

Abstract
Technical lignins are bulk feedstocks. They are generated as byproducts from pulping or cellulosic ethanol production. Since lignin undergoes significant structural changes in the chemical and physical treatments, all technical lignins are unique in terms of chemical structure, molecular weight, polydispersity, and impurity profile.
Kraft lignin is potentially the largest source of technical lignin as new isolation technologies have been implemented on industrial scale in recent years. Lignosulfonate has been an integral product in sulfite pulping biorefinery. It has a well-established market in construction industry. Organosolv-like lignin production is increasing as cellulosic ethanol has been promoted as the substitute of fossil fuel. It may have unique applications because it has low molecule weight and is free from sulfur. Technical lignin application is expected to expand as the characteristics are improved with fractionation or chemical modification.
The application of technical lignin has been focusing on developing products equivalent to those made by petroleum chemicals. The recent development in technical lignin supply should increase its market share as additives in polyurethanes and as the substitute of phenol-formaldehyde adhesives. Quality improvement of technical lignin may also encourage the study of lignin as an alternative feedstock for carbon fiber. In addition, technical lignin depolymerization has been extensively explored to provide renewable aromatic chemicals. Starting from controlled pyrolysis and thermal liquefaction as the baseline technologies, many different chemical depolymerization have been invented with a wide range of underlying chemical principles.
Keywords: Lignocellulose, lignin, Kraft Process, lignosulfonate, biorefinery
Introduction
Lignin is the largest repository of renewable aromatic chemicals on earth. It makes up 20–35% of the biomass. Therefore, lignin valorization is expected to play an essential role in the bio-based economy. There has been a rapid increase in the number of publications to isolate, modify, and expand the use of lignin in recent years. To evaluate the potential of emerging technologies, it is desirable to have a knowledge framework with which stakeholders can characterize the supply of technical lignin, identify the critical issues in lignin transformation processes, benchmark new options with equivalent technologies, and project impacts of an innovation.
Over the history, technical lignin is a byproduct from pulping industry. Also, lignin from the upcoming cellulosic ethanol industry is becoming significant. Both chemical and physical treatments cause changes in lignin in terms of structure, molecular weight distribution and dispersity. The impurity profile is influenced not only by the process, but also by the isolation method. The quality of technical lignin sums up all its experience from feedstock to production isolation. Therefore, it is important to understand the pathway from which a lignin product is generated. The quality of lignin is critical for applications in polymeric form, particularly when as-received lignin is needed for applications with low profit margin. There is also a vigorous effort to depolymerize lignin for monomers as platform chemicals. The quality of lignin feedstock is also important as it influences the yield of depolymerization.
Contextual information of technical lignin production is highly valuable. The production scale and location reflect the local biomass supply and established market. The history of technology development demonstrates how new technologies have been adopted to address the most critical issues in the highly integrated processes. The product portfolio evolution exemplifies the approaches to maximize the value of a biorefinery. All the information would help to identify realistic opportunities to improve efficiency with new technologies, minimize capital investment, identify cross-sector collaborators for new product development, and build flexible and adaptive product portfolio.
These topics are organized by underlying chemical principles with highlights on the efficiency and sustainability. We hope this review will serve as a subject reference in a broad knowledge framework for cross functional collaboration to prioritize research or product development.
Main components of lignocellulosic biomass
Lignocellulosic biomass is the most abundant terrestrial plant material. From energy point of view, biomass is the most condensed form of energy from sunlight on earth surface. Therefore, it is the ultimate renewable source of energy. Biomass is a composite material that is built as plants grow. The property of biomass is unique to the plant and influenced by growth condition. It is made up with three main polymeric components; including cellulose, hemicellulose, and lignin.1 Its physical character can be fine-tuned by changing the composition and organization of the three components. The components are different in chemical structure and physical property. They play complimentary roles in building biomass (Figure 1).
Figure 1.

The organization of three components in lignocellulosic biomass
Cellulose (Figure 2) is the main component in plant cell wall. It is a linear homopolymer consisting of 7000 – 15000 units of β-(1–4) linked D-glucose. The highly regular structure allows cellulose to form crystalline region through hydrogen bonds within and between molecules. Cellulose is highly stable as it has crystalline lattice energy at 20 kcal mole−1 of glucose.2 It is insoluble in water or nearly all traditional solvents. The crystalline structure also renders resistance to enzymatic or chemical hydrolysis.
Figure 2.

Chemical composition and structure of cellulose
Hemicellulose (Figure 3) is a two-dimensional copolymer with lower degree of polymerization (n = 500–3000). It is comprised of different hexoses and pentoses, including D-Glu, D-Gal, D-Man, D-Xyl, and L-Ara. The hydroxyl groups on the branches may also form acetate or glucuronides. Hemicellulose is structurally irregular with several different regional motifs. It is soluble in water and easier to hydrolyze.
Figure 3.

Chemical composition and structure of hemi-cellulose
Lignin contributes to the strength of plant cell wall by gluing cellulose and hemicellulose together. It is a three-dimensional heterogeneous copolymer of three phenylpropanoid monomers, including coniferyl alcohol (G), coumaryl alcohol (H), and sinapyl alcohol (S). Each monomer has several polymerization sites, allowing many different linkages between the monomers (Figure 4). The content of lignin depends on the species of a plant. For example, softwood is comprised of 45–50% of cellulose, 25–35% of hemicellulose, and 25–35% of lignin. Monomer composition of the lignin is also dependent on the plant species.3
Figure 4.

Monomers and linkages in lignin
For structure determination, lignin needs to be separated from the other two components. Native lignin structure is difficult to determine as it is changed in harsh chemical and physical treatment. As the result, much of lignin chemistry is derived from the study on model compounds. With isolated lignin, the structure is characterized in terms of aromatic units (GHS), interunit linkages, functional groups (aliphatic alcohols, aromatic alcohols, and carboxylic acids), side chains, and molar mass distribution.4
Kraft process is potentially the largest source of lignin feedstock
Lignin for chemical recovery in Kraft pulping process
Wood pulp industry provides cellulose fiber for paper and paper board commodity. In a survey by The Food and Agriculture Organization of the United Nations, the total capacity of dry wood pulp in the US has been 53,830 – 54,280 million tons between years 2014 – 2019.5 As the side stream, lignin has been generated at approximately 25% of the scale, but most is burned for energy. The survey also shows that 87% of the product is produced by chemical pulping, which involves thermal chemical digestion of woody feedstock.
The Kraft process (Figure 5) is the primary pulping process.6 The chemicals to digest lignocellulose is a mixture of Na2S and NaOH (White Liquor). At high temperature (150–180 ͦC), ether bonds in lignin are broken through an episulfide intermediate (Figure 6). Cellulose is separated by filtration, leaving lignin and hemicellulose in the liquid phase (Black Liquor). The most important feature for Kraft process is the recovery of all chemicals for pulping. Sulfur and sodium are both completely recycled through the recovery process (Figure 5). To prepare for the recycling, the Weak Black Liquor is concentrated to give Heavy Black Liquor. Heavy Black Liquor is then combusted to reduce sulfate to sulfide by a thermochemical reduction with lignin. Combusted lignin gives carbonate as the byproduct in Green Liquor. In the next step, lime (CaO) is added to Green Liquor. It reacts with sodium carbonate to give sodium hydroxide and calcium carbonate precipitate. White Liquor is regenerated by removing CaCO3 from the liquid. The CaCO3 is transformed back to lime by heating in a kiln. In Kraft process sodium, sulfur and calcium are essentially catalytic as they are recycled through the recovery process. The theoretical byproducts are water and carbon dioxide.
Figure 5.
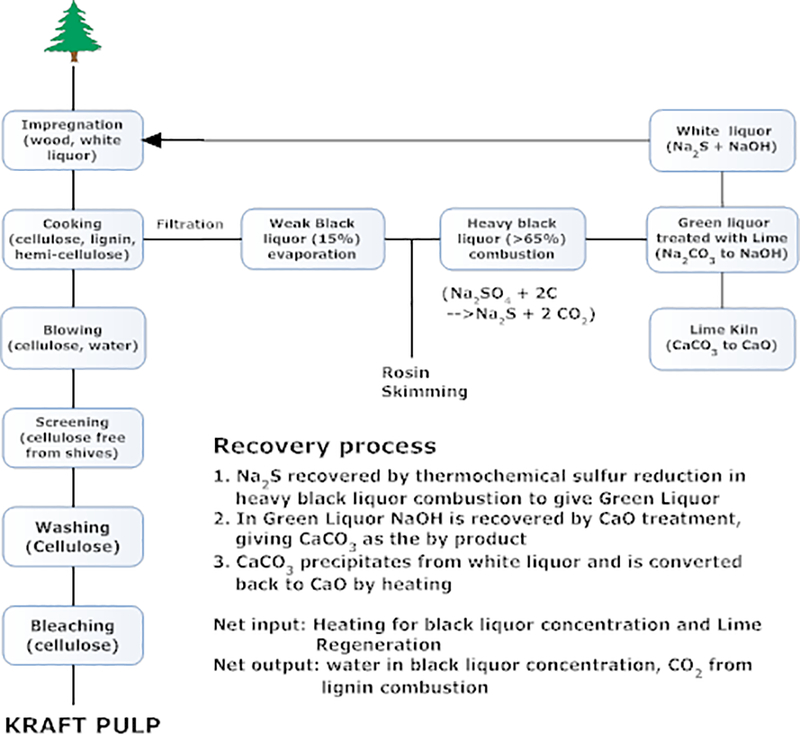
Kraft process involves chemical recovery
Figure 6.

Lignin ether cleavage in Kraft Process (α-O-ether and β-O-4 cleavage)
Energy is also produced in the chemical recovery. In theory, only a fraction of the lignin in Black Liquor is needed for sulfate reduction and energy production. Therefore, it is feasible to remove a portion of lignin form the Black Liquor by rebalancing the chemical and energy allocation in Kraft process.6
Weak Black Liquor is concentrated by evaporation to give Thick Black Liquor to sustain the combustion for chemical recovery. The Thick Black Liquor is sprayed into the boiler and burned to give a smelt of Na2S and Na2CO3. In addition to the chemical products, this step also involves steam generation as the energy product. This step is usually the scale limiting step in Kraft process because recovery boiler is the most expensive unit in Kraft pulping equipment.
The efficiency of the boiler depends on the chemical composition and physical properties of the incoming black liquor. The Heavy Black Liquor contains > 65% of lignin and about 30% of the dissolved salts. The heating value of the black liquor is from the oxidation of the organics, while the boiling point rise is dictated by the inorganics. The smelt product is used to produce green liquor; therefore, a Na/S balance needs to be maintained by adding salts or sulfuric acid before the recovery. Importantly, the non-processing elements (such as potassium, chloride, calcium, aluminum, silicon, and iron) may cause incrustation on the heat exchange surface in boiler and reduce its efficiency. Both thermal and chemical properties need be re-evaluated in the scenario of lignin removal from black liquor.
The composition of black liquor was plant dependent. The lignin contents, average lignin molar masses, and non-processing elements, were quite different among black liquors from eucalyptus, pine, and bamboo.7 However, the heating values were found to be similar among them. The rheological properties were also different among the black liquors.
The technologies to isolate lignin from Kraft Black liquor
Indulin is the technical lignin byproduct from the Kraft black liquor of linerboard pulp. It was marketed by West Virginia Pulp and Paper Company (Now Mead-Westvaco) in 1950’s, with applications in asphalt emulsifiers, or for rubber reinforcement, etc.8, 9 For a long time, this was the only technical lignin from Kraft process. In the last two decades, efforts have been made to improve economic and environmental performance of Kraft pulp mills. Since most Kraft pulp mills were limited by chemical recovery, this step became the target for debottlenecking the mill. Lignin removal would allow higher throughput by reducing heat value in black liquor. Therefore, the pulping scale can be increased without investing on a larger recovery boiler. In addition to improving the efficiency of pulping, the lignin removal can generate technical lignin product for additional revenue.10–12 Kraft lignin can be used either as a biofuel or as a chemical feedstock for value added applications.
In 1989, Uloth and Wearing reported that acid precipitation was more economical than ultrafiltration for Kraft lignin isolation, because its capital and operational costs were lower. Plus, acid precipitation also gave better lignin product.13, 14 Black liquor oxidation was proposed to address the issue of odor hazard. At 15% lignin removal, the heating value and combustibility of the black liquor still met the need for chemical recovery.
Acid precipitation involves protonation of the phenols in lignin. As the pH reaches the pKa range of the phenolic groups, lignin molecules undergo conformation change and start to flocculate.15 In addition to pH, temperature and ionic strength also influence lignin solubility. The molecular weight distribution in precipitated lignin is related to the yield.16 Both molecular weight and the ratio of phenol to aromatic unit contribute to lignin solubility.16, 17
Starting from 1997, Inventia (Formerly STFI-Packforsk, Sweden) and Chalmers Technical University jointly developed a lignin extraction process based on acid precipitation.11 The development focused on maximizing efficiency in lignin isolation and salt removal. As the result LignoBoost was invented in 2002 (Figure 7).18, 19 This process began with adjusting the pH of the black liquor to around 9.5 with carbon dioxide. After aging to flocculate, lignin was isolated by the first filtration. In the next step, the recovered lignin was re-slurried to equilibrate at pH 2. In the last step, the lignin was isolated by filtration, and the liquid from the re-slurry step was displaced by washing with diluted sulfuric acid. In this process, the filtrations were highly efficient. Salts were efficiently removed at low pH; therefore the ash content was low in the product.20 Later, flocculation was further improved by adding lignin germ particles.21
Figure 7.

LignoBoost Process to isolate Kraft lignin
Lignin removal reduced the heat value and changed the physical properties of black liquor. At a constant ratio of air to mass unit of black liquor, the removal of lignin caused reduction of heat transfer rate in recovery boiler, therefore making room for more input in terms of dry solid load or heat load.22 Integrating LignoBoost process into Kraft pulp mill involved re-balancing material and energy.23 To maintain the balance of Na/S, the later streams were sent back to black liquor or for re-slurry. The sodium sulfate in Electro Static Precipitator Dust was also sent back to make up for salt loss.24
Debottlenecking the mill decreased the operational cost. The isolated Kraft lignin was used as a fuel in lime kiln, in which the sulfur in lignin was captured by the calcium. The lignin was also used for other combustion applications with certain benefits. For example, the sulfur in the Kraft lignin (1–3%) was expected to reduce the corrosion caused by KCl when combusted with biomass.23, 25
The LignoBoost process was first scaled up to 8000 tons year−1 in Nordic Paper Bäckhammer (Sweden) for demonstration in 2007. In 2013, Domtar Plymouth Mill in North Carolina (USA) installed the process to produce lignin at about 27,300 tons year−1, and the product was marketed as Bio-Choice Lignin. Comparing with Indulin, Bio-Choice Lignin had higher phenol hydroxyl, catechol, and enol ether; and lower methoxy and β-O-4 contents.26 In 2015, Stora Enso installed the second LignoBoost at Sunila Mill (Finland) to produce LignoBoost lignin at 50,000 tons year−1.
When mill capacity is not limited, lignin isolation has to be justified by high value applications and low isolation cost. FPinnovations, a Canadian non-profit organization surveyed the cost and benefit of isolating lignin from Kraft process.27 Initial evaluation showed that the operating cost of lignin isolation was mainly from the CO2 for acidification. Hazard from sulfur compounds was also a significant concern.28 In Kraft black liquor, totally reduced sulfur (TRS) compounds include hydrogen sulfide, methyl mercaptan, dimethyl sulfide, and dimethyl disulfides. They produce unpleasant odor and are poorly soluble in water. For non-fuel applications, the isolated lignin ought to be free from the malodorous hazard. Although most of the TRS compounds could be removed by repeated wash, they were much easier to remove by oxidation. Based on this observation, the FPinnovations group invented LignoForce process through collaboration with NORAM (Figure 8).29
Figure 8.

LignoForce Process to isolate Kraft lignin
The LignoForce process30 started with sparging oxygen to a black liquor containing 30–40% of solid. The oxidation was carried out at 75 °C until the sulfide concentration was reduced a specific level. The solution was then acidified by CO2 to pH around 10. The lignin slurry was coagulated at 65–70 °C and the lignin particle was isolated with filtration. In the final step, the lignin cake was washed with diluted sulfuric acid. Only one filtration was needed to produce high quality lignin. In this process, the TRS compounds were transformed into more acidic species such as sulfate, sulfonate, or sulfone,28 hence reduced the need for CO2 to adjust pH in lignin precipitation. Lignin formed larger particle in the coagulation step, therefore the filtration was faster. Hazard from TRS compounds was removed from the process and lignin product. This process was demonstrated with feedstocks from hardwood, softwood and eucalyptus. In 2015, West Fraser built a lignin plant based on LignoForce technology in Hinton pulp mill in Alberta, Canada.31 The scale was 30 tons per day, with removal rate at 0.15 ton of lignin per ton of pulp. The integration also involved rebalancing energy, water, steam, chemicals and Na/S ratio.10, 31 Since the mill operated below its maximum capacity, the lignin product was expected to be used in specialty chemical applications such as replacing phenol in phenol formaldehyde resin.31, 32
Process intensification is a less explored, but highly desirable technology for lignin isolation from black liquor. In 1950’s, West Virginia Pulp & Paper Co. patented a coagulating technology involving turbulent mixing in a tubular reactor of black liquor that had been acidified with stack gas. The fusion of lignin particles was better managed in a continuous process.33 In 2010, Finnish company UPM-Kymmene Co. disclosed a method by integrating acidification with pressurized CO2 and turbulent mixing in one tubular reactor plus a rapid pressure release at the exit.34 In the continuous process, lignin precipitation was much faster, therefore black liquor residence time was less than 300s.
In a different approach, the Sequential Liquid-lignin Recovery and Purification (SLRP™) technology employs reactive extraction to facilitate lignin isolation based on the fact that a lignin-rich liquid phase is formed from black liquor upon carbonation.35 After phase-split separation, lignin is precipitated from the raffinate by acidification with sulfuric acid. The reactive extraction was continuous. The H2S was efficiently recovered in the White Liquor, and CO2 consumption was reduced by 30%. This process has been tested on pilot scale with black liquors from different wood feedstocks.
Currently, only press filter has been used for solid lignin separation and wash. Belt filter was tested in LignoForce process, but the solid content was only 34% in product, indicating much lower efficiency for water removal.32
Lignosulfonate is an integral member in the product portfolio from sulfite pulping biorefinery
Sulfite pulping processes and spent sulfite liquor
Sulfite process was the predominant pulping process from late 19th century to early 20th century. The process employs sulfite or bisulfite to digest biomass. It is characterized by the flexibility to operate over a wide pH range. Depending on the pH, the base for pulping is a cation from Ca2+, Mg2+, Na+ and NH4+. Calcium can only be used in acid bisulfite pulping, as it will precipitate when pH is ≥ 3. Magnesium is used in the processes when pH ≤ 5. At higher pH, only ammonium and sodium are feasible. Different degrees of delignification can be achieved by fine tuning the cooking liquor, the ratio of chemicals to biomass, and physical conditions. The pulping can also be carried out in multiple stages with different cooking liquors.36 Major types of sulfite pulping include Acid Sulfite Process (pH 1–2), Neutral Sulfite Process (Neutral Sulfite Semi Chemical, NSSC, pH 5–7), and Alkaline Sulfite Process (pH 9–13.5).
In Acidic Sulfite process with softwood, the main reaction involves the formation of an α-C cation from the α-O-4 ether, followed by a sulfonation (Figure 9). Unlike the lignin depolymerization in Kraft process, the lignin is solubilized through sulfonate formation. For carbohydrate, hemi-cellulose is degraded under acidic condition to give corresponding mono-, oligo saccharides, acetic acid, and other derivatives. As the pH increases, both reactions are slower, and the intensity of pulping is decreased.36
Figure 9.

Lignin sulfonation in acidic sulfite process with softwood feedstock
The spent sulfite liquor (SSL) is largely composed of lignosulfonate, acetic acid, sugars from hemicellulose, inorganics, and small quantities of derivatives from sugar dehydration. Based on the oven dried ton of pulp (ODT), the organic is in the range of 595 – 1224 kg/ODT. Lignin is in the range of 410–892 kg/ODT, representing 64–76% of the organics. Sugar is in the range of 129 – 288 kg/ODT, accounting for 17–25% of the organics. The chemical oxygen demand (COD) of SSL is typically 120–220 g/L.37 In sulfite mill, many products are extracted from the organics in the SSL to improve the economic performance and reduce the burden of waste disposal.
Biorefinery based on sulfite process
When the concept “biorefinery” emerged in the 1990’s,38 sulfite pulping had already advanced to a platform with a multiproduct-portfolio. It offers important experience in maximizing value of a biomass waste with integrated processes. The flexibility of sulfite process allows it to generate many types of pulp, with potential products from both sugar and lignosulfonate in SSL (Figure 10).
Figure 10.

Principles to build a biorefinery based sulfite process
Hardwood gives xylose, while softwood gives mannose and glucose as the main products.39 Higher temperature and longer cooking time increase the amount of dissolved sugars as well as the byproducts from them.40 The analysis of Borregaard (Sarpsborg, Norway) samples showed that the property of lignosulfonates was also dependent on biomass feedstock. Hardwood lignosulfonate had a weighted average molecular weight (Mw with UV detection) of 7.2–11 kDa with polydispersity values of 3.0–5.3 (with MALLS detection), while the softwood lignosulfonate had Mw of 35–57 kDa with polydispersity values of 8.2–12.3. The degrees of sulfonation were similar between hardwood and softwood, but were inversely correlated to the molecular weight. The Mw and degree of sulfonation were both reduced in the oxidation to generate vanillin.41
Lignosulfonate is highly soluble in a wide pH range. Before 1970’s Howard process was the most practical technology to separate lignosulfonate from SSL.42 In this process, lime suspension was added to the SSL. Both lignosulfonate and sulfite were precipitated by forming calcium salts. The calcium lignosulfonate formed a low density colloidal phase as a mixture with air, while the crystalline calcium sulfite settled as solid sediment. They were separated by the difference in density. The isolated calcium lignosulfonate was low in ash and free from carbohydrates. The isolated calcium lignosulfonate was used in tanning. It was also a flexible feedstock, as it could be converted to other salts by exchanging the cation with the corresponding carbonate.43
The calcium lignosulfonate was used in the preparation of vanillin.44 In a process invented in Marathon Paper Mills Company (Wisconsin, US), calcium lignosulfonate was converted to the sodium salt through an exchange reaction. After calcium sulfate precipitate was removed, the sodium lignosulfonate was oxidized to generate vanillin under basic condition and high steam pressure. The reaction mixture was acidified with sulfurous acid, converting vanillin to the bisulfite adduct and precipitating the remaining lignosulfonate. After the remaining lignosulfonate precipitate was removed by filtration, vanillin was regenerated by decomposing the bisulfite adduct and isolated by extraction. Since softwood lignin is rich in guaiacyl unit, it is more suitable for lignin production.
An alternative strategy started from converting sugar to a product separable from lignosulfonate. In 1903, It was found ethanol could be made by fermenting the hexoses in SSL.45 In 1938, Borregaard started to produce ethanol from SSL. The ethanol fermentation step was later integrated with the production of vanillin. The SSL from softwood was fermented to produce ethanol. After ethanol removal, the lignosulfonate in the stillage liquor was used for vanillin production. In 1945, a plant was built in Thorod mill (Ontario, Canada) to produce 227 tons of lignin vanillin per year with this technology.46
The ethanol process is more suitable for softwood SSL because most of the sugars are hexoses. Since most mills used both hardwood and softwood in pulping, this option was not practical. In 1940’s, Candida utilis (torula yeast) was selected to produce single cell protein from SSL because it used both hexose and pentose. Rhinelander Paper Company (Wisconsin, US) employed a fermentation-first process to produce torula yeast from SSL.47 Prior to fermentation, inorganic sulfite was removed with lime treatment or stripping. Nitrogen, potassium, and phosphorus were also added to the SSL for medium preparation. In fermentation, yeast was produced in 50% yield from the fermentable materials. The yeast was isolated and sold as a food or animal feed additive. The lignosulfonate-rich liquor was evaporated to 50% thick and the lignosulfonate was sold as Toranil. It was further processed to a dry powder and sold as Toranil B. An important development of single cell protein from SSL was the commercial production with Pekilo process in Finland.48 In 1973, Jämsä River Mill (Jämsäkoski, Finland) built a single cell protein facility with SSL as the carbohydrate feed. The fast-growing fungus Paescilomyces varioti was produced for animal feed at the scale of 10,000 tons per year.
Membrane technology in biorefinery based on sulfite process
As mature equipment became available in early 1970’s, membrane technology was introduced into pulping industry.49 In 1972, Green Bay Packaging Inc. (WI, USA) used reverse osmosis to treat effluent, resulting in a nearly closed water system.50 The feed was a neutral sulfite hardwood pulping waste with 4–6% dissolved solids. The equipment employed cellulose acetate membrane with a total area of 446 m2 to treat waste water at the rate of 340.6 L min−1. The new system reduced the biological oxygen demand (BOD) of the effluent from 9072 kg day−1 to 454 kg day−1. Later reverse osmosis was used in US, Sweden, and Japan for the treatment of bleaching effluents with polysulfone membrane in larger scales. In 1981, two Japanese companies built membrane separation for effluent treatment. Taio Paper Co. treated 3600 m3 day−1 effluent with 1480 m2 membrane, and Sanyo Kokusaku treated 2500 m3 day effluent with 672 m2 membrane.51 The membrane technology improved the efficiency of waste treatment,52 and the concentrate was incinerated for energy recovery.53
Concentration and fractionation are basic needs to manipulate SSL. There has been an expanding effort to address these needs with membrane technologies including ultrafiltration, nanofiltration, diafiltration, reverse osmosis, and electrolysis.51, 54, 55
A survey demonstrated that sugar and lignosulfonate were separable with a polyethersulfone membrane (UP010 from Microdyn-Nadia) with a molecular weight cut-off (MWCO) at 10 kDa.56 Ideally, all lignosulfonate is rejected by the membrane while all sugar passes through at practical flux. In practice, this was carried out by two or three consecutive filtrations with decreasing MWCO, 57 thereby most lignosulfonate was rejected in retentate while most sugar stayed in permeate.
Membrane technology offered the most effective lignin fractionation processes by combining ultrafiltration (UF) and diafiltration (DF).58 The process involved concentrating SSL to a conversion ratio of 0.75–0.8 with ultrafiltration, followed by small molecule removal with diafiltration until a specific purity of lignin was reached. With molecular weight cut-off at 10 kDa in diafiltration, the process gave a lignosulfonate solution with 25% total solid content. The yield of lignosulfonate was 68%, and the purity was 93%. The capacity of the UF-DF process was dependent on the content of SSL. When the stream was from Pekilo process, the capacity of lignosulfonate fractionation increased by 50% as most sugars had been consumed in the fermentation.59
In late 1970s, several Scandinavian mills and Reed Paper (Quebec, Canada) integrated membrane technology to concentrate SSL,59 thereby to reduce the energy cost and improve product quality. With hyperfiltration (Reverse Osmosis), the SSL could be concentrated from 11 to 22% total solid with a flux of 40L m−2 h−1.52 The technology reduced energy cost by about 50% from that for water reduction by evaporation.59
Lignosulfonate has been used as a dispersant for concrete, but the function was impaired by the presence of impurities such as carbohydrates, inorganic salts, and resins. In addition, the high molecular weight lignosulfonate did not contribute to the dispersant capacity. Therefore, ultrafiltration was used to remove the impurities as well as high molecular weight lignosulfonates.60 The process involved two consecutive ultrafiltration steps with MWCO = 60 kDa at the first step and MWCO = 5 kDa at the second. Ceramic membranes were used so that the both steps could be carried out at 95 °C. High molecular weight lignosulfonate was rejected along with the resins in the retentate of the first filtration. The low molecular weight impurities, such as salts and sugars, were removed in the permeate of the second filtration. The resulting lignosulfonate fraction shortened the settlement time in concrete making and gave a product with lower air content.
For vanillin production, Borregaard 61 employed membrane fractionation to separate lignosulfonate with molecular weight in the range of 6 – 65 kDa. In a batch reaction with copper sulfate as the catalyst, this fraction offered 14.9% vanillin yield on a lignin base. For fraction with molecular weight < 6 kDa and the one with molecular weight > 65kDa, the yields were 9.8% and 9%, respectively. The fractionation step not only improved the yield and throughput of vanillin production, it also reduced the consumption of other reactants. The cost of equipment maintenance was also lower because scaling and crusting incidents were reduced or eliminated.
For sugar conversion in SSL, the liquor needs to be prepared to improve fermentation efficiency because microbial growth is inhibited by such chemicals as acetic acid, furfural, hydroxymethylfurfural, and phenolics.62, 63 These chemicals were typically removed by over-liming and evaporation of SSL. Alternatively, fermentation was carried out with sugars isolated from SSL by membrane technology.56, 57. The latter is more attractive as the separation can be installed before chemical transformation, so that product development can be more flexible from either lignosulfonate or sugar. The process efficiency is also improved because the two streams can be handled in parallel.
Succinic acid could be produced from xylose and hexoses by fermentation with Actinobacillus succinogenes and Basfia succinociproducens.64 To prepare for the fermentation, lignosulfonate was removed from a hardwood SSL with either ultrafiltration or nanofiltration. Nanofiltration was the best pretreatment for the fermentation with Basfia succinociproducens. Comparing to non-treated stream, nanofiltration increased final product concentration by 3 folds and product yield (g of succinic acid/g of consumed sugar) by 29%. By product formation was reduced by 43%. With nanofiltration, 95.6% of the lignosulfonate was rejected in the retentate. The benefit of nanofiltration was also demonstrated in fermentation with immobilized microorganisms for succinic acid production.65
In biorefinery design, sugar fraction from SSL can be converted to many products such as xylitol, ethanol, polyhydroxyalkanoates.66–70 They are the most flexible candidates for biorefinery design, therefore critical for maximizing the profitability of lignosulfonate value chain.71
Many methods have been developed to extract lignosulfonate from SSL.72 A recent method featuring selective adsorption of lignosulfonate with XAD-7 exemplified many desirable features for analytical extraction. 73 The method recovered lignosulfonate quantitatively with a simple protocol. The product had only < 1% of carbohydrate. The method was quick, robust, reproducible, and easy to scale up to prepare grams of material. Size exclusion chromatography (SEC) is the most commonly used method to fractionate lignin.74 Sodium lignosulfonate (4.8–14.2 kDa) was fractionated in 0.5 M NaOH with Toyopearl HW-55.74 With a combination of Sephacryl-100 (2 – 20kDa) and Sephadex LH20 (2–6 kDa), lignosulfonate could be purified to fractions with polydispersity between 1.76 and 1.07.75 Ultrafiltration has been compared with amine extraction76 and ethanol precipitation77 to fractionate lignosulfonate. Ultrafiltration gave products with polydispersity between 1.3–2.4,76 or 1.25–1.98.77 Both were much lower than the values by amine extraction or ethanol precipitation.
Membrane separation is highly flexible for SSL fractionation because the selectivity can be adjusted by the choice of membrane and operational conditions.54, 55, 78 It also has the important advantage of straightforward scale up from analytical fractionation to process technology. When combined with lignin characterization, membrane technology will be a very effective tool for product development.79
Organosolv pulping generates sulfur-free lignin byproduct
Organosolv pretreatment of lignocellulose involves a biomass extraction in a mixture of solvent and water under high pressure.80 The most common solvent is ethanol. In the treatment, lignin is fragmented and dissolved in organic solvent, and hemicellulose is depolymerized into soluble saccharides, leaving cellulose as the insoluble ingredients. Since all solvent is recycled, the environmental impact is small. Smaller mills are economically feasible with Organosolv process because there is no capital investment on chemical recovery boiler and kiln.
In 1989, Repap Enterprise Inc. built a commercial scale mill based on Alcell process in New Castle, New Brunswick, Canada.81 The process used recovered EtOH/water mixture from later steps as the solvent for the prior steps to minimize the amount of ethanol (Figure 11).82 The pulp from the process was easy to bleach and exhibited similar properties to those in Kraft pulp. Lignin was an important by product. In an example (Figure 11),83 the process started from cooking the wood chips in ethanol and water (60:40) mixture under pressure at 190–200 °C for 30 – 90 min. After the treatment, ethanol content was reduced to 12–21% by flash distillation and cellulose was separated. To the black liquor, water and strong acid were added to precipitate lignin. After settling, lignin was isolated by centrifugal filtration, leaving digested hemicellulose and small lignin fragments in the liquid. In the next step, ethanol was stripped from the liquid phase, leaving saccharides from digested hemicellulose in the remaining syrup. Xylose could be recovered from the syrup as a product. In a recent development, lignin was precipitated by a fed-batch precipitation, in which ethanol was removed continuously from the incoming liquor stream.84 The precipitation was optimized based on the solubility profile of the lignin. Overall, spent liquor was reduced by 50% since no water dilution was needed, and filterability was improved by particle size optimization.
Figure 11.

Alcell process based on organosolv pulping
Alcell lignin had low molecular weight (Mw < 900) with low polydispersity, no sulfur, and low ash content.85 Comparing to Kraft lignin and Lignosulfoante, Alcell lignin was more hydrophobic, and had lower glass transition temperature (90–100 °C). After lignin isolation, the xylose was converted to furfural and easily isolated as a fraction in the distillation for solvent recovery. Since lignin was not combusted for energy, all energy was from external source.81
The main mechanism of lignin depolymerization of the original Alcell process involves α-O-4 ether cleavage, while other reactions are involved when additional chemicals are added to increase the severity.86 The lignin also undergoes condensation. In the absence of sulfide, delignification was slower in Alcell process. Indeed, the original Alcell process could only remove less than 60% lignin from softwood.87 In pulping of softwood or mixed hardwood, sulfite, bisulfite, and caustic could be added to improve the efficiency.88
For hardwood pulping with Alcell process, ethanol was more efficient than dioxane and diglyme in delignification.89 The molecular weight distributions of degraded lignin were similar under different digestion conditions. About 60–80% of the lignin had a Mn of 900, which was precipitated in the dilution step. The rest were low molecular lignins remaining in the mixture with depolymerized hemicellulose. From the inception of Alcell process to 2005, 3500 tons of Alcell lignin was sold as the technical lignin.90
Characterizations showed that Alcell lignin had about 2% moisture, and purity > 96%. The main impurities include 0.2% carbohydrates, < 0.1% ash.79, 91–93 Depending on the analytical methods, the Mn was from 600 to approximately 1000, with polydispersity values between 2.4–6.3.91, 93 Alcell lignin had softening points around 138–147 °C. The heating value was 26, 700 J/g. 91 Organosolv lignins are sulfur- and ash- free, therefore they are more suitable for catalytic transformation into value-added material or chemicals.93
Cellulosic ethanol industry is an emerging source of lignin
The background of lignocellulosic ethanol for transportation fuel
Ethanol was used as an oxygenate to replace tetraethyl lead to boost octane value of transportation fuel. Although ethanol has lower energy value, it offers important benefits such as higher combustion efficiency at shorter combustion duration. Gasoline blended with ethanol generates lower emission of carbon monoxide, hydrocarbons, and NOx etc. Gasoline with < 10% ethanol can be used in most modern vehicles and are available in many nations worldwide.94 Methyl tetrabutyl ether was another oxygenate for transportation fuel, but it caused significant environmental issues as a persistent pollutant in water and air.95 Comparing to methyl tetrabutyl ether, ethanol is a safer oxygenate as it is readily biodegradable. As such, ethanol has a stable basic role in transportation fuel market.
In the first oil crisis in 1973, ethanol was evaluated as a primary fuel source when energy security became a legitimate concern. In 1975, Brazil established the National Program of Alcohol (PróAlcool) with ethanol from the sugarcane sucrose for transportation.94 In less than 10 years, sugarcane ethanol established a significant role in transportation fuel.96 From 1976 to 2004, the total amount of ethanol used in transportation was equivalent to 1440 million barrels of gasoline. In 2004, 29.4% of Brazilian energy consumption was from Biomass.97 In 2008, 25 billion liters of ethanol was produced, of which 2.8 billion liters were exported to the US and 2.36 million liters were exported to other nations.96
Since 2000s, replacing fossil fuel with renewable fuel became a global effort as the impact of Greenhouse Gas on climate change was widely recognized. In the US, the first-generation bioethanol is made by fermentation of the glucose from corn starch. This is only a solution in transition because fuel and food are competing for the feedstock. Since cellulose in biomass is the largest source of low cost feedstock, cellulosic ethanol became a highly desirable candidate for transportation fuel.1, 98, 99 Waste biomass is the low-cost byproducts of agriculture and forestry; including wheat straw, rice straw, sugarcane bagasse, and corn stover. The global yield of waste biomass was estimated between 10 and 50 billion tons per year.100 They are ideal feedstocks for ethanol production because they are generated as crop byproduct and must be disposed.
Unlike fossil fuel, biomass is widely distributed on earth surface. Therefore, collecting and shipping costs in supply chain are the limiting factors for feedstock inventory management. The feed stock of a biorefinery can only be collected in a limited area. It is usually packaged and shipped to the biorefinery directly. However, the supply chain can be more resilient by installing an optimized network of biomass depots.101
Lignocellulose waste is abundant in many parts of the US. The National Renewable Laboratory (NREL) characterized the available biomass in the United States by county.102 Web-based tools were put online for users to search and study for general understanding or specific applications. A quick search showed that crop residue is most abundant in the Midwest (Figure 12), where corn, soybeans and wheat are the predominant crops.
Figure 12.

The crop residue available for value-added applications in Midwest (Assuming 35% of the total residue could be collected)102
In the past two decades, US legislation passed a series of laws to support the R&D or scale up in biomass based technologies and subsidize biofuel. The use of biofuel (mainly ethanol) in transportation increased as the result of enforcing National Renewable Fuel Standard rule (RSF, and RSF2) set forth by The Energy Policy Act of 2005, and Energy Independence and Security Act of 2007.103 In the US, National Renewable Energy Laboratory (NREL) has been the leading institution to develop renewable fuel through either fermentation or syngas route. In 2013 the cellulosic ethanol cost by fermentation was reduced to $2.15 per gallon.99
In 2006, Canadian Federal Government initiated a 5% ethanol mix (E5) mandate as guided by the federal environmental policy to reduce greenhouse gas. Tax exemption was applied to E5 gasoline. Different provinces applied additional incentives to promote ethanol use in gasoline mix. To meet this need, fuel ethanol has been produced from starch of corn and wheat.98 For long term supply, bioethanol was expected to be produced from lignocellulose biomass from agriculture residue, forest and mill residues, disturbance wood, and energy crops. Together, the total supply was estimated to be 24–86.7 million dry tons year−1. The ethanol from biomass would meet 6.4 −61.1% of national gasoline need in 2006. The progress of R&D in government and academia in Canada was summarized in two recent reviews. 104, 105
In Europe, lignocellulosic ethanol for transportation was governed by a more complex legal framework.106 For European Union (EU) nations, the Fuel Quality Directive requires its member to reduce greenhouse intensity by 6% in 2020, while the Renewable Energy Directive requires that ≥ 10% of transportation fuel should be from renewable sources by 2020. Each nation in EU needs to provide energy targets and an action plan. The lignocellulose feedstock potential was evaluated with Geographical Information System modeling for 27 nations in Europe, Switzerland, Norway, and Ukraine. In total, 42.7 million hectares of land was available for feedstock production. Within a nation, the government could mandate the use of biofuel or provide tax exemptions on carbon dioxide emission or road use. A 2017 study with the Norwegian case found that the viability of biofuel companies was directly dependent on policy support.107 Another study focused on cellulosic ethanol producers showed that even though mandate for bioethanol created a national market, fuel companies could import the ethanol, or outsource the technology to places where cost of production was lower.108 Basically, all the producers needed to have stable customers to stay in business. Building a cellulosic ethanol industry is highly risky as ethanol is a commodity product with low profit margin. Cellulosic ethanol faces severe competition in the global market while the support for developing biorefinery industry is national.
The unit operations in a lignocellulosic ethanol process
The lignocellulosic ethanol process involves eight essential operation units as shown in Table 1. 104, 105, 109 A certain percentage of biomass is gathered, packaged in the field, and delivered to the biorefinery. The quality of the feedstock needs to be managed by standardized protocols. Agriculture residue could have high non-processing impurities. Indeed, Italian plant Crescentino needed to moving away from wheat straw due to high impurities.108 The quality of the incoming biomass is stabilized in the next step involving cleaning and size reduction. Size-reduced biomass is treated under severe conditions to de-assembly lignocellulose for cellulose hydrolysis. The saccharification of cellulose and hemicellulose is generally carried out by enzyme digestion. The efficiency of enzyme catalysis is dependent on the substrates and impurity profile. In fermentation, hexose and pentose are converted to ethanol. The ethanol product is distilled and dehydrated for transportation fuel application. The lignin has been a spectator species and can be separated from the stream before fermentation or after ethanol separation. Currently, the isolated lignin is burnt for energy recovery. The remaining waste is treated with anaerobic fermentation to generate methane as well as reduce organics in the waste.
Table 1.
The unit operations in lignocellulosic ethanol production and challenges
| Step | Operation Unit | Objectives |
|---|---|---|
| 1 | Feed handling | Efficient biomass gathering Packaging and densification for transportation Efficient delivery to biorefinery Steady inventory maintenance |
| 2 | Cleaning, size reduction | Removal of non-processing impurities. Pelletization, comminution, decreasing the size of lignocellulose waste to improve pretreatment |
| 3 | Pretreatment | Disruption of plant cells Separating hemicellulos and lignin from cellulose Decreasing cellulose crystallinity Minimizing fermentation inhibitor formation. |
| 4 | Saccharification | Hydrolyzing cellulose and hemicellulose into monomers for fermentation Maximizing fermentable sugar generation |
| 5 | Fermentation | Hexose and pentose fermentation to produce ethanol |
| 6 | Ethanol separation | Distillation and dehydration for fuel application |
| 7 | Lignin isolation | Separation for use in energy recovery or further upgrade |
| 8 | Waste treatment | Bio-digestion for methane generation |
Pretreatment is the most critical step in cellulosic ethanol production because a compromise needs to be reached to maximize the efficiency while minimize the impurity generation. Several pretreatment methods are available based on different mechanisms.105 The most popular method is acid hydrolysis in which hemicellulose is depolymerized and ether bonds in lignin are efficiently cleaved. Other approaches emphasize delignification. Lignin is redistributed and depolymerized when heated. This process can be facilitated by including organic solvents in Organosolv, ammonia in Ammonia Fiber Expansion, or ionic liquid solvation.110 However, the recalcitrance of lignocellulosic biomass is a multi-scale issue.111 Chemical treatment alone is not adequate to overcome recalcitrance at nano and meso scales. Intensified physical factors have been added to overcome recalcitrance at higher scales, leading to the increase in porosity and specific area for enzyme digestion. Microwave causes multiscale changes in lignocellulosic biomass by delivering energy directly into the biomass and causing molecule friction. It has been used with chemical pretreatment to enhance biomass degradation, but its application on industrial scale is limited by the concern of energy efficiency.112 A more practical physical treatment is steam explosion.113 The process involves building up energy in biomass with pressurized vapor, followed by a quick pressure release to generate shear force to disrupt cell walls. The severity of steam explosion is mainly dependent on temperature and retention time. The technology has been used on industrial scale, and can be combined with chemical pretreatments.
Woody biomass has higher density, therefore needs to be milled into workable particles before pretreatment. The energy cost of mechanic size reduction can be as high as 42% of that from the ethanol obtainable from woody biomass.114 Sulfide dioxide steam explosion has been applied for woody biomass pretreatment, but pressurized sulfur dioxide is corrosive and toxic. This issue was addressed by changing the order of treatments in SPORL (sulfite pretreatment to overcome recalcitrance of lignocellulose).115 The wood chip was cooked with 8–10% bisulfite and 1.8–3.7% sulfuric acid at 180 °C for 30 min. The cooked chip was then milled for enzyme digestion in the next step. As the result of the mild treatment, the cellulose hydrolysis yield was > 90% and the formation of byproducts was reduced by 53 – 73%. Ethanol from woody biomass became attractive with SPORL because energy consumption in milling was reduced by > 90%. This process was successfully tested with spruce and red pine for ethanol production.115, 116
In saccharification, cellulose and hemicellulose are generally hydrolyzed with enzyme cocktails. A suite of enzymes is needed to break different bonds in carbohydrates. Statistical analysis were applied to understand the need for digestion of different carbohydrates and optimize the ratio of enzymes in the cocktail.117 The saccharification can be consolidated with ethanol fermentation with engineered microorganisms.118 Microorganism selection and metabolic engineering have been used to integrate other desirable features such as robustness for fermentation, low energy demand in cooling, better tolerance of ethanol, etc.119 Metabolic engineering has also been used to enable xylose fermentation, improve electron flux to ethanol, and block the formation of byproducts. 119, 120 Membrane separation has also been integrated with fermentation. In pervaporation-coupled fermentation, ethanol was selectively separated in situ as a gaseous permeate. The product removal increased ethanol production by removing solvent inhibition and improving microorganism activity.121 In a continuous and closed-circulation fermentation with Saccharomyces cerevisiae, integrating pervaporation increased time and space productivity for ethanol when ethanol was recovered with separation factors of 8.8 and 9.5 in two repeats.122 The membrane does not need to be in direct contact with the broth. When vapor permeation was used instead of pervaporation with fermentation, the rate of ethanol production was further increased by 15%.123
Cellulosic ethanol industry is potentially the largest source of industrial lignin. The lignin products from the processes without using sulfite are expected to be sulfur-free, a desirable feature found in Organosolv lignin.
Organosolv ethanol biorefinery with corn stover as the feedstock
Corn stover is the most abundant agricultural residue in the US, with a yield of 64 million tons in 2012. Up to 30% of corn stover can be removed without affecting soil quality. The greenhouse gas emission of using ethanol from corn stover would be 70% lower than that of using US gasoline.124
Corn stover features high content of xylose. The potential yield of ethanol from corn stover was estimated to be 493 L (equivalent to 130.2 gallon) per dry ton. Pentose fermentation contributed 39% of the estimated ethanol due to high content of xylan.125 In an evaluation to compare technology options,126 dilute acid pretreatment was the cheapest one for corn stover. Enzyme for saccharification and feedstock were the main contributors to the operating cost. There was an energy surplus for the scenario involving burning lignin, and the capital cost contributions from combustor, boiler and turbo-generator were high.
The progress of cellulosic ethanol production in the US has been below expectation. According to a 2016 survey, there were five commercial scale biorefineries in the US in operation. They all used fermentation technology to convert lignocellulose into ethanol.127 In total the output of ethanol was 0.73 million gallons in 2014, 2.2 million gallons in 2015, and 3.3 million gallons in 2016. The productivity of lignin byproduct can be estimated with the following two assumptions:128
Biomass conversion efficiency: 70 gal ethanol per ton of biomass
Lignin in Biomass: 25% by weight
Based on the assumption, it takes 14.3 kg of lignocellulose biomass to produce one gallon of ethanol. The amount of lignin byproduct is 3.57 kg. In 2016, the theoretical amount of lignin from biorefinery is 11.8×103 tons. All the lignin was combusted for energy.
In 2011, NREL published a technoeconomic analysis for ethanol production from corn stover.129 The reported case was highly representative as it adopted the latest design with validated key technologies. The scale was set at 7.73×105 US tons year−1 of biomass. Corn stover was the most practical option as it was the most stable lignocellulose waste in the US Corn Belt. The lignin content in corn stover is low. According to the sampling study, the lignin content was between 11.2–17.8% (n = 508). The study used lignin content at 15.76% in the modeling. Correspondingly, the lignin byproduct was less than those from other feedstocks.
The process design integrated several critical innovations as outlined in Table 2. The most significant change involved direct cellulose digestion and sugar fermentation in a heterogeneous mixture. Lignin was removed after ethanol recovery (Figure 13). This design eliminated lignin separation step prior to digestion and fermentation. Another change was co-fermentation of xylose with glucose by an engineered bacteria Zymomonas mobilis strain 8b. With this process, the yield of ethanol was 79.0 gal/dry US ton corn stover. The Minimum Selling Price (MESP) of EtOH was $2.15/gal, equivalent to $3.27/gal gasoline.
Table 2.
Critical feature in the NREL bioethanol process
| Critical feature | Details |
|---|---|
| Pretreatment Intensification | Milled corn stover was treated with H2SO4 in a flow reactor for pretreatment to break down lignocellulose. No organic solvent was used in the pretreatment. In the second step, the oligomer of xylose was completely hydrolyzed. |
| Lignocellulose digestion stream telescoping | The pH was adjusted with NH4OH, which was the nitrogen source in fermentation. Subsequent steps (cellulose hydrolysis and fermentation) was not influenced by the presence of lignin. |
| Cellulose hydrolysis | Efficient enzyme cocktail was prepared by fermentation of Trichoderma reesei. These enzymes include endoglucanases, exoglucanases, β-glucosidase, hemicellulase, and accessory enzymes such as ferulic acid esterase |
| Co-fermentation | Genetically engineered Zymomonas mobilis strain 8b could ferment glucose and xylose to give ethanol. There were improvements in ethanol tolerance, and ability to ferment minor hemicellulose sugars (e.g., arabinose, galactose, and mannose). |
Figure 13.

A lignocellulose ethanol process with corn stover as the feedstock
In the process, the isolated lignin (Stillage solid, Figure 13) was combusted to generate electricity. For each gallon of Ethanol, 5.8 kWh electricity was generated. Only 3.9 kWh was needed for plant use. The rest (1.8 kWh, or 31%) of the electricity was sent back to the grid.
The process of corn stover to ethanol is put to test in Project Liberty
In Late 2000’s POET initiated “Project Liberty” in collaboration with DSM (Netherland) to build a corn stover-to-ethanol process with the key steps involving acid-hydrolysis, saccharification, and fermentation. The site was selected at Emmetsburg, Iowa. It is co-located with a corn starch ethanol facility so that all the equipment after cellulosic ethanol fermentation could be shared.130 Standard protocol was developed to collect 770 bone-dry ton corn stover per day, representing 25% of the above ground plant from the farms in 35 mile radius.131 Non-processable impurities were minimized in the standardized baling procedure. In the plant, the corn stover underwent three stages of size reductions to prepare for acidic pretreatment as well as further impurity removal.132 In the last stage, the feedstock was a slurry of particles with sizes < 0.5 inch and free from dirt, sand, rocks, tramp metals, glass, etc. The biomass was treated with 1–1.6% H2SO4 at 120–150 ͦC to depolymerize xylan while leaving lignin with cellulose in the solid phase.133 The acidic liquid was separated from the solid and reused in the next batch of pretreatment. The cellulose in the solid was converted to glucose in a continuous saccharification at 50 °C.134 The fermentation was carried out with yeast to convert hexoses to ethanol. After fermentation, lignin was separated, dried and pelleted.135 The waste stream was treated in anaerobic digestion for methane generation. Currently, all the lignin was combusted for energy.
The plant capacity was designed to produce 25 million gallons of cellulosic ethanol each year. The realization of this objective is strongly dependent on the implementation of renewable fuel standard (RFS) in the US.131 Since the inception of RFS in 2005, corn ethanol industry expanded significantly. In the period of 2006 and 2014, the consumption of corn rose from 2.1 billion bushels to 5.2 billion bushels, representing an increase from 17.6 to 43.7% of total domestic corn supply.103 Clearly, the successful industrialization of corn stover-to-ethanol process will change the paradigm in corn industry.
The BALI-process is a practical extension of sulfite pulping technology
Borregaard has been producing ethanol by fermenting the hexoses in the SSL stream of sulfite pulping since 1938.136 When transportation ethanol market emerged, a cellulose to ethanol process (BALI, or Borregaard Advanced Lignin Process) was developed in 2000’s based on the delignification with sulfite process.137 The BALI process is the most straightforward approach to convert cellulose to ethanol. It started with cooking the biomass with sulfite to solubilize lignin. Cellulose and a portion of hemicellulose were isolated from the SSL as a pulp. The pulp was enzymatically digested and fermented to give ethanol. Hexoses were converted to ethanol with Saccharomyces cerevisiae in the initial version. Bagasse was extensively tested as the feedstock because it did not require other treatments than sulfite cooking. Sulfite cooking was tested in a wide range of conditions to adjust the extent of delignification and hemicellulose removal. Based on pulp hydrolysis test, the best was weak alkaline sulfite cook, involving treating bagasse with 6% Na2CO3 and 16% Na2SO3 with liquid to solid ratio of 6 to 1 at 160 °C for 180 min. The small amount of lignin remained with cellulose pulp did not inhibit enzymes for saccharification. The glucose yield of the resulting pulp was almost 100%.137 The enzymes for saccharification was expected to be recyclable. Practical cellulose pulp could be generated from several other feedstocks including straw, willow, and spruce with BALI pretreatment.136 Co-fermentation of pentose was planned in the future versions of BALI. The BALI process was put on test at one-ton dry matter day−1 scale in 2012.45 As of the July of 2016, Borregaard were working on the scale up of BALI process after 800 tons of biomass had been successfully processed.138
Application of lignin in polymeric form
For application in polymeric form, a technical lignin needs to be specified by its plant source and process condition. It is also important to characterize the lignin in terms of purity, and chemical and physical properties.79 The heterogeneity in technical lignin mixture is multiscale: the lignin content and impurity profile are process dependent, the lignin may have different molecular weight and polydispersity, and the functional groups are different.139 Despite the fact that feedstock contributes to lignin property, technical lignins are classified based on the chemical processes they are from.140 They include Kraft lignin, Soda Lignin, Lignosulfonate, Organosolve Lignin, Hydrolysis Lignin, and Ionic Liquid Lignin. Only the first four have been commercialized and extensively studied for applications.141 Minimizing the impact of heterogeneity is a critical issue to address for functional specification.
The applications of lignosulfonate
With an annual production of 1.8 million tons per year, lignosulfonate accounts for 90% technical lignin in the world market.142 Four main functions of lignosulfonate have been explored for industrial application. These functions include adhesive properties for a binder, plastic properties for a dispersant, surface stabilizing properties for an emulsifier, and chelation properties for a sequestrant. Major applications of lignosulfonate are limited to those in civil engineering, where multiple functions are employed and specifications are not stringent (Table 3).6, 142
Table 3.
Lignosulfonate application in civil engineering infrastructure
| Application | Key function |
|---|---|
| Soil stabilization | Maintain soil moisture, increase soil particle cohesion, increase cohesive strength, cementing, increase soil density, reduce optimum moisture content, improve oil shear strength and plasticity, water proofing |
| Dust control on unpaved roads | Cement soil, improve surface strength, reduce road surface fines, reduce surface erosion caused by rainfall and temperature change, improve road stability |
| Asphalt stabilization | Decrease asphalt oxidation aging |
| Cement additives | Concrete admixture to reduce the need for water in cement pouring, prevent cavity formation |
Lignosulfonate has also been used for other minor applications, such as component of coal briquettes, components of linoleum paste, dispersant for dyes and pigments, dispersant in oil drilling mud, metal sequestrant for water treatment for boilers and cooling systems, flocculants, metal absorbents, etc.142
The application of lignin as phenol substitute in phenol formaldehyde resin
There have been efforts to find applications for Kraft lignin and Organosolv lignin since Indulin and Alcell lignin became available. While both are insoluble in water under acidic condition, they are very different in structures and composition.93 Alcell lignin had a lignin content of 96.2%, and impurities including 0.2% of carbohydrates and < 0.1% of ash. The aromatic units in Alcell was composed of 50% of S, 37% of G, and 11% of H. The total ethers (β-O-4, β−5, and β-β) was 8.9 per 100 aromatic units, the highest among all technical lignins. Alcell lignin had Mw of 2580 g mol−1 with polydispersity of 4.3. Indulin had a lignin content of 92.2%, and impurities including 1.4% of carbohydrates, 2.6% of Ash, and 1.7% of sulfur. The aromatic units in Indulin was 97% of G and 3% of H. The total ether was 7.4 per 100 aromatic units. Indulin had Mw of 4290 g mol−1 with polydispersity of 8.1. Apparently, Alcell lignin was purer in composition, underwent less structural change in the process, was smaller in size, and was more homogeneous. Organosolv lignins from feedstocks other than mixed hardwood were similar in composition, molecular weight, and polydispersity.
Phenol formaldehyde (PF) resin is a potential application for technical lignin. In PF resin formation, formaldehyde reacts with phenol at ortho or para position to give methylol groups, which can crosslink to other phenols via methylene bridges (Figure 14).143 The largest application of PF resins is wood adhesive in the fabrication of plywood, particle board, waferboard, and fiberboard. It is also used in molding compounds, abrasives, coatings, etc. The aromatic moieties in lignin may react with formaldehyde to form the resin that resembles PF polymer. However, the reactivity of the aromatic units in lignin is low and only a part of reactive sites is accessible. It was reported that the amount of formaldehyde that could be incorporated in each mol aromatic unit was 1.6 moles for Alcell lignin, 1.6–2.1 moles for sulfite lignin, and 0.1–0.5 moles for Kraft lignin.91 Initial evaluation showed that Alcell lignin could replace 30% of the PF resin in waferboard,91 and ultra-filtered Kraft lignin could replace 20% of the PF resin in particle board.144 Alcell lignin was also used in the PF resin for automotive brake pad on commercial scale. The inclusion of 10 – 20% of the lignin stabilized the friction coefficient to temperature change and improved the wear behavior.145
Figure 14.

Formation of resole resin from phenol and formaldehyde
Many methods have been explored to improve the reactivity of lignin to substitute phenols in PF resin preparation.146 Demethylation would create additional phenols in G and S moieties, resulting in aromatics with more active sites for formaldehyde. Methylolation (hydroxymethylation) would generate additional sites for ring-joining reaction in curing. These reactions were mostly tested for Kraft lignin activation.
Plywood manufacture uses resole-type PF adhesive (Figure 14). The adhesive is used as a glue to bind veneers. Filler can be added to the adhesive in glue formulation. In the final stage, the plywood is hot pressed at 140 – 150 °C to cure the glue by finalizing the crosslinking in PF polymer.143 Replacing phenol with lignin caused changes both in adhesive synthesis and in the curing.147 When a portion of phenol was replaced with lignin, the polymerization was faster to form the adhesive, but higher temperature was needed for curing. The lignin impregnated glue was tested in plywood manufacture. It was found 20% of the PF resin could be substituted with pine Kraft lignin without changing the mechanical properties of the plywood samples. The corn stover lignin from POET cellulosic ethanol process was also evaluated for the PF application.148 The POET lignin was purified by dissolution and acid precipitation. The ash content was reduced and structure was changed as indicated by FTIR analysis. The purified lignin had a Mw of 1176 g mol−1 with a polydispersity of 2.1. The adhesive prepared by 100% lignin-based resin had higher viscosity, and required higher temperature for curing. The mechanical properties of the plywood made by 100% lignin resin were similar to those of the plywood made by PF resin.
The application of lignin as polyol substitute in polyurethane synthesis
The available OH group in technical lignin can now be conveniently quantified by 31P NMR after phosphitylation. These groups include the aliphatic alcohols, phenols in different aromatic units, and carboxylic acids. Alcell lignin was found to have 1.10–1.04 mmol g−1 aliphatic OH and 3.30–3.61 mmol/g of total phenols.93, 149 Indulin was found to have 1.79–2.34 mmol/g of aliphatic OH and 2.23 – 3.87 mmol g−1 of total phenols.79, 93, 149 The polyol feature of lignin enabled its direct use as a macromonomers for polyurethane synthesis.149
Polyurethane is formed by the polymerization of diisocyanates with polyols to give urethane polymers via carbamate links (Figure 15). Diol extender may be added to form spacers between neighboring poly isocyanates. Polyurethane polymers are easy to prepare and are remarkably flexible for material design. They can form flexible foam, rigid foam, and CASE (coating, adhesives, sealants, and elastomers) polyurethanes.150
Figure 15.

Polyurethane formation from the condensation of 4,4’-methylenebis-(phenylisocyanate) (MDI) with ethylene glycol
Lignin has been used mostly to synthesize rigid polyurethane foams.145, 151 Rigid polyurethane foams are structural material with low thermal conductivity and high strength-to-weight ratio. They are mostly used for insulation in construction and transportation. Replacing a portion of polyol with Kraft lignin could improve the mechanical property of rigid polyurethane foams.152 However, the use of non-modified technical lignin is limited to less than 30% replacement. Higher percentage replacement resulted in brittle polyurethane foam. It was postulated that the reaction mixture could be heterogeneous because the high molecule weight fraction of Kraft lignin was insoluble. The insoluble lignin would form particles and generate mechanically weak points in polyurethane.
Oxypropylation is an important strategy to convert lignin into liquid polyol monomers. This transformation was applied to modify both Organosolv lignin and Kraft lignin.153 The lignin polyol product had moderate increase in hydroxyl groups through the reaction with propylene oxide. The viscosity of lignin polyol increased rapidly as the content of lignin increased from 10 to 40%. The reaction of Kraft lignin with propylene oxide in 1:4 ratio under basic condition caused favorable changes for urethane synthesis.154 Kraft lignin was depolymerized under the reaction condition. The Mw and polydispersity were both reduced in the transformation. All the phenols were converted to phenoxy ether 2-ols. The hydroxyl index (mg KOH/g), a measure of total hydroxyl group, was increased by 23%. The lignin polyol was more reactive than Kraft lignin in condensation with toluene-2.4-diisocyanate and could be used as the only polyol in polyurethane synthesis. The rigid polyurethane product with lignin polyol had better mechanical properties than the commercial counterpart.
Depolymerization of lignin involves ether fragmentation, thus creating more hydroxyl groups for polyurethane synthesis. Treating a softwood lignin with HBr generated 28% more hydroxyl groups as the result of demethylation and ether cleavage.155 The polyurethane prepared with HBr-treated lignin had dramatic improvement in mechanical strength. Depolymerization based on other mechanisms were also explored with the objective to minimize the molecular weight while creating more hydroxyl groups.151 A lignin from lignocellulosic ethanol process was depolymerized by hydrothermal liquefaction, giving a product with Mw around 1000 g mol−1 and hydroxyl index of 442 mg KOH g−1 in 70% yield. The liquefied lignin was soluble in acetone and tetrahydrofuran. It could replace 50% of sucrose polyols in polyurethane preparation.156
The use of lignin in carbon fiber synthesis
For more than 50 years, there has been persistent R&D to develop carbon fiber with lignin as the feedstock.139 Kayacarbon, a lignin based carbon fiber was commercialized by Nippon Kayaku Company in the 1960s and 1970s. This product was removed from the market in the 1970s because better carbon fibers based on different precursors became available.157
Currently carbon fiber is manufactured with polyacrylonitrile (PAN) as the starting material.158 It is a copolymer made by condensation of acrylonitrile, methyl methacrylate, and itaconic acid. It has a molecular weight of 70,000 – 260,000 g mol−1 with a polydispersity of 1.5 – 3.5. The fiber precursor is made from PAN by wet spinning. The fibers are finished and wound into tow before heat treatment. In the next step, the precursor fiber is oxidized at 200–300 ͦC. The side chains and the backbone undergo cyclization and dehydrogenation to form heteroaromatic structures. This reaction prepares the fibers for carbonization at higher temperature because the stabilized fibers are more heat resistant. In carbonization, the temperature is ramped up to approximately 2000 ͦC. Almost all oxygen, hydrogen, and nitrogen atoms are rejected as HCN, H2O, O2, H2, CO, NH3, and CH4. The resulting carbon fiber is composed of > 98% of graphenated carbon in highly ordered turbostratic structure. The carbon fibers from PAN have tensile strengths of up to 7 GPa and moduli up to 900 GPa. However, PAN based carbon fiber is expensive; therefore it is used only in high-end applications.
Lignin based carbon fiber became an attractive objective as the need for low cost carbon fiber emerges for making major products such as automobile and wind turbines.158 Other than low cost and renewability, lignin is expected to offer additional benefits including high carbon yield and elimination of toxic substances involved in carbon fiber preparation with PAN.
The preparation of carbon fiber from lignin also follows the three-step protocol involving spinning, thermostabilization, and carbonization.139 The most common methods to prepare precursor from lignin are melt-spinning and solvent-assisted spinning. The spinning temperature is dependent on the type of lignin and other components in the blend. According to Oak Ridge National Laboratory, the lignin feedstock has to be highly purified, with ash content < 0.1%, low in sulfur, and free from particulate matter. The spinning should not generate additional impurities through side reactions such as charring. Several strategies have been explored to improve the spinnability. These strategies include lignin fractionation,159 blending with a plasticizer such as polyethylene oxide,160 or partial acetylation.161 However, improving spinnability by these strategies may decrease the lignin softening temperature (Ts) and reduce the thermal resistance of the fiber precursor.158 In the thermostabilization step, the temperature is increased to the range of 200 – 300 ͦC to promote a series of reactions such as rearranging the ether bonds to C-C bonds through radical coupling, dehydration, elimination of CO or CO2, additional crosslinking, etc. Ultimately, the fiber precursor is converted from a thermoplastic to a thermoset in thermostabilization. The cross section of the fiber is expected to become homogeneous in this step. However, impractical slow heating rate was required to minimize fiber fusion in oxidative thermostabilization of lignin precursor,.162 Applying UV reduced the heating time from 40 h to 4 h by promoting crosslinking.163 Embedding desirable reactions in thermostabilization was also explored. One example involved converting a portion of the phenols in softwood lignin to the propargyl ethers. The resulting structure underwent cyclization to give benzopyrans, which created new courses of polymerization in thermostabilization.164 Finally, the stabilized fiber is carbonated at temperatures above 1000 ͦC to eliminate all elements other than carbon. The heating needs to be optimized to minimize defection while facilitating the formation of graphitic structure. The best lignin carbon fiber only reached tensile strength of 1.07 GPa and Modulus 82.7 GPa.157 Analysis showed that the lignin carbon fiber did not have ordered turbostratic structure,158 most likely due to the fact that the lignin-based precursor was poorly oriented. Overcoming heterogeneity remains to be a critical challenge to use lignin in carbon fiber manufacture.
There is much more to explore for lignin applications in polymeric forms. Many options for lignin functionalization have been explored.165, 166 The purified lignin can be functionalized through electrophilic aromatic substitution (Lederer-Manasse reaction with formaldehyde, Kolbe-Schmitt reaction), urethane reaction via hydroxyl groups (with isocyanate), esterification (with ε-caprolactone, maleic anhydride, and succinic anhydride) of the hydroxyl groups, demethylation of the methoxy group to create additional phenols, and radical polymerization, etc. Olefin and epoxy groups can also be introduced via some of these reactions for further derivatization. Although technical lignin is cheap, its derivatives can be expensive due to low efficiency in functionalizing lignin and product isolation. As such, the derivatization process development, and product specification are both critical for product development.
Expanding the use of lignin through depolymerization
Preparing monomeric chemicals from lignin is potentially profitable. The basic cost for technical lignin was €110/ton, while the corresponding phenol monomer price was roughly €1220/ton. The large profit margin has been a main driver for research into lignin depolymerization. In addition, lignin depolymerization is strategically important for sustainable development, as the phenol products may be used as the drop-in replacement for those from petroleum-based BTX (€876/ton in 2010) stream.141
Controlled pyrolysis and hydrothermal liquefaction as the baseline examples
Through combustion, lignin was completely transformed into CO2 and H2O at the temperature between 800–1200 °C. This was the last option to treat the residue. Lignin was degraded into monomers by controlled heating under special reaction conditions. Pyrolysis involved heating in the absence of oxygen. It was a valuable tool to study bond cleavage of lignin as the temperature rose. The degradation of lignin by pyrolysis was divided into two stages (Figure 16). Between 200 – 400 °C, most of the reactions involved heterolytic ether (mainly α-O-4, β-O-4) cleavage. In this temperature range, homolytic cleavage of ether only happened when the radical product was stabilized by aromatic substitution pattern. When temperature rose to > 400 °C, radical reaction mechanism predominated in the degradation and caused extensive rearrangement. Significantly, the C-C bonds were broken and monomers started to form. However, the severe reaction condition also promoted rearrangement and re-polymerization. There was also char formation through some of the intermediates. In practice, tar or coke formation was the main issue in thermal process development.167
Figure 16.

Intermediates and reactions involved in lignin pyrolysis
The mass loss of lignin was slow and continuous in the pyrolysis when temperature was increased from ambient temperature to 900 °C, suggesting the degradation involved many non-specific reactions. In differential scanning calorimetry study, lignin pyrolysis gave a significant exothermal peak at 365 °C as the result of massive char formation. The pyrolysis was endothermic when temperature reached > 400 °C, indicating the predominant reactions were bond breaking. In this temperature range, significant amount CH4 and H2 formed, presumably through the quenching of the free radicals involved in the homolysis.168
There were physical changes involved in char formation. Lignin became a melt at 250 °C. The char formation started around 350 – 400 °C, then underwent solidification and carbonization at higher temperature. The char product was a crosslinked and refractory solid residue.169
At Energy Research Centre of the Netherlands (ECN), continuous pyrolysis was tested with different technical lignins including Alcell lignin, herbaceous lignin, and Kraft lignin from softwood. The pyrolysis was carried out with a fluidized bed reactor at 500 °C. The products were typically composed of 20% gas, 45% oil, and 35% char. The gas was a mixture of CO, CO2, and CxHy. The oil was a mixture of methanol, acetic acid, oligomeric phenols and oligomeric phenols. The phenolic chemicals might generate significant revenue but fractionation of the oil was expected to be challenging.170
The activation energy of char formation was estimated to be 76 kJ/mol. In char solid, there was an increase of aromatic carbons, a decrease of H/C ratio, and a decrease of O/C ratio. The yield of char was reduced from 80% to 40% when pyrolysis temperature was raised from 300 °C to 750 °C. Removing sodium and potassium from lignin by acid wash also decreased char yield. Nevertheless, char formation always preceded complete depolymerization in lignin pyrolysis.169
Thermal liquefaction has been extensively explored to improve the efficiency of lignin depolymerization because the β-O-4 bonds in lignin can be broken in alternative pathways.171 The reactions were typically carried in a solvent at temperatures between 250 – 500 °C in a solvent with a catalyst or an additional reactant. Although thermal liquefaction also suffered from char formation through repolymerization, the yields of oil product were improved to > 80% in many cases.
Most of the thermal liquefactions have been carried out in hot compressed water. The conditions are divided into supercritical water or subcritical water depending on their location in relation to the critical point (347 °C at 22.1 MPa) in the phase diagram.172 In general, liquefaction predominates in the temperature window of 200 – 350 °C (5–28 MPa). At higher temperatures, gasification becomes significant.172, 173 Subcritical water has densities between 0.692 – 0.958 and low viscosities, therefore allows faster mass transfer. It is also unique in promoting certain reactions involving ionic intermediates. The dielectric constant of subcritical water is balanced for polar and non-polar compounds. The Kw (ionization constant) of water rises from 10−12 at 100 °C to 10−11 at 300 °C and quickly decreases to 10−20 at 400 °C. These two qualities of subcritical water are more compatible with the cleavage of β-O-4 bonds by the more selective retro-aldol mechanisms.174
It was shown that many factors could influence char formation in hydrothermal liquefaction.175 In a liquefaction of Organosolv lignin at 500 °C, THF insoluble product (char) increased over the course of 60 min. Adding 10 to 20 folds of phenol reduced char formation and also changed the products. Apparently, phenol prevented repolymerization by quenching the monomeric intermediates. Char formation was 20 – 50% when the ratio of water to lignin was < 10, but can be reduced when water to lignin ratio was increased to 25.
In subcritical regime, increasing temperature from 200 to 350 °C had different impact.176 With Kraft lignin/water ratio at 1:10 and fixed heating time of 40 min, the yield of oil increased from 28.9 to 44.6%, while the yield if solid residue decreased from 40.5 to 27.4%. The gas product remained around 30% in the temperature span. Prolonging time from 0 to 60 min increased the yield of water soluble fraction in oil product, but did not show clear trend on other products. Decreasing lignin/water ratio from 1:10 to 1: 80 also increased the yield of water soluble fraction of oil product.
Phenol and base catalysts greatly improve the yield of liquid product in subcritical regime.177 The impact of temperature was evaluated in the liquefaction of LignoBoost Lignin with 5.5 wt % phenol as the capping agent, and 4.1 wt % of K2CO3 and solid ZrCO2 as the catalysts. The ratio of Lignin/water was 1: 18. The reaction was carried out in a steady state with pressure at 25 MPa and the resident time of 10 −13 min. Temperature increase from 290 to 370 ͦC showed pronounced impact on product composition: the char increased from 16 to 22%, while water soluble organic increased from 5 to 11% and oil decreased from 87 to 69%. Importantly, the total carbon balance was 97 – 103%, suggesting gas formation was insignificant.
Heating rate has been identified as an important operating factor because liquefaction is a kinetically controlled process.172 One way to increase the heating rate involved injecting lignin slurry to a preheated medium.178 With a batch reactor equipped with injection function, the impacts of subcritical temperature and phenol were evaluated on the liquefactions for 15 min. The ratio of Indulin/water was 1: 16.7. Potassium carbonate (wt 1.7%) was used as the catalyst. At 300 ͦC and 20 MPa, phenol was highly efficient to improve the liquefaction. When phenol concentration was increased from 3.4 to 9.7 wt%, char formation was reduced from 11.3% to 1.4%. Correspondingly, the yield of depolymerization product was increased to nearly 100%. Temperature increase from 300 to 350 ͦC showed negative impact. The char formation increased from 11.3% to 15.9%, while total yield of depolymerization product decreased from 81.7 to 53.5%.
Biocrude from lignin liquefaction contains phenols such as syringol, isoeugenol, cresols, guaiacols, vanillin, catechol, and many other phenolic dimers or oligomers.176, 177, 179 In a test, Biocrude from corn stalk lignin was used to replace phenol in phenol formaldehyde resin synthesis.179 Up to 65% of the phenol could be replaced by the biocrude. The product from biocrude had higher viscosity and adhesive strength than the counterpart from phenol.
Chemical depolymerization and metallic catalytic depolymerization
Pulping processes employed acid (Sulfite Process) or alkaline treatment (Kraft, or Soda Processes) to degrade lignin. The mechanism of pulping study provided insights into lignin depolymerization. Under these conditions, ether bonds (α-O-4 or β-O-4) were sequentially cleaved by starting from benzyl ether (α position).180, 181
Under acidic conditions (Figure 17), benzyl ether was heterolytically cleaved to release the aromatic ether and generate a carbon cation. The carbon cation underwent a series of tautomerizations on the propene side chain, and led to β-O-4 either cleavage via a hemiacetal intermediate. In Sulfite Process, the carbon cations were quenched by sulfite to give lignosulfonate. This reaction not only increased the solubility of lignin, but also prevent the carbon cations reaction with neighboring electron-rich aromatic moieties.
Figure 17.

Cleavage of α-O-4 and β−4-O bonds under acidic condition (Sulfite Process)
Under alkaline condition, the depolymerization required the presence of free 4-phenol group (Figure 18). The phenoxy anion triggered the elimination of the phenol at α-O-4 motif through a quinone-methide intermediate. A nucleophile (OH- or SH-) added to the α-carbon in quinone-methide, causing additional fragmentation at β-O-4 motif (Figure 6, and 18). When γ-hydroxyl group was free, the quinone-methide also underwent side chain cleavage, giving formaldehyde trough a retro-aldol reaction.
Figure 18.

Cleavage of α-O-4 and β-O-4 bonds under alkaline condition (Soda or Kraft Process)
In pulping, lignin always underwent unintended structural changes in addition to fragmentation. This is because the active intermediates, including carbon cations in Sulfite pulping (acidic) and quinone-methide intermediate in Kraft Process (basic), tend to react with electron-rich positions or undergo rearrangements. A recent study showed Kraft lignin had molecular weights around 4290, corresponding to 25 aromatic units, while soda lignin had molecular weight around 3270, corresponding to 19 aromatic units.93 Technical lignin was more recalcitrant to further depolymerization due to additional C-C bond formation in pulping process.
Additional physical, chemical, or biological factors were introduced to better control lignin depolymerization or liquefaction. Based on these factors, depolymerization can be classified into thermal, chemical, metallic catalytic, and biological processes (Table 4).
Table 4.
Classification of lignin depolymerization
| Depolymerization | Subtype | Objective |
|---|---|---|
| Thermal | Pyrolysis and hydrothermal liquefaction | Controlled thermal degradation to give bio-crude |
| Gasification | Controlled degradation of aromatics to give combustible gas | |
| Combustion | Oxidative energy generation | |
| Chemical | Base/acid catalysis | Ether bond/side chain cleavage under milder condition |
| Oxidation/Reduction | Bond cleavage coupled with further conversion to minimize re-polymerization | |
| Capping depolymerization | Minimize re-polymerization by temporary protection | |
| Ionic liquid/Supercritical fluid, etc | Solubilizing lignin to improve selectivity/mass & energy transfer | |
| Metallic Catalytic | Selective bond cleavage and coupling with redox reactions | |
| Biological | Enzymatic, growing cell | Oxidative depolymerization or catabolizing lignin |
Oxidation is a facile choice for lignin depolymerization as lignin is made up with electron-rich aromatics.182 Metal oxides and metalloporphyrins were tested to oxidize either lignin model compounds or lignin. In most cases, the reaction involved electron transfer or hydrogen atom extraction from lignin, causing a broad range of reactions including phenol oxidation to benzoquinone, benzylic oxidation, demethylation, ring opening reaction, hydroxylation of aromatic rings, side chain fragmentation, etc. Importantly, oxidative depolymerization was effective to break C-C bonds (5–5’, α−5’, diphenylmethane, etc.), therefore suitable for high C-C content Kraft lignin. Oxidation mediators were used to improve accessibility in lignin, as they have better mass transfer in polymer.
Lignin was also depolymerized by reductive fractionation. The primary mechanism involved hydrogenolysis of aryl ether bonds. The most frequently used metal catalysts were nickel or ruthenium. The chemical pathway involved insertion followed by reductive elimination by H2 (Figure 19) to give aromatic and alkyl alcohol products.183 In native lignin (protolignin), ethers represent 52 – 83% of the inter-unit bonds in lignin. By selectively breaking ether bonds, hydrogenolysis is potentially an ideal approach to depolymerize lignin with minimal side reactions.
Figure 19.

Plausible pathway for hydrogenolysis of Alkyl-Aryl ether by Nickel Catalyst
The efficiency of reductive catalytic fractionation was demonstrated in a “lignin-first approach” with corn stover as the substrate.184 The milled corn stover was digested with H2 and Ni or Ru catalyst in MeOH. Under 30 bar H2 at 200 – 250 ͦC, the lignin was depolymerized for 6 h to give 20 – 38 wt % of monomer and a mixture of oligomers. The compounds in the monomer mixture were propenyl phenols, hydroxycinnamates, and ethylene phenols. In the hydrogenation, more than 90% of enzyme digestible cellulose and hemicellulose were retained.
The situation with technical lignin is more complicated. For Kraft Lignin, the β-O-4 ether was only 6.1% (number per 100 aromatic units), down from 35–65% in milled/cellulase treated lignin. It also contained 1.7% of sulfur, which might cause metal catalyst inactivation.93 In a recent example, Kraft lignin was fragmented with a sulphided NiW at 320 ͦC and 35 bar H2 in methanol.185 After 8 h, the reaction generated a MeOH-miscible oil product in 82% yield. However, the oil was a complex mixture of alkyl phenols, guaiacols, and lignin oligomers. The surface area of the catalysts was reduced by 81% in the reaction.
In n-hexadecane, Organosolv lignin was fully converted to saturated hydrocarbons by a Ni-Zeolite catalyst at 320 ͦC and 20 bar H2. The reaction underwent hydrogenolysis, hydrogenation, and then Brönsted acid catalyzed dehydration, giving liquid oil product in 70% yield.186 In these catalytic reductive fragmentation, the product from technical lignin was highly complex, therefore not yet suitable for aromatic feedstock preparation.
To inhibit benzylic cation formation in pretreatment under acidic condition, Luterbacher’s group used formaldehyde to protect the α and γ hydroxyls by forming a 1,3 dioxane (Figure 20). The extracted lignin was readily depolymerized by hydrogenolysis at lower temperature (150 – 250 ͦC, 40 bar H2), giving guaiacyl and syringyl monomer products with much simpler composition and a yield of 78%.187
Figure 20.

Formaldehyde protection to preserve lignin structure in extraction
Lignin depolymerization with microorganisms or enzymes
Biological oxidative depolymerization is uniquely useful for lignin valorization as the catalysts are more selective and concurrent reactions can be integrated into one step. The catalysts were either growing cells or enzymes. A recent example demonstrated a “funneling” or catabolism approach to convert heterogeneous lignin into homogenous products.188 In the report, Pseudomonas putida KT2440 transformed lignin or lignin model compounds by breaking the aromatic rings to give suitable substrates for β-ketoadipate pathway. The carbon intermediates were subsequently converted into polyhydroxyalkanoates, which could be a feedstock chemical for hydroxyacid monomers. Laccase and peroxidase are important lignin modifying enzymes. They catalyze the oxidation of lignin via single electron transfer mechanism, giving phenoxy radical or carbon centered radical intermediates that promote subsequent reactions. Both laccase and peroxidase have been used to depolymerize lignin either in free or immobilized form. While depolymerization with enzyme did not digest lignin completely into smaller molecules, the reaction appeared to be more specific. In a report, treating Kraft lignin with laccase caused up to 35 wt % solubilization.189 The distribution of soluble and solid lignin products strongly depended on the electron mediators. The soluble lignin had a molecular weight range from 135–646 Da/mol, suggesting monomeric and oligomeric lignin were generated in the treatment. The insoluble part had higher molecular weights with decreased polydispersity.
Producing vanillin from lignosulfonate
Vanillin is the only commercial monomer product from lignin. It is the most important flavoring chemical with a market value around $650 million per year.190, 191 The total vanillin output is 20,000 tons from three major routes including 85% from petroleum based chemicals, 15% from lignin, and < 1% from vanilla bean. Vanillin from lignin is slightly more expensive than that from chemical synthesis, as its aromatic intensity is 1.2 times stronger. Currently, all lignin vanillin is from the biorefinery facility of Borregaard.
The formation of vanillin starts from the typical quinone-methide intermediate, which can be generated from lignosulfonate by copper catalyzed air-oxidation. It undergoes vinyl ether bond cleavage at γ position through a hemiacetal. The resulting aldehyde is cleaved at Cα-Cβ bond by a retro-aldol reaction to give vanillin and side-chain products (Figure 21).
Figure 21.

Mechanism to prepare vanillin from the quinone-methide intermediate
Concluding remarks
Lignin valorization is expected to play a strategic role in advancing sustainability because it is the only renewable aromatic polymer on bulk scale. For pulping industry and lignocellulosic industry, lignin production is attractive because it improves throughput and generates additional revenue. As the result, there are vigorous efforts to develop processes for lignin isolation, purification, and fractionation in recent years. To develop a lignin based product, it is imperative to evaluate the quality and supply of the feedstock. The evaluation is based on the knowledge of the production pathway and associated impacts on lignin.
The largest potential source of technical lignin is Kraft Process, in which lignin is depolymerized by sulfide under basic condition. Removing a portion of lignin from the Black Liquor improves the efficiency of Kraft mill, therefore it is affordable to use Kraft lignin as a fuel. Kraft lignin features high C-C bond content, small molecular weight and sulfur contamination. Indulin has been available for decades. LignoBoost and LignoForce represent recent development in Kraft lignin production on commercial scale. Efficient fractionation or transformation of Kraft lignin to functional products will be the key to non-fuel applications.
Lignosulfonate is an important member in the product portfolio of sulfite process. It may also be generated from the BALI process. Lignosulfonate has established market and accounts for 90% of commercial technical lignin. The sugars in SSL can be converted to single cell protein or many chemicals by fermentation. The product portfolio can be designed to maximize the current value by extracting multiple products from SSL. The point to isolate lignosulfonate from SSL stream is dependent on biorefinery configuration.
Cellulosic ethanol industry is yet to be stabilized as the ethanol product needs to be supported by public policy. The lignin by product is currently combusted for energy although there is an energy surplus in this practice. Lignin valorization is critical in the development of an integrated biorefinery for cellulosic ethanol. The scale of lignin from cellulosic ethanol industry may exceed that from pulping. The corn stover lignin from POET will be a stable product as the process matures. It was generated by a pathway involving acid treatment, saccharification, hexose fermentation, ethanol distillation, and precipitation from the stillage. This product is sulfur-free and its property resembles that of Organosolv lignin.
Technical lignins have been used in polymeric form since they became available. The molecular weight and polydispersity have strong impact in lignin applications. Membrane technology has long been used in the biorefinery based on sulfite process. It should be used as the first line option to purify and fractionate technical lignins. Solvent fractionation is also a robust tool to obtain lignin with desired solubility and high purity, although it may not reduce the polydispersity of the product. Lignin is a polyfunctional polymer. It can be chemically derivatized for additional properties. For practical application, a chemical process should be highly efficient, and requires no or minimal effort for product isolation.
Lignin depolymerization is an attractive option for practical application as it may provide renewable substitutes for aromatic petro-chemicals. Technical lignin has been degraded in four general approaches. The difficult to break C-C bonds and repolymerization are the common problems in all options. Additional measures, such as protection of α,γ hydroxyl groups on the side chain before treatment, may help in efficient depolymerization.
Footnotes
Disclaimer
The views expressed in this article are those of the authors and do not reflect the official policy or position of the Unites State Environmental Protection Agency. Mention of trade names, products, or services does not convey official EPA approval, endorsement, or recommendation.
Contributor Information
Tao Li, U. S. Environmental Protection Agency, Office of Research and Development, NRMRL, 26 W. Martin L. King Drive, Cincinnati, OH 45268, United States.
Sudhakar Takkellapati, U. S. Environmental Protection Agency, Office of Research and Development, NRMRL, 26 W. Martin L. King Drive, Cincinnati, OH 45268, United States.
References
- 1.Marriott PE, Gomez LD and McQueen-Mason SJ. Unlocking the potential of lignocellulosic biomass through plant science. New Phytol 209: 1366–1381 (2016) [DOI] [PubMed] [Google Scholar]
- 2.French AD, Miller DP and Aabloo A. Miniature crystal models of cellulose polymorphs and other carbohydrates. Int J Biol Macromol 15: 30–36 (1993) [DOI] [PubMed] [Google Scholar]
- 3.Lebo SE Jr., Gargulak JD and McNally TJ Lignin. Kirk-Othmer Encyclopedia of Chemical Technology (5th Edition), John Wiley & Sons, Inc., p. 1–25. (2005) [Google Scholar]
- 4.Yuan T-Q, Xu F and Sun R-C. Role of lignin in a biorefinery: separation characterization and valorization. J Chem Technol Biotechnol 88: 346–352. (2013) [Google Scholar]
- 5.Food and Agriculture Organization of the United Nations: Pulp and paper capacities, 2016–2021, www.fao.org/forestry/statistics/81757/en [Accessed 20 April 2018]
- 6.Gopalakrishnan K, Kim S and Ceylan H. Lignin recovery and utilization. in Bioenergy and Biofuel from Biowastes and Biomass, American Society of Civil Engineers, p. 247–274. (2010) [Google Scholar]
- 7.Cardoso M, Domingos de Oliveira E and Passos ML. Chemical composition and physical properties of black liquors and their effects on liquor recovery operation in Brazilian pulp mills. Fuel. 88: 756–763. (2009) [Google Scholar]
- 8.Brauns FE, and Brauns DA in The Chemistry of Lignin, Supplement Volume Covering the Literature for the Years 1949–1958. Academic Press, London, UK: pp.742–745. (1960) [Google Scholar]
- 9.Moorer HH. Anionic bituminous emulsions. USA Patent 19763956002 (1976)
- 10.Benali M, Perin-Levasseur Z, Savulescu L, et al. Implementation of lignin-based biorefinery into a Canadian softwood kraft pulp mill: Optimal resources integration and economic viability assessment. Biomass Bioenergy 67: 473–482 (2014) [Google Scholar]
- 11.Axegård P and Ab S-P. The KRAFT pulp mill as a biorefinery. Third ICEP International Colloquium on Eucalyptus Pulp, Belo Horizonte, Brazil (2007) [Google Scholar]
- 12.Gellerstedt G, Tomani P, Axegaard P and Backlund B. Lignin recovery and lignin-based products in RSC Green Chem Series Vol 18, Integrated Forest Biorefineries. pp180–210 (2013) [Google Scholar]
- 13.Uloth VC and Wearing JT. Kraft lignin recovery: acid precipitation versus ultrafiltration. Part II: Technology and economics. Pulp Pap Can 90: 34–37 (1989) [Google Scholar]
- 14.Uloth VC and Wearing JT. Kraft lignin recovery: acid precipitation versus ultrafiltration. Part I. Laboratory test results. Pulp Pap Can 90: 67–71 (1989) [Google Scholar]
- 15.Ragnar M, Lindgren CT and Nilvebrant N-O. pKa-values of guaiacyl and syringyl phenols related to lignin. J Wood Chem Technol 20: 277–305 (2000) [Google Scholar]
- 16.Zhu W and Theliander H. Precipitation of lignin from softwood black liquor: an investigation of the equilibrium and molecular properties of lignin. BioResources 10: 1696–1714 (2015) [Google Scholar]
- 17.Evstigneev EI. Factors affecting lignin solubility. Russ J Appl Chem. 84: 1040–1045 (2011) [Google Scholar]
- 18.Oehman F, Theliander H, Norgren M, Tomani P and Axegaard P. Separating lignin from a lignin containing liquid/slurry, lignin product or an intermediate lignin product, and their use as fuels. World Patent 2006038863 (2006)
- 19.Oehman F, Theliander H, Tomani P and Axegard P. Method for separating lignin from black liquor. World Patent 2006031175 (2006)
- 20.Tomani P The lignoboost process. Cellul Chem Technol 44: 53–58 (2010) [Google Scholar]
- 21.Ohman F, Theliander H, Tomani P and Axegard P. Method for lignin separation from black liquor. World Patent 20150119559 (2015)
- 22.Vakkilainen E and Valimaki E. Effect of lignin separation to Black Liquor and recovery boiler operation 2009 TAPPI Engineering, Pulping, Environmental Conference Memphis, Tennessee, USA (2009) [Google Scholar]
- 23.Tomani P, Axegaard P, Berglin N, Lovell A and Nordgren D. Integration of lignin removal into a kraft pulp mill and use of lignin as a biofuel. Cellul Chem Technol 45: 533–540 (2011) [Google Scholar]
- 24.Oehman F, Theliander H, Tomani P and Axegard P. A method for separating lignin from black liquor, a lignin product, and use of a lignin product for the production of fuels or materials. World Patent 2009104995 (2009)
- 25.Kassman H, Brostroem M, Berg M and Aamand L-E. Measures to reduce chlorine in deposits: Application in a large-scale circulating fluidised bed boiler firing biomass. Fuel 90: 1325–1334 (2011) [Google Scholar]
- 26.Hu Z, Du X, Liu J, Chang H-m and Jameel H. Structural Characterization of Pine Kraft Lignin: BioChoice Lignin vs Indulin AT. J Wood Chem Technol 36: 432–446 (2016) [Google Scholar]
- 27.Paleologou M, Radiotis T, Kouisni L, et al. New and emerging biorefinery technologies and products for the canadian forest industry. J Sci Technol Forest Prod Processes 1: 6–14 (2011) [Google Scholar]
- 28.Kouisni L, Gagne A, Maki K, Holt-Hindle P and Paleologou M. LignoForce System for the Recovery of Lignin from Black Liquor: Feedstock Options, Odor Profile, and Product Characterization. ACS Sustainable Chem Eng. 4: 5152–5159 (2016) [Google Scholar]
- 29.Kouisni L, Holt-Hindle P, Maki K and Paleologou M. The LignoForce System: a new process for the production of high-quality lignin from black liquor. J Sci Technol Forest Prod Processes. 2: 6–10 (2012) [Google Scholar]
- 30.Kouisni L and Paleologou M. Method for separating lignin from black liquor. World Patent 2011150508 (2011)
- 31.Wells K, Pors D, Foan J, Maki K, Kouisni L and Paleologou M. CO2 Impacts of Commercial Scale Lignin Extraction at Hinton Pulp using the LignoForce Process & Lignin Substitution into Petroleum-based Products. PACWEST Conference, June 10−13, 2015, Whistler, BC, Canada (2015) [Google Scholar]
- 32.Kouisni L, Fang Y, Paleologou M, et al. Kraft lignin recovery and its use in the preparation of lignin-based phenol formaldehyde resins for plywood. Cellul Chem Technol. 45: 515–520 (2011) [Google Scholar]
- 33.Ball FJ, Gressang RW and Keilen JJJ. Method of coagulating colloidal lignates in aqueous dispersions. US Patent 2623040 (1952)
- 34.Miettinen M Continuous method for the precipitation of lignin from black liquor. World Patent 2012049375 (2012)
- 35.Lake MA and Blackburn JC. SLRP - an innovative lignin-recovery technology. Cellul Chem Technol. 48: 799–804 (2014) [Google Scholar]
- 36.Ragnar M, Henriksson G, Lindström ME, Wimby M, Blechschmidt J and Heinemann S. Pulp. in Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH, Weinheim, Germany: pp. 1–92 (2014) [Google Scholar]
- 37.Sumathi S and Hung Y-T. Treatment of pulp and paper mill wastes in Waste Treatment in the Process Industries, CRC Press LLC, Boca Raton, Florida, pp. 453–497 (2006) [Google Scholar]
- 38.Clark J and Deswarte F. The biorefinery concept: an integrated approach in Introduction to Chemicals from Biomass, 2nd Ed, John Wiley & Sons Ltd., West Sussex UK, pp. 1–29 (2015) [Google Scholar]
- 39.Thompson NS. Hemicellulose. in Kirk-Othmer Encyclopedia of Chemical Technology., Wiley InterScience, Hoboken, NJ: (2000) [Google Scholar]
- 40.Fatehi P and Ni Y. Integrated forest biorefinery - sulfite process ACS Symp Series vol 1067, Sustainable Production of Fuels, Chemicals, and Fibers from Forest Biomass pp. 409–441 (2011) [Google Scholar]
- 41.Fredheim GE, Braaten SM and Christensen BE. Comparison of molecular weight and molecular weight distributions of softwood and hardwood lignosulfonates. J Wood Chem Technol. 23: 197–215. (2003) [Google Scholar]
- 42.Howard GC. Process of utilizing waste sulphite liquor US Patent 1551882 (1925)
- 43.Howard GC. Utilization of sulfite liquor. Ind Eng Chem. 26: 614–617 (1934) [Google Scholar]
- 44.Sandborn LT, Richter SJ and Clemens HG. Process of making vanillin. US Patent 2057117 (1936)
- 45.Frölander A and Rødsrud G. Conversion of cellulose, hemicellulose and lignin into platform molecules: biotechnological approach, http://www.eurobioref.org/Summer_School/Lectures_Slides/day3/Lectures/L06_A.Frolander.pdf.pdf. EuroBioRef Summer school. Lecce, Italy (2011) [Accessed 20 April 2018] [Google Scholar]
- 46.Hocking MB. Vanillin: synthetic flavoring from spent sulfite liquor. J Chem Educ 74: 1055–1059 (1997) [Google Scholar]
- 47.Holderby JM and Moggio WA. Utilization of Spent Sulfite Liquor. J Water Pollution Control Federation 32: 171–181 (1960) [Google Scholar]
- 48.Romantschuk H and Lehtomaki M. Operational experiences of first full scale Pekilo SCP-Mill application. Process Biochem. 13: 16–17,29 (1978) [Google Scholar]
- 49.Kofod Nielsen W, Frik Madsen R and Jentoft Olsen O. Experience with plate-and-frame ultrafiltration and hyperfiltration systems for desalination of water and purification of waste water. Desalination. 32: 309–326 (1980) [Google Scholar]
- 50.Walraven GO, Nelson WR, DeRossi PE and Wisneski RL. Closed process water loop NSSC corrugating medium manufacture, https://cfpub.epa.gov/si/si_public_record_Report.cfm?dirEntryID=40367. Washington D.C,, USA (1977) [Accessed 20 April 2018) [Google Scholar]
- 51.Joensson AS and Traegaardh G. Ultrafiltration applications. Desalination 77: 135–79 (1990) [Google Scholar]
- 52.Joensson AS and Wimmerstedt R. The application of membrane technology in the pulp and paper industry. Desalination 53: 181–96 (1985) [Google Scholar]
- 53.Okamoto H, Mizuhara M, Numata Y, Nakagome K, Ochiumi T and Kuroda T. Treatment of paper-plant wastewater by ultrafiltration. A case history in ACS Symp Series Vol 281: Reverse Osmosis and Ultrafiltration pp. 273–281 (1985) [Google Scholar]
- 54.Adnan S, Hoang M, Wang H, Bolto B and Xie Z. Recent trends in research, development and application of membrane technology in the pulp and paper industry. Appita J. 63: 235–241 (2010) [Google Scholar]
- 55.Humpert D, Ebrahimi M, Czermak P, Czermak P and Czermak P. Membrane Technology for the Recovery of Lignin: A Review. Membranes (Basel). 2016; 6 42, doi: 10.3390/membranes6030042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Restolho JA, Prates A, Norberta de Pinho M and Afonso MD. Sugars and lignosulphonates recovery from eucalyptus spent sulphite liquor by membrane processes. Biomass Bioenergy. 33: 1558–1566 (2009) [Google Scholar]
- 57.Fernández-Rodríguez J, García A, Coz A and Labidi J. Spent sulphite liquor fractionation into lignosulphonates and fermentable sugars by ultrafiltration. Separation Purification Technol 152: 172–179 (2015) [Google Scholar]
- 58.Eriksson P Ultrafiltration for recovery of lignosulfonates from spent sulfite liquor. AIChE Symp Series Vol 76, pp 316–320 (1979) [Google Scholar]
- 59.Claussen PH. Ultrafiltration and hyperfiltration in the pulp and paper industry for by-product recovery and energy savings. in ACS Symp Series Vol 154: Synthetic Membranes: Volume II, pp 361–72. (1981) [Google Scholar]
- 60.Joensson B, Grundberg H and Gustafsson A. Lignosulfonate of a certain quality and method of preparation of lignosulfonate of a certain quality. US Patent 20120267064 (2012)
- 61.Evju H 3-Methoxy-4-hydroxybenzaldehyde. Borregaard Industries Ltd., US Patent 4151207 (1979)
- 62.Guo Z and Olsson L. Characterization and fermentation of side streams from sulfite pulping. Process Biochem 49: 1231–1237 (2014) [Google Scholar]
- 63.Marques AP, Evtuguin DV, Magina S, Amado FML and Prates A. Chemical Composition of Spent Liquors from Acidic Magnesium-Based Sulphite Pulping of Eucalyptus globulus. J Wood Chem Technol. 29: 322–336 (2009) [Google Scholar]
- 64.Pateraki C, Ladakis D, Stragier L, et al. Pretreatment of spent sulphite liquor via ultrafiltration and nanofiltration for bio-based succinic acid production. J Biotechnol 233: 95–105 (2016) [DOI] [PubMed] [Google Scholar]
- 65.Alexandri M, Papapostolou H, Komaitis M, et al. Evaluation of an integrated biorefinery based on fractionation of spent sulphite liquor for the production of an antioxidant-rich extract, lignosulphonates and succinic acid. Bioresource Technology. 214: 504–513 (2016) [DOI] [PubMed] [Google Scholar]
- 66.Lawford HG and Rousseau JD. Production of ethanol from pulp mill hardwood and softwood spent sulfite liquors by genetically engineered E. coli. Appl Biochem Biotechnol 39–40: 667–685 (1993) [DOI] [PubMed] [Google Scholar]
- 67.Harner NK, Wen X, Bajwa PK, et al. Genetic improvement of native xylose-fermenting yeasts for ethanol production. J Ind Microbiol Biotechnol 42: 1–20 (2015) [DOI] [PubMed] [Google Scholar]
- 68.Heikkila H, Nurmi J, Rahkila L and Toyryla M. Xylitol manufacture from xylose-containing liquors with Candida or Debaryomyces. World Patent 09008193 (1990)
- 69.Eroma O-P, Heikkila H, Ojamo H, et al. Process for the simultaneous production of xylitol and ethanol by fermentation of lignocelluloses. US Patent 20020164731 (2002)
- 70.Bertrand JL, Ramsay BA and Chavarie C. Biosynthesis of poly-β-hydroxyalkanoates from pentoses by Pseudomonas pseudoflava. Appl Environ Microbiol 56: 3133–3138 (1990) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rueda C, Calvo PA, Moncalián G, Ruiz G and Coz A. Biorefinery options to valorize the spent liquor from sulfite pulping. J Chem Technol Biotechnol 90: 2218–2226 (2015) [Google Scholar]
- 72.Fatehi P and Chen J. Extraction of Technical Lignins from Pulping Spent Liquors, Challenges and Opportunities In: Fang RLS Z Jr., (ed.). Production of Biofuels and Chemicals from Lignin. Springer, Singapore: pp. 35–54 (2016) [Google Scholar]
- 73.Sumerskii I, Korntner P, Zinovyev G, Rosenau T and Potthast A. Fast track for quantitative isolation of lignosulfonates from spent sulfite liquors. RSC Adv. 5: 92732–9274242 (2015) [Google Scholar]
- 74.Baumberger S, Abaecherli A, Fasching M, et al. Molar mass determination of lignins by size-exclusion chromatography: towards standardisation of the method. Holzforschung. 61: 459–468 (2007) [Google Scholar]
- 75.Ouyang X, Zhang P, Qiu X, Deng Y and Chen P. Lignosulfonate Separation Using Preparative Column Chromatography. Ind Eng Chem Res 50: 10792–10799 (2011) [Google Scholar]
- 76.Ringena O, Saake B and Lehnen R. Characterization of electrolyzed magnesium spent-sulfite liquor. Holzforschung 59: 604–611 (2005) [Google Scholar]
- 77.Duval A, Molina-Boisseau S and Chirat C. Fractionation of lignosulfonates: Comparison of ultrafiltration and ethanol solubility to obtain a set of fractions with distinct properties. Holzforschung 69: 127–134 (2015) [Google Scholar]
- 78.Bhattacharya PK, Todi RK, Tiwari M, Bhattacharjee C, Bhattacharjee S and Datta S. Studies on ultrafiltration of spent sulfite liquor using various membranes for the recovery of lignosulphonates. Desalination 174: 287–297 (2005) [Google Scholar]
- 79.Ahvazi B, Cloutier E, Wojciechowicz O and Ngo T-D. Lignin Profiling: A Guide for Selecting Appropriate Lignins as Precursors in Biomaterials Development. ACS Sustainable Chem Eng. 4: 5090–5105 (2016) [Google Scholar]
- 80.Zhao X, Cheng K and Liu D. Organosolv pretreatment of lignocellulosic biomass for enzymatic hydrolysis. Appl Microbiol Biotechnol 82: 815–827 (2009) [DOI] [PubMed] [Google Scholar]
- 81.Pye EK and Lora JH. The Alcell process. A proven alternative to kraft pulping. Tappi J 74: 113–118 (1991) [Google Scholar]
- 82.Diebold VB, Cowan WF and Walsh JK. Solvent pulping process US Patent 4100016 (1978)
- 83.Lora JH, Katzen R, Cronlund M and Wu CF. Recovery of lignin US Patent 4764596 (1988)
- 84.Schulze P, Seidel-Morgenstern A, Lorenz H, Leschinsky M and Unkelbach G. Advanced process for precipitation of lignin from ethanol organosolv spent liquors. Bioresour Technol 199: 128–134 (2016) [DOI] [PubMed] [Google Scholar]
- 85.Lora JH, Creamer AW, Wu LCF and Goyal GC. Industrial scale production of organosolv lignins: Characteristics and applications In Cellulose: Chemical Biochemical Material Aspects, Ellis Horwood, Chichester, UK, pp. 251–256 (1993) [Google Scholar]
- 86.McDonough TJ. The chemistry of organosolv delignification. Tappi J 76: 186–193 (1993) [Google Scholar]
- 87.Liu Y, Carriero S, Pye K and Argyropoulos DS. A comparison of the structural changes occurring in lignin during Alcell and kraft pulping of hardwoods and softwoods in ACS Symp Ser. Vol 742: Lignin: Historical, Biological, and Materials Perspectives, pp 447–464 (2000) [Google Scholar]
- 88.Lora JH, Winner SR and Goyal GC. Modified organosolv pulping. US Patent 4764596 (1996)
- 89.Hergert HL, Goyal GC and Lora JH. Limiting molecular weight of lignin from autocatalyzed organosolv pulping of hardwoodin ACS Symp Ser. Vol 742: Lignin: Historical, Biological, and Materials Perspectives, pp 265–77 (2000) [Google Scholar]
- 90.Arato C, Pye EK and Gjennestad G. The lignol approach to biorefining of woody biomass to produce ethanol and chemicals. Appl Biochem Biotechnol. 121–124: 871–882 (2005) [DOI] [PubMed] [Google Scholar]
- 91.Lora JH, Wu CF, Pye EK and Balatinecz JJ. Characteristics and potential applications of lignin produced by an organosolv pulping process in ACS Symp Ser 397, Lignin, pp 312–323 (1989) [Google Scholar]
- 92.Gosselink RJA, Abacherli A, Semke H, et al. Analytical protocols for characterisation of sulphur-free lignin. Ind Crops Prod. 19: 271–281 (2004) [Google Scholar]
- 93.Constant S, Wienk HLJ, Frissen AE, et al. New insights into the structure and composition of technical lignins: a comparative characterisation study. Green Chem. 18: 2651–2665 (2016) [Google Scholar]
- 94.Awad OI, Mamat R, Ali OM, et al. Alcohol and ether as alternative fuels in spark ignition engine: A review. Renewable and Sustainable Energy Rev 82: 2586–2605 (2018) [Google Scholar]
- 95.Deeb RA, Chu K-H, Shih T, et al. MTBE and other oxygenates: Environmental sources, analysis, occurrence, and treatment. Environ Eng Sci. 20: 433–447 (2003) [Google Scholar]
- 96.Soccol CR, Vandenberghe LPdS, Medeiros ABP, et al. Bioethanol from lignocelluloses: Status and perspectives in Brazil. Bioresour Technol 101: 4820–4825 (2010) [DOI] [PubMed] [Google Scholar]
- 97.Macedo IdC. Share in the use of fossil energy, in Sugar Cane’s Energy – Twelve studies on Brasilian sugar cane agribusiness and its sustainability.: Berlendis Editores Ltda, UNICA – São Paulo Sugar Cane Agroindustry Union, Sao Paulo, Brazil, pp 48–64 (2007) [Google Scholar]
- 98.Mabee WE and Saddler JN. Bioethanol from lignocellulosics: Status and perspectives in Canada. Bioresour Technol. 101: 4806–4813 (2010) [DOI] [PubMed] [Google Scholar]
- 99.Laboratory NRE. Sustainable Transportation, https://www.nrel.gov/continuum/sustainable_transportation/cellulosic_ethanol.html. Golden Colorado (2013) [Accessed 20 April 2018] [Google Scholar]
- 100.Taha M, Foda M, Shahsavari E, Aburto-Medina A, Adetutu E and Ball A. Commercial feasibility of lignocellulose biodegradation: possibilities and challenges. Current Opinion in Biotechnol 38: 190–197 (2016) [DOI] [PubMed] [Google Scholar]
- 101.Ng RTL and Maravelias CT. Design of Cellulosic Ethanol Supply Chains with Regional Depots. Ind Eng Chem Res 55: 3420–3432 (2016) [Google Scholar]
- 102.Laboratory NRE. Geospatial Science Data and Tools, Biomass Maps. https://www.nrel.gov/gis/biomass.html. National Renewable Energy Laboratory; (2014)[Accessed 20 April 2018] [Google Scholar]
- 103.Cedeño W What happened to ethanol producer prices after passage of the Renewable Fuel Standard? Beyond the Numbers 5: 1–9 published by US Department of Labor; (2016) [Google Scholar]
- 104.Mupondwa E, Li X, Tabil L, Sokhansanj S and Adapa P. Status of Canada’s lignocellulosic ethanol: Part II: Hydrolysis and fermentation technologies. Renewable and Sustainable Energy Rev 79: 1535–1555 (2017) [Google Scholar]
- 105.Mupondwa E, Li X, Tabil L, Sokhansanj S and Adapa P. Status of Canada’s lignocellulosic ethanol: Part I: Pretreatment technologies. Renewable and Sustainable Energy Reviews. 72: 178–190 (2017) [Google Scholar]
- 106.Gnansounou E Production and use of lignocellulosic bioethanol in Europe: Current situation and perspectives. Bioresource Technol 101: 4842–4850 (2010) [DOI] [PubMed] [Google Scholar]
- 107.Fevolden AM and Klitkou A. A fuel too far? Technology, innovation, and transition in failed biofuel development in Norway. Energy Res Social Sci 23: 125–135 (2017) [Google Scholar]
- 108.Gregg J, Bolwig S, Hansen T, et al. Value Chain Structures that Define European Cellulosic Ethanol Production. Sustainability 9: 118, doi 10.3390/su9010118 (2017) [DOI] [Google Scholar]
- 109.Wyman CE. What is (and is not) vital to advancing cellulosic ethanol. Trends Biotechnol 25: 153–157 (2007) [DOI] [PubMed] [Google Scholar]
- 110.Li H and McDonald AG. Fractionation and characterization of industrial lignins. Ind Crops Prod 62: 67–76 (2014) [Google Scholar]
- 111.McCann MC and Carpita NC. Biomass recalcitrance: a multi-scale, multi-factor, and conversion-specific property. J Exp Bot 66: 4109–4118 (2015) [DOI] [PubMed] [Google Scholar]
- 112.Bundhoo ZMA. Microwave-assisted conversion of biomass and waste materials to biofuels. Renewable Sustainable Energy Rev. 82: 1149–1177 (2018) [Google Scholar]
- 113.Jacquet N, Maniet G, Vanderghem C, Delvigne F and Richel A. Application of Steam Explosion as Pretreatment on Lignocellulosic Material: A Review. Ind Eng Chem Res 54: 2593–2598 (2015) [Google Scholar]
- 114.Zhu JY and Pan XJ. Woody biomass pretreatment for cellulosic ethanol production: Technology and energy consumption evaluation. Bioresour Technol 101: 4992–5002 (2010) [DOI] [PubMed] [Google Scholar]
- 115.Zhu JY, Pan XJ, Wang GS and Gleisner R. Sulfite pretreatment (SPORL) for robust enzymatic saccharification of spruce and red pine. Bioresource Technol 100: 2411–2418 (2009) [DOI] [PubMed] [Google Scholar]
- 116.Shuai L, Yang Q, Zhu JY, et al. Comparative study of SPORL and dilute-acid pretreatments of spruce for cellulosic ethanol production. Bioresource Technol 101: 3106–3114 (2010) [DOI] [PubMed] [Google Scholar]
- 117.Banerjee G, Car S, Scott-Craig JS, Borrusch MS, Aslam N and Walton JD. Synthetic enzyme mixtures for biomass deconstruction: production and optimization of a core set. Biotechnol Bioeng 106: 707–720 (2010) [DOI] [PubMed] [Google Scholar]
- 118.Olson DG, McBride JE, Joe Shaw A and Lynd LR. Recent progress in consolidated bioprocessing. Current Opinion in Biotechnol 23: 396–405 (2012) [DOI] [PubMed] [Google Scholar]
- 119.Jiang Y, Xin F, Lu J, et al. State of the art review of biofuels production from lignocellulose by thermophilic bacteria. Bioresour Technol. 245: 1498–1506 (2017) [DOI] [PubMed] [Google Scholar]
- 120.Shaw AJ, Podkaminer KK, Desai SG, et al. Metabolic engineering of a thermophilic bacterium to produce ethanol at high yield. Proc Natl Acad Sci U S A. 105: 13769–13774 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vane LM. A review of pervaporation for product recovery from biomass fermentation processes. J Chem Technol Biotechnol. 80: 603–629 (2005) [Google Scholar]
- 122.Fan S, Xiao Z, Zhang Y, et al. Enhanced ethanol fermentation in a pervaporation membrane bioreactor with the convenient permeate vapor recovery. Bioresour Technol 155: 229–234 (2014) [DOI] [PubMed] [Google Scholar]
- 123.Sun W, Jia W, Xia C, Zhang W and Ren Z. Study of in situ ethanol recovery via vapor permeation from fermentation. J Membr Sci. 530: 192–200 (2017) [Google Scholar]
- 124.Trivedi P, Malina R and Barrett SRH. Environmental and economic tradeoffs of using corn stover for liquid fuels and power production. Energy Environ Sci. 8: 1428–1437 (2015) [Google Scholar]
- 125.Sassner P, Galbe M and Zacchi G. Techno-economic evaluation of bioethanol production from three different lignocellulosic materials. Biomass and Bioenergy 32: 422–430 (2008) [Google Scholar]
- 126.Kazi FK, Fortman JA, Anex RP, et al. Techno-economic comparison of process technologies for biochemical ethanol production from corn stover. Fuel 89: S20–S28 (2010) [Google Scholar]
- 127.Warner e, Schwab a and Bacovsky d. Survey of Non-Starch Alcohol and Renewable Hydrocarbon Biofuels Producers, https://www.nrel.gov/docs/fy17osti/67539.pdf. NREL, Golden, Colorado: (2016) [Accessed 20 April 2018] [Google Scholar]
- 128.Allen J, Soare A, Udupa S, Kozarsky R and Vicari A. Finding Untapped Value: converting lignin to higher value chemicals. Lux Research Inc. (2014) [Google Scholar]
- 129.Humbird D, Davis R, Tao L, et al. Process design and economics for biochemical conversion of lignocellulosic biomass to ethanol https://www.nrel.gov/docs/fy11osti/47764.pdf. NREL, Golden, Colorado, (2011)[Accesssed 20 April 2018] [Google Scholar]
- 130.Narendranath NV, McDonald WF and Bootsma JA. Systems and methods for hydrolysis of biomass. US Patent 20140234911 (2014)
- 131.Ward L 2015 DOE Bioenergy Technologies Office(BETO) IBR Project Peer Review, POET-DSM Project LIBERTY https://www.energy.gov/sites/prod/files/2015/04/f22/demonstration_market_transformation_ward_3433.pdf. Department of Energy; (2015)[Accessed 20 April 2018] [Google Scholar]
- 132.Radigan DS II, Geraets JM, Heikes JR and Pierson RD. Methods of reducing the size of lignocellulosic material, and related systems. World Patent 2016183336 (2016)
- 133.McDonald WF, Urban SS and Martin JL. Systems and methods for acid recycle in pretreatment of lignocellulosic biomass. World Patent 2013006856 (2013)
- 134.Carlson DC. System for the treatment of biomass. US Patent 20130065289 (2013)
- 135.Bootsma J Composition of lignin pellets and system for producing. US Patent 8852301 (2014)
- 136.Rodsrud G, Lersch M and Sjoede A. History and future of world’s most advanced biorefinery in operation. Biomass Bioenergy. 46: 46–59 (2012) [Google Scholar]
- 137.Sjoede A, Froelander A, Lersch M and Roedsrud G. Lignocellulosic biomass conversion. US Patent 20110250638 (2011)
- 138.Borregaard,12 million more for Borregaard research, https://www.borregaard.com/News/12-million-more-for-Borregaard-research (2016) [Accessed 20 April 2018]
- 139.Chatterjee S and Saito T. Lignin-Derived Advanced Carbon Materials. ChemSusChem 8: 3941–3958 (2015) [DOI] [PubMed] [Google Scholar]
- 140.Vishtal A and Kraslawski A. Challenges in industrial applications of technical lignins. BioResources 6: 3547–3568 (2011) [Google Scholar]
- 141.Strassberger Z, Tanase S and Rothenberg G. The pros and cons of lignin valorization in an integrated biorefinery. RSC Adv. 4: 25310–25318 (2014) [Google Scholar]
- 142.Aro T and Fatehi P. Production and Application of Lignosulfonates and Sulfonated Lignin. ChemSusChem 10: 1861–1877 (2017) [DOI] [PubMed] [Google Scholar]
- 143.Kopf PW. Phenolic resins. in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc, pp. 756–802 (2006) [Google Scholar]
- 144.Olivares M, Guzmán JA, Natho A and Saavedra A. Kraft lignin utilization in adhesives. Wood Sci Technol 22: 157–165 (1988) [Google Scholar]
- 145.Lora JH and Glasser WG. Recent Industrial Applications of Lignin: A Sustainable Alternative to Nonrenewable Materials. Journal of Polymers and the Environment 10: 39–48 (2002) [Google Scholar]
- 146.Hu L, Pan H, Zhou Y and Zhang M. Methods to improve lignin’s reactivity as a phenol substitute and as replacement for other phenolic compounds: a brief review. BioResources 6: 3515–3525 (2011) [Google Scholar]
- 147.Ghorbani M, Liebner F, van Herwijnen HWG, et al. Lignin phenol formaldehyde resoles: the impact of lignin type on adhesive properties. BioResources 11: 6727–6741 (2016) [Google Scholar]
- 148.Kalami S, Arefmanesh M, Master E and Nejad M. Replacing 100% of phenol in phenolic adhesive formulations with lignin. J Appl Polym Sci 134: 451–424 (2017) [Google Scholar]
- 149.Cateto CA, Barreiro MF, Rodrigues AE, Brochier-Salon MC, Thielemans W and Belgacem MN. Lignins as macromonomers for polyurethane synthesis: a comparative study on hydroxyl group determination. J Appl Polym Sci 109: 3008–3017 (2008) [Google Scholar]
- 150.Ulrich H Urethane polymers in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc; pp. 454–485 (2007) [Google Scholar]
- 151.Mahmood N, Yuan Z, Schmidt J and Xu C. Depolymerization of lignins and their applications for the preparation of polyols and rigid polyurethane foams: A review. Renewable Sustainable Energy Rev. 60: 317–329 (2016) [Google Scholar]
- 152.Yoshida H, Morck R, Kringstad KP and Hatakeyama H. Kraft lignin in polyurethanes. II. Effects of the molecular weight of kraft lignin on the properties of polyurethanes from a kraft lignin-polyether triol-polymeric MDI system. J Appl Polym Sci 40: 1819–1832 (1990) [Google Scholar]
- 153.Cateto CA, Barreiro MF, Rodrigues AE and Belgacem MN. Optimization Study of Lignin Oxypropylation in View of the Preparation of Polyurethane Rigid Foams. Ind Eng Chem Res 48: 2583–2589 (2009) [Google Scholar]
- 154.Li Y and Ragauskas AJ. Kraft Lignin-Based Rigid Polyurethane Foam. J Wood Chem Technol 32: 210–224 (2012) [Google Scholar]
- 155.Chung H and Washburn NR. Improved Lignin Polyurethane Properties with Lewis Acid Treatment. ACS Appl Mater Interfaces 4: 2840–2846 (2012) [DOI] [PubMed] [Google Scholar]
- 156.Mahmood N, Yuan Z, schmidt J and Xu C. Vaolorization of hydrolysis lignin for polyols and rigid polyurethane foam. J Sci Technol Forest Prod Process 3: 26–31 (2013) [Google Scholar]
- 157.Baker DA and Rials TG. Recent advances in low-cost carbon fiber manufacture from lignin. J Appl Polym Sci. 130: 713–728 (2013) [Google Scholar]
- 158.Frank E, Steudle LM, Ingildeev D, Spoerl JM and Buchmeiser MR. Carbon Fibers: Precursor Systems, Processing, Structure, and Properties. Angew Chem, Int Ed. 53: 5262–5298 (2014) [DOI] [PubMed] [Google Scholar]
- 159.Baker DA, Gallego NC and Baker FS. On characterization and spinning of organic-purified lignin toward manufacture of low-cost carbon fiber. J Appl Polym Sci 124: 227–234 (2012) [Google Scholar]
- 160.Kadla JF, Kubo S, Venditti RA, Gilbert RD, Compere AL and Griffith W. Lignin-based carbon fibers for composite fiber applications. Carbon 40: 2913–2920 (2002) [Google Scholar]
- 161.Zhang M and Ogale AA. Carbon fibers from dry-spinning of acetylated softwood kraft lignin. Carbon 69: 626–629 (2014) [Google Scholar]
- 162.Braun JL, Holtman KM and Kadla JF. Lignin-based carbon fibers: oxidative thermostabilization of kraft lignin. Carbon 43: 385–394 (2005) [Google Scholar]
- 163.Zhang M, Jin J and Ogale AA. Carbon fibers from UV-assisted stabilization of lignin-based precursors. Fibers 3: 184–196 (2015) [Google Scholar]
- 164.Sadeghifar H, Sen S, Patil SV and Argyropoulos DS. Toward Carbon Fibers from Single Component Kraft Lignin Systems: Optimization of Chain Extension Chemistry. ACS Sustainable Chem Eng 4: 5230–5237 (2016) [Google Scholar]
- 165.Matsushita Y Conversion of technical lignins to functional materials with retained polymeric properties. J Wood Sci 61: 230–250 (2015) [Google Scholar]
- 166.Upton BM and Kasko AM. Strategies for the Conversion of Lignin to High-Value Polymeric Materials: Review and Perspective. Chem Rev 116: 2275–2306 (2016) [DOI] [PubMed] [Google Scholar]
- 167. Kawamoto H Lignin pyrolysis reactions. J Wood Sci 63: 117–132 (2017) [Google Scholar]
- 168.Yang H, Yan R, Chen H, Lee DH and Zheng C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 86: 1781–1788 (2007) [Google Scholar]
- 169.Sharma RK, Wooten JB, Baliga VL, Lin X, Geoffrey Chan W and Hajaligol MR. Characterization of chars from pyrolysis of lignin. Fuel 83: 1469–1482 (2004) [Google Scholar]
- 170.De Wild PJ, Huijgen WJJ and Gosselink RJA. Lignin pyrolysis for profitable lignocellulosic biorefineries. Biofuels, Bioprod Biorefin 8: 645–657 (2014) [Google Scholar]
- 171.Kozliak EI, Kubátová A, Artemyeva AA, et al. Thermal Liquefaction of Lignin to Aromatics: Efficiency, Selectivity, and Product Analysis. ACS Sustainable Chem Eng 4: 5106–5122 (2016) [Google Scholar]
- 172.Maria M, Peter N, Falk H and Uwe S. Subcritical Water as Reaction Environment: Fundamentals of Hydrothermal Biomass Transformation. ChemSusChem 4: 566–579 (2011) [DOI] [PubMed] [Google Scholar]
- 173.Fang Z, Sato T, Smith RL, Inomata H, Arai K and Kozinski JA. Reaction chemistry and phase behavior of lignin in high-temperature and supercritical water. Bioresource Technol 99: 3424–3430 (2008) [DOI] [PubMed] [Google Scholar]
- 174.Lui Matthew Y, Chan B, Yuen Alexander KL, Masters Anthony F, Montoya A and Maschmeyer T. Unravelling Some of the Key Transformations in the Hydrothermal Liquefaction of Lignin. ChemSusChem 10: 2140–2144 (2017) [DOI] [PubMed] [Google Scholar]
- 175.Saisu M, Sato T, Watanabe M, Adschiri T and Arai K. Conversion of Lignin with Supercritical Water−Phenol Mixtures. Energy & Fuels 17: 922–928 (2003) [Google Scholar]
- 176.Islam MN, Taki G, Rana M and Park J-H. Yield of Phenolic Monomers from Lignin Hydrothermolysis in Subcritical Water System. Ind Eng Chem Res 57: 4779–4784 (2018) [Google Scholar]
- 177.Nguyen TDH, Maschietti M, Åmand L-E, et al. The effect of temperature on the catalytic conversion of Kraft lignin using near-critical water. Bioresource Technol 170: 196–203 (2014) [DOI] [PubMed] [Google Scholar]
- 178.Arturi KR, Strandgaard M, Nielsen RP, Søgaard EG and Maschietti M. Hydrothermal liquefaction of lignin in near-critical water in a new batch reactor: Influence of phenol and temperature. The Journal of Supercritical Fluids. 123: 28–39 (2017) [Google Scholar]
- 179.Yan L, Cui Y, Gou G, et al. Liquefaction of lignin in hot-compressed water to phenolic feedstock for the synthesis of phenol-formaldehyde resins. Composites Part B: Engineering 112: 8–14 (2017) [Google Scholar]
- 180.Santos RB, Hart PW, Jameel H and Chang H-M. Wood based lignin reactions important to the biorefinery and pulp and paper industries. BioResources 8: 1456–1477 (2013) [Google Scholar]
- 181.Gratzl JS and Chen C-L. Chemistry of pulping: lignin reactions ACS Symp Ser. Vol 742: Lignin: Historical, Biological, and Materials Perspectives pp392–421 (2000) [Google Scholar]
- 182.Lange H, Decina S and Crestini C. Oxidative upgrade of lignin - Recent routes reviewed. Eur Polym J. 49: 1151–1173 (2013) [Google Scholar]
- 183.Zaheer M and Kempe R. Catalytic hydrogenolysis of aryl ethers: A key step in lignin valorization to valuable chemicals. ACS Catal 5: 1675–1684 (2015) [Google Scholar]
- 184.Anderson EM, Katahira R, Reed M, et al. Reductive Catalytic Fractionation of Corn Stover Lignin. ACS Sustainable Chem Eng 4: 6940–6950 (2016) [Google Scholar]
- 185.Narani A, Chowdari RK, Cannilla C, et al. Efficient catalytic hydrotreatment of Kraft lignin to alkylphenolics using supported NiW and NiMo catalysts in supercritical methanol. Green Chem. 17: 5046–5057 (2015) [Google Scholar]
- 186.Kasakov S, Shi H, Camaioni DM, et al. Reductive deconstruction of organosolv lignin catalyzed by zeolite supported nickel nanoparticles. Green Chem 17: 5079–5090 (2015) [Google Scholar]
- 187.Shuai L, Amiri MT, Questell-Santiago YM, et al. Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization. Science 354: 329–333 (2016) [DOI] [PubMed] [Google Scholar]
- 188.Linger JG, Vardon DR, Guarnieri MT, et al. Lignin valorization through integrated biological funneling and chemical catalysis. Proc Natl Acad Sci USA 111: 12013–12018 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Xie S, Sun Q, Pu Y, et al. Advanced Chemical Design for Efficient Lignin Bioconversion. ACS Sustainable Chem Eng 5: 2215–2223 (2017) [Google Scholar]
- 190.Fache M, Boutevin B and Caillol S. Vanillin Production from Lignin and Its Use as a Renewable Chemical. ACS Sustainable Chem Eng 4: 35–46 (2016) [Google Scholar]
- 191.Leffingwell J and Leffingwell D. Flavours & fragrances: Recent advances in biotechnology. Speciality Chem Mag 32–34 (2015) [Google Scholar]