

Abstract
The cannabinoid receptor 1 (CB1) is a G protein-coupled receptor (GPCR) that is located primarily in the central nervous system. CB1 is a therapeutic target which may impact pathways to mediate pain, neurodegenerative disorders, hunger, and drug-seeking behavior. Despite these benefits, development of orthosteric therapeutic compounds, which target the endogenous ligand-binding site of CB1, has been challenging due to detrimental side effects including psychoactivity, depression, and suicidal thoughts. However, CB1 also has an allosteric binding site(s), which is topographically distinct from the orthosteric site. Allosteric modulation of CB1 has a number of potential advantages including providing a mechanism for more precise control of downstream pathways and circumventing these side effects. In this review, we summarize the concept of allosteric modulation and focus on the structure activity relationship studies of the well-characterized allosteric modulators, ORG27569 and PSNCBAM-1 and their derivatives, and a few other recent modulators. We review studies on the properties of these modulators on CB1 signaling in cells and their effects in vivo. While many current allosteric modulators also produce complex outcomes, they provide new advances for the design of CB1 centered therapeutics.
Keywords: GPCR, functional selectivity, CB1, allosteric modulation, allosteric ligands, ligand binding
Introduction
The cannabinoid receptor 1 (CB1) is an attractive drug target for the management of pain (Donvito et al. 2017; Woodhams et al. 2017), neurodegenerative disorders (Fernández-Ruiz et al. 2015), obesity (Silvestri and Di Marzo 2013; Simon and Cota 2017), and substance abuse (Maldonado et al. 2006; Smith et al. 2010). Traditional approaches to designing therapeutics for targeting CB1 have focused on the orthosteric site of the receptor; the site where the endogenous cannabinoids such as anandamide (AEA) and 2-arachidonylglycerol (2-AG) bind, and where the phytocannabinoid Δ9-tetrahydrocannabinol (Δ9-THC) binds (Pertwee 2008). However, ligands targeting the orthosteric site on CB1 can cause unwanted psychoactive effects, tolerance and dependence. Allosteric ligands, which bind to site(s) topographically distinct on CB1 have been identified. These novel CB1 ligands exhibit new mechanisms of action and hold promise for discovery of CB1-targeting drugs which may have less side effects than the drugs developed by targeting the CB1 orthosteric site (Nguyen et al. 2016).
Defining allostery and allosteric parameters
Allosteric ligands can impact receptor activity in several ways, including: (1) modulate the binding affinity of the orthosteric ligands to regulate potency; (2) control the signaling pathways of the orthosteric ligands including in a biased manner; and (3) elicit signaling responses independent of orthosteric ligand binding (Figure 1) (Gentry et al. 2015). Using equilibrium binding, an allosteric modulator can be characterized by KB, the equilibrium dissociation constant, of the allosteric ligand in the presence of an orthosteric ligand (Conn et al. 2009) and by α, the cooperativity factor, which defines the extent of the allosteric interaction (Christopoulos and Kenakin 2002). At the receptor binding level, an α greater than 1 represents a positive allosteric modulator (PAM), which increases the binding of the orthosteric agonist, and decreases the binding of an orthosteric inverse agonist. In contrast, a ligand with an α less than 1 represents a negative allosteric modulator (NAM) for agonist binding. Binding of NAMs impacts orthosteric compounds in an opposite manner to a PAM; that is, a NAM enhances inverse agonist binding and decreases agonist binding. Both a competitive orthosteric inverse agonist and a NAM can show a decrease in binding of an orthosteric agonist and its corresponding receptor activity. However, this can be differentiated from one another in kinetic binding analysis; dissociation rates will change in the presence of an allosteric modulator due to conformational changes caused by modulator binding to a different site on the orthosteric ligand-receptor complex (Christopoulos and Kenakin 2002; Gregory KAREN J et al. 2010). Some allosteric modulators can themselves behave as allosteric agonists, eliciting signaling by binding to an allosteric site on a receptor without any orthosteric ligand (Schwartz and Holst 2007). Furthermore, some allosteric modulators can be both, and act with or without an orthosteric ligand (Schwartz and Holst 2006).
Figure 1.
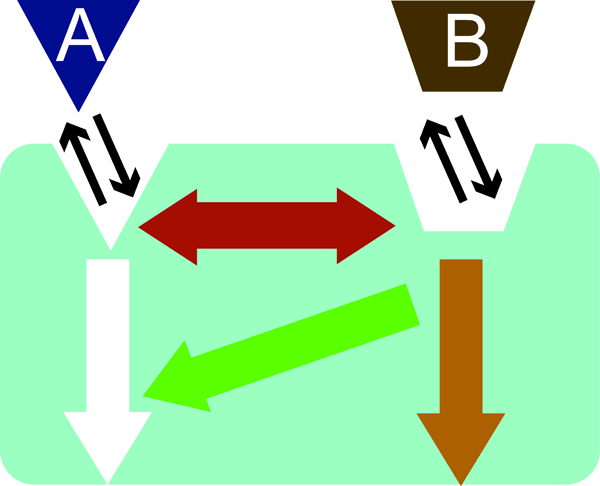
Scheme of allosteric modulation. An orthosteric agonist (blue triangle) binds to the orthosteric site and elicit its effects (white arrow). An allosteric agonist binds to a separate site on the receptor (brown trapezoid), and can influence binding (red arrow) and efficacy (green arrow) of the orthosteric agonist, or cause its own signaling effects (orange arrow) separate from the effects of the orthosteric ligand (Conn et al. 2009).
Advantages of targeting an allosteric site
Ligands targeting an allosteric site have multiple benefits relative to those which target the orthosteric site. Allosteric compounds have the potential to be more selective for subtypes of a receptor due to higher sequence variation between subtypes in the allosteric binding site than the orthosteric site. Allosteric modulators often promote cooperativity between orthosteric and allosteric sites that is exclusive to a receptor subtype (Conn et al. 2009). In CB1, the orthosteric ligand binding site(s) are in the more conserved transmembrane domain (Hua et al. 2016; Shao et al. 2016; Hua et al. 2017), while the allosteric sites are proposed to be near more variable regions such as the external or internal loops (Fay and Farrens 2013; Shore et al. 2014; Stornaiuolo et al. 2015). Allosteric modulators can also allow for improved spatiotemporal regulation of an endogenous ligand. A positive allosteric modulator targets its receptor, enhancing the effects of the orthosteric ligand only during the time and location in which the endogenous ligand is active. An orthosteric ligand competes with the endogenous ligand and may target all receptors continually until it is metabolized (Burford et al. 2015). Pathway selectivity is another advantage of allosteric modulators. Whereas an orthosteric ligand may cause therapeutic and unwanted side effects by affecting multiple pathways, an allosteric modulator may circumvent side effects through biased regulation of signaling pathways. Moreover, allosteric binding alone may cause signaling which is unique to the allosteric modulator (Gao and Jacobson 2013). Allosteric modulators can elicit responses in a saturable manner with a ceiling on their effect once the receptors are occupied. This characteristic is beneficial because it makes effects of the compound more predictable, and reduces the risk of overdose (Gregory Karen J. et al. 2013).
FDA approved allosteric modulators
Basic research investigating allosteric modulators has led to the development and FDA approval of two medications that mechanistically function as allosteric modulators of G protein coupled receptors (GPCRs), Cinacalcet and Maraviroc (Dorr et al. 2005; Harrington and Fotsch 2007). Cinacalcet functions as a PAM of the calcium sensing receptor (CSR), a receptor in the parathyroid gland that monitors calcium levels in the blood to control the release of parathyroid hormone (PTH) by monitoring calcium levels in the blood. Cinacalcet binds the allosteric site of the CSR which leads to increased sensitivity of the receptor to calcium in serum, and lowers the serum concentration of PTH, in turn lowering the calcium levels in the blood. These actions are beneficial for treating hyperparathyroidism (Harrington and Fotsch 2007). Maraviroc is a NAM used to treat HIV infection. It binds to the allosteric site on the co-receptor CC chemokine receptor 5 (CCR5) that elicits a conformational change of CCR5, which prevents HIV entry into a cell (Dorr et al. 2005).
Scope of this review
CB1 is a GPCR with one or more allosteric sites (Fay and Farrens 2013; Shore et al. 2014; Stornaiuolo et al. 2015). There are several compounds that have been discovered that target allosteric sites on CB1 such as ORG27569 (Price et al. 2005), PSNCBAM-1 (Horswill et al., 2007), RTI-371 (Navarro et al. 2009), lipoxin A4 (Pamplona et al. 2012), pepcans (Bauer et al. 2012), ZCZ011 (Ignatowska-Jankowska et al. 2015) and GAT211 (Nguyen et al. 2016; Laprairie et al. 2017). This review will focus on the research centered on the allosteric modulators of CB1, ORG27569 and PSNCBAM-1, and their derivatives, and a few other recent compounds.
Allosteric Modulators of CB1: Binding and SAR studies
The indole-2-carboxamides
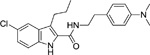
The indole-2-carboxamides were first described by Price and colleagues in 2005 as a series of compounds identified by Organon Research (Price et al. 2005). The general structure of indole-2-carboxamide includes a bicyclic aryl fragment and an amide fragment (Khurana et al. 2014). The compounds include Org27759 (Table 1), Org29647, and Org27569. When tested in equilibrium binding assays, these compounds caused an increase in binding of the CB1 agonist CP 55,940, and a decrease in binding of the CB1 inverse agonist SR141716A. Org27569 (Table 1) had the greatest effect on the increase in CP 55,940 binding, with the lowest pKB value (pKB = 6) of the three compounds tested. In addition, the cooperativity factors (α) for all three of the Org compounds were greater than 1 when the CB1 agonist CP 55,940 was employed, indicating a positive cooperativity, while the cooperativity factors for all three of the Org compounds were less than 1, when the CB1 inverse agonist SR141716A was employed, indicating negative cooperativity. This indicates Org27759, Org29647, and Org27569 promote an active conformation of the receptor. Org27569 had the highest α value (α = 14) out of the three compounds tested. Dissociation kinetic studies performed with CP 55,940 provided additional evidence of the allosteric properties of these compounds as they caused a decrease in the rate of orthosteric ligand dissociation (Price et al. 2005).
Table 1.
Structures of select compounds from the SAR studies of Org27569
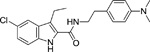
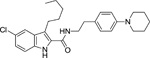
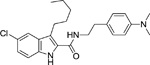
Identified with the names given in:
To further study Org27569, Ahn and colleagues (Ahn et al. 2012) tested its binding properties on two mutated forms of CB1: T210A, an inactive form of the receptor, and T210I, a constitutively active form of the receptor. Wild-type CB1 is intermediate between the two mutant receptors in terms of its biochemistry and cellular activity. Using these receptors, Org27569 promotes a receptor state which increases affinity for an agonist, while decreasing affinity for an inverse agonist. This is consistent with the active conformation of the receptor. While Org27569 had little additional effect on CP 55,940 binding to the constitutively active T210I receptor, it had a profound effect on the inactive receptor T210A, and its effect on wild-type CB1 was intermediate. That is as expected if Org27569 promotes an active form of the receptor. T210I is already highly active, while Org27569 promotes activity of wild-type CB1 and even more so of T210A. Conversely, in the presence of inverse agonist SR141716A and Org27569, wild type CB1 and inactive T210A had weaker KD values than when presented with SR141716A alone. The constitutively active receptor T210I did not have saturable binding of SR141716A. One would expect weaker binding of the inverse agonist if Org27569 promoted an active state. Furthermore, the rank order is as anticipated of the impact of Org27569 on the T210A, wild-type CB1, and T210I receptors. (Ahn et al. 2012).
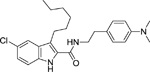
To develop more robust CB1 allosteric modulators than first generation compounds, a number of structure-activity relationship (SAR) studies on the scaffold of Org27569 (indole-2-carboximide) were carried out. Piscitelli and colleagues (Piscitelli et al. 2012) have identified several key SAR factors. The carboxamide function is pivotal. When the amide group was replaced by an ester group, the allosteric modulation of agonist binding was drastically altered and became inhibitory. The piperidin-1-yl group of Org27569 can be optimized by a dimethyl amino group, which led to an increase in allosteric modulation of the CB1 receptor. Additionally, the alkyl group at the C3 position of the indole ring is quite influential. Within this series of compounds, compounds 13 (i.e. 5-chloro-N-(4-(dimethylamino)phenethyl)-3-ethyl-1H-indole-2-carboxamide) and 21 (i.e. 5-chloro-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide) (Table 1) were highly potent, with an EC50 in the nanomolar range (50 nM and 90 nM, respectively) (Piscitelli et al. 2012). The importance of the C3 alkyl group was quickly confirmed by Ahn and colleagues (Ahn, Mahmoud, Samala, et al. 2013) through the compound ICAM-b (5-chloro-3-pentyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide) (Table 1). This compound possesses an n-pentyl group at the C3 position of the indole ring. It exhibited a KB of 470 nM and an α value of 18, which is significantly improved from the binding cooperativity of ORG27569 (α = 7, tested in the same binding assay). Following this, a series of analogs of Org27569 with various substitution at C3 of the indole ring were synthesized and assessed for CB1 allostery (Mahmoud et al. 2013; Khurana et al. 2014). It was found that the linear alkyl group is superior to cyclic groups at the C3 position (Mahmoud et al. 2013). The length of the alkyl group at the C3 position of the indole ring can impact both the binding affinity (KB) and binding cooperativity (α), but the influences are not parallel. However, binding cooperativity was generally enhanced with the alkyl group ranging from n-propyl to n-pentyl groups. A key finding in these series of analogs is that the ethylene linker between the amide group and the 4-aminophenyl group is essential for the allosteric effects. Several compounds identified in these series showed improved allostery in comparison with the lead compound Org27569. For instance, 12d (5-chloro-N-(4-(dimethylamino)phenethyl)-3-propyl-1H-indole-2-carboxamide) (Table 1) exhibited a KB of 259 nM and an α of 25 (Khurana et al. 2014). Compound 12f (5-chloro-N-(4-(dimethylamino)phenethyl)-3-hexyl-1H-indole-2-carboxamide) (Table 1) of this series, which had an n-hexyl group at the C3 position, exhibited the lowest KB value in the series at 89 nM, and an α comparable to Org27569 (Khurana et al. 2014). With regard to the optimization of the substitution on the indole ring, it was demonstrated that an electron-withdrawing group at the C5 position such as a chloro or an isothiocyanate group is preferred (Khurana et al. 2014; Kulkarni et al. 2015). The C5-isothiocyanato analog of Org27569 (20, Table 1) displayed a potent negative allostery in functional assays (Kulkarni et al, 2015). Furthermore, it appears that the presence of the indole moiety is critical for the allosteric effects since replacing the indole ring with a benzofuran ring abolished the allosteric binding to the CB1 receptor (Mahmoud et al. 2013). In an attempt to develop photoactivatable ligands that behaved like Org27569, Qiao and colleagues (Qiao et al. 2016) found that addition of an aliphatic azide, phenyl azide, or benzophenone on the scaffolds related to Org27569 were tolerated and showed similar binding properties to their parent compounds.
To date, several structural features have been elucidated as critical for the indole-2-carboxamides to retain or improve allosteric modulation of CB1. These include the presence of an indole ring, having a short-chain alkyl group at the C3 position of the indole ring and an electron-withdrawing group at the C5 position of the indole ring, maintaining an ethylene linker between the amide group and the aminophenyl group, and introducing a dimethylamino group in lieu of the piperidinyl group (Piscitelli et al. 2012; Ahn, Mahmoud, Samala, et al. 2013; Khurana et al. 2014). These preliminary SAR will guide the future development of more potent CB1 allosteric modulators using the indole-2-carboxamide scaffold.
Diarylureas
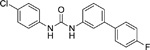
PSNCBAM-1 (Table 2), a diarylurea allosteric modulator of CB1, was first described and characterized by Horswill and colleagues (Horswill et al. 2007). Similar to ORG27569, PSNCBAM-1 increased the binding of the CB1 agonist CP 55,940, while reducing the binding of the inverse agonist SR141716A. This indicated that PSNCBAM-1 promotes an active conformation of CB1 (Horswill et al. 2007).
Table 2.
Structures of select compounds from SAR studies on PSNCBAM-1.
Identified with the names given in:
Following its identification, several SAR studies have been focused on PSNCBAM-1 to improve the allosteric modulation effects of this class of compounds. German and colleagues have found that the pyrrolidinyl ring is not essential since it can be replaced by a dimethylamino group (German et al. 2014). In addition, the chloro group can be replaced by other halogens and electron-withdrawing groups. The most potent compounds from this series were 29 (1-(4-cyanophenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridin-2-yl)phenyl)urea) and 11 (1-(4-chlorophenyl)-3-(3-(6-(dimethylamino)pyridin-2-yl)phenyl)urea) (Table 2) (German et al. 2014). Similarly, Kulkarni and colleagues showed that some electron-withdrawing groups were tolerated in replacement of the chloro group (Kulkarni et al. 2015).
To develop new scaffolds based on the structure of PSNCBAM-1, Khurana and colleagues (Khurana et al. 2017) replaced the pyridine ring with a pyrimidine ring, which contains two nitrogen atoms that possibly form two scaffolds containing pyrimidin-2-yl and pyrimidin-4-yl. Some compounds derived from the new scaffolds retained the positive modulation of CP 55,940 binding and their binding affinity (KB) and cooperativity (α) were similar to PSNCBAM-1. The structural optimization of the two new scaffolds demonstrated that the substituent on the pyrimidinyl ring is instrumental and a 1-pyrrolidinyl group is optimal. Similar to an early finding (German et al. 2014), replacing the chloro group with a cyano group is favorable. Two compounds (Table 2), 7d (1-(4-cyanophenyl)-3-(3-(2-(pyrrolidin-1-yl)pyrimidin-4-yl)phenyl)urea) and 8d (1-(4-cyanophenyl)-3-(3-(4-(pyrrolidin-1-yl)pyrimidin-2-yl)phenyl)urea) were the most effective allosteric modulators from this study (Khurana et al. 2017). Another effort to replace the pyridine ring of PSNCBAM-1 was made by Bertini and colleagues (Bertini et al. 2017) who replaced the pyridine ring with a phenyl ring, with which the allosteric effects seen in PSNCBAM-1 were maintained. Further efforts to explore the diarylurea scaffold include replacing the urea functionality with an amide, and introducing an NH group between the center phenyl and the pyridine ring. It was found that replacing the urea group with an amide group abolished the allosteric effects on CP 55,940 binding. Compounds SN15b (1-(4-chlorophenyl)-3-(3-((6-(pyrrolidin-1-yl)pyridin-2-yl)amino)phenyl)urea) and SC4a (1-(4-chlorophenyl)-3-(3’-(pyrrolidin-1-yl)-[1,1’-biphenyl]-3-yl)urea) (Table 2) were the most successful compounds from this study, with EC50 values of 1.3 μM and 0.043 μM in augmentation of CB1 agonist binding. Although compound SN15b is weaker than PSNCBAM-1 (EC50 = 0.27 μM) in modulation of CB1 agonist binding, it behaved as potent as PSNBAM-1 in modulation of the agonist–induced MAPK/ERK phosphorylation.(Bertini et al. 2017). Recently, Nguyen and colleagues have found that the 2-pyrrolidinyl pyrimidinyl moiety of PSNCBAM-1 is not essential for CB1 allostery, and it can be replaced with a para-fluoro substituted phenyl ring, such that the compound (34, Table 2) can be an allosteric modulator of CB1 comparable to the prototypical PSNCBAM-1, but with significantly improved half-life (t½ ) (Nguyen et al. 2017).
Collectively, current SAR studies indicate several key factors for future optimization of the diaryl urea scaffold. These include maintaining the urea functionality, introducing a cyano or other strong electron-withdrawing group to replace the chloro group and incorporating non-heteroaryl or other heteroaromatic rings to replace the pyridine ring. By far, this class of compounds offers more opportunities for structural optimization than the scaffold of indole-2-carboxamide does.
Allosteric modulators of CB1 and their effects on CB1 signaling
The indole-2-carboxamides
Org27569 was tested for its effects on G protein coupling of the wild-type CB1, the inactive T210A, and the constitutively active T210I (Ahn et al. 2012). In general, Org27569 inhibited G protein coupling, consistent with the observation of others (Price et al. 2005). Org27569 without CP 55,940 caused a decrease in the basal GTPγS binding of wild-type CB1 and T210I receptor; however, it had no effect on GTPγS binding of the inactive T210A receptor since it is inactive and already does not bind G protein. Org27569 was also able to decrease GTPγS binding in the presence of the agonist CP 55,940, showing that its effects on GTPγS binding, and thus G protein coupling activity, was inhibited in both basal and CP 55,940-induced activated states. The cellular internalization of CB1 with Org27569 was studied, since CB1 typically is internalized upon prolonged activation. It was found that Org27569 promoted cellular internalization of the wild-type CB1 receptor (Ahn et al. 2012). Since some wild-type CB1 receptor is internalized prior to treatment (Leterrier et al. 2004), however (perhaps due to constitutive activity), the inactive T210A receptor that remains on the cell surface until activated was evaluated (Ahn et al. 2012). When either Org27569 alone, or Org27569 plus CP 55,940 were introduced, the T210A receptors were internalized. This is further suggestive of receptor activation induced by Org27569. Signaling studies involving ERK1/2 and JNK were also performed. Org27569 prevented the CP 55,940-induced phosphorylation of JNK isoforms 1 and 2, while Org27569 alone caused an increase in ERK1/2 phosphorylation deceptively similar to CP 55,940 treatment alone; however, it was not pertussis toxin sensitive, rather it was β-arrestin siRNA sensitive. Org27569 with CP 55,940 also showed an increase in ERK1/2 phosphorylation, though not to the same degree as when the cells were treated with Org27569 alone. The results of these experiments indicated that Org27569 displayed functional selectivity (Ahn et al. 2012).
Additionally, Ahn and colleagues (Ahn, Mahmoud, Shim, et al. 2013) observed that Org27569 treatment caused phosphorylation of c-src, MEK1/2, and ERK1/2. In addition, Org27569 caused co-localization of CB1 and β-arrestin 1 consistent with its role in signaling. Further, it was found that β-arrestin 2 mediates the internalization of the receptor after treatment with either CP 55,940 or Org27569 (Ahn, Mahmoud, Shim, et al. 2013).
Four compounds from the study by Mahmoud and colleagues (Mahmoud et al. 2013) were evaluated for their effects on G protein coupling with the GTPγS binding assay. While all compounds decreased CP 55,940-induced G protein coupling to CB1, compound 11j had an especially robust effect on both agonist-induced GTPγS binding, and basal levels of GTPγS binding (Mahmoud et al. 2013). Two compounds (12d and 12f) from the study by Khurana and colleagues (Khurana et al. 2014) were evaluated in the GTPγS binding assay, and both caused a decrease in CP 55,940-induced GTPγS binding. Compounds 12d and 12f were additionally tested for ERK1/2 phosphorylation with CP 55,940, and induced ERK1/2 phosphorylation in a G protein-independent manner. β-arrestin 1 siRNA knockdown caused a loss of this ERK1/2 phosphorylation, indicating that these compounds induced ERK1/2 phosphorylation via the β-arrestin 1 pathway (Khurana et al. 2014). Similarly, ICAM-b negatively modulated CP 55,940-induced G-protein-coupling of CB1 and induced ERK1/2 phosphorylation via the β-arrestin pathway (Ahn, Mahmoud, Samala, et al. 2013).
Diarylureas
PSNCBAM-1 antagonized CB1 signaling of CP 55,940 in a dose-dependent manner in yeast reporter assays (Horswill et al. 2007). PSNCBAM-1 was also shown to antagonize CB1 signaling in response to other CB1 agonists such as AEA, 2-AG, and WIN55212–2, but the IC50 values for the different agonists varied greatly, suggesting that the antagonistic effect of PSNCBAM-1 in yeast reporter assays might be dependent on the agonist (Horswill et al. 2007). PSNCBAM-1 also decreased GTPγS binding stimulated by either CP 55,940 or AEA. In cyclic AMP (cAMP) accumulation assays, PSNCBAM-1 reduced the accumulation of cAMP due to forskolin stimulation in cells treated with agonists CP 55,940 or AEA (Horswill et al. 2007). Analogs of PSNCBAM-1 from Bertini and colleagues (Bertini et al. 2017) showed an inhibition of CP 55,940-induced serum response element (SRE) expression, which was measured by luciferase luminescence. Compounds SC4a and SN15b were the most potent in this assay. PSNCBAM-1 was tested for its effects on electrophysiology, particularly on miniature inhibitory postsynaptic currents (mIPSCs). PSNCBAM-1 alone did not have an effect on mIPSCs, but pretreatment of the neurons with PSNCBAM-1, followed by treatment with CP 55,940 showed that PSNCBAM-1 was capable of antagonizing the mIPSC frequency reduction caused by CP 55,940. In addition, PSNCBAM-1 reduced agonists WIN55212–2 and CP 55,940-induced GTPγS stimulation in rat cerebellar membranes, further indicating G protein coupling is reduced (Wang et al. 2011). A similar reduction in agonist-induced GTPγS stimulation was seen in CB1 transfected HEK293 cells (Horswill et al. 2007). Derivatives of PSNCBAM-1 7d and 8d previously described (Khurana et al. 2017) also maintained G-protein coupling antagonism. The compounds also stimulated ERK1/2 phosphorylation via the β-arrestin pathway, and based on its pertussis toxin sensitivity, ERK1/2 phosphorylation did not occur via G protein coupling (Khurana et al. 2017).
Allosteric modulators of CB1 and their effects in preclinical models
While substantial advances have been made in the development of CB1 allosteric modulators and the understanding of their cellular mechanisms, pharmacodynamics and pharmacokinetic factors represent significant challenges in translating this basic information to the whole animal and ultimately for the development of candidate medications. While in vivo studies investigating CB1 PAMs or NAMs may yield significant pharmacological effects that are consistent with the effects of CB1 orthosteric agonists or antagonists, this type of evidence is insufficient to exclude the possibility that non CB1 targets mediated the pharmacological effects. Thus, studies investigating CB1 allosteric modulators in rodent behavioral assays must not only demonstrate in vivo activity, but also that the ligand elicits pharmacological effects consistent with an allosteric modulator that require CB1 receptors. Complementary pharmacologic and genetic approaches provide useful tools to determine this important criterion. Accordingly, the availability of CB1 knockout mice render this species more amenable than rats in demonstrating that an allosteric ligand elicits its pharmacological effects through a CB1 dependent manner. As described below, only two CB1 allosteric modulators have been demonstrated to elicit in vivo pharmacological effects through a CB1 mechanism of action, ZCZ011 (Ignatowska-Jankowska et al. 2015) and GAT211 (Slivicki et al. 2017).
Jing and colleagues (Jing et al. 2014) explored the effect of Org27569 on drug-seeking behavior in rats. Rats were trained to self-administer either methamphetamine or cocaine by pressing a lever for 14 days, after which the drug treatments were stopped for 7 days. Rats treated with Org27569 10 minutes before the reintroduction of either cocaine or methamphetamine showed a dose-related reduction in drug-seeking behavior reinstatement; similar results were seen if the rats were treated with the CB1 inverse agonist SR141716A, indicating that the compounds have similar effects on drug-seeking behavior (Jing et al. 2014). In addition, Ding and colleagues (Ding et al. 2014) report that rats exhibited decreased food intake of both plain and palatable food, decreased body weight, as well as a reduction in CP 55,940-induced hypothermia with Org27569 treatment (Ding et al. 2014). However, neither of these studies demonstrated that these pharmacological effects were CB1 dependent. Indeed, another study conducted by Gamage and colleagues (Gamage et al. 2014) using mice failed to establish that Org27569 acts a CB1 allosteric modulator. Although they too found that Org27569 reduced food intake, this anorectic effect occurred in both CB1 knockout and wild type mice, indicating a CB1 independent mechanism of action. Moreover, they found that Org27569 did not alter common in vivo pharmacological effects (i.e., antinociception, catalepsy, and hypothermia) of orthosteric CB1 agonists (i.e., anandamide, CP55,940, and Δ9-tetrahydrocannabinol) and did not alter the discriminative stimulus effects of anandamide in FAAH-deficient mice or Δ9-tetrahydrocannabinol in wild-type mice in the drug discrimination paradigm. Thus, it remains to be determined whether Org27569 decreases methamphetamine or cocaine seeking behavior through a CB1 mechanism of action or through another target PSNCBAM-1 has also been shown to elicit pharmacological effects in rat behavioral assays. Specifically, it reduced food intake and decreased body weight, while vehicle-treated rats gained weight within the same time period. Adverse effects on behavior and toxicity were not seen during the course of the experiments (Horswill et al. 2007). Additionally Gamage and colleagues (Gamage et al. 2017) demonstrated that PSNCBAM-1 attenuated THC-induced antinociception in mice, but it did not reduce THC-induced hypothermia, catalepsy, or hypomotility in mice (Gamage et al. 2017).
Other allosteric modulators of CB1
Aside from PSNCBAM-1 and Org27569 and their derivatives, other allosteric modulators of CB1 have been reported, including endogenously produced pepcans and lipoxin A4, and the racemic compound GAT211 (Bauer et al. 2012; Pamplona et al. 2012; Laprairie et al. 2017). Some endogenously expressed peptides such as Pepcan-12 (RVD hemopressin; RVDPVNFKLLSH) act as negative allosteric modulators of CB1 by causing a loss in agonist binding, negative modulation of agonist-induced accumulation of cAMP, and by abolishing agonist-induced GTPγS binding (Bauer et al. 2012). Lipoxin A4 (LXA4) caused an increase in CP 55,940 binding, and enhances AEA-triggered cAMP inhibition (Pamplona et al. 2012). Additionally, the CB1 allosteric modulator ZCZ011 has been shown to reduce nociceptive behavior in the chronic constriction sciatic nerve injury model of neuropathic pain and carrageenan model of inflammatory pain through a CB1 dependent mechanism (Ignatowska-Jankowska et al. 2015). Moreover, ZCZ011 augmented the pharmacological effects of the orthosteric agonists anandamide and CP55,940 in the drug discrimination and tetrad assays, without causing cannabimimetic psychoactive effects on its own or eliciting rewarding effects in the conditioned place preference paradigm (Ignatowska-Jankowska et al. 2015).
GAT211, structurally similar analog of ZCZ011, has also been demonstrated to elicit in vivo effects that are consistent with actions of a CB1 PAM. The two enantiomers, GAT228 (R) and GAT229 (S) of this racemic compound have been successfully separated and studied (Laprairie et al. 2017). GAT211 is effective in enhancing agonist CP 55,940 binding, and in reducing inverse agonist SR141716A binding. Taken together, these data indicate that GAT211 may shift CB1 to a more activated conformation. An increase in recruitment of β-arrestin 1 was seen in treatment of both GAT211 alone, and GAT211 plus an orthosteric agonist (based on orthosteric agonists 2-AG, AEA, and CP 55,940). GAT228, the R enantiomer of GAT211, acts as an allosteric agonist; it does not require orthosteric binding, and elicits its own signaling. GAT228 had no effect on CP 55,940 binding to CB1, but in signaling assays, it increased ERK1/2 and PLCβ3 phosphorylation, and enhanced β-arrestin 1 recruitment. The S enantiomer of GAT211, GAT229, is a positive allosteric modulator, and enhanced CP 55,940 binding at CB1. GAT229 did not have its own intrinsic activity, instead acting as a positive allosteric modulator for orthosteric agonists of CB1 (Laprairie et al. 2017). In vivo, the racemic mix as GAT211 suppressed Freund’s adjuvant and paclitaxel-stimulated allodynia in wild-type mice. Further, this suppression of allodynia was not present in CB1 knockout mice. Similar to ZCZ011, GAT211 did not alter rotarod performance or body temperature, indicating that it did not elicit cardinal signs of CB1 activation. Furthermore, mice treated repeatedly with GAT211 and challenged with a CB1 antagonist did not show withdrawal symptoms. Collectively, these findings provide proof of principle that a CB1 allosteric agonist can produce pain-relieving effects, while avoiding unwanted effects of CB1 agonists (Slivicki et al. 2017).
Future considerations
The development of allosteric modulators for GPCRs, and CB1, specifically, has shown promising though complex results thus far. The modulators have proven to have unique properties such as pathway specificity and the ability to affect binding of orthosteric ligands. Further exploration could result in therapeutic compounds with less side effects than their orthosteric counterparts. However, it remains important to demonstrate the in vivo pharmacological effects of candidate compounds produce their actions at CB1 receptors. Therapeutic applications of such compounds could include analgesics or weight loss compounds which would avoid psychoactive and mood-altering side effects.
Acknowledgments
Funding
This research was supported in part by NIH grant DA039942 (to D.A.K, A.H.L. and D.L.).
Footnotes
Disclosure statement
The authors do not have a conflict of interest.
References
- Ahn KH, Mahmoud MM, Kendall DA. 2012. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. Journal of biological chemistry. 287(15):12070–12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn KH, Mahmoud MM, Samala S, Lu D, Kendall DA. 2013. Profiling two indole‐2‐carboxamides for allosteric modulation of the CB1 receptor. Journal of neurochemistry. 124(5):584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn KH, Mahmoud MM, Shim J-Y, Kendall DA. 2013. Distinct roles of β-arrestin 1 and β-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1). Journal of biological chemistry. 288(14):9790–9800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Chicca A, Tamborrini M, Eisen D, Lerner R, Lutz B, Poetz O, Pluschke G, Gertsch J. 2012. Identification and quantification of a new family of peptide endocannabinoids (pepcans) showing negative allosteric modulation at CB1 receptors. Journal of biological chemistry. 287(44):36944–36967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini S, Chicca A, Gado F, Arena C, Nieri D, Digiacomo M, Saccomanni G, Zhao P, Abood ME, Macchia M. 2017. Novel analogs of PSNCBAM-1 as allosteric modulators of cannabinoid CB1 receptor. Bioorganic & medicinal chemistry. [2017 Nov 20]:[8 p.]. doi: 10.1016/j.bmc.2017.10.015. [DOI] [PMC free article] [PubMed]
- Burford N, Traynor J, Alt A. 2015. Positive allosteric modulators of the μ‐opioid receptor: a novel approach for future pain medications. British journal of pharmacology. 172(2):277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. 2002. G protein-coupled receptor allosterism and complexing. Pharmacological reviews. 54(2):323–374. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. 2009. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature reviews Drug discovery. 8(1):41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Qiu Y, Jing L, Thorn DA, Zhang Y, Li JX. 2014. Behavioral effects of the cannabinoid CB1 receptor allosteric modulator ORG27569 in rats. Pharmacology research & perspectives. 2(6):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donvito G, Nass SR, Wilkerson JL, Curry ZA, Schurman LD, Kinsey SG, Lichtman AH. 2017. The endogenous cannabinoid system: a budding source of targets for treating inflammatory and neuropathic pain. Neuropsychopharmacology. [2017 Nov 21]:[28 p.]. doi: 10.1038/npp.2017.204. [DOI] [PMC free article] [PubMed]
- Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C et al. 2005. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrobial agents and chemotherapy. 49(11):4721–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JF, Farrens DL. 2013. The membrane proximal region of the cannabinoid receptor CB1 N-terminus can allosterically modulate ligand affinity. Biochemistry. 52(46):8286–8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Ruiz J, Romero J, Ramos JA. 2015. Endocannabinoids and neurodegenerative disorders: Parkinson’s disease, Huntington’s chorea, Alzheimer’s disease, and others Endocannabinoids. Springer; p. 233–259. [DOI] [PubMed] [Google Scholar]
- Gamage TF, Farquhar CE, Lefever TW, Thomas BF, Nguyen T, Zhang Y, Wiley JL. 2017. The great divide: separation between in vitro and in vivo effects of PSNCBAM-based CB1 receptor allosteric modulators. Neuropharmacology. 125:365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamage TF, Ignatowska-Jankowska BM, Wiley JL, Abdelrahman M, Trembleau L, Greig IR, Thakur GA, Tichkule R, Poklis J, Ross RA. 2014. In-vivo pharmacological evaluation of the CB1-receptor allosteric modulator Org-27569. Behavioural pharmacology. 25(2):182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z-G, Jacobson KA. 2013. Allosteric modulation and functional selectivity of G protein-coupled receptors. Drug discovery today: technologies. 10(2):237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry PR, Sexton PM, Christopoulos A. 2015. Novel allosteric modulators of G protein-coupled receptors. The journal of biological chemistry. 290(32):19478–19488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German N, Decker AM, Gilmour BP, Gay EA, Wiley JL, Thomas BF, Zhang Y. 2014. Diarylureas as allosteric modulators of the cannabinoid CB1 receptor: structure–activity relationship studies on 1-(4-Chlorophenyl)-3-{3-[6-(pyrrolidin-1-yl) pyridin-2-yl] phenyl} urea (PSNCBAM-1). Journal of medicinal chemistry. 57(18):7758–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Noetzel MJ, Niswender CM. 2013. Chapter two - pharmacology of metabotropic glutamate receptor allosteric modulators: structural basis and therapeutic potential for CNS disorders In: Kenakin T, editor. Progress in molecular biology and translational science. Academic Press; p. 61–121. [DOI] [PubMed] [Google Scholar]
- Gregory KJ, Valant C, Simms J, Sexton PM, Christopoulos A. 2010. The emergence of allosteric modulators for G protein-coupled receptors GPCR molecular pharmacology and drug targeting: shifting paradigms and new directions (Gilchrist A, ed). New York: John Wiley; p. 61–87. [Google Scholar]
- Harrington PE, Fotsch C. 2007. Calcium sensing receptor activators: calcimimetics. Current medicinal chemistry. 14(28):3027–3034. [DOI] [PubMed] [Google Scholar]
- Horswill J, Bali U, Shaaban S, Keily J, Jeevaratnam P, Babbs A, Reynet C, Wong Kai In P. 2007. PSNCBAM‐1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. British journal of pharmacology. 152(5):805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, Pu M, Korde A, Jiang S, Ho J-H. 2017. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature. 547:468–471. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y, Zhao S, Shui W, Li S, Korde A. 2016. Crystal structure of the human cannabinoid receptor CB 1. Cell. 167(3):750–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Baillie GL, Kinsey S, Crowe M, Ghosh S, Owens RA, Damaj IM, Poklis J, Wiley JL, Zanda M. 2015. A cannabinoid CB1 receptor-positive allosteric modulator reduces neuropathic pain in the mouse with no psychoactive effects. Neuropsychopharmacology. 40(13):2948–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L, Qiu Y, Zhang Y, Li J-X. 2014. Effects of the cannabinoid CB 1 receptor allosteric modulator ORG 27569 on reinstatement of cocaine-and methamphetamine-seeking behavior in rats. Drug and alcohol dependence. 143:251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana L, Ali HI, Olszewska T, Ahn KH, Damaraju A, Kendall DA, Lu D. 2014. Optimization of chemical functionalities of indole-2-carboxamides to improve allosteric parameters for the cannabinoid receptor 1 (CB1). Journal of medicinal chemistry. 57(7):3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana L, Fu B-Q, Duddupudi AL, Liao Y-H, Immadi SS, Kendall DA, Lu D. 2017. Pyrimidinyl biphenylureas: identification of new lead compounds as allosteric modulators of the cannabinoid receptor CB1. Journal of medicinal chemistry. 60(3):1089–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni PM, Kulkarni AR, Korde A, Tichkule RB, Laprairie RB, Denovan-Wright EM, Zhou H, Janero DR, Zvonok N, Makriyannis A. 2015. Novel electrophilic and photoaffinity covalent probes for mapping the cannabinoid 1 receptor allosteric site (s). Journal of medicinal chemistry. 59(1):44–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Kulkarni PM, Deschamps JR, Kelly ME, Janero DR, Cascio MG, Stevenson LA, Pertwee RG, Kenakin TP, Denovan-Wright EM. 2017. Enantiospecific allosteric modulation of cannabinoid 1 receptor. ACS chemical neuroscience. 8(6):1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier C, Bonnard D, Carrel D, Rossier J, Lenkei Z. 2004. Constitutive endocytic cycle of the CB1 cannabinoid receptor. Journal of biological chemistry. 279(34):36013–36021. [DOI] [PubMed] [Google Scholar]
- Mahmoud MM, Ali HI, Ahn KH, Damaraju A, Samala S, Pulipati VK, Kolluru S, Kendall DA, Lu D. 2013. Structure–activity relationship study of indole-2-carboxamides identifies a potent allosteric modulator for the cannabinoid receptor 1 (CB1). Journal of medicinal chemistry. 56(20):7965–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado R, Valverde O, Berrendero F. 2006. Involvement of the endocannabinoid system in drug addiction. Trends in neurosciences. 29(4):225–232. [DOI] [PubMed] [Google Scholar]
- Navarro HA, Howard JL, Pollard GT, Carroll F. 2009. Positive allosteric modulation of the human cannabinoid (CB1) receptor by RTI‐371, a selective inhibitor of the dopamine transporter. British journal of pharmacology. 156(7):1178–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, German N, Decker AM, Langston TL, Gamage TF, Farquhar CE, Li J-X, Wiley JL, Thomas BF, Zhang Y. 2017. Novel diarylurea based allosteric modulators of the cannabinoid CB1 receptor: evaluation of importance of 6-Pyrrolidinylpyridinyl substitution. Journal of medicinal chemistry. 60(17):7410–7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Li JX, Thomas BF, Wiley JL, Kenakin TP, Zhang Y. 2016. Allosteric modulation: an alternate approach targeting the cannabinoid CB1 receptor. Medicinal research reviews. 37(3):441–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamplona FA, Ferreira J, de Lima OM, Duarte FS, Bento AF, Forner S, Villarinho JG, Bellocchio L, Wotjak CT, Lerner R. 2012. Anti-inflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proceedings of the National Academy of Sciences. 109(51):21134–21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. 2008. Ligands that target cannabinoid receptors in the brain: from THC to anandamide and beyond. Addiction biology. 13(2):147–159. [DOI] [PubMed] [Google Scholar]
- Piscitelli F, Ligresti A, La Regina G, Coluccia A, Morera L, Allarà M, Novellino E, Di Marzo V, Silvestri R. 2012. Indole-2-carboxamides as allosteric modulators of the cannabinoid CB1 receptor. Journal of medicinal chemistry. 55(11):5627–5631. [DOI] [PubMed] [Google Scholar]
- Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, McLean A, McIntosh L, Goodwin G, Walker G et al. 2005. Allosteric modulation of the cannabinoid CB1 receptor. Molecular pharmacology. 68(5):1484–1495. [DOI] [PubMed] [Google Scholar]
- Qiao C-J, Ali HI, Ahn KH, Kolluru S, Kendall DA, Lu D. 2016. Synthesis and biological evaluation of indole-2-carboxamides bearing photoactivatable functionalities as novel allosteric modulators for the cannabinoid CB1 receptor. European journal of medicinal chemistry. 121:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz TW, Holst B. 2006. Ago-allosteric modulation and other types of allostery in dimeric 7TM receptors. Journal of receptors and signal transduction. 26(1–2):107–128. [DOI] [PubMed] [Google Scholar]
- Schwartz TW, Holst B. 2007. Allosteric enhancers, allosteric agonists and ago-allosteric modulators: where do they bind and how do they act? Trends in pharmacological sciences. 28(8):366–373. [DOI] [PubMed] [Google Scholar]
- Shao Z, Yin J, Chapman K, Grzemska M, Clark L, Wang J, Rosenbaum DM. 2016. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature. 540(7634):602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore DM, Baillie GL, Hurst DH, Navas F, Seltzman HH, Marcu JP, Abood ME, Ross RA, Reggio PH. 2014. Allosteric modulation of a cannabinoid G protein-coupled receptor binding site elucidation and relationship to G protein signaling. Journal of biological chemistry. 289(9):5828–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri C, Di Marzo V. 2013. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell metabolism. 17(4):475–490. [DOI] [PubMed] [Google Scholar]
- Simon V, Cota D. 2017. Mechanism in endocrinology: endocannabinoids and metabolism: past, present and future. European journal of endocrinology. 176(6):309–324. [DOI] [PubMed] [Google Scholar]
- Slivicki RA, Xu Z, Kulkarni PM, Pertwee RG, Mackie K, Thakur GA, Hohmann AG. 2017. Positive allosteric modulation of cannabinoid receptor type 1 suppresses pathological pain without producing tolerance or dependence. Biological psychiatry. [2017 Nov 20]:[12 p.]. doi: 10.1016/j.biopsych.2017.06.032. [DOI] [PMC free article] [PubMed]
- Smith TH, Sim‐Selley LJ, Selley DE. 2010. Cannabinoid CB1 receptor‐interacting proteins: novel targets for central nervous system drug discovery? British journal of pharmacology. 160(3):454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornaiuolo M, Bruno A, Botta L, La Regina G, Cosconati S, Silvestri R, Marinelli L, Novellino E. 2015. Endogenous vs exogenous allosteric modulators in GPCRs: a dispute for shuttling CB1 among different membrane microenvironments. Scientific reports. 5:15453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Horswill JG, Whalley BJ, Stephens GJ. 2011. Effects of the allosteric antagonist 1-(4-chlorophenyl)-3-[3-(6-pyrrolidin-1-ylpyridin-2-yl) phenyl] urea (PSNCBAM-1) on CB1 receptor modulation in the cerebellum. Molecular pharmacology. 79(4):758–767. [DOI] [PubMed] [Google Scholar]
- Woodhams SG, Chapman V, Finn DP, Hohmann AG, Neugebauer V. 2017. The cannabinoid system and pain. Neuropharmacology. 124:105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]