

Abstract
Read-across is a popular data gap filling technique used within analogue and category approaches for regulatory purposes. In recent years there have been many efforts focused on the challenges involved in read-across development, its scientific justification and documentation. Tools have also been developed to facilitate read-across development and application. Here, we describe a number of publicly available read-across tools in the context of the category/analogue workflow and review their respective capabilities, strengths and weaknesses. No single tool addresses all aspects of the workflow. We highlight how the different tools complement each other and some of the opportunities for their further development to address the continued evolution of read-across.
Keywords: Category approach; analogue approach; data gap filling; Read-across; (Q)SAR; trend analysis, nearest neighbours
1. Introduction
1.1. Background context
Read-across as a data gap filling technique has garnered considerable attention in recent years as a result of the changing regulatory landscape worldwide. The most significant regulations have been in Europe with Registration Evaluation Authorisation and Restriction of Chemicals (REACH) (EC, 2006) and the Cosmetic Regulation (EC, 2009). These regulations have mandated the use of non-animal approaches to address information needs for hazard and risk assessment. Concurrently, there has been a shift for toxicity testing itself to move towards a mechanistic basis exploiting high throughput screening (HTS) and high content (HC) in vitro approaches (NRC, 2007). The context of how these in vitro approaches can be interpreted is still evolving, though examples using adverse outcome pathways (AOPs) have started to be developed (Patlewicz et al., 2015, Delrue et al, 2016; NRC, 2017). Read-across is also undergoing a transformation with increasingly efforts to exploit High Throughput/High Content (HT/HC) screening data as a means of substantiating biological similarity (Low et al., 2013; Pradeep et al., 2015; Shah et al., 2016; Zhu et al., 2016). An OECD work programme under the auspices of the Task Force of Hazard Assessment (TFHA)1 has published several examples of AOP informed Integrated Approaches to Testing and Assessment (IATA) that have been based on read-across where data generated as part of the EPA ToxCast program have been utilised (see http://www.oecd.org/chemicalsafety/risk-assessment/iata-integrated-approaches-to-testing-and-assessment.htm for a list of case studies both published and under review).
Although there has been a wealth of technical guidance developed (OECD 2014; ECHA, 2008) which describe the workflow of category/analogue development and associated read-across, many challenges still remain. The consistency in how read-across predictions are made and the level of evidence required to substantiate a read-across prediction and document its justification persist, thus, thwarting greater acceptance of read-across for regulatory purposes (Patlewicz et al., 2014; 2015; Ball et al, 2016). Many researchers are working to address these challenges. Industry, through the European Centre for Ecotoxicology and Toxicology of Chemicals (ECETOC) and the European Chemistry Industry Council’s Long Range Initiative (Cefic-LRI), sponsored a task force and a workshop respectively to characterise the state of the art in read-across (ECETOC, 2012; Patlewicz et al., 2013a,b; 2015). Cefic-LRI has also invested in the development of software tools notably AMBIT to facilitate read-across predictions in particular for REACH (see http://cefic-lri.org/lri_toolbox/ambit/). The European Chemicals Agency (ECHA) developed a Read-Across Assessment Framework (RAAF) (ECHA, 2016) to establish a consistent set of principles for evaluating read-across justifications submitted under REACH. ECHA continues to sponsor the development of the OECD QSAR Toolbox (http://www.oecd.org/chemicalsafety/risk-assessment/oecd-qsar-toolbox.htm), a software tool for the development, justification and documentation of chemical categories (Dimitrov et al., 2016). The Center for Alternative to Animal Testing (CAAT) initiated a cross stakeholder workgroup including representatives from academia, industry and governmental agencies to summarise the available read-across guidance, and in particular to illustrate the extent to which HT/HC screening data could be useful in capturing biological similarity in conjunction with the traditional chemical similarity approaches (Ball et al., 2016; Zhu et al., 2016). Two additional CAAT workshops were also held, one in Europe and a second in the US to disseminate the learnings gained (Maertens et al, 2016). In addition, several of the most recent EU research programmes have been aimed at moving away from traditional animal testing – the SEURAT-1 programme is a particular example that included a significant read-across component and published templates for the structuring and reporting of read-across predictions (Berggren et al, 2015; Schultz et al., 2015).
Hence, there has been a wealth of activity exploring ways of refining and improving the manner in which read-across is performed. There have also been a number of software tools aimed at facilitating read-across prediction. Some of these tools have been in existence for many years, others have been developed more recently in response to regulatory drivers. Keeping abreast of these different tools, understanding their capacities and limitations, and where they might best be exploited within the context of the category/analogue workflow (as outlined in the OECD technical guidance (OECD, 2014)) is less clear. This article has attempted to clarify some of these aspects. To do so: we describe a workflow of category/analogue development (adapted from that described in the OECD grouping guidance (OECD, 2014)) and associated read-across including common terms of reference; we describe several of the publicly available tools and indicate their capacities with respect to this workflow in order to provide context of where these different tools offer their greatest value. We then propose how a combination of these tools address specific research and regulatory questions. Finally, we suggest what refinements in these read-across tools would be most constructive in the near term.
1.2. Terms of reference
It is worth defining various terms as they are pertinent for the comprehension of the remainder of the article. The terms category approach and analogue approach are used to describe means of grouping chemicals together that are similar in some context or another. The term read-across is reserved for a technique of filling data gaps in either approach. Analogue approach refers to the grouping of a target and source analogue together whereas a category approach refers to the grouping of a target and at least 2 or more source chemicals. The target is denoted as the chemical of interest whereas source analogues refer to similar chemicals to the target where similarity, typically structural similarity, is used as the criterion. Within an analogue or category approach, there is usually one or more rationale underpinning the selection of the source analogues. This is captured in the definition of a category as described within the OECD grouping guidance (OECD, 2014) as:
“A chemical category is a group of chemicals whose physicochemical and human health and/or ecotoxicological properties and/or environmental fate properties are likely to be similar or follow a regular pattern, usually as a result of structural similarity.
The similarities may be based on the following:
a common functional group (e.g. aldehyde, epoxide, ester, specific metal ion);
common constituents or chemical classes, similar carbon range numbers;
an incremental and constant change across the category (e.g. a chain-length category);
the likelihood of common precursors and/or breakdown products, via physical or biological processes, which result in structurally similar chemicals (e.g. the metabolic pathway approach of examining related chemicals such as acid/ester/salt)”.
Three main data gap filling techniques are used in characterising the hazard2 profile of the target chemical. These are: read-across, trend analysis and QSARs. Read-across represents the application of data from a source chemical(s) for a particular property or effect to predict the same property or effect for the target chemical. Trend analysis refers to the development of an internal QSAR model using the data from the category members (selected source analogues) to predict the specific property or effect for the target chemical (Note: This can be likened to a local QSAR model). QSAR as a data gap filling technique refers to the application of a QSAR model developed independently from the source analogues and used to predict a specific property or effect of interest. These distinctions are important to bear in mind as some of the “read-across” tools have one or more of these data gap filling techniques implemented.
1.3. The Category/analogue workflow
There are a number of steps in the development of a category or analogue approach. Slight variations of the exact number and name of these steps depends on which technical guidance and publication is referenced (see OECD, 2014; ECHA, 2008; ECETOC, 2012). It is also important to note that this workflow only considers a discrete organic chemical as the target. The workflow may well vary when considering mixtures, polymers, inorganics or nanomaterials (see ECETOC, 2012).
The seven key steps in the workflow are as follows:
Decision context
Data gap analysis
Overarching similarity rationale
Analogue identification
Analogue evaluation
Data gap filling
Uncertainty assessment
1.3.1. Decision context
The first step is a consideration of the decision context. Decision contexts can take many forms including prioritisation, screening level hazard assessment, or risk assessment. The type of decision will dictate the level of uncertainty that can be tolerated with the read-across prediction being made. For example, a prioritisation decision for a target chemical can tolerate more uncertainty than a risk assessment outcome considering the downstream consequences of the decision context.
1.3.2. Data gap analysis
This step refers to a data collection exercise for the target chemical to understand what is known from a hazard perspective in order to be able to prioritise next steps and to determine whether any read-across approach should be broadly based in scope or limited to a specific endpoint. Patlewicz et al. (2014; 2015a) discussed the number and type of data gaps pertinent for the type of non-testing approach3 used. This step identifies what is known about the hazard profile of the chemical that informs the number of data gaps and the subsequent analogue identification and selection strategy.
1.3.3. Overarching similarity rationale for the category/analogue approach
The data gap analysis for a target chemical should inform the most practical and pragmatic means of identifying source analogues. For example, according to the OECD guidance (OECD, 2014) for what a category represents, if the overarching rationale is a common functional group or structural similarity, this will focus the tactical approach of identifying analogues. If, for example, the data gap analysis shows that the only gap is for a single endpoint, such as skin sensitisation, then a more targeted search strategy might be applied to identify analogues on the basis of their common reaction mechanistic domains (Aptula et al, 2005; Roberts and Aptula, 2008). Other rationales could also be based on types of toxicity effects such as nephro or hepato-toxicants or process of manufacture.
1.3.4. Analogue identification (Analogue searching)
Analogue identification is the process of searching for analogues similar to the target chemical. The overarching similarity rationale dictates how this search is conducted practically. A search on the basis of structural similarity where a similarity index such as the Jaccard distance (Tanimoto coefficient) is used as a convenient threshold to limit the number of source analogues retrieved would be categorised as an ‘unsupervised’ approach. Whereas, a search that is informed by parameters relevant to the endpoint (e.g. a specific structural alert) would be categorised as a supervised approach.
Willett et al (1998) provide a comprehensive description of the different similarity indices for chemical searching and their respective calculation formulae. Many web-based tools that permit structure searching typically include an algorithm to search for structurally similar chemicals with a Tanimoto similarity cut off. Similarity searches for chemicals are provided in common web based tools such as ChemID plus (https://chem.nlm.nih.gov/chemidplus/), Chemspider (http://www.chemspider.com/StructureSearch.aspx) as well as in commercial applications such as Scifinder (http://www.cas.org/products/scifinder) and Leadscope (http://www.leadscope.com/).
1.3.5. Analogue evaluation
After a search of source analogues has been performed, a critical step is to evaluate the validity and relevance of these analogues. This is particularly important if the initial search is an unsupervised one since no assumptions would have been made to limit the analogue search on the basis of properties or parameters pertinent to a specific endpoint. Evaluation entails gathering associated property and effect information for the source analogues identified (see data gap filling). Source analogues with limited data, and particularly for the endpoint(s) of interest required for the target chemical, are not viable candidates for further consideration. Source analogues should be evaluated in terms of their similarity relative to the target chemical specifically with respect to their general physicochemical characteristics, metabolic profile and reactivity (Wu et al, 2010; Patlewicz et al, 2013a; Patlewicz et al, 2015a). A preliminary indication of the relative similarity can also be made by reference to existing (Q)SAR tools (ECETOC, 2012; Patlewicz et al, 2013a; 2015a). QSAR tools can be particularly helpful to provide an estimate of physicochemical characteristics such as LogKow, molecular weight (MW) and vapour pressure, all of which will be informative in assessing bioavailability. Tools that can identify structural alerts will be helpful to judge whether the toxicity profile of the source analogues relative to the target chemical are likely to be similar e.g. Derek Nexus (https://www.lhasalimited.org/products/derek-nexus.htm); OECD QSAR Toolbox. Some of these structural alerts are also informative of chemical reactivity. For example, protein binding alerts within the Toolbox mimic features indicative of electrophilic reactivity. Other tools exist that are able to make predictions of likely metabolites which provide an indication of whether metabolic pathways diverge or converge to any extent. Tools that can make prediction of metabolism include expert systems such as TIMES (http://oasis-lmc.org/products/models/metabolism-simulators.aspx), Meteor Nexus (https://www.lhasalimited.org/products/meteor-nexus.htm). Freely available tools include MetaPrint2D (Boyer et al, 2007; see also http://www-metaprint2d.ch.cam.ac.uk/) or the Cytochrome P450 predictor (Rydberg et al, 2010a,b) that exists as a module in Toxtree, a software platform developed by IdeaConsult Ltd as part of a JRC contract (see https://eurl-ecvam.jrc.ec.europa.eu/laboratories-research/predictive_toxicology/qsar_tools/toxtree). Considerations for evaluating analogue suitability are discussed in more depth in other publications including Wu et al, (2010), and Patlewicz et al (2013a; 2015a).
1.3.6. Data gap filling
This step requires a subjective evaluation of the validity of the analogues with respect to their actual experimental data, and judging the concordance and consistency of their effects across the members and across the endpoints (Blackburn et al, 2011; Blackburn and Stuard, 2014; Patlewicz et al, 2015a). The data evaluation itself is largely expert driven though can be facilitated by data quality assessment tools such as ToxRTool (Schneider et al., 2009; Segal et al., 2015) to assign scores as described by Klimisch et al., (1997). This evaluation is best informed by construction of a data matrix to readily identify which data gaps need to be filled. Each data gap is then filled based on the available data for the source analogues. In an analogue approach, a simple extrapolation between source and target chemical is performed for specific effects or properties. In a category approach, the prediction made is either based on expert judgement using one or more of the source analogues, or objectively estimated by mathematical calculation using the source analogues. Depending on the type of property data under consideration, the read-across prediction could be qualitative or quantitative. Other data gap filling techniques such as trend analysis or QSARs might also be exploited.
1.3.7. Uncertainty assessment
Although this step has not been systematically or consistently performed in practice, there are two main approaches – expert-driven or data driven. Expert-driven approaches rely on the judgement of domain scientists/experts to evaluate the relevance of the analogues as well as their underlying data. A framework was proposed by Blackburn and Stuard (2014) which describes potential areas of uncertainty, and provides a questionnaire to help assign a level of uncertainty using qualitative scores. This framework was adapted and extended by Schultz et al (2015) whereby templates were proposed to assist in assessing similarity in the context of chemistry, toxicokinetics and toxicodynamics as well as to guide the systematic characterisation of uncertainty both in the context of the similarity rationale, the read across data, and overall approach and conclusion. Similar scientific confidence considerations were outlined in Patlewicz et al. (2015a). The RAAF presents a complementary framework for structuring the read-across justification and articulating the different sources of uncertainty (ECHA, 2016). All these frameworks allow a read-across prediction and its associated justification to be evaluated, however, the assessment is subjective expert driven and qualitative in nature. Efforts are currently being investigated to determine to what extent the performance of the prediction can be objectively evaluated to enable a quantitative assessment of the uncertainty associated with the prediction possible – i.e. data -driven. For example, Shah et al (2016) have devised a means of quantifying the uncertainty associated with a read-across prediction based on a data-driven approach named GenRA, generalised read-across. The prediction accuracy of each toxicity outcome across all chemicals in a local neighbourhood was evaluated by a receiver operating characteristic (ROC) for the range of similarity indices (s) and nearest neighbours (k). The area under the curve (AUC) was then taken as a measure of performance for a given k and s.
2.1. Available “read-across” tools
There have been a number of software tools developed in the last decade to assist in category/analogue development and associated data gap filling. Here we review a representative selection of publicly available tools. Table 1 summarises the key features of each tool. The descriptions are presented in order of when the software was first developed.
Table 1:
Summary of key features of selected publicly available read-across tools
| AIM | ToxMatch | Ambit | OECD Toolbox | CBRA | ToxRead | CIIPro | |
|---|---|---|---|---|---|---|---|
| Development timeline | Java based version is dated 2012. Initial development of web version was 2005. | First public version released in Dec 2006 | Original AMBIT tool was developed in 2004–2005 | Proof of concept released in 2008 | Implementation of the Low et al (2013) article | Implementation of Gini et al (2014) | Implementation described in Russo et al (2017) |
| Type of Tool | Standalone | Standalone | Web-based and standalone | Standalone or Client/Server | Standalone | Standalone | Web-based |
| Latest Version | 1.01 (Nov 2013) Static |
1.07 (Jan 2009) Static |
3.0.3 Ongoing Enhanced in 2013–2015 |
3.4 (July 2016) Version 4 released April 2017 Ongoing |
0.75 First release |
0.11 BETA Ongoing |
First release – still prone to bugs |
| Developed by | SRC Inc | Ideaconsult Ltd | Ideaconsult Ltd | LMC, Bourgas | Fourches Lab at North Carolina State University | Istituto di Ricerche Farmacologiche Mario Negri | Zhu Research Group at Rutgers University |
| Available from | https://www.epa.gov/tsca-screening-tools/analog-identification-methodology-aim-tool | https://eurl-ecvam.jrc.ec.europa.eu/laboratories-research/predictive_toxicology/qsar_tools/toxmatch | http://cefic-lri.org/lri_toolbox/ambit/ | www.qsartoolbox.org | http://www.fourches-laboratory.com/software | http://www.toxread.eu/ | http://ciipro.rutgers.edu/ |
| Accepted Chemical Input | CAS, Name, SMILES, structure drawing/import | CAS, Name, SMILES, InChI | Name, identifiers, SMILES, InChI | CAS, Name, SMILES, structure drawing, MOL, sdf | Mol file, descriptors as txt | SMILES | PubChem CID, CAS, IUPAC, SMILES, InChI |
| Endpoint Coverage | N/A | Any based on user provided. | IUCLID8 5-supported endpoints (43 total) | Any as per the regulatory endpoints | Any based on user input | Mutagenicity and Bioconcentration Factor (BCF) | Any based on user input |
| Analogue Identification Approach | fragment matching | Distance and correlation based similarity indices based on descriptors or fingerprints | Substructure or similarity searching using structure, name, SMILES, InChI | Category definition followed by subcategorisations | Tanimoto distance using chemical and biological descriptors | VEGA similarity algorithm | Weighted Estimated Biological Similarity |
| Neighbour Selection | Automatic | Automatic | Manual | Automatic + Manual Filter | Automatic | Automatic | Automatic + Manual Filter |
| Data Source | Tool provides inventory index | User provided or tool provided | User and tool provided | User provided or tool provided | User provided | Tool provided as a result of the EU ANTARES project | User provided but tool provides PubChem in vitro data |
| Quantitative vs Qualitative | N/A | Both | User determined - Qualitative | Both | Qualitative | Qualitative for mutagenicity, quantitative for BCF | Qualitative |
| Visualisation | None | Standard 2D plots, histograms and similarity matrix | None | Standard 2D Plots | Radial plot of neighbours | Interactive Neighbour plot | Activity Plot |
| Output/Export | Output reports in the form of HTML, pdf or Excel | sdf or txt files of data, image files of plots | assessment report as docx or xlsx, data matrix as xlsx | IUCLID format, pdf and rtf files of prediction report, text files of data, image files of plots etc | NA | image file of plot | ND |
Analog Identification Methodology (AIM)
Software development
The Analog Identification Methodology (AIM) was developed by SRC Inc for the EPA’s OCSPP (Office of Chemical Safety and Pollution Prevention). The current version 1.01 was released in November 2013 (https://www.epa.gov/tsca-screening-tools/analog-identification-methodology-aim-tool) as a freely available standalone tool to help identify potential analogues for read-across. Previous iterations of the AIM methodology were web-based and date back at least a decade.
Description of tool
A target chemical is entered into the software on the basis of an identifier such as CAS, Name, SMILES or a structure (Figure 1). The structure can be drawn using the drawing palette embedded in the tool or imported as a standard chemical MOL file.
Figure 1:
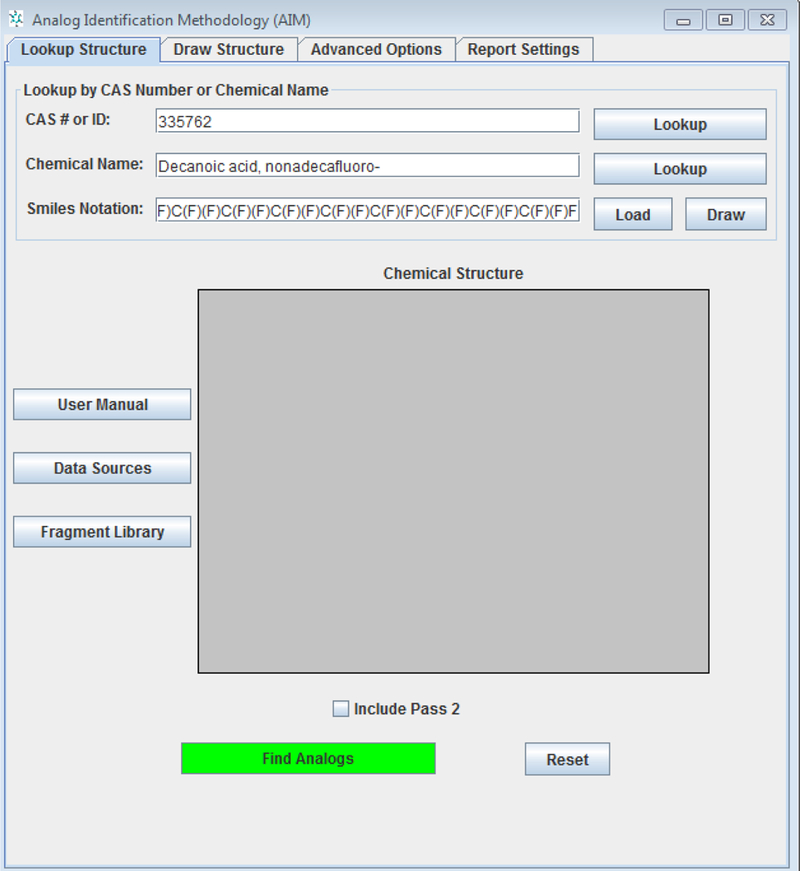
User interface of AIM
Analogues are searched using over 700 structural features (atoms, groups and super fragments) as characteristics and matched against an inventory of source analogues with available experimental data (in total the inventory comprises over 86,000 analogues pre-indexed with publicly experimental data and links to data sources). The software provides hyperlinks to the experimental data sources available but does not actually provide the underlying data themselves. AIM uses a two-tiered system for identifying analogues. The default approach is for analogues to be selected if all fragments/atoms and super fragments in the target chemical are contained in the source analogues proposed. This type of query assumes a one to one match and is the most stringent means of identifying analogues. If no analogues are identified that satisfy these criteria, a second tier is performed. Many of the large super fragments specify orientation of atoms and these types of criteria are relaxed in the subsequent search. Other rules such as combining acrylates and methacrylates, allowing metal substitutions, treating primary, secondary and tertiary amines equivalently or adjacent halogens as the same are then permitted. Specific user defined rules can also be encoded in the tool and stored for subsequent searches. AIM does not assign any relevance metric on the analogues identified to prioritise them such as a similarity index, instead it provides the end-user with a list of potential analogues and their associated data sources links. The end-user must apply subjective judgement to determine the validity of any of the suggested analogues for the decision context of interest.
Data Sources
There are a number of different data sources indexed. In AIM, descriptions of each of these sources and links are provided in a pdf document that can be accessed by clicking on the Data Sources button within the user interface. Data sources include chemicals whose data have been submitted under the Toxic Substances Control Act (TSCA); chemicals that have been indexed within the following databases: the Aggregated Computational Toxicology Resource Database (ACToR), the ECOTOXicology database (ECOTOX), the Hazardous Substances Data Bank (HSDB), the Integrated Risk Information System (IRIS), the National Toxicology Program (NTP), the Agency for Toxic Substances and Disease Registry (ATSDR), the High Production Volume Information System (HPVIS), the OECD High Production Volume (HPV) chemical database, the Canadian Chemicals Management Programme (CMP), the OPP Pesticide Ecotoxicology database, the Distributed Structure-Searchable Toxicity Database Network (DSSTox), the Registry of Toxic Effects of Chemical Substances (RTECS), the International Uniform Chemical Information Database (IUCLID) and the Acute Exposure Guideline Levels (AEGLs). It should be noted that some of these sources have since been updated or have become superseded by new developments. For example, the DSSTox library is now part of the US EPA’s National Center for Computational Toxicology (NCCT) Chemistry dashboard (https://comptox.epa.gov/dashboard) which is indexed to a number of different data sources. IUCLID referenced in AIM is IUCLID v4 which has since been superseded by later releases of IUCLID and the data that have been submitted for the old EU legislation has since been re-submitted in more detail under the REACH regulation (EC, 2006). AIM has not as yet accommodated these updated resources.
Output
AIM provides an output report file in the form of a html, pdf or excel file. This presents the user with a summary of the target chemical and presents the source analogues starting with the exact target match if available as well as other related analogues. These are ordered on the basis of the number of data sources available. Hyperlinks to the data sources are presented in the report to enable closer inspection. No evaluation of the quality of the data sources is provided in the report.
Toxmatch
Software development
Toxmatch is an open source application which encodes a variety of chemical similarity indices to facilitate the grouping of chemicals into categories for the purposes of performing objective endpoint specific read-across. It was developed by IdeaConsult under the terms of a EU JRC contract (https://eurl-ecvam.jrc.ec.europa.eu/laboratories-research/predictive_toxicology/doc/Toxmatch_user_manual.pdf). Several case studies outlining its functionality were described by Gallegos Saliner et al (2008) and Patlewicz et al (2008). The current version of the software is 1.07, which was released in January 2009.
Description of tool
A dataset comprising structures, activity information and descriptors is either introduced into the software as a training set, or one of the pre-defined datasets can be selected for analysis (see data sources). Pairwise similarity measures are then calculated for this dataset and a similarity matrix is created to visualise the structural diversity within the dataset. There are a range of different similarity indices that can be computed such as distance like similarity indices or correlation like similarity indices. The type of similarity index will depend on the type of chemical descriptors used to characterise the dataset. In Toxmatch, the following measures are included: Euclidean distance, Cosine similarity, Hodgkin-Richards Index, Hellinger distance and Tanimoto distance. The first four of these are implemented as descriptor based methods. The latter two are structure-based using atom environments or fingerprints. In the simplest use case, a dataset is introduced, depending on whether a descriptor or structure-based approach characterises the dataset, one of the different similarity index method will be used to derive pairwise similarity measures. Toxmatch provides two options for a similarity assessment: either an average similarity between a query chemical and its nearest neighbours, where by default, the number of neighbours is 10 (though a user can change the setting) or a similarity between a query chemical and the composite fingerprint or fragment set. The latter approach is limited to the Tanimoto distance and Hellinger distance. The similarity information is then used to derive a prediction of the endpoint (activity) of interest. The way in which this prediction is calculated depends on the dataset in question and whether the endpoint or activity is categorical or continuous in nature. If the activity is continuous in nature, the read-across prediction is effectively a prediction of activity based on the weighted average of the activity values of the k nearest neighbours i.e. the activity of the most similar (closest) chemicals are averaged proportionately and used to estimate the activity of a chemical of interest. If the activity is categorical, the read-across prediction is a classification problem. In this case, the read-across is slightly different as the source analogues are binned into groups and the similarity measure provides the means to define the likelihood that the target chemical falls into one or other group. The procedure also relies on k nearest neighbours and classifies the target chemical into the group where most of the k most similar chemicals belong.
For a de nuovo chemical, the target chemical is introduced into the test set domain of the software interface. A similarity with respect to the training set will identify the 10 nearest neighbours in the training set (by default) in order to calculate a similarity weighted average of the activity. A prediction of the categorical activity is made based on which activity the nearest neighbours to the target chemical belong to. Within the similarity matrix view (Figure 2), a user can browse the similarity matrix created and filter the view to identify the most similar analogues from the training set that meet a specific similarity threshold. A view of the respective source analogues and their associated experimental values, predicted values and similarity index can be viewed and exported by selecting the option to calculate a similarity with respect to the test (target) chemical.
Figure 2:
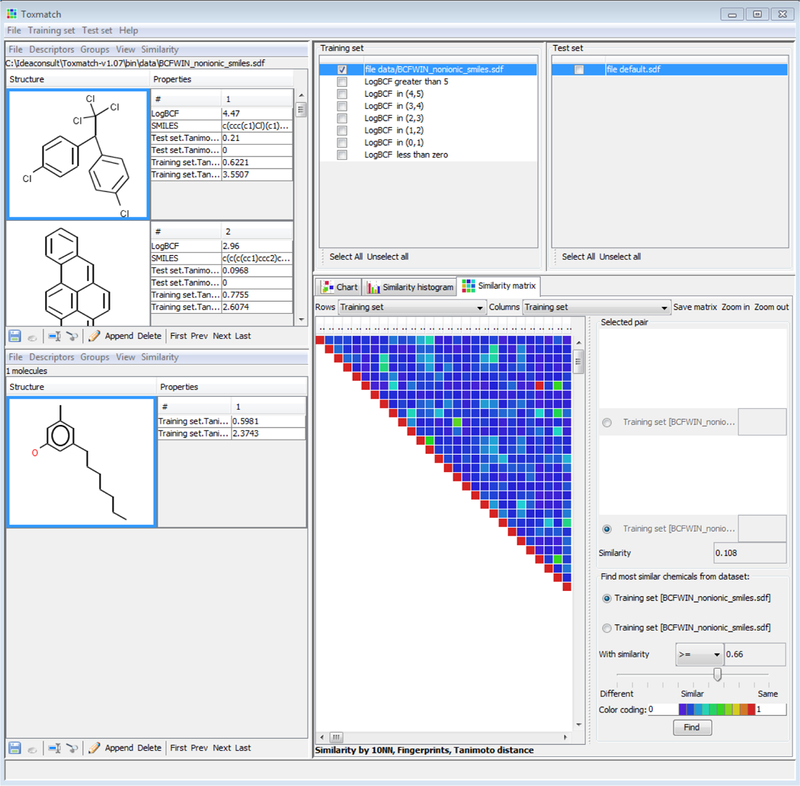
Similarity matrix view from within Toxmatch
Data sources
Several pre-defined training sets are provided in the software including aquatic acute toxicity (fish), bioconcentration factor, skin sensitisation, skin irritation, carcinogenicity and mutagenicity. The aquatic toxicity dataset is a copy of the DSSTox EPA Fathead Minnow Acute toxicity (April 2006 update). The BCF dataset is taken from the EPA’s EPIsuite software. The skin sensitisation dataset is from Gerberick et al (2005), a collection of published LLNA data. Two skin irritation datasets are included; one a reference bank from ECETOC and a second a compilation from several other sources. The carcinogenicity and mutagenicity datasets are those provided by ISSCAN that were made available as DSSTox files. The intent with these datasets is help a user familiarise themselves with the functionalities and features of the tool itself rather than be an exhaustive repository of datasets. The sources are described in more detail in the Toxmatch user manual. A user can import their own dataset of toxicity information together with their own descriptor information – typically chemical descriptors or use the descriptor calculators within Toxmatch to generate chemical information.
Output
Scatter plots of the similarity index as a predictor of the activity or plots to explore the correlations between descriptors and activity can be generated and exported. Table exports of the similarity information and predicted activities can also be generated. A picture of the pairwise similarity matrix can also be copied and pasted or saved as a png file.
AMBIT
Software development
AMBIT is a cheminformatics software tool that was first developed by Ideaconsult Ltd and sponsored by Cefic LRI in 2004. The current version incorporates specific functionality to more efficiently support category formation and read-across to address the specific needs for REACH (http://cefic-lri.org/lri_toolbox/ambit/). Under REACH, industry registrants have to use IUCLID as means of submitting their registration dossiers to ECHA. IUCLID is structured to capture endpoint data and substance composition. Although IUCLID can be queried on the basis of a specific substance (e.g. by name or ID) or endpoint data, it cannot be searched on the basis of structure. The functionality within the current version of AMBIT enables both structure and data searches with IUCLID which in turn facilitate category development and read-across. Using AMBIT for read-across analysis creates a chemical assessment aimed at producing a report/document suitable for regulatory submission.
Description of tool
The AMBIT system comprises a database and functional modules to enable different search queries to be performed. Search queries can either be performed on the basis of chemical identifiers (structure or IDs such as CAS) or on the basis of endpoint data (Figure 3).
Figure 3:
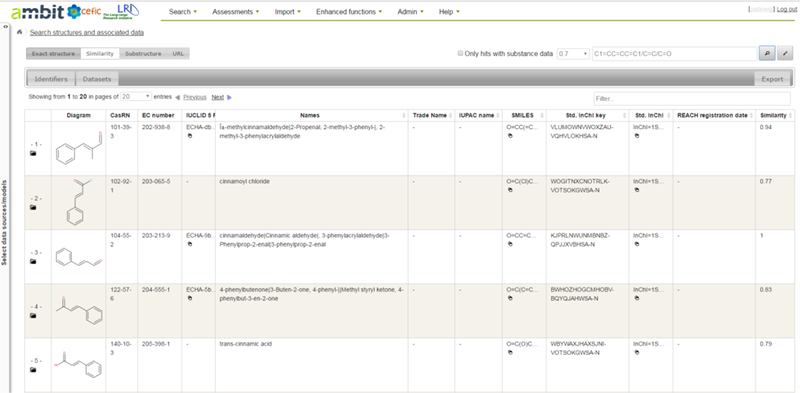
User interface of AMBIT following structural similarity search
AMBIT was updated as part of a followup Cefic LRI project in 2013 to handle substance4 IDs, including structures, composition of mono- and multi-constituent substances, and, special cases of UVCBs (unknown or variable compositions, complex reaction products and biological materials), addressing constituents, impurities and additives in the same way as IUCLID is structured. In addition to search queries, a category assessment workflow was developed to enable a user to create a read-across prediction and its associated report. The workflow consists of the following steps: assessment identifier (this helps to document and track the assessment being developed), collect structures (this includes identifying the target chemicals and associated source analogues), endpoint data used (selecting what endpoint data to be queried, depending on the scope of the read-across intended), assessment details (creation and revision of the data matrix), and report. The workflow begins with populating specific fields in an assessment identifier form with user information and name of the assessment being created. This information is used later to prepopulate an assessment report. The next step is to select a target substance using the “collect structures’ tab. A target substance can be searched on the basis of various identifiers including exact structure, CAS, EINECS, InChI, SMILES or name. Structurally similar analogues to that target can then be identified using Tanimoto as a similarity index or on the basis of substructure searching. Analogues identified can be limited to only those with some associated data. At this point the target and associated analogues are tagged as target and source substances and a rationale for their inclusion can be added. The next step is to collect the resulting list of substances and select endpoint data of interest. The available endpoint data will only be visible if the specific substances which are associated with a data record are checked. This is done by opening the structures in turn within the “endpoint data used” tab and inspecting to see what information is available within the IUCLID database e.g. if the structure is associated with information on particular impurities or other constituents, this will be reflected here. The constituent substances selected which update the “selection of endpoints” tab with the number of data records for all the available endpoints. A user can select all endpoints of interest or select a handful of endpoints of interest depending on the scope of the intended read-across. Clicking on the next tab – assessment details presents the analogues and data in an initial data matrix. This enables an assessment of data gaps for the target which are to be filled through read-across from the remaining source analogues. Outlier data can also be readily identified. Predictions from Toxtree5 modules are also made automatically where feasible to supplement any data gaps. The read-across prediction and justification are user derived based on subjective expert judgement. The read-across predictions are recorded by launching the working matrix and adding new records within the relevant endpoint cells. Information on the read-across approach, the rationale, the source analogue used for the prediction, the toxicity value being used as the read-across value can be annotated by the end user as a record. Any outlier data can be deleted within the working matrix or missing records not reflected in the database can be added in the relevant endpoint cell (see Figure 4).
Figure 4:
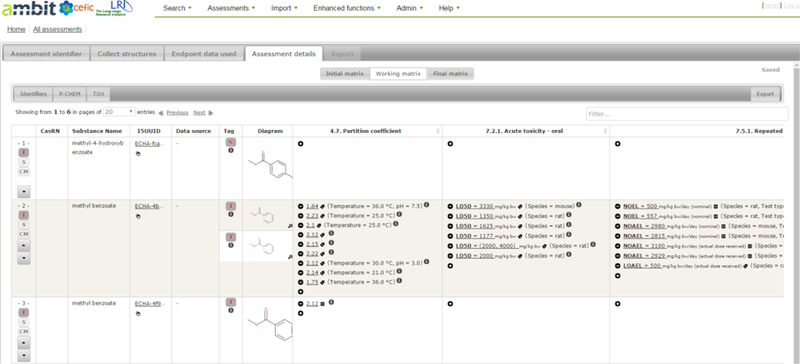
Screenshot of the working matrix within the AMBIT assessment workflow
The last step is to finalise the matrix by saving any edits made. This will enable creation of an assessment report which mimics the category and analogue reporting formats that are described in the ECHA and OECD grouping guidance documents (ECHA, 2008; OECD, 2014). The report can be exported as a word document and the associated data matrix files will be able for download. An audit trail of data records that were deleted are also tracked in the report.
Data sources
The AMBIT database stores more than 450,000 chemical structures and their identifiers such as CAS, EINECS. ECHA also provided access to the entire non-confidential REACH dataset, amounting to information on 14,570 substances for a range of physicochemical, environmental fate, ecotoxicity and toxicological information.
Output
The main outputs that AMBIT provides are exports in various formats of the data matrices created and the assessment report.
OECD QSAR Toolbox
Software development
The OECD QSAR Toolbox6 is a software application intended for use by government agencies, chemical industry and other stakeholders in filling gaps in ecotoxicity, environmental fate and toxicity data needed for assessing the hazards of chemicals. The Toolbox was developed through a series of phases, starting with a proof of concept which was released in 2008. The version of the OECD Toolbox described herein is v3.4 (see Figure 5), this third generation contains many advanced features and was released in July 2016. The Toolbox is developed by the Laboratory of Mathematical Chemistry (LMC), University As Zlatorov, and its development is managed by OECD with funding from ECHA. A new version on a different software platform was released 4th April 2017, version 4. Training and documentation is still in development though the principles and main functionalities described here will be also applicable to version 4.
Figure 5:
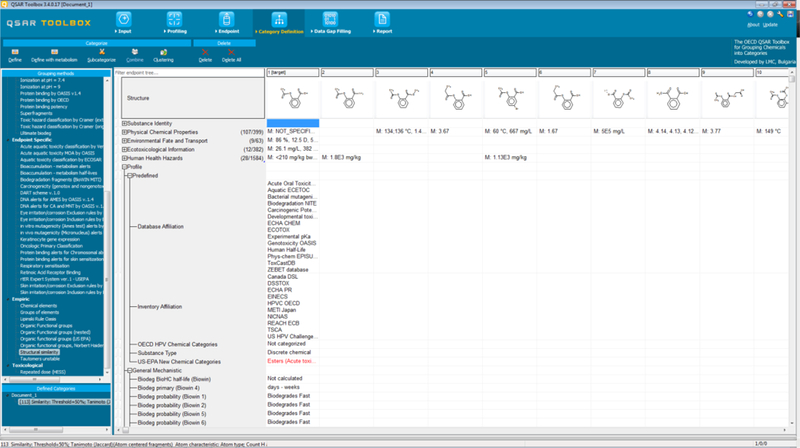
Screenshot of the OECD Toolbox v3.4
Description of tool
The OECD Toolbox incorporates information and tools from various sources into a workflow. The workflow mimics that described in the OECD grouping guidance (OECD, 2014). Dimitrov et al (2015) have described the workflow and the major functionalities in more detail. Here we describe the main features and use. A target chemical is introduced into the Toolbox using a chemical identifier such as a name, a CAS registry number or by drawing a chemical structure using the inbuilt drawing tool. The target can then be “profiled”. There are several types of ‘profilers’ in the Toolbox namely: predefined, general mechanistic, endpoint specific, empiric and toxicological. Each type is described to help clarify what is meant by the term ‘profiler’, though they might also be considered as structural matching or indexing rulebases. Figure 5 shows a screenshot of the interface.
Predefined profilers capture affiliations to the databases or inventories contained within the Toolbox. There are also a handful of profilers within this set that flag whether a chemical is a member or a potential member7 of the EPA New Chemical Categories (EPA, 2010) or the OECD High Production Volume (HPV) categories (see http://webnet.oecd.org/hpv/ui/Default.aspx) as well as define its substance type (discrete organic, salt, inorganic, mixture etc). In the Toolbox, the general mechanistic profilers are in many cases collections of structural alerts that may or may not be substantiated by experimental data. For example, there are DNA binding profilers that are pertinent to genotoxicity endpoints, but a subset of these alerts may be more theoretical in nature in that the alerts are based on chemical reasoning and not necessarily supported by experimental genotoxicity data. These general mechanistic profilers also include the Cramer structural classes that are used in the application of the Toxicological Threshold of Concern (TTC) (Cramer et al, 1978). Endpoint specific profilers are tailored for specific endpoints and are underpinned by experimental data. For example, the DNA binding profiler will be the same profiler as is reflected in the general mechanistic profiler but certain alerts that have a lower confidence due to the lack of supporting data will not be copied across i.e. the endpoint specific DNA binding profiler is a subset of the general mechanistic profiler for DNA binding. Other profilers in the Toolbox such as the ‘aquatic toxicity classification by ECOSAR’ rely on the SAR classes within the EPA’s ecotoxicity prediction tool, ECOSAR (https://www.epa.gov/tsca-screening-tools/ecological-structure-activity-relationships-ecosar-predictive-model). Empiric profilers comprise unsupervised approaches to help in the identification of analogues – these profilers include identifying what chemical elements or functional groups are present in the target chemical. An example of the functional group profiler is the Organic functional groups (US EPA) which consists of the 645 structural fragments and correction factors taken from the LogKow model KOWWIN that is part of EPISuite. The toxicological profiler contains only one toxicity rulebase for repeated dose toxicity (HESS). This profiler was developed by the National Institute of Technology and Evaluation (NITE) in Japan. The profiler contains categories that are expected to induce similar toxicological effects in repeated dose oral toxicity studies. These categories were developed using repeated dose toxicity test data collected as part of Hazard Evaluation Support System (HESS) (see http://www.nite.go.jp/en/chem/qsar/hess-e.html).
The next step of the workflow involves gathering available endpoint data from multiple sources that have been provided to the Toolbox. Some datasets are focused on a specific endpoint whereas other sources are more encompassing. Examples include the COLIPA Dendritic cell collection – data from the h-CLAT and MUSST assays which are both in vitro sensitisation tests donated by the Cosmetics Industry’s trade association, whereas the ECHA CHEM presents the data that were submitted under REACH that have been since disseminated publicly through the ECHA website and via the OECD Chemportal (http://www.echemportal.org/). There are underlying templates – the OECD harmonised templates that structure the available data into specific fields based on standardised ontologies. These are essential to enable any subsequent data gap filling to be performed using data generated through the same protocol, species etc. The endpoint experimental gathering step is critical to focus how the category should be defined for the subsequent data gap filling. The Toolbox is really intended to facilitate the endpoint specific data gap filling rather than developing a category of analogues to address more than one endpoint at the same time. After data are collected for the target, the user selects the endpoint of interest to focus the subsequent evaluation. This is a data gap analysis step to identify and prioritise efforts to fill them. A cell is selected, which denotes the specific endpoint of interest at the appropriate level of detail, to start the next step in the workflow – the category definition step. This category definition step directs a user to select one or more of the profilers to identify source analogues and subcategorise the analogues retrieved so that the set of final analogues identified are similar with respect to all the profiling outcomes chosen. The profiling to identify these analogues can factor in potential metabolites although the decision to select this will depend on what the initial profiling outcomes are for the target chemical and whether activation is considered to be an important factor for the endpoint of interest. After subcategorising the source analogues, the next step is to perform the data gap filling for the endpoint of interest, using one or more of the data gap filling approaches. Both the number of analogues with data and the type of data will determine which data gap filling technique is most appropriate. A number of QSARs and their predictions are implemented into the OECD Toolbox – including the Danish EPA QSAR models and the EPA EPISuite models. If a user has a licence for the OECD Toolbox developer’s commercial tools such as TIMES (discussed earlier), these will be integrated and docked automatically into the Toolbox to enable predictions to be made and surfaced in the Toolbox data matrix interface. Trend analysis will only be invoked if the property data are quantitative in nature and a minimum of 3 data points are available for the source analogues. The trend analysis permits a regression model to be derived on the basis of a single parameter. A quadratic or averaging approximation can be selected as alternative trend analysis options. The default correlating parameter is LogKow but other chemical descriptors and endpoint tree descriptors are encoded to facilitate other correlations to be explored and trends to be derived. Endpoint tree descriptors represent any other experimental information to be used as a descriptor – e.g. a correlation between a specific ToxCast assay and the endpoint of interest can be evaluated. A level of significance can be set with the prediction made which by default is a confidence level of 95%. Read-across can be performed quantitatively or qualitatively, and various approaches are included in the prediction calculation to enable a prediction to be an average, minimum value, maximum, mode or median value on the basis of 5 neighbours. The number of neighbours can also be changed by the end-user. The prediction is still reliant on a single descriptor as a means of scaling the prediction made and this is by default LogKow.
The last step in the workflow is documenting the prediction made. Prediction templates that follow a similar structure to the QSAR Prediction reporting format (QPRF) as reported in the ECHA guidance (ECHA, 2008) can be created which document the logic and steps a user has made in deriving the prediction. Export files in IUCLID can also be generated which is particularly pertinent for Industry users submitting registration dossiers to ECHA.
Data Sources
The OECD Toolbox contains many data sources covering many different endpoints of regulatory interest. The data sources cover physical chemical properties, environmental fate and transport, ecotoxicological information, and human health hazards. Physical chemical property data comprise the phys-chem data that exist within the EPA EPISuite tool as well as data collected as part of REACH submissions. Other data include chemical reactivity data and pKa information. The e-fate and ecotox data comprise datasets from Cefic LRI, LMC, REACH data and EPISUITE data. The human health dataset is by far the largest set including datasets for specific endpoints or the REACH compilation. Users can also import their own datasets or databases into the Toolbox.
Outputs
There are many outputs that can be exported and extracted from the Toolbox depending on the step of the workflow. Profiling outcomes can be exported as text files together with their structural identifiers such as SMILES strings. Endpoint data can be exported to an extent: the REACH data and certain other datasets are not permitted to be exported. Within the data gap filling, scatter plots generated of the predictions made are exportable. Reports summarising the category derived and the predictions derived can be exported as reporting formats consistent with ECHA and OECD guidance. IUCLID file exports are also possible, which is useful for users submitting REACH dossiers.
CBRA
Software development
Chemical Biological Read-Across (CBRA) was a term coined by Low et al (2013) in research aimed to extend the chemical similarity principle to predict toxicity by incorporating biological activity data in an effort to account for biological similarity. This hybrid approach of using both chemical and biological activity data was expected to be more predictive of in vivo toxicity. The actual toxicity prediction was a similarity weighted average of the activities of nearest neighbours visualised as a radial plot. A software implementation of the approach was developed and is freely available from http://www.fourches-laboratory.com/software.
Description of tool
Users need to introduce three different input files: 1) a file of chemical structural descriptors, 2) biological activity information which are structured as descriptors; and 3) a file of toxicity information – namely the activity to be predicted. In the software download, sample files of these input files are provided to illustrate the format of the files and the way in which the software functions. Once files have been imported, information is provided to summarise the number of chemicals included in the input file, as well as the number of chemical and biological descriptors. Options are available to modify the number of neighbours or the Tanimoto similarity threshold. The default values are 5 and 0.4 respectively. To run a prediction, the “Compute CBRA” button is clicked and a radial plot is automatically generated. CBRA radial plots and the associated predictions for each of the chemicals in the input file can be viewed in turn by clicking on the arrows to move from one chemical to the next. In the details window of the tool, information is provided to indicate what the nearest neighbours are for a specific target chemical and their associated similarity thresholds. The radial plot is structured to reflect 2 sets of neighbours – those on the basis of biological similarity and those on the basis of chemical similarity. The neighbours identified are not necessarily the same but may overlap. Chemical neighbours are reflected on the right hand side of the plot whereas biological neighbours are shown on the left hand side of the plot. The toxicity activity and the prediction is reflected by colour – red for active and green for inactive. The target chemical is represented as the centroid in the radial plot and its colour will indicate its read-across prediction outcome. In the screenshot (Figure 6), the centroid is red, indicating that the prediction of the target chemical is active.
Figure 6:
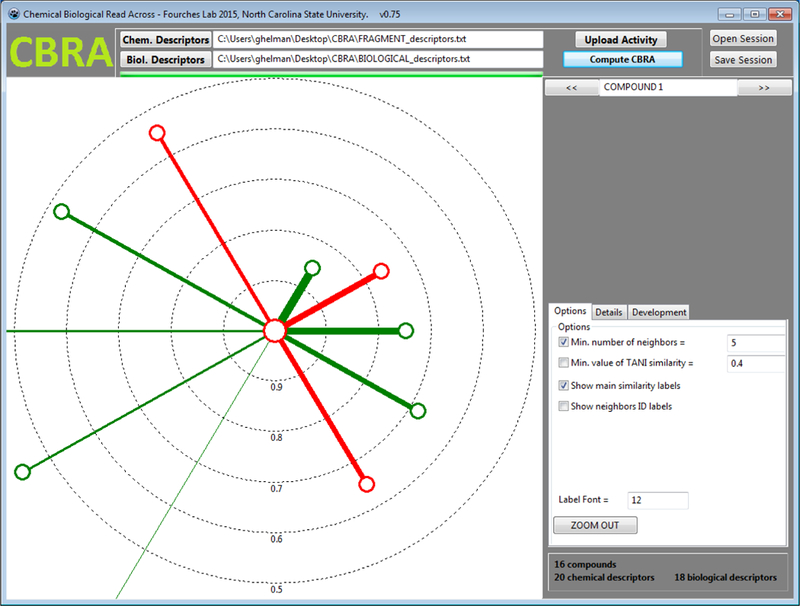
Read-across prediction with the CBRA tool
Data sources
There are no data sources available within the tool. End users provide their own datasets and upload them in the tool itself.
Outputs
The main output is the radial plot summarising the prediction (the centroid colour represents the overall prediction for the target chemical) and the neighbours. There are no export options in the current version of the software.
ToxRead
Software development
ToxRead was developed by Gini et al (2014) as a standalone Java tool to help in the assessment of Ames mutagenicity. The research was funded by two EU projects CALIEDOS (http://www.caleidos-life.eu/) and PROSIL (http://www.life-prosil.eu/). The current version of the tool is v0.11 (http://www.toxread.eu/) and includes modules to make read-across predictions of both Ames mutagenicity and Bioconcentration factor (BCF).
Description of tool
An end-user inputs a structural identifier in the form of a SMILES string and chooses the number of nearest neighbours (source analogues) and endpoint in order to run a read-across prediction (Figure 7). By default, three analogues are typically presented. The application relies on the VEGA core library (http://www.vega-qsar.eu/index.php) that implements a similarity index. The VEGA library also provides other features, such as parsing of the SMILES string, SMARTS matching and molecule depiction. The read-across prediction approach relies on accessing a local internal database within the ToxRead application (see later), and extracting the source analogues with such information in order to construct a read-across plot.
Figure 7:
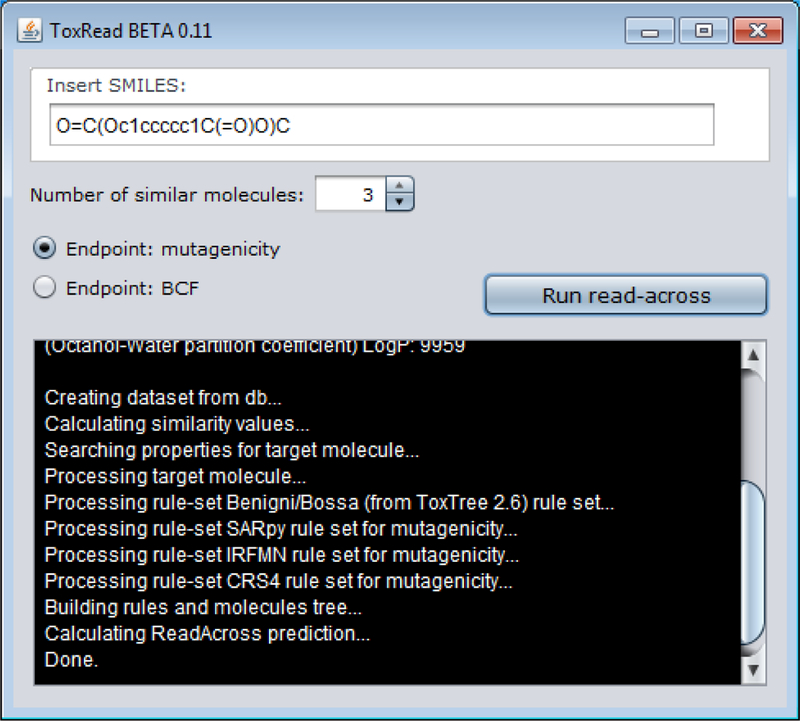
User interface of ToxRead
The output from ToxRead produced takes the form of a plot where the centre represents the target chemical with various outgoing links to the identified source analogues. The target and source analogues are represented in different colours and by different shapes. Source analogues that are identified by the VEGA similarity algorithm rely on a similarity value that is calculated as the weighted combination of a fingerprint, three structural keys based on molecular descriptors and a series of other descriptors (constitutional, hetero-atoms and specific functional groups) all of which are described in more detail in Gini et al (2014). Figure 8 presents the chart for the read-across prediction for chemical (2E)-2,3-dichloroprop-2-enal [SMILES string ClC(=CCl)C=O]. The chart for this chemical is discussed in more detail later.
Figure 8:
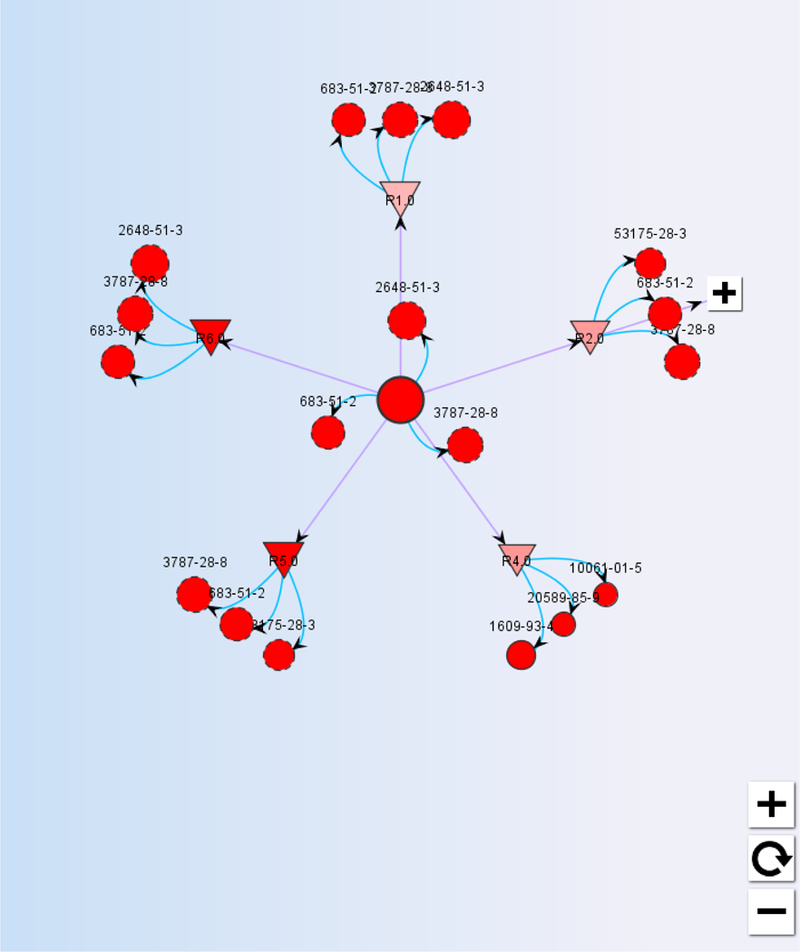
Read-across prediction chart from ToxRead
The target chemical of interest is represented as the centroid. Surrounded by it are 3 red circles labelled by CAS numbers. These are source analogues identified on the basis of the VEGA similarity algorithm. The size of the circles of these source analogue is proportional to their similarity index. The smaller the circle, the less similar the analogue. Double clicking on each of the 3 analogues will reveal the Tanimoto similarity index of the source analogue relative to the target, its identity by CAS and SMILES string, as well and its experimental mutagenicity outcome and any other experimental information that might be available e.g. LogKow, BCF. The source analogues identified in this manner rely on the same algorithm that exist in the other VEGA QSAR tools where QSAR predictions are made for an endpoint and the “similar” analogues to the chemical of interest are extracted to enable an end-user to evaluate the validity of the prediction made (Manganelli and Benfenati, 2016). The other links (arising from the triangles as shown in Figure 8) identify source analogues that share a mechanistic similarity – in this case analogues that share one or more common structural alerts for mutagenicity. ToxRead includes 4 main libraries for mutagenicity comprising some 759 rules in total. The structural alerts are taken from the SARpy rulebase developed within the VEGA program developers (Ferrari et al, 2013), the Benigni-Bossa rulebase that is implemented in both the OECD Toolbox and Toxtree platforms (Benigni et al 2008) as well as 281 alerts that were manually extracted by experts and rules empirically extracted as part of the LIFE PROSIL project (www.life-prosil.eu). Some of the alerts are negative alerts for mutagenicity – i.e. flagging the absence of a mutagenicity outcome rather than its presence. The structural alerts are represented by triangles and their interpretation differs depending on their colour and orientation upwards or downwards. Those pointing indicate downwards are coloured red and reflect a mutagenic outcome whereas those pointing upwards are green and reflect a non-mutagenic outcome. The confidence of the alert is reflected in the colour shading of the triangle – a darker colour represents an alert where there is a greater proportion of actives to inactives for the alert. Clicking on a triangle reveals more information about the alert – its structural definition and source, the accuracy of the alert (expressed from 0–1), and the confidence associated with it, expressed as a Fisher test p-value. It is also possible to view and export a list of up to 100 similar chemicals presenting that alert, together with their similarity indices and mutagenicity outcomes. The three most similar analogues that present the alert in common with the target are reflected in the chart (as illustrated in Figure 8). The read-across prediction is readily summarised for the target chemical by the colour – red for mutagenic and green for non-mutagenic. Double clicking on the target chemical reveals the summary prediction information. A numeric score between 1 and 0 is reflected together with a text assessment for the read-across. QSAR model predictions from ISS, Caesar, SARPy and KNN models are also provided with reliability scores and a consensus score of all 4 models. An overall assessment for the target chemical is also provided that integrates both the QSAR and read-across predictions. An example of how this assessment output is represented for the same target chemical is shown in Figure 9.
Figure 9:
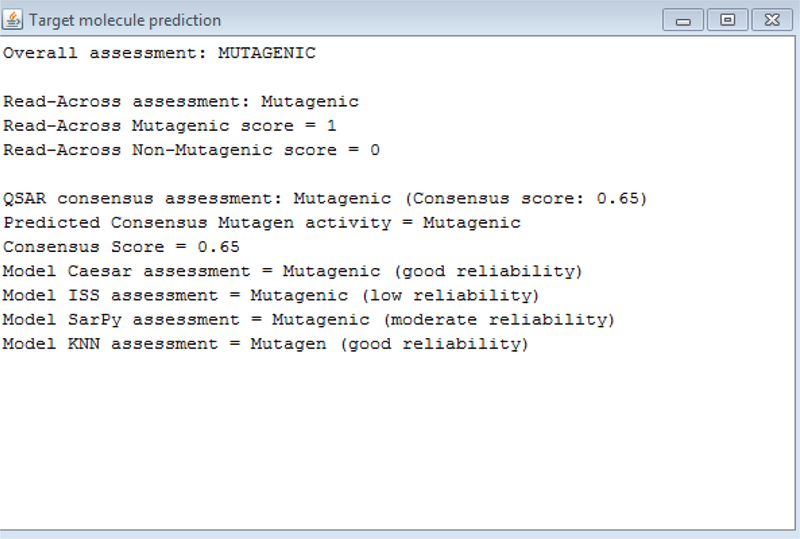
Overall target prediction outcome
In the example shown in Figure 8 – there are 3 similar analogues on the basis of “structural” similarity that are all mutagenic experimentally. Four structural alerts are identified as relevant for the target chemical – three of these have lower validity as reflected by the lesser density of their red shading, but the similar analogues that fire these alerts are all mutagenic experimentally. The analogues identified may appear more than once and be linked to different rules. These are reflected as circles with dashed lines. This is true for this example as at least 3 analogues are identified more than once. The overall evaluation is straightforward if there is no conflicting information from the different sources, in cases where this is not the case, an objective overall assessment is still derived but this can be potentially overruled by the end-user based on their own knowledge and expertise. More detailed examples are presented in Manganelli and Benfenati (2016) who demonstrate the value of an integrated approach of combining both QSAR and read-across predictions to derive an overall assessment.
Data sources
The underlying database of ToxRead contains 6065 chemicals with experimental data for mutagenicity. The mutagenicity data are taken from several well-known sources included CCRIS, Helma et al (2004), Kazius et al (2005), Feng et al (2003), VITIC (2005) and GeneTox databases (Matthews et al, 2006). Other experimental data also include BCF data in fish for 857 chemicals, carcinogenicity outcomes for 784 chemicals and LogKow values for 9959 chemicals.
Output
The main output is a read-across plot which can be copied or saved as an image file. Analogues that present specific structural alerts can also be viewed and exported, up to 100 chemicals.
CIIPro
Software development
CIIPro is a cheminformatics web portal freely available at http://ciipro.rutgers.edu/. It is intended to facilitate read-across predictions of a target on the basis of chemical and/or biological similarity. The prediction result can be visualised by a similarity chart along with associated similarity and confidence values. The novelty of the approach lies in taking advantage of the wide array of bioassay data publicly available from PubChem (https://pubchem.ncbi.nlm.nih.gov/). Updates to the underlying bioassay database are made on a monthly basis by the CIIPro developers.
Description of tool
The starting point in using the portal is to upload a training and test set of chemicals on the basis of their chemical identifiers (PubChem compound identifier, CID). The training set of chemicals is used to create a biological profile using the CIIProfiler tab. This is created by extracting relevant bioassay data from Pubchem. As stated by the website’s tutorial, the biological profile can be optimised by requiring a minimum number of active responses per assay, the default is otherwise set to 6. The biological profile derived is represented as a heatmap (not shown). The density of the colour will dictate whether a response is active (dark blue = 1), inconclusive (grey = 0) or inactive (light blue = −1). CIIP Predictor is then used to calculate a Weighted Estimated Biological Similarity (WEBS) between the chemicals in the test set and the chemicals in the training set. The WEBS tool calculates 2 values for each chemical pair, the biological similarity (from 0–1) and its confidence score. The confidence score is an estimate of the reliability of the calculated biological similarity, the higher the score, the more reliable the biological similarity value. The confidence score represents the number of assays that have results for both chemicals in a given pair but gives less weight to the assays that only have inactive results for both chemicals (discussed in more detail in Russo et al., 2017). Biological nearest neighbors are then calculated by the WEBS tool by setting suitable parameter cutoffs for both the biological similarity and the confidence scores. The biological similarity cutoff is the minimum biological similarity score for a chemical to be considered as a nearest neighbor to the target chemical. The confidence score cutoff is the percentage of assays in the biological profile that both chemicals need to have responses in for a biological similarity calculation to be meaningful. The number of biological nearest neighbors (from 1 to 5) to be used for predictions is also selected by the end-user. The activities of each test chemical’s biological nearest neighbours’ are averaged together to predict the target chemical’s activity. Chemicals that do not have enough biological nearest neighbors to make a prediction are labelled as ‘N/A’. Results are presented in a table listing the chemicals in the test set, the chemicals’ in vivo activity, and the activity for each chemical predicted by CIIP Predictor. The biological nearest neighbours and the chemical nearest neighbors (i.e. chemicals in the training set structurally similar to the target chemical in the test set on the basis of MACCS keys) of the predicted chemicals can be visualised in a similarity plot (Figure 10 reflects an example). Biological nearest neighbours are presented on the right hand side of the plot whereas chemical nearest neighbours are on the left of the target chemicals’ predicted activity (Russo et al, 2017).
Figure 10:
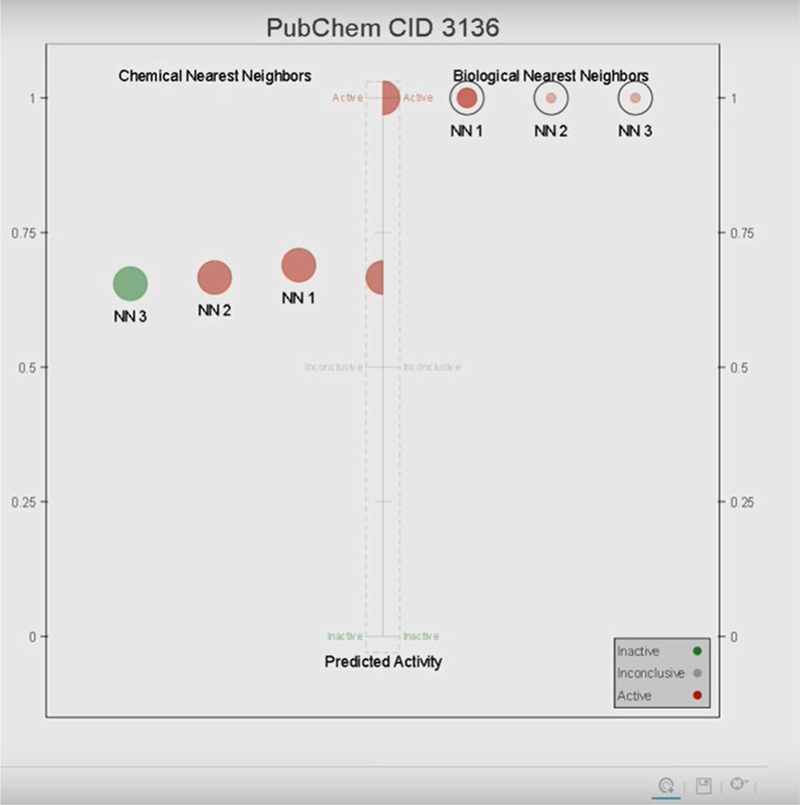
Example plot from CIIPro
Data sources
The main data source is the PubChem data that is accessed from within the portal. The remaining data inputs are all user provided.
Output
The main outputs are the read-across predictions in a tabular form and the similarity plot.
3. Putting the tools into the context of the category/analogue workflow
Figure 11 outlines the main steps in the category/analogue workflow and highlights where the different tools overlap with respect to these steps.
Figure 11:
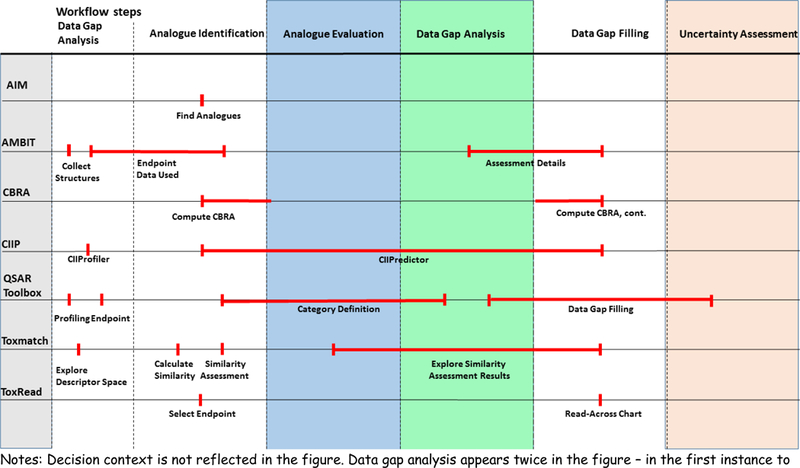
Selected read-across tools in the context of the category/analogue workflow
Notes: Decision context is not reflected in the figure. Data gap analysis appears twice in the figure – in the first instance to do a data gap analysis of the target chemical and a second time to help evaluate the data coverage for the source analogues. The red lines are aimed to indicate what parts of the workflow are captured explicitly or implicitly in each of the tools.
AIM
The AIM tool performs the analogue identification step of the workflow. It identifies analogues using its database of atoms, groups, and super fragments and matches the target chemical to its in built inventory. It is not designed to address any of the remaining steps in the workflow.
Toxmatch
Toxmatch does not have a proposed workflow in its design. Instead, the tool divides its functionality into different windows that all occupy the same screen. Users are instructed to upload their own data or use one of the predefined data sets. Similarity can then be calculated on the basis of fingerprints or descriptors, which can themselves be calculated by the tool. Toxmatch also includes robust charting and similarity matrix querying for exploring the results of a similarity assessment. The data matrix is not visible to gauge data gaps. Indirectly Toxmatch addresses the data gap filling, analogue identification and evaluation steps of the workflow but this is incumbent on the end-user to supply the data sources from which analogue identification is performed and to make a determination of what endpoint to focus the subsequent assessment on.
AMBIT
AMBIT performs several of the steps in the workflow – identifying analogues (on the basis of structural similarity), constructing and displaying a data matrix of available data for target and source analogues to documenting the read-across prediction as part of an assessment report. Any read-across predictions themselves as well as the justification of the validity of those predictions are determined by the end-user. The main advantage of AMBIT is the ability to browse in some level of detail the available data from its sources and present these data in a matrix view for browsing and exporting. The ability to search for analogues is currently restricted to a structural similarity search using a Tanimoto index as a threshold. This makes AMBIT a good starting point to identify preliminary source analogues which can be evaluated on the basis of their reported empirical data but offers limited ability to evaluate their validity by reference to reactivity, metabolic, mechanistic profiles. Toxtree modules are implemented in the AMBIT workflow so that certain profiling capabilities exist to evaluate these contexts of similarity to a limited extent. There is no particular objective means of data gap filling. The data currently captured in AMBIT are focused on regulatory endpoint data collected as part of REACH. Approaches to exploit other data that are not necessarily anchored to an apical endpoint have not yet been addressed within the tool.
QSAR Toolbox
The QSAR Toolbox is probably the most comprehensive read-across tool currently available in terms of the aspects of the workflow that it addresses. The Toolbox compartmentalises its workflow into six modules: input, profiling, endpoint, category definition, and data gap filling. The category definition is particularly unique among read-across tools as a way of ensuring the most valid source analogues relative to the target. Categories can be defined on general mechanistic properties, empiric properties, or specific endpoints. The Toolbox comes prepackaged with a number of databases, as well as allowing the user to import their own data. Notably, the Toolbox is not just limited to read-across as a data gap filling technique, but it can also perform trend analysis and fit QSAR models for filling quantitative data gaps. The data gap analysis step is explicit and a data matrix is visible throughout the workflow to allow the end-user to evaluate the consistency and concordance of different endpoints at the same time, not just the endpoint being predicted. The Toolbox mimics the regulatory workflow most closely. However, the Toolbox limits read-across and other data gap filling approaches to be performed on the basis of one correlating descriptor at a time (e.g. LogKow by default is the correlating descriptor used to scale any prediction made). Hence the ability of deriving predictions on the basis of more than one correlating factor is not afforded. The Toolbox has begun implementing AOPs into its infrastructure, the current example is that of skin sensitisation for which a linear workflow is outlined and where one or more assay characterises a key event in the AOP. The Toolbox possesses the most comprehensive of read-across functionality within a tool but its workflow means that all analysis is restricted to within the Toolbox environment. There is limited ability to integrate any functionality of the Toolbox with other tools. Docking of third party tools is possible but this must be requested formally as part of the ongoing development of the Toolbox and addressed by the Toolbox developers through the OECD work programme.
CBRA
CBRA evaluates the (in vivo) activity of chemicals using the chemical-biological read across approach. ‘Compute CBRA’ is the only step in CBRA’s workflow, and the tool is otherwise completely automated. The output is expressed as a radial plot visualisation of the target chemical surrounded by its nearest neighbours. Indirectly it addresses the analogue identification and data gap filling steps but the source of those analogues are dependent on user input. The tool does not make explicit the algorithm used to derive the prediction. The data matrix is not explicit and there is no reporting mechanism to readily export the predictions derived. Having said that, it is novel in the manner that an integration of different sources of information (both biological and chemical) can be made and summarised in an objective manner to derive a reproducible prediction.
ToxRead
ToxRead is capable of deriving read-across predictions for two endpoints: mutagenicity Ames test and BCF. While this makes it not as all-encompassing as other tools, it does allow for endpoint-specific rule sets and structural alerts to be applied to the target chemical in an automated fashion, leaving little room for user error. When the read-across is run, the tool returns an interactive chart where it is possible to examine the resulting rules applied and similar molecules found. However, the identification of analogues, their evaluation and prediction is all performed automatically by the tool. Although the chart allows for an end-user to inspect the analogues and interrogate the validity of their selection, it offers no explicit explanation of how the overall assessment is computed. The data matrix is also latent from the end-user. The underlying data are not accessible within the tool, only the summary call as taken from one of the datasets sourced. There is no flexibility in the operation of the tool, the only input an end-user can make is to alter the number of neighbours used in the prediction.
CIIPro
CIIPro uses PubChem data to construct a biological profile and then perform a read-across using those biological descriptors. The workflow is divided into two parts: the CIIProfiler and the CIIP Predictor. The user need only enter a list of chemical identifiers as a training set for the CIIProfiler to create a biological profile. This is used in the CIIP Predictor for similarity calculation with regard to the test set. The similarity calculation also includes a confidence score, which is a measure that can be used to assess the validity of the comparison. While structural information is not used in the read-across prediction, structural neighbours are presented in the tool’s activity plot based on the same training set data used in the construction of the biological profile. Indirectly many of the steps in the category workflow are addressed with exception to a transparent data matrix.
4. Practical insights and scope for refinement
Each of the tools have been designed for different uses, hence each has its own advantages and limitations (as outlined in Table 2).
Table 2:
Advantages and limitations of selected publicly available read-across tools
| AIM | ToxMatch | Ambit | OECD Toolbox | CBRA | ToxRead | CIIPro | |
|---|---|---|---|---|---|---|---|
| Pros | Good for identifying analogues with potential data | • Provides sample datasets to make
categorical and continuous reproducible read-across predictions for a
selection of endpoints. • Provides the ability to use a number of different similarity indices • Provides the ability to calculate certain chemical descriptors • Predictions are endpoint specific • Flexibility to consider other data such as HT as descriptors • Open source software |
• Ability to search for specific
chemicals or structurally similarity chemicals • Access to a large body of data from ECHA as part of the REACH submissions • Data matrix view to allow consistency and concordance of data across analogues to be viewed and evaluated • Report produced that can be useful in regulatory submission • Open source software |
• Ability to search for specific
chemicals, structurally similar, mechanistically similar
etc • Data matrix view to allow consistency and concordance of data across analogues to be viewed and evaluated • Access to a large body of data from ECHA as part of the REACH submissions • Report produced that can imported as part of REACH regulatory submission • Developer tools if licenced provide greater functionality to evaluate analogues and provide supplementary predictions • Ability to qualitatively evaluate analogues on the basis of their simulated metabolites • Quantitative assessment of predictions feasible |
• Objective, reproducible read-across
predictions • Ability to use multi descriptor sets to make predictions • Open source software |
• Objective, reproducible read-across
predictions • Open source software |
• Objective, reproducible read-across
predictions • Ability to exploit PubChem assay information • Open source software |
| Cons | • User needs to independently extract
any data for the target and associated source analogues • Freeware software – no source code available |
No data matrix view available | • No objective read-across prediction
is made • Limited in silico tools are integrated to help in the evaluation of analogues |
• Objective read-across predictions can
be made but analogue selection is subjective hence read-across
predictions are not necessarily consistent and
reproducible • Comparing analogue sets to address more than 1 endpoint is not feasible • Actual read-across predictions are anchored by 1 descriptor |
• Dependent on user to provide all data
files from which any read-across is made • No clarity on the format of the files • No user manual to provide further interpretation of predictions made • No export functionality in current format • No data matrix view |
• Only 2 endpoints
covered • Read-across chart is only exportable |
Limited documentation and use case information
available |
| Decision context | Addresses the initial step of identifying potential analogues only | • Screening level predictions for
specific endpoints • Useful as a means of identifying similar analogues from specific sets of chemicals |
Useful for identifying potential source analogues within published REACH dossiers and exploring the breadth of data for those analogues relative to a specific target | • Useful for evaluating the analogue
validity for specific endpoints • Endpoint specific read-across predictions that can be readily exported to IUCLID to support REACH regulatory submissions |
Useful to consider biological and chemical similar neighbours at the same time and evaluate their impact of the overall prediction for screening level predictions | Useful as supporting information in a regulatory read-across or use in screening level profiling of potential mutagenicity or bioconcentration predictions | Useful to consider biological and chemical similar neighbours at the same time and evaluate their impact of the overall prediction for screening level predictions |
Making a determination of which tool or combination of tools to use however really depends on the decision context. For a preliminary ‘internal’ screening level assessment, AMBIT, the OECD Toolbox and AIM all provide a means of identifying potential analogues for which data can be searched within the tools themselves or using other databases or the literature. The data collected for the available analogues provide the starting point in inferring the likely toxicity profile of the target chemical and what effects require further consideration. If the use case was to explore a set of chemicals and tease out pragmatic chemical groupings based on their similarities in chemical and biological data and make objective predictions – Toxmatch, CIIPro and CBRA would be fit for purpose as these all rely on user provided input data. ToxRead is unique since it has been developed to make objective predictions of Ames mutagenicity and bioconcentration factor only – hence it is best used to complement assessments of these endpoints.
For current regulatory purposes, none of the tools address the requirements necessary to construct a robust read-across justification such as is expected under REACH. AIM, AMBIT and the OECD Toolbox all provide a means of identifying preliminary analogues which is one of the first steps. AIM and AMBIT both require the end-user to make a determination of which analogues identified might be most relevant to carry forward into the assessment. Their unsupervised approach of identifying analogues makes them most useful in the scenario of category/analogue approaches where the same analogues are intended to address a number of different endpoints as appropriate. The OECD Toolbox’s strengths are really in terms of developing an endpoint specific category from which data gaps are filled. This means the profilers it contains are particularly useful to assess the validity of analogues identified. At the same time the structure of the Toolbox means that a comparison of analogue sets identified based on different endpoints cannot be readily performed. Analogues can be filtered not only on the basis of structural similarity but can be also subcategorised on the basis of their physicochemical characteristics, reactivity and metabolism/degradation similarity. None of the tools have any functionality to assess the similarity of bioavailability across source analogues relative to their targets.
AMBIT and the OECD Toolbox offer particular appeal with their explicit data matrix views which afford an inspection of the data availability as well as its consistency and concordance across the source analogues and target chemical at the same time. However, it should be noted for a regulatory submission, the end user would still need to explore gaining access to the data including the full study report information in order to evaluate the data quality more closely and to navigate any hurdles associated with data ownership and rights of use. These profilers and associated QSARs that the Toolbox contains play a useful role in augmenting a read-across justification by providing other information to substantiate the similarity rationale with respect to the endpoints being read across. For specific endpoints, such as Ames mutagenicity, skin irritation, aquatic toxicity, skin sensitisation; this information can take the form of actual predictions generated using one of the data gap filling approaches that can be helpful to complement the read-across justification narrative. Additionally, the mechanistic justification information underpinning the profilers e.g. the DART profiler within the Toolbox can be useful in strengthening the read-across justification. ToxRead exploits both chemical structural and mechanistic similarity together to provide objective read-across predictions. Its main shortcoming is that the tool is restricted to 2 endpoints and the underlying data sources, data matrix or the ability refine predictions made are all lacking. Whilst this thwarts its use in addressing a data gap on its own, the predictions it provides can be useful to augment the read-across justification made for these two endpoints.
Use of HT data, either user provided or automatic extraction from PubChem is best exploited using the CBRA and CIIPro tools which rely on weighted average nearest neighbour approaches. These tools enable an objective prediction to be made using both chemical and biological descriptors and do so in an unsupervised manner. The Toolbox allows HT data to be added which is not tied to a specific apical endpoint within its hierarchy but the algorithms to compute a read-across prediction only allow for a single descriptor to be used which is a significant limitation. For HTS, a single assay is unlikely to be meaningful to be used in vacuo to predict an in vivo endpoint. Although Toxmatch was never designed to consider HT biological data as descriptors, it is sufficiently flexible to derive hybrid predictions. The advantage of these three tools is that they are able to exploit novel data to enhance predictions and perform them in a reproducible and objective manner. Within the context of a regulatory application, these predictions are probably best used to augment the read-across justification.
The ability of quantifying the performance in read-across and its associated uncertainties is a key challenge going forward. Some of the tools described have the potential to help address such needs. The exception is really AMBIT which lacks a read-across algorithm. This is a shortcoming in terms of ensuring consistency in prediction outcomes.
As indicated earlier, no single tool offers all capabilities, meaning that a combination of the different tools is merited to address a particular decision context depending on the data available. A significant limitation is the incompatibility of the different tools to be used seamlessly together. All tools employ different platforms and most are open sourced. However, some such as the OECD Toolbox whilst being freely available, do not make the source code available. Lack of source code inhibits collective use. Each tool is really intended to be used in vacuo from the rest.
Another challenge is how the predictions from these different read-across tools could be combined. Some efforts have been made to address the issue of interpretation and combination of predictions from multiple QSAR tools (Mansouri et al., 2016; Pradeep et al., 2016; Hewitt et al, 2007). Such approaches could be in principle extended for read-across tools.
An ideal workflow requires a data gap analysis to be performed in order to identify priority endpoint(s) on which to focus the assessment. Currently such an analysis is best carried out using a tool such as the OECD Toolbox and/or AMBIT. If the data gaps are such that an endpoint specific approach was appropriate, the OECD Toolbox would probably be the most convenient in terms of evaluating analogue validity with respect to the specific profilers. In any case, even on the basis of an unsupervised search, the Toolbox profilers allow for an assessment of the similarity between sources analogues on the basis of common structural alerts as well as empirical data viewable in the data matrix format. These data could be exported and combined with other sources of data such as from HTS assays in order to investigate the feasibility of performing parallel endpoint specific and objective read-across within such platforms such as CBRA or Toxmatch.
The current landscape of read-across tools is quite rich in diversity with the OECD Toolbox most advanced in terms of its functionality to address some of the current regulatory needs. Many opportunities still remain in the tool landscape including efforts to integrate existing tools together, extend the scope of tools to capture more of the category workflow steps explicitly or provide more user intervention to refine predictions made based on different threshold criteria such as toxicokinetic similarity, reactivity similarity or metabolic similarity. The utility of HT is only starting to be exploited, and integrating QSAR and read-across predictions together to derive overall assessments are yet to be investigated in more detail. As read-across continues to evolve to incorporate more mechanistic data in a systematic manner, more targeted tool development may need to occur to meet such needs. In addition, efforts to compare and contrast the utility of these different tools to evaluate specific chemicals of interest (such as that performed by Benfenati et al., 2016) would be helpful to better understand the relative strengths and weakness of the different approaches used and the consistency and reproducibility of any predictions made.
Highlights.
Category/analogue workflow outlined
Read-across tools have been described in the context of this workflow
Strengths and limitations of the tools have been described
Footnotes
Disclaimer The views expressed in this article are those of the authors and do not necessarily reflect the views or policies of the U.S. Environmental Protection Agency. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
TFHA has since been renamed to the Working Party on Hazard Assessment (WPHA)
We use the term hazard broadly to encompass physicochemical, ecotoxicological, environmental fate or toxicological properties. In practice, data gap filling could be performed for a number of different decision contexts. For the purposes of this article, we have opted to place data gap filling in the context of properties pertinent for current regulatory purposes.
Non-testing approaches is a term that encompasses the search and retrieval of existing data, the application of structural alerts, the grouping of chemicals for read-across as well as Q)SARs.
IUCLID stands for International Uniform Chemical Information Database. IUCLID is a software program for the administration of data on chemical substances first developed to fulfill EU information requirements under REACH
The term substance is used here deliberatively to acknowledge that a ‘chemical’ registered under REACH is characterised at a detailed level to account for its composition.
Toxtree was developed by Ideaconsult Ltd under the terms of a JRC contract to develop a software tool of decision tree approaches including the Cramer structural classes, structural alerts for different endpoints. It can be downloaded as a standalone tool from the JRC website (https://eurl-ecvam.jrc.ec.europa.eu/laboratories-research/predictive_toxicology/qsar_tools/toxtree). The algorithms have also been implemented into AMBIT.
OECD QSAR Toolbox will be referenced as the OECD Toolbox or Toolbox throughout this article
Potential member means a chemical obeys the structural definitions of the category as opposed to be a member actually named in the categories assessed by EPA or OECD
References
- Aptula AO, Patlewicz G, Roberts DW. 2005. Skin sensitization: reaction mechanistic applicability domains for structure-activity relationships. Chem. Res. Toxicol. 18: 1420–1426. [DOI] [PubMed] [Google Scholar]
- Ball N, Cronin MT, Shen J, Blackburn K, Booth ED, Bouhifd M, Donley E, Egnash L, Hastings C, Juberg DR, Kleensang A, Kleinstreuer N, Kroese ED, Lee AC, Luechtefeld T, Maertens A, Marty S, Naciff JM, Palmer J, Pamies D, Penman M, Richarz AN, Russo DP, Stuard SB, Patlewicz G, van Ravenzwaay B, Wu S, Zhu H, Hartung T. 2016. Toward Good Read-Across Practice (GRAP) guidance. ALTEX. 33(2): 149–166. doi: 10.14573/altex.1601251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati E, Belli M, Borges T, Casimiro E, Cester J, Fernandez A, Gini G, Honma M, Kinzl M, Knauf R, Manganaro A, Mombelli E, Petoumenou MI, Paparella M, Paris P, Raitano G. 2016. Results of a round-robin exercise on read-across. SAR QSAR Environ Res. 27(5):371–384. doi: 10.1080/1062936X.2016. [DOI] [PubMed] [Google Scholar]
- Benigni R, Bossa C, Jeliazkova NG, Netzeva TI, Worth AP. 2008.The Benigni / Bossa rulebase for mutagenicity and carcinogenicity - a module of Toxtree. EUR 23241 EN. 2008.
- Berggren E, Amcoff P, Benigni R, Blackburn K, Carney E, Cronin M, Deluyker H, Gautier F, Judson RS, Kass GE, Keller D, Knight D, Lilienblum W, Mahony C, Rusyn I, Schultz T, Schwarz M, Schüürmann G, White A, Burton J, Lostia AM, Munn S, Worth A. 2015. Chemical Safety Assessment Using Read-Across: Assessing the Use of Novel Testing Methods to Strengthen the Evidence Base for Decision Making. Environ Health Perspect. 123(12): 1232–1240. doi: 10.1289/ehp.1409342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn K, Bjerke D, Daston G, Felter S, Mahony C, Naciff J, Robison S, Wu S. 2011. Case studies to test: A framework for using structural, reactivity, metabolic and physicochemical similarity to evaluate the suitability of analogs for SAR-based toxicological assessments. Regul. Toxicol. Pharmacol. 60(1): 120–135. doi: 10.1016/j.yrtph.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Blackburn K, Stuard SB. 2014. A framework to facilitate consistent characterization of read across uncertainty. Regul. Toxicol. Pharmacol. 68: 353–362. [DOI] [PubMed] [Google Scholar]
- Boyer S, Hasselgren Arnby C, Carlsson L, Smith J, Stein V, Glen RC. 2007. Reaction Site Mapping of Xenobiotic Biotransformations. J. Chem. Inf. Model. 47(2): 583–590. doi: 10.1021/ci600376q/ [DOI] [PubMed] [Google Scholar]
- Cramer GM, Ford RA,Hall RL. 1978. Estimation of Toxic Hazard - A Decision Tree Approach. Food Cos Toxicol. 16: 255–276. [DOI] [PubMed] [Google Scholar]
- Delrue N, Sachana M, Sakuratani Y, Gourmelon A, Leinala E, Diderich R. 2016. The adverse outcome pathway concept: A basis for developing regulatory decision-making tools. Altern Lab Anim. 44(5): 417–429. [DOI] [PubMed] [Google Scholar]
- Dimitrov SD, Diderich R, Sobanski T, Pavlov TS, Chankov GV, Chapkanov AS, Karakolev YH, Temelkov SG, Vasilev RA, Gerova KD, Kuseva CD, Todorova ND, Mehmed AM, Rasenberg M, Mekenyan OG. 2016. QSAR Toolbox - workflow and major functionalities. SAR QSAR Environ Res. 19: 1–17. [DOI] [PubMed] [Google Scholar]
- EC – European Commission. 2006. Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC. Off J Eur Union L396/1 of 30.12.2006. Commission of the European Communities.
- EC. 2009. Regulation (EC) No 1223/2009 of the European Parliament and the Council of 30 November 2009 on cosmetic products. Off. J. Eur. Union L 342, 59–209. [Google Scholar]
- ECETOC. 2012. Technical Report 116 Category approaches, read-across, (Q)SAR available at http://www.ecetoc.org/technical-reports.
- ECHA. 2008. Guidance on information requirements and chemical safety assessment. Chapter R.6: QSARs and grouping of chemicals. Available at http://echa.europa.eu/documents/10162/13632/information_requirements_r6_en.pdf
- ECHA. 2015. Read-across Assessment Framework (RAAF). ECHA-15-R-07-EN
- Feng J, Lurati L, Ouyang H, Robinson T, Wang Y, Yuan S, Young SS. 2003. Predictive toxicology: Benchmarking molecular descriptors and statistical methods. J. Chem. Inf. Comput. Sci. 43: 1463–1470. [DOI] [PubMed] [Google Scholar]
- Ferrari T, Cattaneo D, Gini G, Bakhtyari NG, Benfenati E, Manganaro A. 2013. Automatic knowledge extraction from chemical structures: The case of mutagenicity prediction. SAR QSAR Environ. Res. 24: 365–383. [DOI] [PubMed] [Google Scholar]
- Gallegos Saliner A, Poater A, Jeliazkova N, Patlewicz G, Worth AP. 2008. Toxmatch - A Chemical Classification and Activity Prediction Tool based on Similarity Measures. Regul. Toxicol. Pharmacol. 52: 77–84. [DOI] [PubMed] [Google Scholar]
- Gerberick GF, Ryan CA, Kern PS, Schlatter H, Dearman RJ, Kimber I, Patlewicz GY, Basketter DA. 2005. Compilation of historical local lymph node data for evaluation of skin sensitization alternative methods. Dermatitis. 16(4), 157–202. [PubMed] [Google Scholar]
- Gini G, Franchi AM, Manganaro A, Golbamaki A, Benfenati E. 2014. ToxRead: A tool to assist in read across and its use to assess mutagenicity of chemicals. SAR QSAR Env. Res. 25: 999–1011. [DOI] [PubMed] [Google Scholar]
- Helma C, Cramer T, Kramer S, De Raedt L 2004. Data mining and machine learning techniques for the identification of mutagenicity inducing substructures and structure activity relationships of noncongeneric compounds. J. Chem. Inf. Comput. Sci. 44: 1402–1411. [DOI] [PubMed] [Google Scholar]
- Hewitt M, Cronin MT, Madden JC, Rowe PH, Johnson C, Obi A, Enoch SJ. 2007. Consensus QSAR models: do the benefits outweigh the complexity? J. Chem. Inf. Model. 47(4): 1460–1468. [DOI] [PubMed] [Google Scholar]
- Kazius J, McGuire R, Bursi R. 2005. Derivation and validation of toxicophores for mutagenicity prediction. J. Med. Chem. 48: 312–320. [DOI] [PubMed] [Google Scholar]
- Klimisch HJ, Andreae M, Tillmann U. 1997. A systematic approach for evaluating the quality of experimental toxicological and ecotoxicological data. Regul Toxicol Pharmacol. 25(1): 1–5. [DOI] [PubMed] [Google Scholar]
- Low Y, Sedykh A, Fourches D, Golbraikh A, Whelan M, Rusyn I, Tropsha A. 2013. Integrative chemical-biological read-across approach for chemical hazard classification. Chem. Res. Toxicol. 26(8): 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens A, Hubesch B, Rovida C. 2016. Two Good Read-across Practice workshops. Making it work for you! ALTEX. 33(3): 324–326. doi: 10.14573/altex.1605301. [DOI] [PubMed] [Google Scholar]
- Manganelli S, Benfenati E. 2016. Use of Read-Across Tools. Methods Mol Biol. 1425: 305–322. doi: 10.1007/978-1-4939-3609-0_13. [DOI] [PubMed] [Google Scholar]
- Mansouri K, Abdelaziz A, Rybacka A, Roncaglioni A, Tropsha A, Varnek A, Zakharov A, Worth A, Richard AM, Grulke CM, Trisciuzzi D, Fourches D, Horvath D, Benfenati E, Muratov E, Wedebye EB, Grisoni F, Mangiatordi GF, Incisivo GM, Hong H, Ng HW, Tetko IV, Balabin I, Kancherla J, Shen J, Burton J, Nicklaus M, Cassotti M, Nikolov NG, Nicolotti O, Andersson PL, Zang Q, Politi R, Beger RD, Todeschini R, Huang R, Farag S, Rosenberg SA, Slavov S, Hu X, Judson RS.2016. CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ Health Perspect. 124(7):1023–33. doi: 10.1289/ehp.1510267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews EJ1, Kruhlak NL, Cimino MC, Benz RD, Contrera JF.2006. An analysis of genetic toxicity, reproductive and developmental toxicity, and carcinogenicity data: I. Identification of carcinogens using surrogate endpoints. Regul. Toxicol. Pharmacol. 44(2): 83–96. doi: 10.1016/j.yrtph.2005.11.003 [DOI] [PubMed] [Google Scholar]
- National Research Council (NRC). 2007. Toxicity Testing in the 21st Century: A Vision and a Strategy Washington, DC: National Academies Press. [Google Scholar]
- National Research Council (NRC). 2017. Science and Decisions: Advancing Risk Assessment, Washington, DC: National Academies Press. [PubMed] [Google Scholar]
- OECD. 2012. The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins Part 1: Scientific Evidence. Series on Testing and Assessment No. 168 ENV/JM/MONO(2012)10/PART1
- OECD. 2014. Guidance on grouping of chemicals. OECD Series on Testing and Assessment No. 194. Organisation for Economic Co-operation and Development, Paris, France. [Google Scholar]
- Patlewicz G, Jeliazkova N, Gallegos Saliner A, Worth AP. 2008. Toxmatch – a new software tool to aid in the development and evaluation of chemically similar groups. SAR QSAR Environ. Res. 19(3–4): 397–412. [DOI] [PubMed] [Google Scholar]
- Patlewicz G, Ball N, Booth ED, Hulzebos E, Zvinavashe E, Hennes C. 2013a.Use of category approaches, read-across and (Q)SAR: general considerations. Regul. Toxicol. Pharmacol. 67(1): 1–12. doi: 10.1016/j.yrtph.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Patlewicz G, Roberts DW, Aptula A, Blackburn K, Hubesch B. 2013b.Workshop: use of “read-across” for chemical safety assessment under REACH. Regul. Toxicol. Pharmacol. 65(2): 226–228. doi: 10.1016/j.yrtph.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Patlewicz G, Ball N, Becker RA, Booth ED, Cronin MTD., Kroese D, Steup D, Van Ravenzwaay B, Hartung T 2014aFood for thought….. Read-across approaches - misconceptions, promises and challenges ahead. ALTEX 31: 387–396. [DOI] [PubMed] [Google Scholar]
- Patlewicz G, Ball N, Boogaard P, Becker RA, Hubesch B 2015a.Building scientific confidence in the development and evaluation of read-across. Regul. Toxicol. Pharmacol. 72: 117–133. [DOI] [PubMed] [Google Scholar]
- Patlewicz G, Simon T, Rowlands JC, Budinsky R, Becker RA. 2015b.Proposing a scientific confidence framework to support application of Adverse Outcome Pathways for regulatory purposes. Regul. Toxicol. Pharmacol. 72: 17–25. [DOI] [PubMed] [Google Scholar]
- Roberts DW., Aptula AO 2008. Determinants of skin sensitisation potential. J. Appl. Toxicol. 28(3): 377–387. [DOI] [PubMed] [Google Scholar]
- Pradeep P, Povinelli RJ, Merrill SJ, Bozdag S, Sem DS. 2015. Novel Uses of In Vitro Data to Develop Quantitative Biological Activity Relationship Models for in Vivo Carcinogenicity Prediction. Mol. Inf. 34: 236–245. doi: 10.1002/minf.201400168 [DOI] [PubMed] [Google Scholar]
- Pradeep P Povinelli RJ, White S, Merrill SJ 2016. An ensemble model of QSAR tools for regulatory risk assessment. J. Cheminform. 8: 48. doi: 10.1186/s13321-016-0164-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo DP, Kim MT, Wang W, Pinolini D, Shende S, Strickland J, Hartung T, Zhu H. 2017. CIIPro: a new read-across portal to fill data gaps using public large-scale chemical and biological data. Bioinformatics 33(3): 464–466. doi: 10.1093/bioinformatics/btw640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydberg P, Gloriam DE, Zaretzki J, Breneman C, Olsen L. 2010a.SMARTCyp: A 2D Method for Prediction of Cytochrome P450-Mediated Drug Metabolism, ACS Med. Chem. Lett. 1(3): 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydberg P, Gloriam D, Olsen L. 2010b.The SMARTCyp cytochrome P450 metabolism prediction server. Bioinformatics 26: 2988–2989 [DOI] [PubMed] [Google Scholar]
- Schneider K, Schwarz M, Burkholder I, Kopp-Schneider A, Edler L, Kinsner-Ovaskainen A, Hartung T, Hoffmann S. 2009. “ToxRTool”, a new tool to assess the reliability of toxicological data. Toxicol. Lett. 189(2): 138–144. doi: 10.1016/j.toxlet.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Schultz TW., Amcoff P, Berggren E, Gautier F, Kalric M, Knight DJ, Mahony C, Schwarz M, White A, Cronin MTD. 2015. A strategy for structuring and reporting a read-across prediction of toxicity. Regul. Toxicol. Pharmacol. 72: 586–601. [DOI] [PubMed] [Google Scholar]
- Segal D, Makris SL, Kraft AD, Bale AS, Fox J, Gilbert M, Bergfelt DR, Raffaele KC, Blain RB, Fedak KM, Selgrade MK, Crofton KM. 2015. Evaluation of the ToxRTool’s ability to rate the reliability of toxicological data for human health hazard assessments. Regul. Toxicol. Pharmacol. 72(1): 94–101. doi: 10.1016/j.yrtph.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Shah I, Liu J, Judson RS, Thomas RS, Patlewicz G. 2016. Systematically evaluating read-across prediction and performance using a local validity approach characterized by chemical structure and bioactivity information. Regul. Toxicol. Pharmacol. 79: 12–24. doi: 10.1016/j.yrtph.2016.05.008. [DOI] [PubMed] [Google Scholar]
- US EPA. 2010. TSCA New Chemicals Program (NCP) Chemical Categories. Office of Pollution Prevention and Toxics; 2010. Accessible from https://www.epa.gov/sites/production/files/2014-10/documents/ncp_chemical_categories_august_2010_version_0.pdf [Google Scholar]
- Willett P, Barnard J, Downs G. 1998. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 38: 983–996. [Google Scholar]
- Wu S, Blackburn K, Amburgey J, Jaworska J, Federle T. 2010. A framework for using structural, reactivity, metabolic and physicochemical similarity to evaluate the suitability of analogs for SAR-based toxicological assessments. Regul. Toxicol. Pharmacol. 56(1): 67–81. doi: 10.1016/j.yrtph.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Zhu H, Bouhifd M, Donley E, Egnash L, Kleinstreuer N, Kroese ED, Liu Z, Luechtefeld T, Palmer J, Pamies D, Shen J, Strauss V, Wu S, Hartung T. 2016. Supporting read-across using biological data. ALTEX. 33(2): 167–182. doi: 10.14573/altex.1601252. [DOI] [PMC free article] [PubMed] [Google Scholar]