

Abstract
The development of new knowledge around the genetic determinants of variable drug action has naturally raised the question of how this new knowledge can be used to improve the outcome of drug therapy. Two broad approaches have been taken: a point-of-care approach in which genotyping for specific variants(s) is undertaken at the time of drug prescription, and a pre-emptive approach in which multiple genetic variants are typed in an individual patient and the information archived for later use when a drug with a “pharmacogenetic story” is prescribed. This review will address the current state of implementation, the rationale for these approaches, and barriers that must be overcome. Benefits to pharmacogenetic testing are only now being defined and will be discussed.
One important goal of the broad field of pharmacogenomics is an improved understanding of the mechanisms underlying variability in drug action. However, in addition to knowledge for knowledge’s sake, the great promise of pharmacogenetics is that the information can be used to improve the outcome of drug therapy: reduce variability in drug actions by dose adjustment, develop new drugs or targeted to specific disease mechanisms, and avoid certain drugs altogether in subjects at high risk for serious adverse drug reactions. It has been argued that drug selection or dose adjustment based on pharmacogenetic factors should be as self-evident as dose adjustment based on pharmacokinetic factors, such as dose reduction for renally excreted drugs in the face of renal dysfunction. An opposing view is that pharmacogenetic factors may only account for a small proportion of variability in drug response, and rigorous evidence demonstrating improved clinical outcomes, preferably from randomized clinical trials (RCTs), should be required before pharmacogenetic testing is widely implemented. This ongoing controversy has motivated attention to how evidence should be generated to support tailoring drug therapy based on pharmacogenetics. The rationale for a pre-emptive, multiplexed approach is outlined below. It is clear that this strategy is still in its infancy and the first data on process and clinical outcomes are only now being reported.
Lessons from individual drugs
For some drugs, clinical trials and other data are available that can guide individualization of treatment, and lessons learned with specific drugs can then be applied to the development of implementation programs. Initial implementation programs focused on delivery of genetic information at the point of care when a specific drug was prescribed (sometimes termed “reactive”), while more recently multiplexed, pre-emptive approaches have been developed at a number of centers as described below.
Vitamin K antagonists:
One drug that has been extensively studied is warfarin, because metrics of drug effect, such as time in therapeutic range or time to therapeutic range or steady-state dose requirement to achieve target INR values are readily obtained; the genetic basis for variability in steady-state dose requirements is well supported; the drug is widely used; inter-individual variability in dose requirement is substantial; and a narrow therapeutic window exists to achieve effective anticoagulation and avoid bleeding complications. Indeed, warfarin was one of the first drugs for which specific genetic variants (in this case CY2C9*2 and CYP2C9*3) were associated with reduced dose requirement in Caucasian subjects.1–3 Discussions to conduct trials to determine the efficacy of CYP2C9 genotyping on warfarin dose requirement in the early 2000s were shelved when it was discovered that a common promoter variant in VKORC1, encoding the warfarin target,4 contributes at least as importantly as variants in CYP2C9 to warfarin dose requirements, particularly during drug initiation.5,6
Four large trials have randomized subjects to a pharmacogenetically-guided dosing strategy versus a clinical dosing strategy.7 The COAG trial8 randomized patients to a dose defined by a complex clinical algorithm that included variables such as interacting drugs and gender, or an algorithm that included these factors plus pharmacogenetic determinants of drug dose. COAG failed to demonstrate any difference in primary outcome (time in therapeutic range for the first 30 days of therapy) and in fact the pharmacogenetic arm performed less well than the clinical only arm in African-American subjects. The latter result likely reflects the different variants driving warfarin dose requirements among African Americans: CYP2C9*2 and *3 are much less common; other variants (CYP2C9*5, *6, *8, and *11) are more frequent but were not included in the COAG dosing algorithm.9,10 Like COAG, a European group randomized patients receiving the vitamin K antagonists acenocoumarol or phenprocoumon to a clinical + genetic algorithm versus a clinical algorithm only and found no difference in time in therapeutic range during the first 12 weeks of treatment.11 On the other hand, the EU-PACT study group randomized patients to a standard dosing regimen or a genetically-guided one – one that may highlight the “real-world” impact of genetic variation – showed an improvement to time in therapeutic range during the first 3 months of treatment.12 Most recently,13 GIFT (the Genetic Informatics Trial) demonstrated a statistically significant improvement in an adverse event-focused primary outcome (a composite of death, major bleeding, INR≥4, or venous thromboembolism) in subjects randomized to a clinical algorithm + genotype information versus clinical algorithm after orthopedic surgery. Time in therapeutic range during the first 30 days was slightly but not significantly (P=0.06) better in the genotype-guided group. The genotypes assessed included not only the common VKORC1 promoter and CYP2C9*2 and *3, but also CYP4F2 V433M (a determinant of vitamin K metabolism that also affects warfarin dose14). GIFT stratified by ancestry and included fewer African-American subjects (6.4%) compared to COAG (27%). In GIFT, the primary endpoint was driven mainly by cases of INR≥4; 10/1597 subjects had a major bleeding event, similar to the rate observed in COAG (14/1015). The genetic determinants of major bleeding during warfarin have also been assessed by accumulating cases from administrative or electronic health record (EHR) databases.15,16 While these were not randomized studies, they did include far more bleeding cases (n=250–265), and implicated CYP2C9*315 and CYP4F2 V433M16 as risk factors. The major lessons for pharmacogenetic implementation (Table 1) are that it is important to define as completely as possible the pharmacogenetic determinants of variable drug response; inclusion of subjects of diverse ancestries is important and requires further consideration of specific variants to be tested; and the diseases to be studied, the comparator arms, and the primary outcome definitions are important. Whether prospective pharmacogenetic testing is routinely applied to warfarin therapy will further depend on payer attitudes, professional society opinions, and the extent to which newer anticoagulants replace warfarin in the marketplace.
Table 1:
Major lessons learned from studies of single drug pharmacogenetic outcomes
| Drug | Major lessons learned |
|---|---|
| Warfarin | • Define multiple genetic pathways and specific variants modulating drug action prior to mounting trials • Include multiple ancestries • Specific endpoints chosen (e.g. efficacy versus adverse events) determine ability to detect an effect |
| Abacavir, carbamazepine | • Pre-clinical data may align with RCT outcomes • Endpoints should be defined as precisely as possible • Practitioner education is critical to successful implementation • Include multiple ancestries |
| Clopidogrel | • Include multiple ancestries • Variant allele frequency determine the likelihood of defining a pharmacogenetic outcome effect |
| Azathioprine | • A potentially beneficial outcome of pharmacogenetic testing is confined to patients with variant alleles • Equipoise can change as genotypes become available and used |
HLA variants – abacavir and carbamazepine:
Early studies with the antiretroviral abacavir implicated HLA-B*57:01 as a critical determinant of drug-related Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN).17 Subsequently, a double-blind prospective clinical trial randomized 1,956 patients starting antiretroviral therapy to be genotyped, and to exclude abacavir in the treatment arm if HLA-B*57:01 was present.18 No genotyping was performed in the controls, and cases of possible SJS/TEN underwent skin testing for confirmation. The trial demonstrated abolition of immunologically-proven SJS/TEN in the genetically-guided group and a 2.7% incidence of SJS in the control group. These data led to endorsement by professional societies of HLA testing in subjects starting abacavir, inclusion of this recommendation in a black box in the drug’s FDA approval label, and thus widespread clinical use of this pharmacogenetic test. Similarly, initial studies implicated HLA-B*15:02 as a risk factor for carbamazepine-related SJS/TEN in Asians.19 Subsequent studies replicated this signal in Asians20 and identified HLA-A*31:01 as a risk factor in Caucasians.21 National efforts to implement HLA-B*15:02 testing in Southeast Asia have met with variable outcomes. In Taiwan, there was a striking decrease in carbamazepine-related SJS/TEN.20 However, in Hong Kong, the incidence of carbamazepine-related SJS/TEN fell but this was attributable to wholesale avoidance of the drug and prescription of other anticonvulsants (which also can cause SJS/TEN): the overall incidence of the adverse drug reaction did not change.22 Lessons here include the ability of pre-RCT data to predict the outcome of the RCT, the power and resource expense of mounting and RCT, the need for precise diagnosis of endpoints, the need for studies across ancestries, and a need for practitioner education during introduction of an implementation program.
Clopidogrel:
Multiple retrospective analyses of very large clinical trials comparing clopidogrel and comparator treatments (generally placebo) have shown that CYP2C19 genetic variants (predominately *2, as well as *3 in Asian populations) confer an increased risk of thrombotic events (i.e. failure of drug efficacy due to reduced bioactivation23) in patients presenting with acute coronary syndrome and treated with coronary stenting.24–26 In other settings, such as atrial fibrillation, in which clopidogrel efficacy is less compelling, genetic variants have not been shown to play a role in determining outcome.27 Most recently, a large trial in China showed decreased efficacy among CYP2C19 variant carriers treated with clopidogrel for stroke or TIA.28 Reasons that this trial could demonstrate efficacy whereas previous studies in this indication did not29 may include the large number of subjects studied, and the fact that variants are more common in Asian subjects, increasing the likelihood of detecting a genotype-dependent effect.
Azathioprine:
The theme that the likelihood that a trial can demonstrate efficacy across a large population when only a portion of that population carries genetic variants is further highlighted by a trial of TPMT testing in patients with inflammatory bowel disease treated with azathioprine.30 Excess bone marrow suppression (hematologic toxicity) is a risk in patients with TPMT loss of function variants. The trial randomized 783 patients to undergo genetic testing and subsequent dose adjustment, or to be treated with standard doses. Importantly, there was no difference in disease outcomes in the two groups, nor was there a difference in overall hematologic toxicity, the primary endpoint, across the two groups. However, in a planned secondary analysis, there was a striking difference in incidence of hematologic toxicity among 74 TPMT carriers randomized to genetically-guided therapy (1/39, 2.2%) compared to those randomized to standard therapy (8/35, 22.9%). An earlier British trial was limited by slow recruitment thought to reflect in part the fact that TPMT genotyping was already clinically available.31 The lessons learned here, therefore, are that genetic testing for a single drug in a broad population is likely to benefit only a subset of that population, the size of that subset is determined by the frequency of risk alleles, and that equipoise with respect to clinical trials can change with availability and dissemination of genotype information.
Regulatory agency response to pharmacogenomic risks
Pharmacogenetic variants can be used to identify subjects at high risk for serious drug toxicity, and therefore to avoid those drugs in those patients. The US Food and Drug Administration (FDA) has since 2007 included pharmacogenetic information, sometimes with dosing recommendations, in drug labels.32 In some instances, however, the FDA has chosen to issue dosing recommendations that downplay a potential role of pharmacogenetics in adverse drug reactions. Two prominent examples are codeine and simvastatin. In the case of codeine, the ultra-rapid metabolizer (UM) genotype for CYP2D6 confers increased biotransformation to the active metabolite morphine, and thus respiratory depression; this has been a particular problem in children treated for postoperative pain after tonsillectomy as well as a cause of respiratory depression in neonates being breastfed by UM mothers.33–35 In the case of simvastatin, a specific variant in SLCO1B1 has been associated with a dramatically increased risk of biochemically proven myopathy among patients receiving high-dose (80 mg per day) simvastatin.36 In both of these settings, therefore, pre-emptive genotyping could be useful (as suggested in the current CPIC guideline37), and indeed for over a decade, the FDA has included pharmacogenetic data in drug labels,38,39 However, in the case of codeine and simvastatin, the FDA took the view that public interest is better served by relabeling the drugs to recommend avoidance in at-risk populations. The black box warnings now recommend avoiding codeine in all children under 12 and limiting simvastatin doses to 40 mg per day, or even lower in the presence of potentially interacting drugs. Others have argued that a genetically-based approach would allow effective pain management in the pediatric population.40
Beyond point of care gene testing
Rationale:
Performing genetic testing at the time of prescription of a target drug represents the point of care approach evaluated in the studies outlined above. A “pre-emptive” approach represents an alternative model in which pharmacogenetic testing is performed for multiple variants that are thought to affect drug response (either efficacy or toxicity); these are saved for later use in an EHR system, and deployed with appropriate clinical decision support (CDS) when a target drug is prescribed.
The rationale for preemptive multiplexed testing has been supported through retrospective studies of electronic health records. Schildcrout et al41 examined 52,942 subjects followed for long term care at Vanderbilt University Medical Center (VUMC) and determined the frequency with which 56 medications with outcomes known to be influenced by variant pharmacogenes were prescribed. Over the course of five years, they estimated that 64.8% of subjects were exposed to at least one such medication and over 12% were exposed to four or more such medications. Their modeling suggested that at least 383 serious adverse drug events (associated with only 6 medications) would have occurred over 5 years in these subjects, and therefore their frequency could be reduced or abolished with an effective pre-emptive pharmacogenetic genotyping program. This modeling study illustrates many of the potentially intuitively obvious advantages of pre-emptive testing: pharmacogenomic data can be used over long periods of time, data can be reused, the risk of serious adverse outcomes across multiple drugs can be mitigated, and the incremental cost to obtain multiple pharmacogenomic genotypes is small. It is also been argued that many patients require multiple drug trials before settling on an effective therapy and that use of pharmacogenetic data embedded in EHRs should shorten this “therapeutic odyssey”.42
Choosing a target population:
While it is convenient to envision a future delivery model in which all common pharmacogenetic variants have been cataloged and embedded across all EHRs, pre-emptive pharmacogenetic testing programs generally have launched with a focus on specific “high risk” patients, in order to demonstrate use of the data and ultimately utility.43,44 Examples of populations that have been initially tested or proposed for testing include algorithmically defined high-risk subjects being seen in medicine clinics,41,45 subjects with specific diseases such as childhood leukemia and other cancers that mandate therapy with many drugs with known pharmacogenetic variation,46 or patients being seen for initiation of one drug with known pharmacogenetic variants.47 Investigators at Indiana identified 7,039 patients who started one or more of 30 target medications within a year at 73 sites in a safety net health care system, and have therefore proposed that this is a target population for pharmacogenetic implementation.48 They interpreted these data as justifying a prospective multi-gene/multi-drug approach to pharmacogenetic testing. Examples of these programs are described further below.
Barriers to a pre-emptive program:
Despite the intuitive appeal of the pre-emptive approach,49 there are obstacles that need to be overcome. One is the problem of evidence, outlined above for single drugs which still holds for multiple drugs. Indeed, demonstrating the value of a pre-emptive approach may be more difficult given that the drugs and genes tested represent a spectrum of variable risk allele frequencies and variable efficacy and susceptibility to serious adverse drug events over time. A second issue is assay validity. Some variants, such as CYP2C19*2 or CYPC9*2 or *3, represent single nucleotide polymorphisms (SNPs) and are relatively simple to assay in a reliable fashion. However, two important pharmacogenes, CYP2D6 and HLAB, present specific assay difficulties. CYP2D6 has the problems of an adjacent pseudogene CYP2D7, and of copy number variation,50 while the HLA region is one of the most polymorphic in the human genome and calling variant haplotypes is a process that may require either specialized computational methods (whose efficacy can vary by ancestry51) or long read sequencing.52 Increasing enthusiasm over the use of sequencing platforms also raises the issue, common across genomic medicine,53 that rare variants will be discovered but their contribution to variable drug effects will be more difficult to assess than common variants. Further, use of sequence data also raises the problem of reinterpretation; that is, a variant designated of uncertain significance may subsequently be reclassified as pathogenic and thus mechanisms to alert providers and patients need to be developed.
As programs have been designed and implemented, other barriers have been identified and solutions developed. In some programs, the multiplexed genotyping platform deployed includes variants of uncertain significance, so mechanisms have been developed to save these data outside the EHR, and to only deploy variants judged to be clinically important in the EHR environment. Early programs developing pre-emptive genotyping strategies relied on internal data reviews to decide which variants should be clinically deployed; the development of consensus guidelines by the Clinical Pharmacogenetics Implementation Consortium (CPIC) has relieved individual institutions of this burden.44,54,55 The Dutch working group in pharmacogenetics working group has produced a similar set of guidelines.56 A pre-emptive pharmacogenetic program requires the development and implementation of CDS, and again individual early adopter institutions have had to develop their own mechanisms for this portion of the program and it is hoped that consortia and multi-institutional efforts will results in common CDS code and availability, perhaps based in part on CPIC guidelines.44,57 Healthcare providers are in general are unfamiliar with pharmacogenetic data and the way in which it can be used to make clinical decisions, so engaging the provider community and undertaking educational efforts is an important component of any such program.58,59 The payer community has been reluctant to support multiplexed pharmacogenetic testing, but has in some instances been willing to support targeted genotyping during initiation of specific drugs.60
Early adopter implementation approaches:
The pre-emptive model has been implemented in a number of academic centers. A common theme is very strong institutional support since efforts like this apply knowledge across a healthcare system, and thus require engagement of multiple constituencies, notably physicians and other providers, pharmacists, informatics experts, and ethicists. The VUMC program, Pharmacogenomic Resource for Enhanced Decisions In Care and Treatment (PREDICT), was launched in 2010.45 The initial platform was the VeraCode ADME core panel which tested 184 variants and 34 genes. The program started with a focus on patients homozygous for the CYP2C19 *2 loss-of-function variant in whom cardiac catheterization with stenting was anticipated, and has since expanded to 6 drug gene pairs, including CYP2C19 heterozygotes. The majority of the VeraCode variants were thus held outside the EHR; CYP2D6 variants are not called on this platform. More recently the PREDICT program has moved to a QuantStudio platform that includes common CYP2D6 variants. CDS has been developed and validated,61 and PREDICT results are displayed prominently in the EHR and delivered to the patient through the EHR-based patient portal.
St. Jude built on its experience using TPMT and CYP2D6 single gene testing62 to implement the Pharmacogenetics for Kids program in 2011.46 The St. Jude program includes EHR-delivered alerts if a drug is prescribed and pharmacogenetic testing has not been obtained as well as EHR-based CDS once the result is obtained. The University of Florida (UF) personalized medicine program launched in 2011 with a focus on point of care genotyping targeted at clopidogrel use.47 The platform used, from QuantStudio, allowed interrogation of 256 SNPs, most of which are currently held in escrow pending activation of a planned pre-emptive program. The Mayo Clinic “Right drug right dose right time using genomic data to individualize treatment (RIGHT)” uses the PGRN-Seq next generation sequencing platform and a specialized CYP2D6 assay and aims to integrate high-risk pharmacogenetic results in the EHR coupled to point of care CDS.63,64 As in other programs, internal evidence review was necessary and has since been complemented by CPIC. This program also includes testing for HLA-B*15:02 and HLA-B*57:01; both UF and Mayo also plan to implement IFNL3 for hepatitis C treatment.65 The Mount Sinai CLIPMERGE pharmacogenetics program, initiated in 2013, enrolls subjects who are participants in the institution’s DNA biobank and bases implementation decisions on CPIC guidelines.66 The Indiana INGENIOUS trial is using the pre-emptive model to deliver point of care CDS for 28 drugs, focusing on engaging the most expensive patients in a safety net program, anticipating this may represent a cost-savings opportunity.48,67 Other institutions that have reported on development of single drug or multi-drug based on pharmacogenetic testing programs include the Cincinnati Children’s Hospital’s Christine program focusing on epilepsy and attention deficit hyperactivity disorder,68 the University of Chicago’s 1200 patients project,69 and the Ohio State University program that includes a focus on engaging genetic counsellors.70
NIH-supported consortia:
Many of these programs were put in place with support from the Pharmacogenetics Research Network (PGRN), an effort supported by the National Institute for General Medical Sciences Institute and other NIH institutes.71 The National Human Genome Research Institute’s (NHGRI) eMERGE (electronic MEdical Records and GEnomics) network also includes a pre-emptive pharmacogenomics program, eMERGE-PGx,72 that has now genotyped over 9000 subjects on a next generation platform developed by PGRN. NHGRI’s IGNITE network includes sites which have a major focus on pharmacogenomics.73 The PGRN’s Translational Pharmacogenetics Program (TPP) recently reviewed commonalities and differences across eight participating US healthcare systems.44 With time, there has been an increasing reliance on CPIC guidelines and CDS methods common across the sites, with high quality genotype data being delivered. Differences across sites include the point of care versus pre-emptive testing paradigm, platforms being deployed, and clinical sites (inpatient vs outpatient, distinct subspecialties, pediatrics vs adult). A total of 20,258 subjects had been tested and the common drug-gene pairs were CYP2C19-clopidogrel (at 7 sites), TPMT-thiopurine (6) and SLCO1B1-simvastatin (5).
TPP also outlined important advances in education, reimbursement, and informatics development across the sites.44 UF reported 85% reimbursement rate for the point of care testing for CYP2C19-clopidogrel.60 Individual sites created educational materials ranging from online courses to websites to newsletters. St. Jude and UF established American Society for Health System Pharmacist’s-Accredited residency in clinical pharmacogenomics, and other institutions offer fellowships accredited by the American Board of Clinical Pharmacology. Vanderbilt and UF have recently been awarded NHGRI-supported training grants in genomic medicine.
Impact of pharmacogenetic programs on outcomes
Two types of outcomes can be tracked: process outcomes which describe what happens when a program is put place (e.g. How many patients? How often is advice delivered and how often are medications changed?) and clinical outcomes (Do patients have increased efficacy or fewer adverse events?) To date, very few programs have reported on either outcome.
Most patients carry a variant pharmacogene:
The Vanderbilt PREDICT program has tested >14,000 subjects to date and has reported outcomes in a number of areas. An analysis of the first 10,000 patients tested revealed that the frequency of “highly actionable” genetic variants (e.g. CYP2C19*2 homozygotes) varied from 0% (CYP3A5-tacrolimus) to 2.5% (CYP2C19-clopidogrel) while the frequency of other actionable variants (e.g. CYP2C19*2 heterozygotes) varied from 68.7% (VKORC1/CYP2C9-warfarin) to 8.9% (TPMT-thiopurine).74 Overall, 91% of subjects tested had a variant in one of five drug-gene pairs tested including 4.8% with “highly actionable” variants. These data reinforce the appeal of the pre-emptive approach since virtually all patients carry at least one pharmacogenetic variant, and with the pre-emptive approach these data are available at the point of care. Further, modeling indicated that point of care genotyping would have generated 14,656 tests, so the pre-emptive approach, by implementing a multiplexed strategy, saves genotyping test costs. Similar data were reported by the Mayo Clinic RIGHT program which reported that among 1,013 subjects tested, 99% carried an actionable variant in SLCO1B1, CYP2C19, CYP2C9, or VKORC1.64 Analysis of the first 5,000 subjects tested in eMERGE-PGx found that >96% of samples had CPIC-designated high priority actionable variants.75
The Vanderbilt PREDICT program reported that 22% of patients tested (2,676/12,157) were prescribed clopidogrel. In this group, there were 450 carriers of a single loss-of-function variant (intermediate metabolizers) and 64 carriers of two loss-of-function alleles, predominantly CYP2C19*2 (poor metabolizers).76 A year after initiation of clopidogrel CDS, only 8.3% of the normal metabolizer group (those lacking loss-of-function variants) had switched to alternate therapy. By contrast, 33.2% of the intermediate metabolizers and 57.6% of poor metabolizers had been switched to alternate antiplatelet therapy. Factors associated with reduced “conversion” of clopidogrel to alternate therapy included perception of increased risk of bleeding (a problem with alternate antiplatelet treatments such as prasugrel) and delayed delivery of genotype information after initiation of treatment. Interestingly, there was also striking variability in the extent to which high volume practitioners (those caring for at least forty patients within the cohort) adopted alternate antiplatelet therapy, from 23% to 68%. Further questioning of practitioners revealed that virtually all agree that genetic variants influence drug response, and the vast majority (92%) favored immediate notification when a clinically action variant was detected in a patient on a target drug.77 However, there was variability in perception of who was responsible for changing the drug dose, and whether patients should be notified of their results.
The IGNITE Pharmacogenetics Working Group78 has reported on the impact of CYP2C19 point of care testing on the development of major adverse cardiovascular events (cardiovascular events, MI, stroke and instant thrombosis) over six months after initiation of clopidogrel CDS. There was a lower incidence of MACE in subjects with loss-of-function alleles switched to alternate therapies, compared to those with loss-of-functional alleles maintained on clopidogrel. However, there was no significant difference between subjects with loss-of-function alleles prescribed alternate therapy and those without loss-of-function alleles. These outcome data support the use of clopidogrel CYP2C19 testing to reduce the incidence of MACE.79
Cost-effectiveness:
To date, cost-effectiveness analyses have largely examined single-gene scenarios.80 One study suggested a benefit of the strategy of genotyping for CYP2C19 and using alternate antiplatelet therapy instead of clopidogrel in variant carriers compared to just using more expensive alternate therapies (that have also been associated with increased bleeding risk).81,82 Similarly, cost-effectiveness analysis suggested that HLA-B*57:01 testing for abacavir was less expensive and more beneficial than not testing;83 interestingly, this result was reported several years prior to the RCT that demonstrated benefit. In the case of warfarin, cost-effectiveness was suggested only if the cost of testing were low (<$200), if testing were limited only to patients at high risk of bleeding, or if the testing reduced major bleeding by a third and results were available within 24 hours.84 Other studies have similarly suggested only marginal benefit85 or testing would be cost-effective only if it markedly reduced major bleeding complications or substantially increased time in therapeutic range, by >5–9%.86
There are limited data available on the potential cost-effectiveness of multiplexed pre-emptive strategies. Reusing panel results over time would incur near-zero marginal acquisition cost for any additional genes on the panel, but the scale and scope of benefits are dependent on variant frequencies, penetrance and a host of other gene, clinical, and health system factors mediating the clinical impact of reporting each additional genetic variant. Further, the individual with the genotype data must be exposed to the relevant drug to accrue any benefit; for example, as mentioned above, only 22% of patients in the Vanderbilt PREDICT program received clopidogrel. Thus, the use of clinical sequencing in routine clinical care depends on the population selected for genotyping, the ability for front-line clinicians to effectively incorporate genomic data into their clinical decision-making, and the impact of tailored screening or diagnosis to alter patients’ clinical course. In an approach using simulation to project benefits over patients’ lifetimes, one study has shown that screening patients with a broad panel covering all top CPIC drug-gene interactions would approach cost effectiveness (Incremental Cost-Effectiveness Ratio <$150,000 per quality-adjusted life year) assuming the information was used at a high rate by clinicians and pharmacogenomic variants had moderate efficacy at reducing adverse events.87
Future directions
To date, data from individual sites have generated relatively small data sets reporting outcomes using multiplexed approaches. As mentioned above, IGNITE has begun to examine outcomes across sites, and the eMERGE-PGx program has used a common genotyping platform across sites, although CDS varies across sites and the collection of clinical outcome data is still slow. The Ubiquitous Pharmacogenomics (U-PGx) Consortium is a group funded by the European Union to examine pharmacogenetic implementation.88 Their PREPARE (PREemptive Pharmacogenomic testing for prevention of Adverse drug Reactions) project is using a multiplexed pre-emptive genotyping strategy implementing the Dutch Pharmacogenetics working group guidelines across seven European countries. The projected enrollment is 8,100 subjects, randomized to pharmacogenetic-guided therapy or standard of care; 50 variants in 13 pharmacogenes and 43 drug-gene pairs will be studied with standardized outcome measures established. The goal is to focus on prevention of adverse drug reactions across all drug-gene pairs. The study has launched and anticipates a three-year recruitment and follow-up period to end in 2020. Notably, U-PGx, TPP, and other efforts have focused largely on populations of European ancestry, and as with discovery highlighted above, implementation efforts will require studies across diverse ancestries. Thus information from ongoing programs that continue to accrue subjects as well as national and international consortia should start to provide data on process and clinical outcomes in pre-emptive pharmacogenetic programs.
Figure 1:
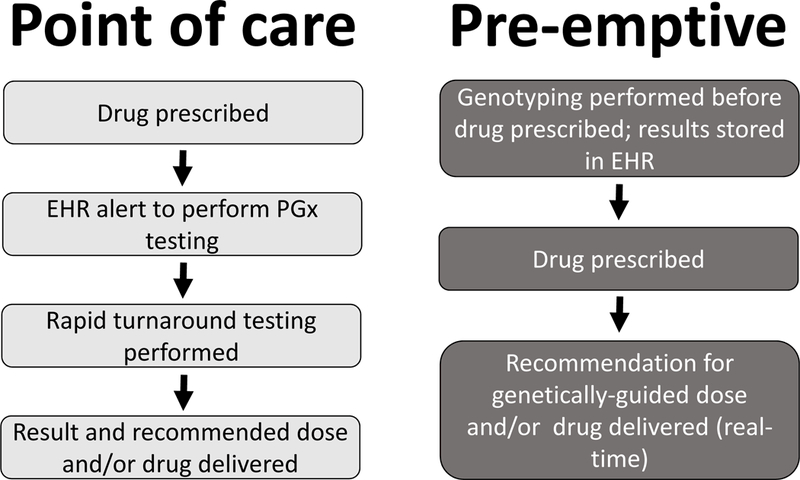
Comparison of the point of care and the pre-emptive approaches to acquiring and delivering pharmaco genetic variant data
Acknowledgments
Supported in part by U01 HG008672, P50 GM1156305, U01 HG007253
References
- 1.Rettie AE et al. Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: a role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. ChemRes Toxicol 5, 54–9 (1992). [DOI] [PubMed] [Google Scholar]
- 2.Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF & Korzekwa KR Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics 4, 39–42 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Aithal GP, Day CP, Kesteven PJ & Daly AK Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 353, 717–9 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Rost S et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 427, 537–41 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Rieder MJ et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. New England Journal of Medicine 352, 2285–93 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Schwarz UI et al. Genetic Determinants of Response to Warfarin during Initial Anticoagulation. The New England Journal of Medicine 358, 999–1008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pirmohamed M, Kamali F, Daly AK & Wadelius M Oral anticoagulation: a critique of recent advances and controversies. Trends in pharmacological sciences 36, 153–63 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Kimmel SE et al. A Pharmacogenetic versus a Clinical Algorithm for Warfarin Dosing. New England Journal of Medicine 369, 2283–93 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Limdi NA et al. Race influences warfarin dose changes associated with genetic factors. Blood 126, 539–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perera MA et al. Genetic variants associated with warfarin dose in African-American individuals: a genome-wide association study. The Lancet 382, 790–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verhoef TI et al. A Randomized Trial of Genotype-Guided Dosing of Acenocoumarol and Phenprocoumon . New England Journal of Medicine 369, 2304–12 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Pirmohamed M et al. A Randomized Trial of Genotype-Guided Dosing of Warfarin. New England Journal of Medicine 369, 2294–303 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Gage BF et al. Effect of Genotype-Guided Warfarin Dosing on Clinical Events and Anticoagulation Control Among Patients Undergoing Hip or Knee Arthroplasty: The GIFT Randomized Clinical Trial. JAMA : the journal of the American Medical Association 318, 1115–24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caldwell MD et al. CYP4F2 genetic variant alters required warfarin dose. Blood 111, 4106–12 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawai VK et al. Genotype and risk of major bleeding during warfarin treatment. Pharmacogenomics 15, 1973–83 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roth JA et al. Genetic Risk Factors for Major Bleeding in Warfarin Patients in a Community Setting. Clin Pharmacol Ther 95, 636–43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mallal S et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 359, 727–32 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Mallal S et al. HLA-B*5701 Screening for Hypersensitivity to Abacavir. The New England Journal of Medicine 358, 568–79 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Chung WH et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature 428, 486 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Chen P et al. Carbamazepine-Induced Toxic Effects and HLA-B*1502 Screening in Taiwan. New England Journal of Medicine 364, 1126–33 (2011). [DOI] [PubMed] [Google Scholar]
- 21.McCormack M et al. HLA-A*3101 and Carbamazepine-Induced Hypersensitivity Reactions in Europeans. New England Journal of Medicine 364, 1134–43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z, Liew D & Kwan P Effects of a HLA-B*15:02 screening policy on antiepileptic drug use and severe skin reactions. Neurology 83, 2077–84 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Hulot JS et al. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood 108, 2244–7 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Collet JP et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 373, 309–17 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Mega JL et al. Cytochrome P-450 Polymorphisms and Response to Clopidogrel. N Engl J Med 360, 354–62 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Simon T et al. Genetic determinants of response to clopidogrel and cardiovascular events. New England Journal of Medicine 360, 363–75 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Holmes MV, Perel P, Shah T, Hingorani AD & Casas JP CYP2C19 genotype, clopidogrel metabolism, platelet function, and cardiovascular events: a systematic review and meta-analysis. JAMA : the journal of the American Medical Association 306, 2704–14 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Wang Y et al. Association Between CYP2C19 Loss-of-Function Allele Status and Efficacy of Clopidogrel for Risk Reduction Among Patients With Minor Stroke or Transient Ischemic Attack. JAMA : the journal of the American Medical Association 316, 70–8 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Hoh BL et al. CYP2C19 and CES1 polymorphisms and efficacy of clopidogrel and aspirin dual antiplatelet therapy in patients with symptomatic intracranial atherosclerotic disease. Journal of neurosurgery, 1–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coenen MJ et al. Identification of Patients With Variants in TPMT and Dose Reduction Reduces Hematologic Events During Thiopurine Treatment of Inflammatory Bowel Disease. Gastroenterology 149, 907–17 e7 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Newman WG et al. A pragmatic randomized controlled trial of thiopurine methyltransferase genotyping prior to azathioprine treatment: the TARGET study. Pharmacogenomics 12, 815–26 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Lesko LJ & Zineh I DNA, drugs and chariots: on a decade of pharmacogenomics at the US FDA. Pharmacogenomics 11, 507–12 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Gasche Y et al. Codeine intoxication associated with ultrarapid CYP2D6 metabolism. New England Journal of Medicine 351, 2827–31 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Kirchheiner J et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. PharmacogenomicsJ 7, 257–65 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Ciszkowski C, Madadi P, Phillips MS, Lauwers AE & Koren G Codeine, Ultrarapid-Metabolism Genotype, and Postoperative Death. New England Journal of Medicine 361, 827–8 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Link E et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med 359, 789–99 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Ramsey LB et al. The clinical pharmacogenetics implementation consortium guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin Pharmacol Ther 96, 423–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lesko LJ & Woodcock J Translation of pharmacogenomics and pharmacogenetics: a regulatory perspective. NatRevDrug Discov 3, 763–9 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Frueh FW et al. Pharmacogenomic biomarker information in drug labels approved by the United States food and drug administration: prevalence of related drug use. Pharmacotherapy 28, 992–8 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Gammal RS et al. Pharmacogenetics for Safe Codeine Use in Sickle Cell Disease. Pediatrics 138, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schildcrout JS et al. Optimizing Drug Outcomes Through Pharmacogenetics: A Case for Preemptive Genotyping. Clin Pharmacol Ther 92, 235–42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lazaridis KN Improving Therapeutic Odyssey: Preemptive Pharmacogenomics Utility in Patient Care. Clin Pharmacol Ther 101, 39–41 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Dunnenberger HM et al. Preemptive clinical pharmacogenetics implementation: current programs in five US medical centers. Annual review of pharmacology and toxicology 55, 89–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luzum JA et al. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: Outcomes and Metrics of Pharmacogenetic Implementations Across Diverse Healthcare Systems. Clin Pharmacol Ther, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pulley JM et al. Operational Implementation of Prospective Genotyping for Personalized Medicine: The Design of the Vanderbilt PREDICT Project. Clin Pharmacol Ther 92, 87–95 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoffman JM et al. PG4KDS: a model for the clinical implementation of pre-emptive pharmacogenetics. American journal of medical genetics Part C, Seminars in medical genetics 166C, 45–55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cavallari LH et al. Institutional profile: University of Florida Health Personalized Medicine Program. Pharmacogenomics 18, 421–6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carpenter JS, Rosenman MB, Knisely MR, Decker BS, Levy KD & Flockhart DA Pharmacogenomically actionable medications in a safety net health care system. SAGE open medicine 4, 2050312115624333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weitzel KW, Cavallari LH & Lesko LJ Preemptive Panel-Based Pharmacogenetic Testing: The Time is Now. Pharmaceutical research 34, 1551–5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hicks JK, Swen JJ & Gaedigk A Challenges in CYP2D6 phenotype assignment from genotype data: a critical assessment and call for standardization. Current drug metabolism 15, 218–32 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Karnes JH et al. Comparison of HLA allelic imputation programs. PloS one 12, e0172444 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pavlos R, Mallal S & Phillips E HLA and pharmacogenetics of drug hypersensitivity. Pharmacogenomics 13, 1285–306 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Manolio TA et al. Bedside Back to Bench: Building Bridges between Basic and Clinical Genomic Research. Cell 169, 6–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Relling MV & Klein TE CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin Pharmacol Ther 89, 464–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caudle KE, Gammal RS, Whirl-Carrillo M, Hoffman JM, Relling MV & Klein TE Evidence and resources to implement pharmacogenetic knowledge for precision medicine. American journal of health-system pharmacy : AJHP : official journal of the American Society of Health-System Pharmacists 73, 1977–85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bank PC et al. Comparison of the Guidelines of the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Group. Clin Pharmacol Ther, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bell GC et al. Development and use of active clinical decision support for preemptive pharmacogenomics. Journal of the American Medical Informatics Association:JAMIA 21, e93–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feero WG, Manolio TA & Khoury MJ Translational research is a key to nongeneticist physicians’ genomics education. Genet Med 16, 871–3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manolio TA & Murray MF The growing role of professional societies in educating clinicians in genomics. Genet Med 16, 571–2 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Weitzel KW et al. Clinical pharmacogenetics implementation: approaches, successes, and challenges. American journal of medical genetics Part C, Seminars in medical genetics 166C, 56–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peterson JF et al. Electronic health record design and implementation for pharmacogenomics: a local perspective. Genet Med 15, 833–41 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crews KR et al. Development and implementation of a pharmacist-managed clinical pharmacogenetics service. American journal of health-system pharmacy : AJHP : official journal of the American Society of Health-System Pharmacists 68, 143–50 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bielinski SJ et al. Preemptive genotyping for personalized medicine: design of the right drug, right dose, right time-using genomic data to individualize treatment protocol. Mayo Clinic proceedings 89, 25–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ji Y et al. Preemptive Pharmacogenomic Testing for Precision Medicine: A Comprehensive Analysis of Five Actionable Pharmacogenomic Genes Using Next-Generation DNA Sequencing and a Customized CYP2D6 Genotyping Cascade. The Journal of molecular diagnostics : JMD 18, 438–45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muir AJ et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for IFNL3 (IL28B) genotype and PEG interferon-alpha-based regimens. Clin Pharmacol Ther 95, 141–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gottesman O et al. The CLIPMERGE PGx Program: clinical implementation of personalized medicine through electronic health records and genomics-pharmacogenomics. Clin Pharmacol Ther 94, 214–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eadon MT et al. Implementation of a pharmacogenomics consult service to support the INGENIOUS trial. Clin Pharmacol Ther 100, 63–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pestian J, Spencer M, Matykiewicz P, Zhang K, Vinks AA & Glauser T Personalizing Drug Selection Using Advanced Clinical Decision Support. Biomedical informatics insights 2, 19–29 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O’Donnell PH et al. Adoption of a clinical pharmacogenomics implementation program during outpatient care--initial results of the University of Chicago “1,200 Patients Project”. American journal of medical genetics Part C, Seminars in medical genetics 166C, 68–75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sturm AC, Sweet K & Manickam K Implementation of a clinical research pharmacogenomics program at an academic medical center: role of the genetics healthcare professional. Pharmacogenomics 14, 703–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Giacomini KM et al. The pharmacogenetics research network: from SNP discovery to clinical drug response. Clin Pharmacol Ther 81, 328–45 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rasmussen-Torvik LJ et al. Design and anticipated outcomes of the eMERGE-PGx project: a multicenter pilot for preemptive pharmacogenomics in electronic health record systems. Clin Pharmacol Ther 96, 482–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weitzel KW et al. The IGNITE network: a model for genomic medicine implementation and research. BMC medical genomics 9, 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van Driest SL et al. Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin Pharmacol Ther, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bush WS et al. Genetic Variation among 82 Pharmacogenes: the PGRN-Seq data from the eMERGE Network. Clin Pharmacol Ther, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peterson JF et al. Physician response to implementation of genotype-tailored antiplatelet therapy. Clin Pharmacol Ther 100, 67–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peterson JF et al. Attitudes of clinicians following large-scale pharmacogenomics implementation. The pharmacogenomics journal, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cavallari LH et al. The IGNITE Pharmacogenetics Working Group: An Opportunity for Building Evidence with Pharmacogenetic Implementation in a Real-World Setting. Clinical and translational science 10, 143–6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cavallari LH et al. Multisite Investigation of Outcomes With Implementation of CYP2C19 Genotype-Guided Antiplatelet Therapy After Percutaneous Coronary Intervention. JACC Cardiovascular interventions, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wong WB, Carlson JJ, Thariani R & Veenstra DL Cost effectiveness of pharmacogenomics: a critical and systematic review. PharmacoEconomics 28, 1001–13 (2010). [DOI] [PubMed] [Google Scholar]
- 81.Reese ES, Daniel Mullins C, Beitelshees AL & Onukwugha E Cost-effectiveness of cytochrome P450 2C19 genotype screening for selection of antiplatelet therapy with clopidogrel or prasugrel. Pharmacotherapy 32, 323–32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kazi DS et al. Cost-effectiveness of genotype-guided and dual antiplatelet therapies in acute coronary syndrome. Ann Intern Med 160, 221–32 (2014). [DOI] [PubMed] [Google Scholar]
- 83.Hughes DA, Vilar FJ, Ward CC, Alfirevic A, Park BK & Pirmohamed M Cost-effectiveness analysis of HLA B*5701 genotyping in preventing abacavir hypersensitivity. Pharmacogenetics 14, 335–42 (2004). [DOI] [PubMed] [Google Scholar]
- 84.Eckman MH, Rosand J, Greenberg SM & Gage BF Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with nonvalvular atrial fibrillation. Ann Intern Med 150, 73–83 (2009). [DOI] [PubMed] [Google Scholar]
- 85.Meckley LM, Gudgeon JM, Anderson JL, Williams MS & Veenstra DL A policy model to evaluate the benefits, risks and costs of warfarin pharmacogenomic testing. Pharmacoeconomics 28, 61–74 (2010). [DOI] [PubMed] [Google Scholar]
- 86.Patrick AR, Avorn J & Choudhry NK Cost-Effectiveness of Genotype-Guided Warfarin Dosing for Patients With Atrial Fibrillation. Circulation: Cardiovascular Quality and Outcomes, CIRCOUTCOMES (2009). [DOI] [PubMed] [Google Scholar]
- 87.Graves JA, Garbett S, Zhou Z & Peterson JF The Value of Pharmacogenomic Information. National Bureau of Economic Research, (2018). [Google Scholar]
- 88.van der Wouden CH et al. Implementing Pharmacogenomics in Europe: Design and Implementation Strategy of the Ubiquitous Pharmacogenomics Consortium. Clin Pharmacol Ther 101, 341–58 (2017). [DOI] [PubMed] [Google Scholar]