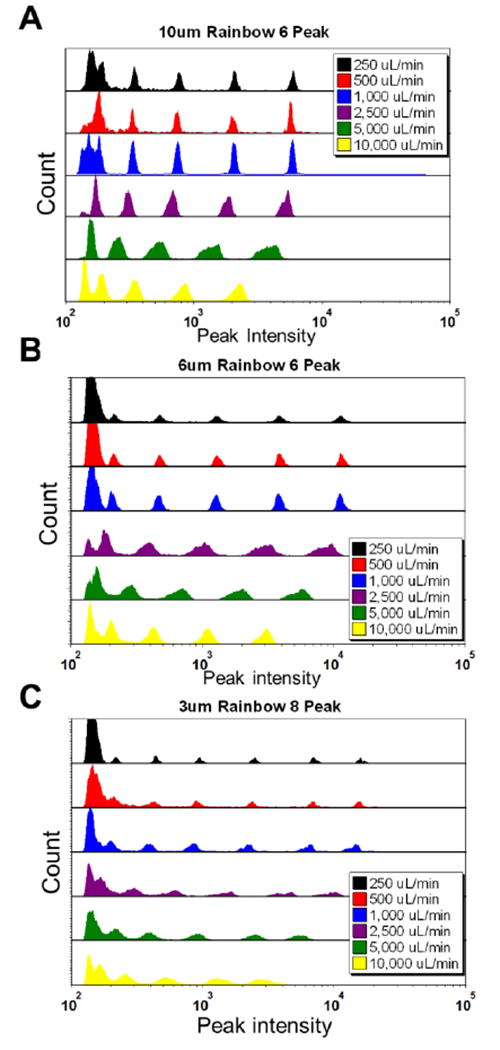
Peak intensity histograms of (A) six peak 10um beads verses flow rate, (B) six peak 6um beads verses flow rate, and (C) 8 peak 3 um beads vs. flow rate.
Despite these two effects (fewer total photons and wider acoustic focusing widths) we still observe strong system performance across a wide range of flow rates and particle sizes. Although the CVs are higher at high flow rates, we see all five fluorescently tagged bead populations for the 10 μm and 6 μm beads at 10 mL/min and achieve moderate performance for the 3 μm 8-peak beads at this flow rate. Additionally, for the constant sample concentration used in these experiments, we observe a linear relationship between event rate and input flow rate (Supporting Information Figure S-5). This result suggests that we detect most beads at all flow rates. Given that linear sensitivity increases with laser power (Supporting Info Figure S-3), performance sufficient for ~500 MEFL sensitivity may be possible even at 10 mL/min flow rates.
4. High Analysis Rates
The ability of the cytometer to run at high analytical event rates is characterized by running 6 μm Nile Red beads at 10 mL/min (Supporting Information Figure S-6). At these high flow rates, 957,366 particles are analyzed within 9.4 seconds, yielding a minimal digital processing rate of ~102k/s. The imperfect acoustic focusing of 6 μm particles at these flow rates may lead to some particles being missed, but this study demonstrates that the data system can process at least ~100k events/s in this proof-of-principle implementation.
DISCUSSION
We demonstrate that line-focused optical excitation of parallel acoustically focused streams provides optical sensitivity and precision comparable to a traditional flow cytometer. While the optical performance is complicated by issues of even light delivery and challenges in high numerical aperture wide-field optical collection, our approach offers the advantage of highly increased volumetric sample delivery rates and the potential to greatly increase analytical collection rates. Our current cytometer design uses three key components: 1) a multi-node acoustic focusing flow cell, 2) a wide line laser excitation profile, and 3) a high-speed imaging detector, all of which are inherently parallelizable. Combining these components into a single platform yields a compact and cost effective instrument for highly parallel analysis. The high event rates and volumetric throughputs of this platform will allow parallel flow cytometry to be applied to a number of traditional areas, as well as new application spaces where throughput is critical or desirable.
Notably, the optical system described here could also be used with any approach that produces parallel streams of flowing particles. There are many excellent examples of such systems using inertial focusing,21 surface acoustic waves,27 dielectrophoresis,28,29 and microfluidics.30,31 Additionally, while other approaches to parallel optics have been developed, which include micro-lens arrays and fiber optic methods, 32,33 the simplicity of our optical system, both in terms of optical alignment and number of components, makes it a compelling choice to include in an instrument.
It is also useful to consider this work in context of excellent examples of image flow cytometry, 25,26 where the highest speed imaging cytometer, which uses acoustic focusing to maintain a single node in the focal plane,26 can use offline analysis to analyze low magnification images of samples at analytical rates of ~200k events/s at reduced volumetric rates of ~1 mL/min. While the low magnification images may offer some ability to examine particle morphology, this approach requires image analysis paradigms that are performed offline. Other image cytometers employ higher resolution optics to extract greater morphological details as well as the location of probes within the cell,12,25 although the throughputs of such higher resolution optical systems are generally limited to <5,000 events/s. Comparatively, our approach offers real time data processing, higher volumetric throughput, and simpler optical hardware. While our approach can give parameters such as event width and time of flight, these can be correlated to particle morphology using established approaches.34 Given the factors discussed above, our system is anticipated to be more amenable to future efforts to create sorting systems and for applications requiring higher throughput analysis.
Applications using large sample volumes containing dilute cell concentrations are already addressable using our approach. As our system can analyze samples at 10 mL/min and process data at rates of 100K events/s, simple sample dilution might already allow our system to analyze some rare cell samples. However, applications such as CTCs and fetal cells in maternal blood are more difficult as they require high volumetric and high analytical rate analysis. For example, CTCs can be present at less than 10 cells/mL in a background of ~109 cells/mL in whole blood.35 Therefore, increasing the analytical rate of our platform will make it a more compelling solution for rare cell analysis, which, in the absence of processing steps to remove background cells, will be limited by the overall analytical rate of the system. Most current approaches use a combination of lysis, centrifugation, and antibody based pull down approaches; however, such steps can result in the loss of rare cells, which has led to many alternative approaches.35 Our platform would provide a comparative advantage as it would provide a method for direct high throughput analysis for rare cells in blood, in the absence of any separations step, which may provide more accurate and affordable assays.
To increase cellular analysis rate, we will explore increasing the number of streams in our system and the data acquisition rate. Each approach poses specific challenges. Increasing the stream number in the flow cell will require either a higher drive frequency or a wider flow cell. A higher frequency drive will result in tighter spacing between streams, which may affect future work on multicolor systems, but will increase the number of analysis points in our system. The increased number of analysis points across the same width of flow cell will allow for higher cellular concentrations to be analyzed by our system due to reduced effects of coincident events at any given analysis point. The analysis of increased cellular concentrations at larger numbers of analysis points will result in an overall increase of analysis rate. A wider flow cell could also have a higher volumetric delivery rate and analyze more streams, which would provide a correspondingly higher analysis rate. However, this would require optical excitation and collection across a wider field of view. Based upon our experience, the current width of high-NA optical collection is limited to ~4 mm using 25 mm optics and an 8 mm field of view is possible using 50 mm optics.
Beyond the acoustic and optical limits, the detector technology is a key factor. Despite our high volumetric and analytical throughputs, linear velocities through the interrogating laser beam are still about an order of magnitude smaller than typical cytometers (0.04m/s at 1mL/min, 0.42 m/s at 10 mL/min: compared to a typical cytometer at 1-10 m/s). Given the current system geometry, these parameters result in ~5 frames collected for every particle, even at the highest flow rates. To increase analytical rates, we are exploring use of as few as 3 frames per event to provide accurate data. Clearly, faster cameras or array detectors would improve performance.
In addition to increased analytical rates, multicolor detection is needed to enable typical flow cytometry assays. To that end, we can either explore the use of multiple detectors (1 per detection color) or methods that discriminate multiple colors on a single detector. Use of multiple detectors would enable detection of more fluorescence wavelengths as well as traditional parameters such as forward and side scatter. Such scatter parameters would be helpful in creating a system that could be directly implemented with the many existing flow cytometry assays. However, due to the acoustic focusing, there is a significant amount of free space on the camera’s chip that is currently unused (e.g., dark regions adjacent to each stream in Figure 1E). Applying additional optical filters and a small spatial displacement, may enable this chip space to acquire multiple optical parameters simultaneously on a single array detector.
In summary, using multi-node acoustic standing waves to precisely focus particles for analysis, we demonstrate the ability to analyze 16 streams in real time at high event rates (100k/s) and flow rates (10 mL/min). Without complex fluidic or optical designs, this system allows high throughput analysis in a robust, compact and relatively inexpensive platform. This new approach to parallel flow cytometry has the potential to open new applications for flow cytometry including extremely rare cell analysis and inherently dilute large volume samples.
CONCLUSION
Using the above approaches, our goal is to construct very high throughput flow cytometry systems based on the new analysis paradigm demonstrated here. Although we have demonstrated an analytical rate of ~100k/s, truly rare blood cell applications would benefit from analytical rates greater than 1M events/s. Within the optical and acoustic bounds described above, we see potential pathways leading to such rates. For example, using a 50 mm optical system, we could drive our standing wave at 5 MHz 128 nodes across an 8 mm field of view to achieve 640k/s. Given this, a simple two-fold increase in detector speed would enable our approach to reach 1.28M events/s, providing a transformational advance in flow cytometry.
Supplementary Material
ACKNOWLEDGMENTS
We thank Alireza Goudarzi for his assistance. This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R21GM107805 and the NSF under award OCE 1131134. JJJ was supported by the Academic Science Education & Research Training (ASERT) program, NIH K12GM088021. Some data were generated in the UNM Shared Flow Cytometry & High Throughput Screening Resource Center supported by the UNM Health Sciences Center and the UNM Cancer Center with funding from NCI 2P30 CA118100-11 “UNM Cancer Center Support Grant”.
TAW, GCM, APS, and SWG declare financial interest in this technology as it is licensed by Eta Diagnostics, Inc., a company they have a financial stake in. GCM, BSE, APS, and SWG declare they are working with Eta Diagnostics, Inc., to commercialize the technology. AS has financial interest in DarklingX, LLC, which develops data systems.
Footnotes
ASSOCIATED CONTENT
Supporting Information supplied in separate file.
REFERENCES
- (1).Givan AL Flow cytometry: first principles; John Wiley & Sons, 2013. [Google Scholar]
- (2).Piyasena ME; Graves SW Lab Chip 2014, 14, 1044–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bacher P; Scheffold A Cytometry Part A 2013, 83, 692–701. [DOI] [PubMed] [Google Scholar]
- (4).Alix-Panabières C; Pantel K Clinical chemistry 2013, 59, 110–118. [DOI] [PubMed] [Google Scholar]
- (5).Jaye DL; Bray RA; Gebel HM; Harris WAC; Waller EK The Journal of Immunology 2012, 188, 4715–4719. [DOI] [PubMed] [Google Scholar]
- (6).Hedley BD; Keeney M International journal of laboratory hematology 2013, 35, 344–350. [DOI] [PubMed] [Google Scholar]
- (7).Saunders MJ; Graves SW; Sklar LA; Oprea TI; Edwards BS Assay and drug development technologies 2010, 8, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kuckuck FW; Edwards BS; Sklar LA Cytometry 2001, 44, 83–90. [PubMed] [Google Scholar]
- (9).Edwards BS; Sklar LA J. Biomol. Screen 2015, 20, 689–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Olson RJ; Sosik HM Limnol. Oceanogr. Methods 2007, 5, 195–203. [Google Scholar]
- (11).Dubelaar GBJ; Gerritzen PL; Beeker AER; Jonker RR; Tangen K Cytometry 1999, 37, 247–254. [PubMed] [Google Scholar]
- (12).Sieracki CK; Sieracki ME; Yentsch CS Marine Ecology Progress Series 1998, 285–296. [Google Scholar]
- (13).Mandal PK; Biswas AK; Choi K; Pal UK American Journal Of Food Technology 2011, 6, 87–102. [Google Scholar]
- (14).Shapiro HM Practical flow cytometry; John Wiley & Sons, 2005. [Google Scholar]
- (15).Goddard G; Martin JC; Graves SW; Kaduchak G cytometry part A 2006, 69, 66–74. [DOI] [PubMed] [Google Scholar]
- (16).Goddard GR; Sanders CK; Martin JC; Kaduchak G; Graves SW Analytical chemistry 2007, 79, 8740–8746. [DOI] [PubMed] [Google Scholar]
- (17).Ward M; Turner P; DeJohn M; Kaduchak G Current Protocols in Cytometry 2009, 1–22. [DOI] [PubMed] [Google Scholar]
- (18).Picot J; Guerin CL; Kim CLV; Boulanger CM Cytotechnology 2012, 64, 109–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wlodkowic D; Darzynkiewicz Z In Recent Advances in Cytometry, Part a: Instrumentation, Methods, Fifth Edition, Darzynkiewicz Z; Holden E; Orfao A; Telford W; Wlodkowic D, Eds.; Elsevier Academic Press Inc: San Diego, 2011, pp 105–125. [Google Scholar]
- (20).Edwards BS; Zhu JS; Chen J; Carter MB; Thal DM; Tesmer JJG; Graves SW; Sklar LA Cytometry Part A 2012, 81A, 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hur SC; Tse HTK; Di Carlo D Lab Chip 2010, 10, 274–280. [DOI] [PubMed] [Google Scholar]
- (22).Lenshof A; Laurell T Chemical Society Reviews 2010, 39, 1203–1217. [DOI] [PubMed] [Google Scholar]
- (23).Piyasena ME; Suthanthiraraj PPA; Applegate RW; Goumas AM; Woods TA; Lopez GP; Graves SW Analytical Chemistry 2012, 84, 1831–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Suthanthiraraj PPA; Piyasena ME; Woods TA; Naivar MA; Lopez GP; Graves SW Methods 2012, 57, 259–271. [DOI] [PubMed] [Google Scholar]
- (25).Basiji DA Imaging Flow Cytometry: Methods and Protocols 2016, 13–21. [DOI] [PubMed] [Google Scholar]
- (26).Zmijan R; Jonnalagadda US; Carugo D; Kochi Y; Lemm E; Packham G; Hill M; Glynne-Jones P RSC Advances 2015, 5, 83206–83216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shi J; Mao X; Ahmed D; Colletti A; Huang TJ Lab Chip 2008, 8, 221–223. [DOI] [PubMed] [Google Scholar]
- (28).Wang L; Flanagan LA; Jeon NL; Monuki E; Lee AP Lab Chip 2007, 7, 1114–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Holmes D; She JK; Roach PL; Morgan H Lab Chip 2007, 7, 1048–1056. [DOI] [PubMed] [Google Scholar]
- (30).McKenna BK; Evans JG; Cheung MC; Ehrlich DJ Nature methods 2011, 8, 401–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ateya DA; Erickson JS; Howell PB; Hilliard LR; Golden JP; Ligler FS Analytical and bioanalytical chemistry 2008, 391, 1485–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kuo J-N; Hsieh C-C; Yang S-Y; Lee G-B Journal of Micromechanics and Microengineering 2007, 17, 693. [Google Scholar]
- (33).Gilbert JR; Sinofsky E; Deshpande M; Google Patents, 2008. [Google Scholar]
- (34).Maltsev VP Review of Scientific Instruments 2000, 71, 243–255. [Google Scholar]
- (35).Nagrath S; Sequist LV; Maheswaran S; Bell DW; Irimia D; Ulkus L; Smith MR; Kwak EL; Digumarthy S; Muzikansky A Nature 2007, 450, 1235–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.