

Abstract
To more accurately study the complex mechanisms behind cancer invasion, progression, and response to treatment, researchers require models that replicate both the multicellular nature and 3D stromal environment present in an in vivo tumor. Multicellular aggregates (i.e., spheroids) embedded in an extracellular matrix mimic are a prevalent model. Recently, quantitative metrics that fully utilize the capability of spheroids are described along with conventional experiments, such as invasion into a matrix, to provide additional details and insights into the underlying cancer biology. The review begins with a discussion of the salient features of the tumor microenvironment, introduces the early work on non-embedded spheroids as tumor models, and then concentrates on the successes achieved with the study of embedded spheroids. Examples of studies include cell movement, drug response, tumor cellular heterogeneity, stromal effects, and cancer progression. Additionally, new methodologies and those borrowed from other research fields (e.g., vascularization and tissue engineering) are highlighted that expand the capability of spheroids to aid future users in designing their cancer-related experiments. The convergence of spheroid research among the various fields catalyzes new applications and leads to a natural synergy. Finally, the review concludes with a reflection and future perspectives for cancer spheroid research.
Keywords: spheroids, cancer, multicellular, aggregates, cell clusters, tumoroids
The table of contents entry
Embedded spheroid models recreate the complex interactions between cells and extracellular matrix and cell types within the contexts of secondary cell types and tumor heterogeneity in cancer. The combination of natural matrices with spheroids lends itself to studies of cancer invasion, progression, and response to treatment.
1. Tumor Microenvironment and Why the Monolayer Does Not Model It
Tumors do not exist as a homogeneous population of malignant cells. Rather a tumor is characterized by changes in the microenvironment resulting from the interplay between a heterogeneous population of malignant cells and their assorted support of various tumor-associated cells that include macrophages, fibroblasts, pericytes, endothelial and immune cells. However, monolayers of artificially polarized cells spread on treated 2-dimensional (2D) polystyrene are routinely used as our first round of testing for research in cancer biology, cancer immunology, drug delivery, and novel drug discovery.
Culturing cells in a 2D monolayer produces a number of downstream effects that differentiate it from cells present in a 3D tumor microenvironment in an in vivo setting. Notably unlike the rounded morphology present in 3D systems, the planar attachment constrains the cell geometry resulting in extreme spreading [1]. The effect of this artificial geometry is a loss of polarity normally present in epithelial cells, for example [2]. This results in cells that are different on a genetic [3] and proteomic [4] level. A review of genetic differences in cells cultured in 2D versus 3D found upregulation in three categories: 1) cell cycling, 2) metabolism, and 3) turnover of macromolecules, thus enabling enhanced proliferation [2]. For applications in drug discovery and delivery, the change from 2D to 3D dramatically increases the robustness of a cell to toxicity in both normal and cancerous contexts, underscoring the need for 3D models [5, 6].
In addition, there are both cellular and acellular elements within the tumor stroma which dynamically interact with the malignant cells. Furthermore, contributions from both stromal and malignant cells can promote cancer initiation, growth, and progression [7]. For example, stroma characterized by chronic inflammation is now recognized as promoting cancer [8]. Conversely, a normalized stroma can revert a malignant phenotype in a 3D system, introducing the concept that treating the microenvironment may reverse malignancy [9, 10]. The progression of a polarized epithelium to a carcinoma can be documented through changes in the surrounding stroma such as stroma activation, degradation of extracellular matrix (ECM) components, increase in immune components, and breakdown of the basement membrane [7]. Therefore, an ideal cancer model must include stromal components.
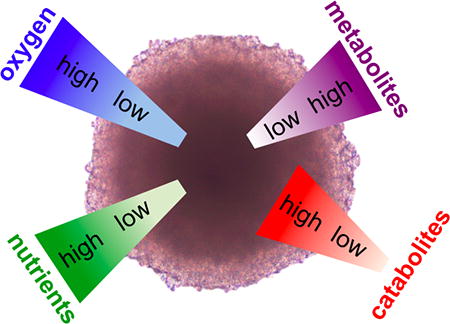
The last factor that differentiates cells in a monolayer from a 3D tumor is the tumor macrostructure and the microenvironment that it supports (Figure 1). An avascular tumor or small micrometastasis under approximately 2 mm3 is characterized by gradients of metabolites, catabolites, and oxygenation, with proliferation at the edges, and necrosis at the core [11]. The macrostructure dictates a microenvironment that differs from normal tissue in terms of oxygenation, perfusion, pH, and metabolic states [12]. After reaching a size of greater than 2 mm3, diffusion supplies insufficient oxygen for the tumor, affording a hypoxic state [13]. Linked to hypoxia, the extracellular pH of a tumor is often lower (6–7) than normal tissue (7.4) due to the use of glycolysis as an energy source for hypoxic cells [14]. Hypoxia triggers upregulation of HIF (hypoxia inducible factor), which initiates the release of pro-angiogenic factors that stimulate angiogenesis [15]. However, the improper development of a mature vasculature system, translates to hypoxic regions even in vascularized tumors. The result is a heterogeneous range of metabolic states where actively cycling cells are adjacent to capillaries, and more distant cells become quiescent and possibly necrotic or apoptotic.
Figure 1. Tumor Microenvironment is Modeled in Spheroids.
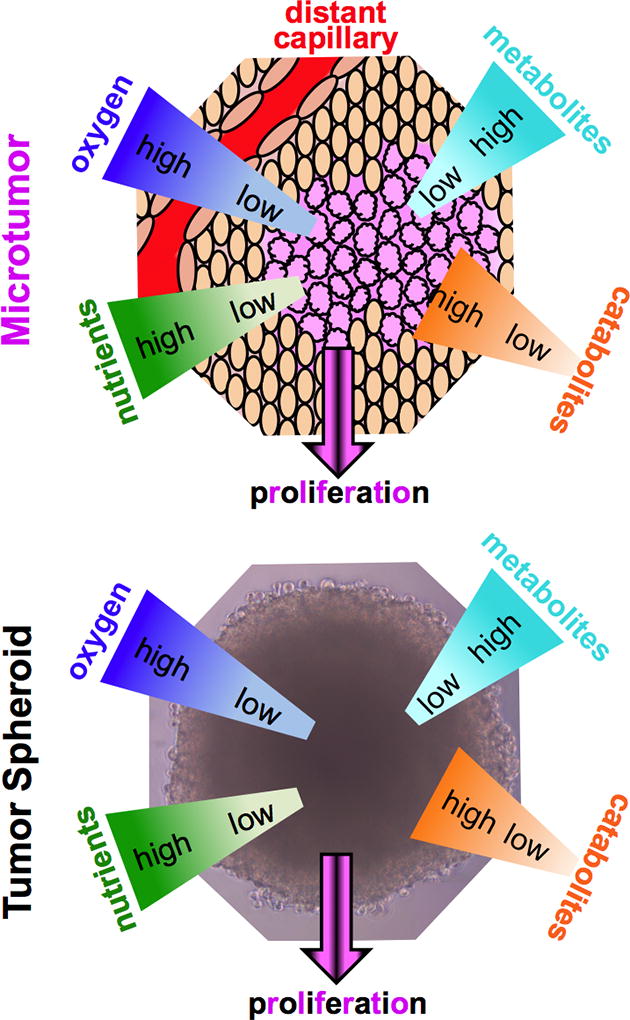
The growth of malignant cells within a tumor leads to a distinct microenvironment that is characterized by gradients of metabolites, oxygen, and nutrients. The multicellular spheroids recapitulate these aspects of tumor growth.
As the scientific community gains insights into the fundamental differences distinguishing a tumor from normal tissue, exciting targets for novel therapeutics are being identified. Prior reviews have highlighted treatments aimed at tumor microenvironment [12], cellular stromal components [16] including tumor associated macrophages (TAMS) [17] and cancer associated fibroblasts (CAFs) [18] in non-spheroid formats. This review begins by discussing non-embedded spheroids and a few representative successes, followed by the advantages of embedded spheroids as cancer models and the contributions to several areas of cancer research including cancer biology, immunology, drug screening, and drug delivery. Next, the utility of analytical methods used on embedded spheroids in other fields is discussed as these methods can enhance ongoing cancer research. Finally, the review concludes with remarks on the field, perspectives on how to best use embedded cancer spheroid models to address both basic and clinical challenges, and opportunities for future research.
2. The Non-embedded Spheroid as a Tumor Model
In 1971 Sutherland et al., first reported spheroids as a model of nodular carcinoma. Spheroids form when cells are denied external attachment sites besides other cells in culture, as shown in Figure 2 [19]. The culture of Chinese hamster V79 lung cells in a rotator flask for 24 days results in spheroids of 150–370 µm in diameter. Upon reaching a diameter of 250 µm, spheroids develop three zones: 1) the outer zone is typically 75 µm in thickness and is composed of rapidly dividing cells in the periphery; 2) an intermediate zone of slower division; and 3) a necrotic zone in the center, which resembles the macrostructure of some human and animal carcinomas (Figure 1). In addition to rotator/spinner flasks, multiple fabrication strategies are routinely used, all of which are based on denying cells attachment sites. The hanging drop method, which was first presented in 1907 in the field of nerve regeneration, consists of inverting a droplet containing cells thus preventing cells from adhering to the culture plate [20]. This technique is prevalent and is commercially available as a specialized plate, to increase the ease of use. Another popular method is the liquid overlay technique where a non-adherent substrate such as an agarose/agar or poly-HEMA coated well induces aggregation [21]. The inclusion of ECM supplements increases the applicable cell types and regularity of spheroid formation [22], while micropatterned substrates increase the control over size and complex shape formation [23–27]. The hepatic field developed tethered spheroids to maintain the hepatic function of primary hepatocytes[28]. Surfaces are treated with galactose and small amounts of RGD peptide, so that the non-binding effect of the galactose dominates, inducing spheroid formation, while the minor contributions of the RGD are sufficient to tether the spheroids to the surface[28, 29]. Lastly, an alternate approach to multicellular aggregates includes the use of microfluidics to form complex 3D structures [30, 31]. Despite the lack of an ECM mimic, spheroids are a robust, scalable model widely used in research. Such non-embedded spheroid models, particularly with mesothelioma, have demonstrated greater resistance to chemotherapeutic agents and are valuable in the investigation of cytotoxic mechanisms, variable survival gene expression profiles, and variations in drug efficacy as a function of delivery methods [26–27].
Figure 2. Spheroids as Model Tumors.
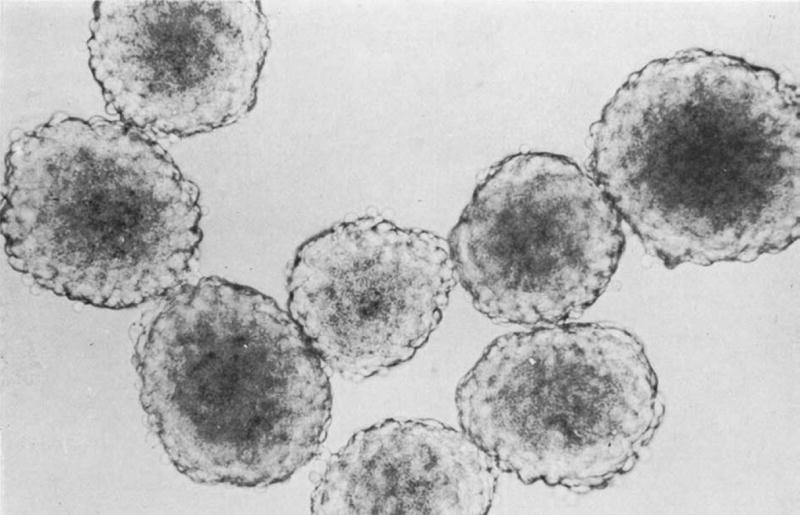
Sutherland first developed spheroids in the 1970s using the rotator flask method. The spheroids imaged are derived from Chinese hamster lung imaged at 120x [19]. Reprinted from reference 19 with permission from Oxford University Press.
3. Translatable Improvements to Non-embedded Spheroid Models
As the predecessor to the embedded spheroid, the non-embedded spheroid is a model in its own right and this model has undergone a number of iterations and advances that increase physiological relevance, ease of use, and serve to inform the embedded spheroid model as well. Therefore improvements in analysis and preparation of non-embedded spheroids will be highlighted in the following section.
3.1. Incorporation of Multiple Cell Types
Within the tumor milieu, there are a number of non-malignant cell types affecting cancer invasion, growth, and metastasis. These stromal cell types are often of significant interest for both understanding cancer biology, and as a target for chemotherapeutics. Ksiazkiewicz et al., use spheroids to separately model the infiltration of tumor associated macrophages (TAMs) into the fibroblast-rich stroma versus a malignant breast carcinoma [32]. Spheroids composed of either immortalized breast cancer cells or breast tumor-derived fibroblasts model the tumor and stroma, respectively. Similarly, multiple iterations of a monocyte or TAM-infiltrated cancer spheroid provide insights into TAM/cancer cell interactions [33, 34]. Hauptman et al., describe the complexity of tumor/macrophage interactions by showcasing the complex effects of different human macrophage phenotypes on colon cancer-derived spheroid migration and proliferation [34]. Using a similar model, Choie et al., report macrophages loaded with gold nanoshells infiltrating a spheroid as a novel cell-based drug delivery system. While monitoring the macrophage invasion into a human breast cancer spheroid as a function of time, the sample is subsequently irradiated at 754 nm leading to macrophage and possibly tumor cell death via photothermal ablation [33]. Pedrersen et al., recapitulate and study the microenvironment of the invading tumor in highly invasive glioblastoma by exposing invasive cells to mature brain aggregates [35, 36]. Specifically, cells from a glioblastoma cell spheroid, cultured next to a spheroid of mature brain aggregates, invade the mature tissue as observed via selective staining [37].
An alternate route to incorporating stromal cells within a spheroid uses two (or more) cell types in the initial suspension prior to a standard spheroid preparation technique such as liquid overlay. For example, a heterospheroid composed of glioblastoma and endothelial cells form a ring of endothelial cells surrounding the center mass of glioblastoma cells [38]. Using this model Ho et al., report selective targeting of endothelial cells by tumstatin-iron oxide nanoparticles. Steven et al., describe an inVERT method that enables preparation of complicated microtissues including different type of heterospheroid architectures [39]. For studying hepatic function, they prepare a heterospheroid of primary hepatocytes and endothelial cells as well as a two-component spheroid where a layer of endothelial cells surrounds a core of hepatocyte cells. These approaches are not limited to stromal cells, but can also be used to model the heterogeneous population, and thus drug responsiveness, invasiveness, and metastatic ability of cancer cells. These multicellular structures highlight the use of multiple cell types with controlled macrostructures that have not been taken advantage of, to the same extent, in embedded cancer models.
3.2. Spheroids Interacting with a 2D System
In order to investigate cell/ECM interactions without using an embedded system, spheroids are often placed upon an ECM coated surface. Beaune et al., describe lymph node cancer prostrate spheroids cultured on fibronectin coated glass to study mechanisms of cell escape from the aggregates with strong cell-cell adhesions [40]. Upon attempted cell escape, membrane tubes form that tether the escaping cell to the spheroid. Membrane rupture or relaxation results in successful cell escape or return to the aggregate, respectively. Spheroids derived from the ascites of a patient with ovarian carcinoma show varying degrees of adhesion on thin layers of either laminin, fibronectin, collagen I, collagen IV or mesothelial cell monolayers as described by Burleson et al., [41]. Spheroids adhere to fibronectin and type I collagen more strongly than laminin and collagen IV. The β1 integrin partially mediates this adherence to both ECM proteins and mesothelial monolayers. These results suggest a mechanism of ovarian cancer dissemination via movement of cells within ascites, consistent with clinical observations.
4. Embedded Spheroid Models
Transitioning the model to a fully embedded system offers further opportunities to mimic a tumor environment, thus facilitating physiological cellular interactions, metabolism, growth, and invasion. In this section, we highlight both novel systems and methods of analysis, with a particular focus on the use of spheroids in the study of cancer. The different models of embedded spheroids are summarized in Figure 3. Specifically, the embedding environments of agarose, matrigel, fibrin, in vivo tissue, synthetic polymers, and the most popular - collagen are discussed.
Figure 3. Spheroid-based Models.
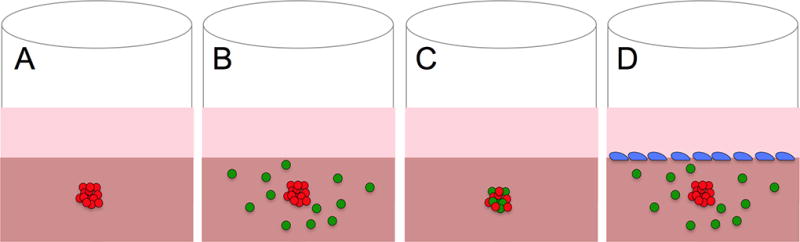
The embedded spheroid provides the opportunity to incorporate multiple cell types either in the collagen matrix (B), within the spheroid (C), or on top of the collagen (D). This modularity has resulted in a number of complex, application-specific models.
4.1. Spheroids Embedded in Agarose
Agarose is traditionally used as a non-adherent material in the liquid overlay spheroid method, however the same non-adhesive property can be useful for embedding. The easily controlled mechanical properties and resistance to cell digestion are ideal characteristics for allowing spheroid growth in 3D without the opportunity for matrix remodeling. For example, human colon adenocarcinoma spheroids, embedded in agarose gels of varying mechanical properties, serve as the experimental basis for modeling the changes in free energy based on the mechanical properties of the agarose gel, glucose consumption, and spheroid growth that results from the biological, chemical and mechanical processes of tumor growth [42]. The agarose-constrained growth of murine carcinoma spheroids enables studying the effects of embedding medium mechanical properties on spheroid proliferation [43]. As the spheroid grows, strain maps of the agarose gel (collected via monitoring the changes in position of embedded fluorescent beads) reveal insight into stress distribution during spheroid growth. Areas of higher compressive stress correlate with decreased proliferation and increased apoptosis, which can be inhibited by elevated BcL-2 activity. These results provide a mechanism of cancer cell escape from a spheroid via areas of lower compressive stress. Although agarose provides a biocompatible matrix, the inability of the cells to remodel it, prevents it from mimicking key features of the cancer microenvironment.
4.2. Spheroids Implanted In Vivo
Although spheroids are often used to mimic the complexities of an in vivo environment while remaining in vitro, they are also used in conjunction with animal models to characterize tumor/stroma interaction. The resistance of agarose to adhesion and remodeling lends itself to controlling spheroid/animal interactions after in vivo implantation. For example, macrophages infiltrating a human breast cancer spheroid produce more VEGF after implantation into a murine model and stimulate greater angiogenesis when compared to spheroids lacking macrophages [44]. In this model, implantation in a dorsal skinfold chamber allows for the constant monitoring of blood vessel growth. The agarose coating around this spheroid prevents autologous macrophage infiltration. Similarly, agarose-coated adipose-derived mesenchymal stem cell spheroids, when placed in a macroencapsulation device, promote angiogenesis and decrease foreign body response [45]. In contrast, human glioblastoma spheroids require embedding directly within murine models to promote invasion into a native tissue [46]. In this study, positron emission topography with 11C-methyl-L-methionine (C-MET) provides vasculature remodeling and proliferation data. However, C-MET remains a limited choice for an invasive, non-angiogenic phenotype.
4.3. Spheroids Embedded in Matrigel
To better replicate in vitro the milieu of ECM proteins present within a tumor, matrigel (i.e, reconstituted basement membrane) is used as both a matrix and additive to aid in spheroid formation. While some cell lines form dense macrocellular structures, others form loose aggregates poorly suited to modeling a tumor. To address this challenge, Ivascu et al., include small amounts of matrigel (2.5 % v/v) in the liquid overlay technique to extend the cell lines suitable for spheroid culture to include MDA-MB 231, MCF7-ADR, and MDA-MB-361 [22]. Although matrigel is not a prevalent choice for embedding, perhaps due to its poor mechanical properties when compared to tissue or other embedding materials, its composition and resultant biological properties promote cell differentiation. In spheroid research, the primary use of matrigel is to aid spheroid formation, although for developmental biology it is widely used as a differentiation-inducing matrix. For example, human pancreatic ductal adenocarcinoma cells form hollow spheroids in vitro when cultured as non-embedded spheroids, but when implanted into matrigel, they form ductal structures which mimic the aggregated structure of pancreatic epithelial cells found in vivo [47]. Spheroids embedded in matrigel are also useful for applications outside the cancer field. The differentiation of spheroids derived from human embryonic stem cells termed embryoid bodies, also benefit from long-term culture in matrigel with an increase in differentiated primate trophoblast markers, (HCG, progesterone, and estradiol-17β) after approximately 20 days in 3D culture with sustained elevation for over 30 days[48]. In contrast, embryoid bodies grown in suspension only demonstrate short-term elevation of these differentiation markers.
4.4. Spheroids Embedded in Collagen for Cancer Research
Collagen is the most prevalent embedding material as it is both a component of the ECM, and is upregulated within a tumor microenvironment. Use is further facilitated due to facile control of mechanical strength, pore size, and fibril fraction by varying the concentration and preparation methods [49]. Typically, spheroids are prepared utilizing one of the earlier methods described (hanging drop, liquid overlay, etc), and transferred into a collagen gel. Tamaki et al., reported the first use of this technique for modeling tumors in 1997 [50]. Analogous non-cancer models for liver [51] and differentiation [52] were reported even earlier.
While embedding increases the utility of the model, it is time intensive, creating a bottleneck for high throughput spheroid formation. One strategy to overcome this limitation is to automate the procedure by microinjecting a mixture of cells and the polymer polyvinylpyrrolidone in collagen gels [53]. The polymer immobilizes the cells at the injection site reducing time for spheroid formation. The spheroids are comparable to those produced by the hanging-drop method. This system is amenable to a variety of cell types (e.g., 4T1, GEβ1, and MDA-MB-231) and includes cells lines that usually require additives (e.g., matrigel) to attain spheroid formation. An alternate strategy consists of labeling cells with magnetic cationic liposomes and directing their growth in a 3D collagen gel with a magnetized iron pin holder array [54]. The center-to-center spacing of the aggregates is 250 µm, limiting the spheroid size (their aggregates had an average of 25 cells) and assay length. A similar array of spheroids uses a PDMS mold to first coat the top of glioma spheroids with collagen, before inverting the mold to embed the basal portion [55]. The PDMS mold offers both control of spheroid size and complex patterns. While these approaches successfully adapt embedding to a high throughput assay, they are limited by their reliance on imaging as the basis for subsequent analysis. Furthermore, arrays impose spatial limits on the ability of the spheroids to grow without spheroid-spheroid interactions that might preclude long-term experiments or migratory cells. These methodologies along with future high throughput approaches will facilitate the study of cancer biology, drug response, differentiation and angiogenesis, and when combined with quantitative methods will provide additional insight. The forthcoming discussion focuses on quantitative methods and the subsequent contributions to cancer-related fields.
4.4.1. Cancer Stroma Interactions
The utility of embedded spheroid models in cancer biology lies in the modularity of the system to evaluate a given element of the microenvironment recreated. For example, the inclusion of fluorescent microbeads for micro-rheology lends itself to both classical rheological data, akin to bulk measurements, and localized matrix properties correlated to invasion of a spheroid [56]. The use of in situ multi-photon imaging of collagen embedded glioma spheroids affords high resolution images of cells during migration [57]. For example, Hwang et al., use GFP-actin, mitochondrial dye, and ratiometric redox imaging of reduced NADH and oxidized FAD to show the creation of pools of actin during migration in 3D. They did acknowledge that the long exposures (of 15 minutes) disrupted 40% of observed migrating cells. Similarly, a quenched-fluorescent IV collagen system allows visualization of the degradation of extracellular proteins in the basement membrane [58]. The presence of fluorescent degradation products indicates the ability of spheroid cells (both migrating and non-migrating populations) to disrupt the basement membrane. Such protein imaging techniques offer new insights into how cells remodel and move within a protein matrix.
The incorporation of phenotypically different cancer lines within a spheroid offers insights into how a heterogeneous tumor grows and invades within a 3D matrix. Carey et al., use confocal reflectance to show matrix remodeling in a mixture of invasive (mesenchymal) and non-invasive (epithelial) breast cancer heterospheroids [59]. Aggressive cells (mesenchymal) remodel the collagen fibers in a radial pattern and create cell scale micro tracks to allow the non-proteolytic invasion of the non-invasive (epithelial) line. Inhibitors of matrix metalloproteases and Rho-associated protein kinase (ROCK) based contractility prevent non-proteolytic invasion of epithelial cells in all matrices, and mesenchymal cell invasion in lower (but not higher) concentration collagen matrices. A similar heterospheroid system shows that the more aggressive cell line initially localizes to the spheroid periphery; however, after extended culture to four days, the less aggressive line becomes the major cell line of the spheroid [60].
The inclusion of multiple cell types is not limited to tumorigenic cells, but extends to secondary cell types as well, typically CAFs and TAMs. There are a few reports of stromal spheroid models that do not include malignant cells such as collagen embedded CAF spheroids [61] or non-embedded TAM-invaded CAF spheroids [32]. However the most prevalent model iteration combines malignant cells with its analogous stromal cells. Mora et al., describe the role of agents aimed at stromal cells to inhibit invasion in a 2:1 glioma and microglia (brain-specific TAM) heterospheroid [62]. Microglia stimulated with lipopolysaccharide and IFN-γ decrease the glioma-led collagen invasion, as it skews the microglia towards an anti-tumor activation pathway. However, the decrease in invasion is not observed with unstimulated microglia or single cell type spheroids regardless of stimulants. A similar study, which used heterospheroids composed of 1:1 primary murine microglia: murine glioblastoma cells, reports that treatment of the microglia with lipopolysaccharide and IFN-γ decreases the overall invasion [63]. A heterospheroid system composed of CAFs and hepatic cancer cells (1:1) documents the effect of stromal fibroblasts on doxorubicin response, showing that heterospheroids are more resistant to treatment than single cell type spheroids or monolayer systems, as measured by the Alamar blue assay [64].
The heterospheroid approach is not the only strategy to incorporate multiple cell types (Figure 3). One limitation of using heterospheroids is that the contributions of indirect versus direct cell-cell contact are not easily separated. The previously mentioned magnetic system, developed by Okachi et al., showcases these differences by including fibroblasts in direct contact with cancer cells in a heterospheroid or indirect contact via fibroblasts homogeneously distributed around the cancer cell spheroid as shown in Figure 3B [65]. Invasion length and area significantly increase in the direct contact cell-cell model, highlighting the need for models that portray such subtle differences. Tevis et al., also report a heterospheroid and a spheroid/diffuse model with macrophages (RAW 264.7) and breast cancer cells (MDA-MB-231). These co-culture model systems contain macrophages dispersed throughout the surrounding collagen gel containing a breast cancer spheroid (Figure 3B) or macrophages as a component of the heterospheroid (Figure 3C) [66]. The macrostructure and interactions imparted by the heterospheroid model are sufficient to encourage EGF-based interactions between the two cell types, increase system metabolism, and promote the cytokine-based TAM phenotype in the macrophages contained within the heterospheroid (Figure 4). The TAM phenotype does not develop when macrophages are diffusely seeded in the collagen matrix surrounding the spheroid. The dissimilarity in cell behavior and phenotype, afforded by the different incorporation methods, offers a useful approach for studying the interactions and progression of the stromal cells that support cancer growth.
Figure 4.
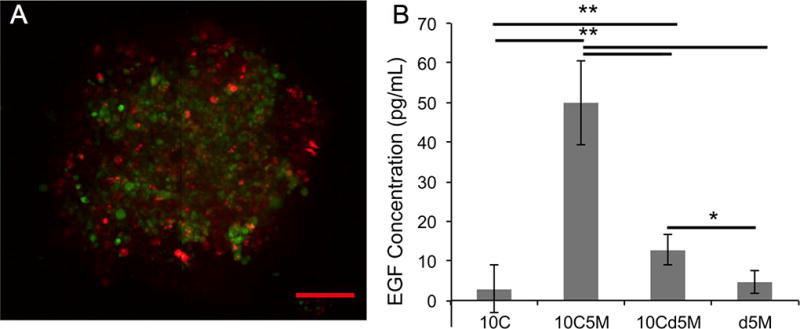
(A) An overlaid optical image of a spheroid with stained MDA-MB-231 cancer cells (green) and RAW 264.7 macrophages (red) shows the two cells types throughout the spheroid. The scale bar is 100 µm for the image. (B) Cytokine based interaction of macrophages and tumor cells as monitored by EGF concentration detected via an ELISA from the four models (10,000 cancer cells (10C); a heterospheroid with 10,000 cancer cells and 5,000 macrophages (10C5M); a 10,000 cancer cell spheroid surrounded by 5,000 macrophages (10Cd5M); n=4; (** p<0.005, *p<0.05). Reprinted from reference 64 with permission from Elsevier.
Systems containing tumor support cells (TAM, CAF, etc) are of significant utility to the development of novel immunotherapies or chemotherapeutics targeted against elements of the tumor stroma. Conversely, models incorporating non-malignant cells allow the demonstration of therapeutic specificity in affecting malignant cells, but leaving non-malignant cells unperturbed. Vorsman et al., describe a multi-cell skin model of melanoma consisting of a top layer of keratinocytes, with a collagen embedded melanoma spheroid surrounded by diffusely seeded primary fibroblasts (Figure 3D) [67]. Treatments with TRAIL and either cisplatin or UV-B as a sensitizer reveal different effects in the 3D model versus single cell 2D culture. Cisplatin and UV-B perform similarly in both models with long duration (10 day) exposures, but the 3D model shows greater potency of cisplatin as a TRAIL sensitizer in the short-term (4 day) treatment experiments. This is evident by reduction in spheroid size and number, which is not observed in 2D culture. In both the TRAIL/cisplatin and TRAIL/UV-B treatment groups, selective induction of apoptosis in the melanoma cells occurs with keratinoctyes while the fibroblasts are largely unaffected in this model. Results such as this support the need for complex 3D models, since 2D models are not (always) faithful predictors of 3D results.
4.4.2. Cancer invasion
The embedded spheroid model is often referred to as the spheroid invasion assay highlighting the utility in studying mechanisms of cancer cell invasion into the ECM. The earliest embedded spheroid research monitored the invasion of glioma cells into collagen I via imaging [50]. The authors quantified the presence and activity of metalloproteases for collagen IV and I within the media, and the subsequent decrease in invasion after MMP inhibitor treatment. The authors propose that the mechanism of invasion occurs through degradation of collagen fibers, which is supported by confocal reflectance images of the ECM [59]. Imaging remains the most prevalent spheroid characterization method to assess invasion distance, area, or individual invading cells as evident from studies on glioma [55, 68–70] and breast [53, 71–74] spheroids. Subsequent modifications that increase the utility of imaging involve the use of other imaging modalities, such as single cell tracking using MRI on iron oxide labeled glioma spheroids [75]. Others improvements focus on removing human bias and increasing analysis throughput by developing an algorithm to map cell densities [63, 76]. Although prevalent and useful, imaging alone does not fully characterize an embedded spheroid.
Embedded spheroids possess two populations, a migratory or invasive population and a non-migratory core. This duality in cell behavior is readily apparent in glioma [63, 77, 78], breast [66], and melanoma [67] spheroids (Figure 5). This is a consequence of the two distinct microenvironments present. Cell-matrix interactions dominate the invasive population and cell-cell contacts dominate the non-migratory core. In order to accurately assess the invasive capabilities of the migratory versus core population, Gole et al., mechanically separate the two populations for characterization [77]. Cathepsin B activity increases in the invading population despite no upregulation at the mRNA (via rt-PCR) or proteomic level (measured in cell extracts by ELISA). Treatment with a chemical inhibitor or siRNA treatment decreases invasion consistent with the role of Cathepsin B. Besides mechanical separation techniques, the invasive or periphery population can also be recovered with collagenase treatment while trypsin treatment provides the core cell population. Reynolds et al., use this approach to demonstrate the susceptibility of the invading population to chemotherapeutics and the increase of cancer stem cells (CSCs) within the core cell population [79]. Such research highlights the ability to separate populations within the model to further elucidate how the microenvironment impacts cell behavior.
Figure 5. Two Distinct Populations in Embedded Spheroids.
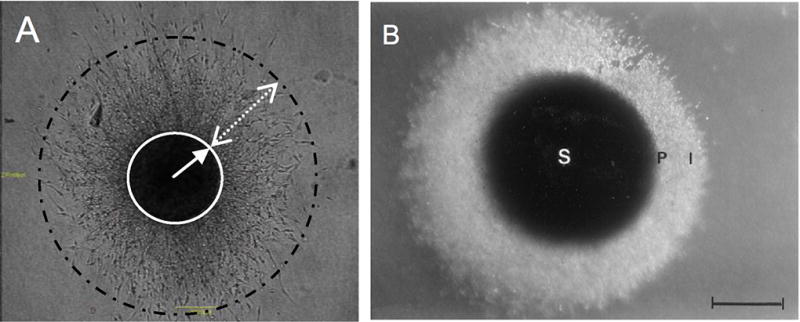
Multiple researchers have observed the presence of two distinct subpopulations within an embedded spheroid. Shown here in a glioma embedded spheroid model, there is a dense core (identified by a circle on the left and an S on the right) and a migrating population (shown with a dashed circle on the left and an I on the right [78, 98]. Reprinted from reference 76 with permission from the JNS Publishing Group. Reprinted from reference 96 with permission from Elsevier
The utility of embedded spheroids as an invasion model extends to complex pathway analysis. Imaging often in conjunction with staining of a spheroid is usually a small part of a larger study detailing the effects of a pathway inhibitor or knockdown on both invasion and viability. In this regard, researchers are able to identify and validate the efficacy of inhibitors with several notable examples reported in the literature including: 1) the SRC pathway in pre-metastatic but not post-metastatic melanoma spheroids [80]; 2) the effect of the MEK pathway on STAT3, in human melanoma spheroids [81]; 3) Rnd3 implication in melanoma spheroid growth [82] and upstream effect of BRAF inhibition on Rnd3 expression [83]; 4) the link between AP-1 components and TGF-β induced invasion in breast cancer [84]; and 5) the role of S100A4 in both epithelial-mesenchymal transition (EMT) and metastasis in squamous carcinoma spheroids [85]. Although the utility of spheroids is evident in this work, there is a significant dependency on additional culture systems due to the lack of characterization methods amenable to embedded spheroid research. Thus, investigators often rely on imaging as the primary tool for analysis of spheroid activity.
As discussed above, image-based analysis of spheroid size and invasion is useful, however the contradictory results observed in 2D versus 3D systems requires the use and development of additional characterization methods for embedded spheroid systems. Pettee et al., use western blots to study the role of RhoA-directed mDia2 invasion in embedded ovarian spheroids [86]. Western blot analyses of spheroid lysates enable measurement of Rock and mDia2 protein levels in 3D during spheroid formation or after knockdown. Results from western blots and stained whole colon cancer spheroids reveal the role of KRS in invasion where it induces intracellular signaling for cancer dissemination [87]. Since western blots do not require whole cells, this technique is well suited for use with embedded spheroids. However, the main limitations are low cell numbers and low protein yields, thus requiring a large number of spheroids for evaluation. This limitation can be addressed with the high throughput systems discussed earlier. The combination of observed invasion and measured protein levels within the same system ensures that 3D cell phenomena are supported by 3D protein expression.
In order to circumvent the limitations of protein yield, quantitative reverse transcription polymerase chain reaction (qrt-PCR) combines the advantages of quantitative characterizations with low RNA requirements. Sodek, et al., use qrt-PCR to investigate the relationship between predisposition towards spheroid formation and an invasive phenotype in ovarian cell lines [88]. The observed positive correlation is likely due to the upregulation of several mesenchymal markers in the lines that form spheroid and invade the collagen. qrt-PCR results can also provide information on other transcription differences including the upregulation of the TGF-β-mediated fibrotic response consistent with the mesenchymal/myofibroblast-like phenotype. Similarly, Reynolds et al., report that qrt-PCR of even the small population of CSCs present within breast cancer spheroids can be assayed and result show an increase in the expression of ALDH1A3 and SOX2 [79].
The Ten Dijke group uses a combination of optical invasion analysis and rt-PCR to study the role of TGF-β induced invasion in breast cancer. The studies employ spheroids of MCF10a derivatives, which modeled tumors of increasing malignant potential [71]. This system allows them to correlate TGF-β induced invasion with malignant potential, implicate BMP-7 as an inhibitor of invasion in the post metastatic, but not pre-malignant MCF10a derivatives, and to quantify the contributions of Smad3, Smad4, MMP-2 and MMP-9 in TGF-β induced invasion [72, 89]. The expression of transcriptional repressors Snail and Slug allows monitoring of the EMT after treatment with TGF- β [89]. Furthermore, the over expression of Snail and Slug enhances TGF- β induced invasion. Although more laborious than a monolayer, determining the expression changes in 3D spheroids is crucial. The spheroid invasion assay captures aspects of cell behavior which are not observable in 2D, and documents how cells in 3D spheroid culture are proteomically and genetically different from their monolayer counterparts. Thus, the use of nucleic acid and protein assays in conjunction with spheroid culture will serve to increase their utility and widespread use.
In contrast to the biological approaches discussed above, Haeger et al., study the invasion of melanoma and fibrosarcoma embedded in a collagen matrix from a mechanical perspective [90]. A number of studies describe different invasion modes in mesenchymal cells namely single cell versus collective invasion [86, 91]. By focusing on the matrix, as opposed to cells, Haegar et al., use confocal reflectance spectroscopy to tease apart the differences in invasion related to pore size and stiffness [90]. To increase pore size with minimal effect on stiffness, collagen is gelled at a lower temperature (21°C versus 37°C). In the high concentration (8 mg/mL) small pore collagen, collective leader/follower invasion occurs [59]. However in the low concentration (2.5 mg/mL) large-pore collagen, invasion ensues with single cells. Interestingly, cell behavior correlates to pore size, and not stiffness, such that invasion with high concentration (8 mg/mL), large pore (37°C gelled) collagen occurs as single cells confirming that collagen pore size governs invasion plasticity. The publication by Ayuso et al., proposes an additional tool for studying invasion, namely combining embedded spheroids with the controlled gradient capabilities of a microfluidic device [92]. Directional invasion of human oral squamous carcinoma and glioblastoma spheroids arises through the use of serum gradients in collective and individual invasion, respectively. This platform enables the facile study of invasion under the influence of chemoattractive or chemorepellant gradients.
4.4.3. Embedded Spheroids to Evaluate Chemotherapeutic Efficacy
The recapitulation of tumor macrostructure and environmental cues of hypoxia, cell-cell, and cell-ECM contacts make spheroids an ideal model of the heterogeneity present in a tumor, and thus useful for evaluating chemotherapeutic efficacy. The synergy between both embedded 3D culture and tumor macrostructure imparts a greater chemotherapeutic resistance than that observed in spheroid culture alone [93, 94]. As with studies focused on pathways affecting tumor growth, there are a number of reports that use stained spheroids combined with a metric of invasion as a measure of drug efficacy. For example, the adjuvant gamma-linolenic acid causes a net increase in glioma spheroid growth and invasion at lower concentrations (<100 µM), but increases the net apoptotic index at higher concentrations (>100 µM) [78]. Furthermore, the effective concentration of gamma-linolenic acid needed to treat spheroids is three times higher than that of a monolayer, as measured by collagen invasion and apoptosis calculated from staining with H&E, TUNEL, and proliferating cell nuclear antigen. As with cancer invasion studies, images of chemotherapy treated spheroids are often part of a larger study such as a study on the role of TM4SF5 in drug resistance and the use of anti TM4SF5 agents to enhance the efficacy of chemotherapeutics [95]. Live/dead staining establishes the susceptibility of a novel subtype of melanoma spheroids characterized by the overexpression of cyclin dependent kinase 4 and KIT to imatinib, which functions through its inhibitory receptor tyrosine kinase activity against KIT [96]. In squamous cell carcinoma spheroids, Basu et al., report the susceptibility of collective migration by E-cadherin positive cells versus infiltrative invasion by mesenchymal-like cells to different anti-neoplastic agents [91]. Treatment with cetuximab or erlotinib increases infiltrative invasion by targeting collective migration by E-cadherin–positive cells while sparing mesenchymal-like cells. In the absence of these mesenchymal like cells, erlotinib inhibits invasion in a dose-dependent fashion. Using a model of human polyoma middle T oncogene-induced breast cancer in mice, Schurigt et al., report that spheroids formed from the primary mouse tumor after treatment with a cysteine cathepsin inhibitor (JPM-OEt) do not exhibit in vitro resistance to the inhibitor.[97]. The evaluation of light based treatments are also described using spheroids, as previously discussed with the multi cell skin model which tested the effect of UV-B and cisplatin [67].
Although invasion and migration are important metrics to be considered, measurements of viability and proliferation are crucial for assessment of drug response. Two studies use the Alamar blue assay as an image-independent metric of spheroid viability after treatment with doxorubicin [64, 94]. This method does not require harsh disaggregation methods, and enables quantification of IC50 concentrations for doxorubicin across various culture models i.e., monolayer (374 ng/mL), spheroid (18962 ng/mL) and embedded spheroid (26687 ng/mL) [94]. Beyond simply quantifying viability, An et al., demonstrate the difference between an effect on invasion versus viability when testing the effect of suberoylanilide hydroxamic (SAHA) on glioma cells. SAHA dramatically reduces invasion into collagen, as quantified via imaging with a concurrent decrease in proliferation as determined using a CyQuant-GR dye on disaggregated spheroids [98].
The use of cell invasion into the matrix/medium as a measure of viability is useful but can bias efficacy results for chemotherapeutics that target rapid division or migration, which inordinately impacts invasion. For example, drugs that function through metabolically independent mechanisms of action might decrease viability, but assert a lesser effect on invasion. This disparity is evident in the response of collagen-embedded MDA-MB-231 breast cancer cells to chemotherapeutics, paclitaxel and cisplatin [79]. Paclitaxel, which targets actively dividing cells, dramatically reduces invasion, while cisplatin, which binds DNA, decreases invasion to a lesser extent. However, when disaggregated and analyzed, the two drugs exhibit similar effects on viability of the non-migratory core and invasive populations as assessed via a colorimetric assay. This discrepancy emphasizes the unreliability of invasion as the sole metric of efficacy. Further examination of the invasive and core populations, when separately assayed after disaggregating and isolation, reveals that both drugs are effective against the invasive population (viability < 34%), but the core population is resistant to treatment (viability >80%). The relative chemoresistance of the core cell population reflects an increased number and enrichment of CSCs in the core compared to the invasive population, as determined via the Aldefluor assay using flow cytometry, the mammosphere assay, and rt-PCR of CSC markers. Furthermore, prior treatment with paclitaxel results in a greater concentration of CSCs in the spheroid core. From a methods perspective, the disaggregation of spheroids to attain viable cells for detailed study (flow, mammosphere) provides a means for further study, with the caveat that a disaggregation step has been used. From a clinical standpoint, these findings underlie the need for agents that can effectively treat the CSCs linked to chemoresistance and recurrence. From a model perspective, the spheroid emulates the heterogeneity present in a tumor, and is more resistant than traditional monolayers or single cell 3D models. To summarize, quantitative assessment of cell death in conjugation with invasion and proliferation studies are providing a greater understanding of drug efficacy.
Embedded spheroids are also ideal to evaluate nanoparticle-based drug delivery systems as spheroids enable assessment of mass transport through a dense multicellular aggregate and an ECM mimic. Nanoparticle penetration and subsequent efficacy has been extensively tested and reviewed in non-embedded spheroids [99]. However, analogous studies performed with embedded spheroids are rare. An et al., report that unloaded PSS/PAH microcapsules exhibit a negligible effect on glioma spheroids invasion after treatment as part of a larger study indicating their promise as a drug delivery system [100]. Using a collagen embedded breast cancer spheroid model, Charoen et al., describe the penetration of paclitaxel-loaded nanoparticles into the entire spheroid with a resultant reduction in spheroid size that is greater than with paclitaxel treatment alone [101]. Notwithstanding the few cases in the literature, the combination of more physiological mass transport barriers and ability to evaluate efficacy demonstrate the potential utility of spheroids to the drug delivery field. However, for bulk delivery devices such as meshes, wafers and patches, the relatively small media volume used in spheroid models can artificially concentrate the released drug likely making the current iterations of spheroid models unsuitable for testing the efficacy of these delivery methods.
4.5. Spheroids Embedded in Collagen and Fibrin in Non-cancer Research Areas
4.5.1. HUVEC Sprouting
This review focuses on spheroid applications in cancer research, however, spheroids are of widespread utility for a number of research areas (Figure 6). Many of the techniques and methods described below not only translate to studies involving cancer spheroids but also can be directly implemented to study the effect of vascular systems on the growth of tumors. The study of endothelial cell migration and sprouting during angiogenesis is one obvious example. Human umbilical vein endothelial cells (HUVECs) or bovine aortic endothelial (BAE) cells form a differentiated surface layer with an unorganized, apoptotic core, when prepared by the liquid overlay method [102]. Using this model, Korff and Augustin report that collagen embedded spheroids spontaneously sprout radial capillary-like protrusions that differentiate to form capillary lumens (Figure 6B) [103]. Adjacent spheroids display directional sprouting and thus multiple spheroid systems form large, complex structures. Mechanically induced sprouting occurs where collagen-transduced mechanical forces direct the sprouting of endothelial cells in the direction that force is applied [103]. This is an example of a system that seamlessly translates to cancer spheroids to study tumor biomechanics. Two quantitative metrics of sprouting are typically used to characterize angiogenesis: the lengths of the three longest protrusions, or the cumulative length of all protrusions [104–112]. As with cancer spheroids, few investigators supplement the quantitative data on sprouting with proteomic expression. However, Haspel et al., report the collection of sprout length data along with western blots to analyze protein expression [113]. An alternative method uses Puramatrix to aid spheroid formation [114]. When the human dermal microvascular endothelial cell toroidal spheroid is embedded in collagen, it maintains a similar behavior to the system by Korff and Augustin, and demonstrates the biphasic effect of the multi kinase inhibitor sorafenib on sprouting, as a model experiment. Spheroids lacking ECs offer insight into the differentiation of vascular smooth muscle cells (SMCs) into myofibroblasts. Spheroids composed of cells derived from chicken embryonic SMCs or human SMCs demonstrate differentiation into myofibroblasts and sprouting without ECs after the addition of VEGF, PDGF-BB and FGF-2 [115].
Figure 6. Spheroids have been Utilized by a Number of Fields.
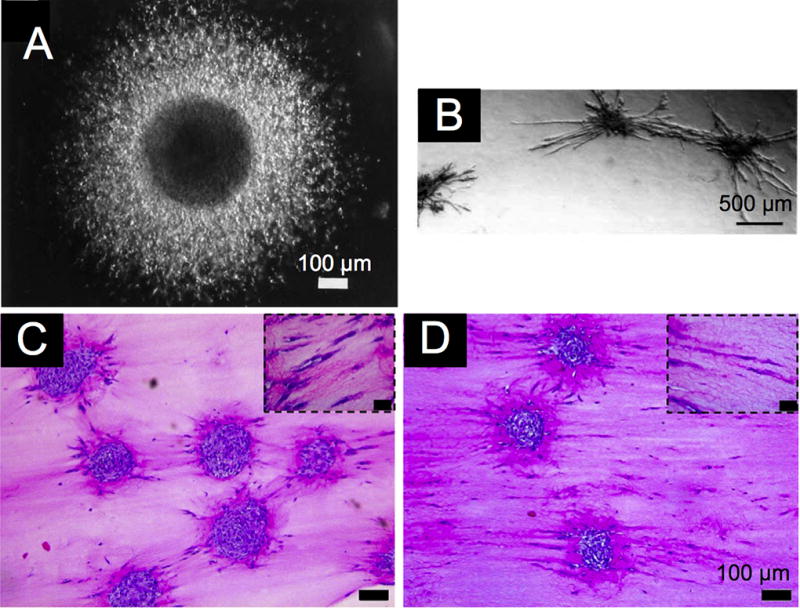
Spheroids have been used in a number of fields including cancer research, (A) angiogenic sprouting of endothelial cells, (B) and artificial tenocytes with embedded tendon cells (C and D) [50, 103, 129]. Despite the diverse applications the similarities of the micron level structures are apparent. Reprinted from reference 48 with permission from the JNS Publishing Group. Reprinted from reference 101 with permission from The Company of Biologists Ltd. Reprinted from reference 127 with permission from Elsevier.
The designers of the endothelial cell spheroid capillary sprouting system, Korff and Augustin, continue to improve the model and explore its applications. For example, embedded spheroids enable determination of IC50 curves for the response of approved drugs on angiogenesis, and have reduced the inherent variability of using primary-derived HUVECs through the use of immortalized HUVECs [116]. To model mature blood vessels, SMCs are incorporated in a (1:1 or 4:1 HUVEC:SMC) heterospheroid, and the two cell types spontaneously form a core of SMCs and a surface layer of HUVECs [117]. The inclusion of SMCs induces a mature EC phenotype supported by an increase in junctional complexes, a decrease in PDGF-β expression, and a decrease in EC apoptosis. Although exogenous VEGF fails to stimulate sprouting in 1:1 heterospheroids, sprouting does occur when the EC content is increased, as in the case of 4:1 heterospheroids. This EC dependence is indicative of crosstalk between ECs and SMCs. Similarly, successful sprouting of 1:1 heterospheroids is achieved with co-stimulation by VEGF and Ang-2, an antagonist of Ang-1, which is expressed in SMCs. Ang-2 destabilizes the EC and SMC interactions, to facilitate VEGF responsiveness [118].
Similar to SMCs, human bone marrow stromal cells (BMSC) or adipose derived stromal cells (ASC) support the formation of prevascular structures by HUVECs when formed as HUVEC-containing heterospheroids [119]. The secretome of both BMSCs and ASCs, as analyzed via ELISA, reveal a number of trophic factors that stimulate EC growth such as HGF, TIMP1, and TIMP2. In contrast, primary human fibroblasts/HUVEC heterospheroids decrease HUVEC apoptosis, but also sprouting, which nevertheless bears promise for vascularizing engineered tissues [120]. An attempt to similarly vascularize collagen-based bone grafts using heterospheroids composed of HUVECs and human osteoblasts found a similar effect where the heterospheroid exhibits decreased sprouting [109]. In an alternate approach to vascularize bone grafts, Stahl et al., suspend single osteoblasts around an endothelial cell spheroid in collagen [121]. Direct contact between osteoblasts and HUVECs inhibits the sprouting of HUVEC spheroids even with exogenous stimulation. However, endothelial progenitor cell (EPC) spheroids sprout in the presence of osteoblasts and exogenous growth factors, making EPCs a better cell prospect for vascularized bone grafts.
4.5.2. HUVEC Sprouting Combined with Cancer Invasion
The merging of the tumor spheroids and EC sprouting systems yields vascularized spheroid models. The inclusion of vasculature in a tumor model enables modeling of late stage tumors as opposed to being limited to avascular early stage tumors. Using a non-embedded melanoma/endothelial heterospheroid, the anti-angiogenic properties of resveratrol become evident unlike in the monolayer system [122]. The inclusion of murine ECs in a non-embedded heterospheroid system sensitizes the resulting murine mammary cells to treatment with paclitaxel, but protects them from radiation [123]. In another model, the addition of fibroblasts stabilizes the endothelial cells spheroids preventing apoptosis in EC/tumor cell models [124]. Others have expanded from two cell systems to spheroids composed of three cell types. Correa et al., report increased sprouting in an embedded heterospheroid composed of a ratio of 2:2:1 (normal human dermal fibroblasts: MDA-MB 231 breast cancer line: HUVECs) where the fibroblasts mimicked mural cells [124]. The incorporation of all three cell types, as opposed to only the stromal cells (fibroblast and ECs), results in different response profiles to anti-angiogenic agents and traditional chemotherapeutics as measured by mean sprout length and tumor cell growth via a luciferase transfected line. Using cell-specific shRNA analysis, this system demonstrates that MT1-MMP regulates endothelial sprouting through expression in fibroblasts and ECs, but not tumor cells. A second three cell system, where the heterospheroids is composed of colon tumor cells and HUVECs with normal human lung fibroblasts embedded in the surrounding a fibrin matrix (Figure 7), reveals robust sprouting with the colon tumor line invading into the capillary lumens [125]. Relative hypoxia in this system encourages greater intravasation by the tumor cells, which is partially regulated by an EMT driver, Slug. Results such as these, advance the spheroid model beyond just tumor cells to the inclusion of multiple cell types and a functioning vasculature. The incorporation of a number of aspects of a tumor microenvironment simultaneously increases the treatment types that the spheroids can effectively evaluate. Furthermore, the selective control either by luciferase, shRNA [124], or siRNA [125] enables monitoring of an individual cell type within such a complex multiple cell system.
Figure 7. The Vascularized Spheroid.
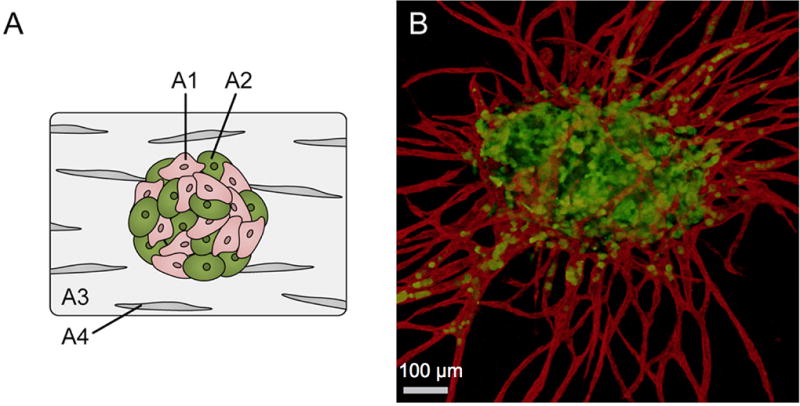
The combination of EC and cancer spheroids has lead to a fully vascularized spheroid. Fibroblasts in the stroma stabilize the formation of vascular networks. Evidence of interaction is apparent as cancer cells (green) can be seen within the vessel lumens (red) [125]. Reproduced from reference 123 with permission from The Royal Society of Chemistry.
4.5.3. Mechanical Matrix Modifications
Based on the effect of tension on directional sprouting reported by Korff et al., [103], Mason et al., describe increasing the stiffness of the collagen gel post-embedding using a non-enzymatic glycation [126]. The stiffness of the 1.5 mg/mL gels increases from 175 Pa to as high as 730 Pa, depending on the ribose concentration added. Importantly, gels possessing a compressive modulus between 175–500 Pa possess similar fibril size and arrangement. The stiffer gels contain slightly larger fibers. Bovine aortic endothelial cell spheroids exhibit higher sprouting in the stiffer collagen gels, indicative of the effect of mechanical environment on angiogenesis. Increased sprouting results from a stiffer environment, and this study decouples the result from contributions of density and fiber structure. This result is easily applicable to cancer invasion, while a similar study on cancer invasion that decoupled collagen pore size from stiffness would be applicable in vascular sprouting [90]. Such approaches are of utility in teasing apart the interconnected relationships between matrix characteristics (stiffness, density, and fiber structure) and cancer invasion.
4.5.4. Maintaining or Influencing Differentiation in Spheroids
Similar to endothelial cell sprouting, embedded spheroids can promote differentiation of stem cells along a preferred lineage or maintain the phenotype of primary cells when treated with the correct mixture of growth factors. In other words, spheroids can mimic the native tissue from which tumors arise. An early attempt at differentiating mammary rabbit cells in a number of culture conditions found that spheroids embedded in collagen, or placed on top of floating collagen gels, recapitulate some aspects of a functioning mammary gland [52]. While both formed fat droplets and differentiated into two cell types, the floating gels exhibit more microfilament rich cells, while the embedded spheroids form large duct-like structures. Drawing inspiration from the EC sprouting assay, similar assays for studying neurite growth are described. The supportive functions of Schwann cells are critical to neurite growth; however, 2D co-culture methods with and without contact do not dramatically increase neurite length [127]. In contrast, in a 3D collagen embedded heterospheroid of 1:10 NG108-15 neurites to neonatal rat Schwann cells, the mean sprout length almost triples in magnitude compared to the results obtained in the 2D experiments [128]. This result highlights the importance of not just cell-cell contacts, but the 3D environment itself in creating and/or maintaining physiological cell interactions.
When seeking to maintain the differentiation of cells, the goal is to recreate the phenotypic environment from which the cell type originated. For example, to model skin, fibroblasts surround an embedded dermal stem cell spheroid in collagen with a layer of keratinocytes on the top. The dermal stem cells differentiate into epidermal melanocytes. The co-culturing of functional healthy tissue whether it be mammary glands or skin, with a tumor spheroid enables studying the interplay between normal and malignant tissue. Similarly, both matrix and growth factors are important for maintaining the differentiation of mature equine tenocytes, as recently reported [129]. Tenocyte spheroids embedded in collagen demonstrate contraction and alignment when treated with low-serum media containing TGF-β1, ascorbic acid and insulin, but not in agarose, or when treated with a high-serum media lacking additional growth factors as shown in Figure 6C.
When the desired application is implantation, the matrix not only supports the function of embedded cells, but it can actively aid in long term viability in vivo. Hou et al., describe a collagen-filled polyurethane foam with a heparin-immobilized VEGF to address the drawback of low vascularization in hepatocyte transplantation [130]. To enhance the viability of hepatocytes in the foam, rat hepatocytes are embedded in a spheroid. The viable spheroids produce albumin which is indicative of hepatocyte specific function [131]. After implantation in a rat model, viability and vascularization improves as a consequence of the synergistic combination of the spheroid macrostructure and application-specific matrix.
Spheroid culture is also applicable to skewing the differentiation of cells along a desired lineage. Recent efforts focus on exploiting the differentiation capabilities of mesenchymal stem cells (MSC). When MSC spheroids, embedded in fibrin and treated with osteogenic media, are cultured in hypoxic, serum-free conditions, they outperform singly embedded cells in terms of metabolism and resistance to apoptosis [132]. Importantly alkaline phosphatase, a marker of osteogenic potential does not significantly decrease, and calcium production is significantly higher. Similarly, MSCs in a fibrin-embedded MSC-HUVEC heterospheroid differentiate along a cardiac pathway, if provided the right encouragement. Subsequent implantation restores function in a post-ischemic rat heart model [133]. In this study, a two-step hanging drop method gives the heterospheroid a layered morphology. First, a spheroid of subamnion cord-lining MSCs (15,000) forms over three days and then on the fourth day newly added HUVECs (2,000) form an outer layer and surround the MSC core. Next, each animal receives a graft containing approximately 150 spheroids embedded in fibrin. Left ventricle remodeling occurs with less scarring and increased vascularization results in an improved ejection fraction. The differentiation of mature cells is also possible by manipulating culture conditions and cytokine exposure in a process called transdifferentiation. Using this approach Nishikawa et al., describe the transdifferentiation of collagen-embedded rat hepatocyte aggregates to bile duct cells [134–136].
4.6. Spheroids Embedded in Synthetic Polymers
Unlike naturally sourced materials, synthetic materials offer unique opportunities to control and modulate spheroid behavior without the complicating interactions between cells and a natural biopolymer – like collagen. For example, PDMS was used to mechanically confine spheroids to study the subsequent effect on mitotic progression[137]. Studies like these are only possible if the mechanical properties are not affected by cellular remodeling. Similarly, synthetic materials that are impervious or very slow to cellular remodeling, such as PLGA or PEG hydrogels, are ideal to study the effects of material pore size, material structure, and mechanical properties on cell migration. Bioactive peptides such as RGD or MMP-sensitive regions can also be selectively incorporated in synthetic polymer matrices to study spheroid-ECM interactions in a modular and controlled manner that is difficult to achieve with native biopolymers. For example, Ho, et al report the effect of 3D culture and spheroid macrostructure on resistance to chemotherapeutics in a collagen-coated PLGA scaffold using an array of methods including confocal microscopy of a fluorescent reagent and measurement of secreted proteins[138]. One potential advantage of working with a synthetic polymer for embedding spheroids is to easily and quickly remove the cells from the polymer for additional analysis. For example, triggered degradation of the polymer scaffold using light would accelerate the collection of live cells and increase the yield without subjecting them to lengthy and harsh biological methods (such as enzymatic degradation). The opportunities provided by the use of synthetic materials to the spheroid field are just beginning to be realized.
5. Perspectives
Embedded spheroids serve as useful models in cancer, angiogenesis, and differentiation research, but a lack of quantitative characterization methods hinders their ultimate utility. While embedding cells increases the physiological relevance, it also increases the time, cost, and complexity of the system. Today, spheroid models are not readily amenable to the many techniques available for 2D systems. There is a heavy reliance on optical imaging methods and subsequent analysis. While improvements in post-imaging analysis are facilitating many studies (e.g., whole cell imaging mosaics/tiles), quantitative non-image based techniques are in demand [63, 76, 139]. However, the quantities of genetic material, protein or cells required for FACs, western blots, viability assays, or mRNA chips analyses is significant and requires the use of multiple spheroids, on the order of several thousand cells. Furthermore, harvest methods to obtain the cells from the spheroid may require harsh conditions or be labor intensive due to the embedding material. Assays relying on protein or nucleic acids may prove easier to adapt, since cell viability is not required for performance of the assay. As the sensitivity of these techniques improves, the potential for performing analyses on a single spheroid will be become more wide-spread. [51, 140]. Importantly, there are a few studies successfully reporting western blots, ELISAs, luciferase expression, oxoplates, or rt-PCR data. The use of non-destructive methods to monitor viability (oxoplates or luciferase expressing lines) are especially promising as they bypass the limitations of low biological material as well as time consuming disaggregation methods. Increasing the spheroids per system, to maximize cell yields may introduce unwanted spheroid/spheroid interaction. Efforts are underway to improve the throughput by using synthetic polymers to direct spheroid formation, magnetically labeling cells to direct growth as aggregates, or using a PDMS mold in a facile method for embedding spheroids [53–55].
Innovations in non-embedded spheroids are being translated to embedded systems and these strategies and techniques are expanding the types of data that spheroids can be used to obtain. Li et al., scaled up drug screening to a high throughput system in non-embedded spheroids by combining viability measurements with a colorimetric assay and evidence of EMT through vimentin expression. The stable transfection of a vimentin promoter in a firefly luciferase reporter plasmid enables facile quantification of vimentin expression [141] and screening of a panel of marine secondary metabolites for small molecules that regulate EMT without causing toxicity. Flowcytometry is used to characterize non-embedded spheroids but only limited reports exist with embedded spheroid counterparts [142]. However adoption of flowcytometric techniques with embedded spheroids will facilitate quantification of uptake of fluorescently labeled drugs or nanoparticles as well as analysis of surface markers. The combination of uptake and viability studies is especially important to the evaluation of nanoparticles, in which studies using non-embedded spheroids are prevalent, but lacking in embedded spheroid models.
Although spheroids are used in a number of fields such as cancer, vascular biology, and stem cell research, there is a significant opportunity for universal advancement with crosstalk between these fields. Importantly, the models and methods used are translatable between fields, where the end goal is recapitulation of a microenvironment to influence cell phenotype. This is most striking when examining the research of both Professor Kulms and Herlyn [67, 143]. Both modeled skin by using spheroids of melanoma or dermal stem cells surrounded by fibroblasts in a collagen gel with a layer of keratinocytes on the top. The melanoma system is used for drug testing, while the dermal stem cell system demonstrated their stemness by giving rise to differentiated epidermal melanocytes. The methods used are the same, but differ in application. Greater crosstalk and adoption of methods between these related fields will ultimately increase the capability of spheroids to offer insights into uptake, metabolic activity, protein regulation, and secondary cell interactions that are poorly understood in cancer.
6. Conclusions
Embedded spheroids are a powerful tool in cancer research, angiogenesis, differentiation, and tissue engineering that enables the recapitulation of in vivo like environments within in vitro settings. This review describes contributions of embedded spheroids to cancer research, and methods of interest. Embedded spheroid models recreate the complex interactions between cell types within the contexts of secondary cell types and tumor heterogeneity in cancer. The combination of natural matrices with spheroids lends itself to studies of how a growing tumor interacts with, migrates through, and shapes the ECM. The integration of tumor macrostructure within a 3D environment also provides a means to evaluate the efficacy and mechanism(s) of novel chemotherapeutics and nanoparticle systems. Although characterization methods and time consuming implementations are limiting, novel methods to improve throughput and ease of use are facilitating widespread adoption. The increasing sensitivity of assays combined with non-destructive evaluations of growth and viability will enable quantitative characterization metrics, and thus increase the utility of this model.
The future of spheroid research rests on the marriage of sensitive instrumentation and a readiness to embrace spheroid-focused tools irrespective of application. One of the weaknesses of the spheroid systems is that the quantity of cells, and, thus, the amounts of proteins and nucleic acids isolated are small. The advent of highly sensitive instrumentation and assays are desperately needed to fuel quantitative single cell analyses. For example single cell measurements of mRNA from different locations within the spheroid could be coupled with computational models of pH, oxygen partial pressure, gel composition, and gel stiffness. This would enable the effect of local environment on genetic expression to be characterized. Many of the spheroid systems are focused on breast cancer or glioblastoma, while none or only a few reports described cancers where tumors are embedded in a fibrous and fibroblast rich matrix as seen with sarcomas. A number of additional opportunities exists for embedded spheroid models including their use in: 1) mechanically active or dynamic materials to further simulate in vivo conditions – such as in lung cancer; 2) systems that enable perfusion of the tumor and ability to study the effects of flow rate and nutrient supply; 3) models employing multiple cell types located at specific sites to study nearest neighbor effects and signaling pathways which may or may not be activated; and 4) microfluidic models of vasculature to explore tumor/vascular interactions. We also encourage researchers to use broad search terms in learning about this area as a wealth of spheroid-related articles are found using key words such spheroids, cell clusters, tumoroids, capillary sprouting systems, and multicellular tumor spheroid (MTS).
In summary, spheroids as multicellular aggregates enable one to more accurately study the complex mechanisms behind cancer invasion, progression, and response to treatment while replicating both the multicellular nature and 3D stromal environment present in an in vivo tumor. We encourage all to explore the field of spheroid research where dimensions (length, width, and depth) are highly coupled to cellular and molecular biological events, and are integral to our further understanding of diseases and the treatments that are needed.
Acknowledgments
The authors would like to acknowledge funding support for this project in part from: The Boston University Nanotechnology Innovation Center (BUnano), the National Institutes of Health (Biomaterials Training Fellowship T32 EB006359 and R01 EB017722), Brigham and Women’s Hospital, Boston University, and the Nanotheranostics ARC, Evans Center for Interdisciplinary Biomedical Research at the Boston University School of Medicine.
Biographies

Dr. Kristie Tevis received her B.S. degree in Biomedical Engineering with a focus in Cell and Tissue Engineering from Johns Hopkins University in 2009. She earned her Ph.D. in Biomedical Engineering at Boston University under the mentorship of Professors Grinstaff and Colson. Her research focused on the design and application of in vitro cancer models to study cellular interactions within a tumor and response to chemotherapeutics.

Dr. Yolonda L. Colon is a Professor of Surgery, the Michael A. Bell Family Distinguished Chair in Healthcare Innovation, a clinically active cardiothoracic surgeon, and Director of the Women's Lung Cancer Program at Brigham and Women’s Hospital (BWH). Her area of clinical expertise and innovation is the development of new approaches to diagnose occult nodal metastases and to prevent and treat tumor recurrence after "curative" surgical resection. She also dedicates time as educator/mentor in national roles focused on fostering young academic cardiothoracic surgeons and surgeon-scientists. She has published more than 120 peer-reviewed manuscripts.

Professor Mark W. Grinstaff is the Distinguished Professor of Translational Research and a Professor of Biomedical Engineering, Chemistry, Materials Science and Engineering, and Medicine at Boston University. He is also the Director of BU’s Nanotechnology Innovation Center, and the Associate Director for Engineering and Science at the BU Cancer Center. His inventive scientific contributions span biomedical engineering, biological engineering, nanotechnology, chemistry, and medicine, producing transformational discoveries and technologies unique to the convergence of these fields. His research has yielded more than 250 peer-reviewed publications, more than 325 oral presentations, and several products used in the clinic to treat patients.
Footnotes
Author Contributions:
Conceived and designed the review and wrote the manuscript: KMT, YLC, and MWG.
References
- 1.Almany L, Seliktar D. Biomaterials. 2005;26:2467. doi: 10.1016/j.biomaterials.2004.06.047. [DOI] [PubMed] [Google Scholar]
- 2.Birgersdotter A, Sandberg R, Ernberg I. Semin. Cancer Biol. 2005;15:405. doi: 10.1016/j.semcancer.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Roschke AV, Tonon G, Gehlhaus KS, McTyre N, Bussey KJ, Lababidi S, Scudiero DA, Weinstein JN, Kirsch IR. Cancer Res. 2003;63:8634. [PubMed] [Google Scholar]
- 4.Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud Ø, Gjertsen BT, Nolan GP. Cell. 2004;118:217. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 5.Loessner D, Stok KS, Lutolf MP, Hutmacher DW, Clements JA, Rizzi SC. Biomaterials. 2010;31:8494. doi: 10.1016/j.biomaterials.2010.07.064. [DOI] [PubMed] [Google Scholar]
- 6.Sun T, Jackson S, Haycock JW, MacNeil S. J. Biotechnol. 2006;122:372. doi: 10.1016/j.jbiotec.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 7.Mueller MM, Fusenig NE. Nat. Rev. Cancer. 2004;4:839. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 8.Blanco D, Vicent S, Fraga MF, Fernandez-Garcia I, Freire J, Lujambio A, Esteller M, Ortiz-De-Solorzano C, Pio R, Lecanda F, Montuenga LM. Neoplasia. 2007;9:840. doi: 10.1593/neo.07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. J.Cell Biol. 1997;137:231. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ingber DE. Semin. Cancer Biol. 2008;18:356. doi: 10.1016/j.semcancer.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedrich J, Ebner R, Kunz-Schughart LA. Int. J. Radiat. Biol. 2007;83:849. doi: 10.1080/09553000701727531. [DOI] [PubMed] [Google Scholar]
- 12.Danhier F, Feron O, Préat V. J. Control. Release. 2010;148:135. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 13.Bergers G, Benjamin LE. Nat. Rev. Cancer. 2003;3:401. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 14.Cardone RA, Casavola V, Reshkin SJ. Nat. Rev. Cancer. 2005;5:786. doi: 10.1038/nrc1713. [DOI] [PubMed] [Google Scholar]
- 15.Carmeliet P. Nat. Med. 2000;6:389. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 16.Anton K, Glod J. Curr. Phamaceutical Biotechnol. 2009;10:185. doi: 10.2174/138920109787315088. [DOI] [PubMed] [Google Scholar]
- 17.Yue Z, Lui Y, Ruan J, Zhou L, Lu Y. Chin. Med. J. (Engl) 2015;125:3305. [PubMed] [Google Scholar]
- 18.Gonda TA, Varro A, Wang TC, Tycko B. Semin. Cell Dev. Biol. 2010;21:2. doi: 10.1016/j.semcdb.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sutherland RM, McCredie JA, Inch WR. J. Natl. Cancer Inst. 1971;46:113. [PubMed] [Google Scholar]
- 20.Harrison RG, Greenman MJ, Mall FP, Jackson CM. Anat. Rec. 1907;1:116. [Google Scholar]
- 21.Yuhas JM, Li AP, Martinez AO, Ladman AJ. Cancer Res. 1977;37:3639. [PubMed] [Google Scholar]
- 22.Ivascu A, Kubbies M. J. Biomol. Screen. 2006;11:922. doi: 10.1177/1087057106292763. [DOI] [PubMed] [Google Scholar]
- 23.Rivron NC, Rouwkema J, Truckenmüller R, Karperien M, De Boer J, Van Blitterswijk CA. Biomaterials. 2009;30:4851. doi: 10.1016/j.biomaterials.2009.06.037. [DOI] [PubMed] [Google Scholar]
- 24.Singh M, Mukundan S, Jaramillo M, Oesterreich S, Sant S. Cancer Res. 2016;76 doi: 10.1158/0008-5472.CAN-15-2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen K, Lee J, Yarmush ML, Parekkadan B. Biomed. Microdevices. 2014;16:609. doi: 10.1007/s10544-014-9863-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lei H, Hofferberth SC, Liu R, Colby A, Tevis KM, Catalano P, Grinstaff MW, Colson YL. J. Thorac. Cardiovasc. Surg. 2015;149:1417. doi: 10.1016/j.jtcvs.2015.02.020. [DOI] [PubMed] [Google Scholar]
- 27.Barbone D, Van Dam L, Follo C, Jithesh PV, Zhang S-D, Richards WG, Bueno R, Fennell DA, Broaddus VC. PLoS One. 2016;11:e0150044. doi: 10.1371/journal.pone.0150044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia L, Sakban RB, Qu Y, Hong X, Zhang W, Nugraha B, Tong WH, Ananthanarayanan A, Zheng B, Chau IY-Y, Jia R, McMillian M, Silva J, Dallas S, Yu H. Biomaterials. 2012;33:2165. doi: 10.1016/j.biomaterials.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, Luo X, Anene-Nzelu C, Yu Y, Hong X, Singh NH, Xia L, Liu S, Yu H. doi: 10.1002/jat.3090. n.d., [DOI] [PubMed] [Google Scholar]
- 30.Ong S-M, Zhang C, Toh Y-C, Kim SH, Foo HL, Tan CH, van Noort D, Park S, Yu H. Biomaterials. 2008;29:3237. doi: 10.1016/j.biomaterials.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 31.Moreira Teixeira LS, Leijten JCH, Sobral J, Jin R, van Apeldoorn AA, Feijen J, van Blitterswijk CA, Dijkstra PJ, Karperien HBJ. Eur. Cell. Mater. 2012;23:387. doi: 10.22203/ecm.v023a30. [DOI] [PubMed] [Google Scholar]
- 32.Ksiazkiewicz M, Gottfried E, Kreutz M, Mack M, Hofstaedter F, Kunz-Schughart La. Immunobiology. 2010;215:737. doi: 10.1016/j.imbio.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 33.Choi MR, Stanton-Maxey KJ, Stanley JK, Levin CS, Bardhan R, Akin D, Badve S, Sturgis J, Robinson JP, Bashir R, Halas NJ, Clare SE. Nano Lett. 2007;7:3759. doi: 10.1021/nl072209h. [DOI] [PubMed] [Google Scholar]
- 34.Hauptmann S, Zwadlo-Klarwasser G, Jansen M, Klosterhalfen B, Kirkpatrick CJ. Am. J. Pathol. 1993;143:1406. [PMC free article] [PubMed] [Google Scholar]
- 35.Pedersen P-H, Ness GO, Engebraaten O, Bjerkvig R, Lillehaug JR, Laerum OD. Int. J. Cancer. 1994;56:255. doi: 10.1002/ijc.2910560219. [DOI] [PubMed] [Google Scholar]
- 36.Engebraaten O, Bjerkvig R, Lund-Johansen M, Wester K, Pedersen P-H, Mork S, Backlund E-O, Laerum OD. Acta Neuropathol. 1990;81:130. doi: 10.1007/BF00334501. [DOI] [PubMed] [Google Scholar]
- 37.Nygaard SJ, Pedersen PH, Mikkelsen T, Terzis AJ, Tysnes OB, Bjerkvig R. Invasion Metastasis. 1995;15:179. [PubMed] [Google Scholar]
- 38.Ho DN, Kohler N, Sigdel A, Kalluri R, Morgan JR, Xu C, Sun S. Theranostics. 2012;2:66. doi: 10.7150/thno.3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stevens KR, Ungrin MD, Schwartz RE, Ng S, Carvalho B, Christine KS, Chaturvedi RR, Li CY, Zandstra PW, Chen CS, Bhatia SN. Nat. Commun. 2013;4:1. doi: 10.1038/ncomms2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beaune G, Winnik FM, Brochard-Wyart F. Langmuir. 2015;31:12984. doi: 10.1021/acs.langmuir.5b02785. [DOI] [PubMed] [Google Scholar]
- 41.Burleson KM, Hansen LK, Skubitz APN. Clin. Exp. Metastasis. 2004;21:685. doi: 10.1007/s10585-004-5768-5. [DOI] [PubMed] [Google Scholar]
- 42.Mills K, Garikipati K. Int. J. Mater. Res. 2011;102:889. [Google Scholar]
- 43.Cheng G, Tse J, Jain RK, Munn LL. PLoS One. 2009;4:e4632. doi: 10.1371/journal.pone.0004632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bingle L, Lewis CE, Corke KP, Reed MWR, Brown NJ. Br. J. Cancer. 2005;94:101. doi: 10.1038/sj.bjc.6602901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang K, Yu L-Y, Jiang L-Y, Wang H-B, Wang C-Y, Luo Y. Acta Biomater. 2015;15:65. doi: 10.1016/j.actbio.2014.12.025. [DOI] [PubMed] [Google Scholar]
- 46.Viel T, Talasila KM, Monfared P, Wang J, Jikeli JF, Waerzeggers Y, Neumaier B, Backes H, Brekka N, Thorsen F, Stieber D, Niclou SP, Winkeler A, Tavitian B, Hoehn M, Bjerkvig R, Miletic H, Jacobs AH. J. Nucl. Med. 2012;53:1135. doi: 10.2967/jnumed.111.101659. [DOI] [PubMed] [Google Scholar]
- 47.Lehnert L, Trost H, Schmiegel W, Röder C, Kalthoff H. Ann. N. Y. Acad. Sci. 1999;880:83. doi: 10.1111/j.1749-6632.1999.tb09512.x. [DOI] [PubMed] [Google Scholar]
- 48.Gerami-Naini B, Dovzhenko OV, Durning M, Wegner FH, Thomson Ja, Golos TG. Endocrinology. 2004;145:1517. doi: 10.1210/en.2003-1241. [DOI] [PubMed] [Google Scholar]
- 49.Harjanto D, Maffei JS, Zaman MH. PLoS One. 2011;6:e24891. doi: 10.1371/journal.pone.0024891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tamaki M, McDonald W, Amberger VR, Moore E, Del Maestro RF. J. Neurosurg. 1997;87:602. doi: 10.3171/jns.1997.87.4.0602. [DOI] [PubMed] [Google Scholar]
- 51.Derwahl M, Studer H, Huber G, Gerber H, Peter HJ. Endocrinology. 1990;127:2104. doi: 10.1210/endo-127-5-2104. [DOI] [PubMed] [Google Scholar]
- 52.Haeuptle MT, Suard YL, Bogenmann E, Reggio H, Racine L, Kraehenbuhl JP. J. Cell Biol. 1983;96:1425. doi: 10.1083/jcb.96.5.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Truong HH, de Sonneville J, Ghotra VPS, Xiong J, Price L, Hogendoorn PCW, Spaink HH, van de Water B, Danen EHJ. Biomaterials. 2012;33:181. doi: 10.1016/j.biomaterials.2011.09.049. [DOI] [PubMed] [Google Scholar]
- 54.Okochi M, Takano S, Isaji Y, Senga T, Hamaguchi M, Honda H. Lab Chip. 2009;9:3378. doi: 10.1039/b909304d. [DOI] [PubMed] [Google Scholar]
- 55.Ma J, Zhang X, Liu Y, Yu H, Liu L, Shi Y, Li Y, Qin J. Biotechnol. J. 2016;11:127. doi: 10.1002/biot.201500183. [DOI] [PubMed] [Google Scholar]
- 56.Jones DP, Hanna W, El-Hamidi H, Celli JP. J. Vis. Exp. 2014:1. doi: 10.3791/51302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hwang Y-J, Kolettis N, Yang M, Gillard ER, Sanchez E, Chung-ho S, Tromberg BJ, Krasieva TB, Lyubovitsky JG. Photochem. Photobiol. 2011;87:408. doi: 10.1111/j.1751-1097.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sameni M, Moin K, Sloane BF. Neoplasia. 2001;2:496. doi: 10.1038/sj.neo.7900116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carey SP, Starchenko A, McGregor AL, Reinhart-King Ca. Clin. Exp. Metastasis. 2013;30:615. doi: 10.1007/s10585-013-9565-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vamvakidou AP, Mondrinos MJ, Petushi SP, Garcia FU, Lelkes PI, Tozeren A. J. Biomol. Screen. 2007;12:13. doi: 10.1177/1087057106296482. [DOI] [PubMed] [Google Scholar]
- 61.Alcoser TA, Bordeleau F, Carey SP, Lampi MC, Kowal DR, Somasegar S, Varma S, Shin SJ, Reinhart-King CA. Cell. Mol. Bioeng. 2015;8:76. doi: 10.1007/s12195-014-0360-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mora R, Abschuetz A, Kees T, Dokic I, Joschko N, Kleber S, Geibig R, Mosconi E, Zentgraf H, Martin-Villalba A, Régnier-Vigouroux A. Glia. 2009;57:561. doi: 10.1002/glia.20785. [DOI] [PubMed] [Google Scholar]
- 63.Castillo LRC, Oancea A-D, Stüllein C, Régnier-Vigouroux A. Nat. Publ. Gr. 2016 doi: 10.1038/srep35099. [DOI] [Google Scholar]
- 64.Yip D, Cho CH. Biochem. Biophys. Res. Commun. 2013;433:327. doi: 10.1016/j.bbrc.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 65.Okochi M, Matsumura T, Honda H. Biosens. Bioelectron. 2013;42:300. doi: 10.1016/j.bios.2012.09.067. [DOI] [PubMed] [Google Scholar]
- 66.Tevis KM, Cecchi RJ, Colson YL, Grinstaff MW. Acta Biomater. 2017;50:271. doi: 10.1016/j.actbio.2016.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vörsmann H, Groeber F, Walles H, Busch S, Beissert S, Walczak H, Kulms D. Cell Death Dis. 2013;4:e719. doi: 10.1038/cddis.2013.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Del Duca D, Werbowetski T, Del Maestro RF. J. Neurooncol. 2004;67:295. doi: 10.1023/b:neon.0000024220.07063.70. [DOI] [PubMed] [Google Scholar]
- 69.Deisboeck TS, Berens ME, Kansal a R, Torquato S, Stemmer-Rachamimov a O, a Chiocca E. Cell Prolif. 2001;34:115. doi: 10.1046/j.1365-2184.2001.00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Werbowetski T, Bjerkvig R, Del Maestro RF. J. Neurobiol. 2004;60:71. doi: 10.1002/neu.10335. [DOI] [PubMed] [Google Scholar]
- 71.Wiercinska E, Naber HPH, Pardali E, Van Der Pluijm G, Van Dam H, Ten Dijke P. Breast Cancer Res. Treat. 2011;128:657. doi: 10.1007/s10549-010-1147-x. [DOI] [PubMed] [Google Scholar]
- 72.Naber HPH, Wiercinska E, Pardali E, Van Laar T, Nirmala E, Sundqvist A, Van Dam H, Van Der Horst G, Van Der Pluijm G, Heckmann B, Danen EHJ, Ten Dijke P. Cell. Oncol. 2012;35:19. doi: 10.1007/s13402-011-0058-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lam S, Wiercinska E, Amina FAST, Lodder K, ten Dijke P, Jochemsen AG. Breast Cancer Res. Treat. 2014;148:7. doi: 10.1007/s10549-014-3147-8. [DOI] [PubMed] [Google Scholar]
- 74.Hildegonda PHN, Drabsch Y, Snaar-Jagalska BE, ten Dijke P, van Laar T. Biochem. Biophys. Res. Commun. 2013;435:58. doi: 10.1016/j.bbrc.2013.04.037. [DOI] [PubMed] [Google Scholar]
- 75.Bernas LM, Foster PJ, Rutt BK. J. Neurosurg. 2007;106:306. doi: 10.3171/jns.2007.106.2.306. [DOI] [PubMed] [Google Scholar]
- 76.Castillo LRC, Oancea A-D, Stüllein C, Régnier-Vigouroux A. Nat. Publ. Gr. 2016 doi: 10.1038/srep28375. [DOI] [Google Scholar]
- 77.Gole B, Durán Alonso MB, Dolenc V, Lah TT. Pathol. Oncol. Res. 2009;15:711. doi: 10.1007/s12253-009-9175-8. [DOI] [PubMed] [Google Scholar]
- 78.Bell HS, Wharton SB, Leaver HA, Whittle IR. J. Neurosurg. 1999;91:989. doi: 10.3171/jns.1999.91.6.0989. [DOI] [PubMed] [Google Scholar]
- 79.Reynolds DS, Tevis KM, Colson YL, Zaman MH, Grinstaff MW. Sci. Rep. doi: 10.1038/s41598-017-10863-4. n.d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smalley KSM, Herlyn M. Ann. N. Y. Acad. Sci. 2005;1059:16. doi: 10.1196/annals.1339.005. [DOI] [PubMed] [Google Scholar]
- 81.Vultur a, Villanueva J, Krepler C, Rajan G, Chen Q, Xiao M, Li L, a Gimotty P, Wilson M, Hayden J, Keeney F, Nathanson KL, Herlyn M. Oncogene. 2014;33:1850. doi: 10.1038/onc.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klein RM, Aplin AE. Cancer Res. 2009;69:2224. doi: 10.1158/0008-5472.CAN-08-3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Klein RM, Higgins PJ. Mol. Cancer. 2011;10:114. doi: 10.1186/1476-4598-10-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sundqvist a, Zieba A, Vasilaki E, Herrera Hidalgo C, Söderberg O, Koinuma D, Miyazono K, Heldin C-H, Landegren U, ten Dijke P, van Dam H. Oncogene. 2012;32:3606. doi: 10.1038/onc.2012.370. [DOI] [PubMed] [Google Scholar]
- 85.Rasanen K, Sriswasdi S, Valiga A, Tang H-Y, Zhang G, Perego M, Somasundaram R, Li L, Speicher K, Klein-Szanto AJ, Basu D, Rustgi AK, Speicher DW, Herlyn M. Mol. Cell. Proteomics. 2013;12:3778. doi: 10.1074/mcp.M113.029587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pettee KM, Dvorak KM, Nestor-Kalinoski AL, Eisenmann KM. PLoS One. 2014;9:e90371. doi: 10.1371/journal.pone.0090371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nam SH, Kim DGD, Lee M, Lee D, Kwak TK, Kang M, Ryu J, Kim H, Song HE, Choi J, Lee G, Kim SS, Park SH, Kim DGD, Kwon NH, Kim TY, Thiery JP, Kim SS, Lee JW. Oncotarget. 2015 [Google Scholar]
- 88.Sodek KL, Ringuette MJ, Brown TJ. Int. J. Cancer. 2009;124:2060. doi: 10.1002/ijc.24188. [DOI] [PubMed] [Google Scholar]
- 89.Naber H, Wiercinska E, ten Dijke P, van Laar T. J. Vis. Exp. 2011:1. doi: 10.3791/3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haeger A, Krause M, Wolf K, Friedl P. Biochim. Biophys. Acta - Gen. Subj. 2014;1840:2386. doi: 10.1016/j.bbagen.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 91.Basu D, Bewley AF, Sperry SM, Montone KT, Gimotty PA, Rasanen K, Facompre ND, Weinstein GS, Nakagawa H, Diehl JA, Rustgi AK, Herlyn M. Mol. Cancer Ther. 2013;12:2176. doi: 10.1158/1535-7163.MCT-12-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ayuso JM, Basheer HA, Monge R, Sánchez-Álvarez P, Doblaré M, Shnyder SD, Vinader V, Afarinkia K, Fernández LJ, Ochoa I. PLoS One. 2015;10:e0139515. doi: 10.1371/journal.pone.0139515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liao O, Hu Y, Zhao Y, Zhou T, Zhang O. Chin. Med. J. (Engl) 2010;123:1871. [PubMed] [Google Scholar]
- 94.Zhang W, Li C, Baguley BC, Zhou F, Zhou W, Shaw JP, Wang Z, Wu Z, Liu J. Anal. Biochem. 2016;515:47. doi: 10.1016/j.ab.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 95.Choi J, Kang M, Nam SH, Lee G-H, Kim H-J, Ryu J, Cheong JG, Jung JW, Kim TY, Lee H-Y, Lee JW. Lung Cancer. 2015;90:22. doi: 10.1016/j.lungcan.2015.06.023. [DOI] [PubMed] [Google Scholar]
- 96.Smalley KSM, Contractor R, Nguyen TK, Xiao M, Edwards R, Muthusamy V, King AJ, Flaherty KT, Bosenberg M, Herlyn M, Nathanson KL. Cancer Res. 2008;68:5743. doi: 10.1158/0008-5472.CAN-08-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schurigt U, Sevenich L, Vannier C, Gajda M, Schwinde A, Werner F, Stahl A, Von Elverfeldt D, Becker AK, Bogyo M, Peters C, Reinheckel T. Biol. Chem. 2008;389:1067. doi: 10.1515/BC.2008.115. [DOI] [PubMed] [Google Scholar]
- 98.An Z, Gluck CB, Choy ML, Kaufman LJ. Cancer Lett. 2010;292:215. doi: 10.1016/j.canlet.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 99.Mehta G, Hsiao AY, Ingram M, Luker GD, Takayama S. J. Control. Release. 2012;164:192. doi: 10.1016/j.jconrel.2012.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.An Z, Kavanoor K, Choy ML, Kaufman LJ. Colloids Surfaces B Biointerfaces. 2009;70:114. doi: 10.1016/j.colsurfb.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 101.Charoen KM, Fallica B, Colson YL, Zaman MH, Grinstaff MW. Biomaterials. 2014;35:2264. doi: 10.1016/j.biomaterials.2013.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Korff T, Augustin HG. J. Cell Biol. 1998;143:1341. doi: 10.1083/jcb.143.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Korff T, Augustin HG. J. Cell Sci. 1999;112(Pt 1):3249. doi: 10.1242/jcs.112.19.3249. [DOI] [PubMed] [Google Scholar]
- 104.Mohan R, Hammers H, Bargagna-Mohan P, Zhan X, Herbstritt C, Ruiz A, Zhang L, Hanson A, Conner B, Rougas J, Pribluda V. Angiogenesis. 2004;7:115. doi: 10.1007/s10456-004-1026-3. [DOI] [PubMed] [Google Scholar]
- 105.Potente M, Urbich C, Sasaki KI, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho Ra, Zeiher AM, Dimmeler S. J. Clin. Invest. 2005;115:2382. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Diehl F, Rössig L, Zeiher AM, Dimmeler S, Urbich C. Blood. 2007;109:1472. doi: 10.1182/blood-2006-08-039651. [DOI] [PubMed] [Google Scholar]
- 107.Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. Circ. Res. 2007;101:59. doi: 10.1161/CIRCRESAHA.107.153916. [DOI] [PubMed] [Google Scholar]
- 108.Scheidegger F, Ellner Y, Guye P, a Rhomberg T, Weber H, Augustin HG, Dehio C. Cell. Microbiol. 2009;11:1088. doi: 10.1111/j.1462-5822.2009.01313.x. [DOI] [PubMed] [Google Scholar]
- 109.Solinas M, Massi P, Cantelmo A, Cattaneo M, Cammarota R, Bartolini D, Cinquina V, Valenti M, Vicentini L, Noonan D, Albini A, Parolaro D. Br. J. Pharmacol. 2012;167:1218. doi: 10.1111/j.1476-5381.2012.02050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Breyer J, Samarin J, Rehm M, Lautscham L, Fabry B, Goppelt-Struebe M. Biochem. Pharmacol. 2012;83:616. doi: 10.1016/j.bcp.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 111.Jankowski V, Tölle M, Tran TNA, van der Giet M, Schuchardt M, Lehmann K, Janke D, Flick B, Ortiz A, Ortiz AA, Sanchez-Niño MD, Sanchez NMD, Tepel M, Zidek W, Jankowski J. PLoS One. 2013;8:e68575. doi: 10.1371/journal.pone.0068575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hawinkels LJAC, Kuiper P, Wiercinska E, Verspaget HW, Liu Z. Cancer Res. 2010;14:4141. doi: 10.1158/0008-5472.CAN-09-4466. [DOI] [PubMed] [Google Scholar]
- 113.Haspel HC, Scicli GM, McMahon G, Scicli a G. Microvasc. Res. 2002;63:304. doi: 10.1006/mvre.2001.2383. [DOI] [PubMed] [Google Scholar]
- 114.Zeitlin BD, Dong Z, Nör JE. Lab. Investig. 2012;92:988. doi: 10.1038/labinvest.2012.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kilarski WW, Jura N, Gerwins P. Lab. Investig. 2005;85:643. doi: 10.1038/labinvest.3700255. [DOI] [PubMed] [Google Scholar]
- 116.Heiss M, Hellstrom M, Kalen M, May T, Weber H, Hecker M, Augustin HG, Korff T. FASEB J. 2015;29:3076. doi: 10.1096/fj.14-267633. [DOI] [PubMed] [Google Scholar]
- 117.Korff T, Kimmina S, Martiny-Baron G, Augustin HG. FASEB J. 2001;15:447. doi: 10.1096/fj.00-0139com. [DOI] [PubMed] [Google Scholar]
- 118.Gluzman Z, Koren B, Preis M, Cohen T, Tsaba A, Cosset F-L, Shofti R, Lewis BS, Virmani R, Flugelman MY. Biochem. Biophys. Res. Commun. 2007;359:263. doi: 10.1016/j.bbrc.2007.05.097. [DOI] [PubMed] [Google Scholar]
- 119.Verseijden F, Posthumus-van Sluijs SJ, Pavljasevic P, Hofer SOP, van Osch GJVM, Farrell E. Tissue Eng. Part A. 2010;16:101. doi: 10.1089/ten.TEA.2009.0106. [DOI] [PubMed] [Google Scholar]
- 120.Wenger A, Kowalewski N, Stahl A, Mehlhorn AT, Schmal H, Stark GB, Finkenzeller G. Cells. Tissues. Organs. 2005;181:80. doi: 10.1159/000091097. [DOI] [PubMed] [Google Scholar]
- 121.Stahl A, Wu X, Wenger A, Klagsbrun M, Kurschat P. FEBS Lett. 2005;579:5338. doi: 10.1016/j.febslet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 122.Trapp V, Parmakhtiar B, Papazian V, Willmott L, Fruehauf JP. Angiogenesis. 2010;13:305. doi: 10.1007/s10456-010-9187-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Upreti M, Jamshidi-Parsian A, a Koonce N, Webber JS, Sharma SK, Asea AA, Mader MJ, Griffin RJ. Transl. Oncol. 2011;4:365. doi: 10.1593/tlo.11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Correa de Sampaio P, Auslaender D, Krubasik D, Failla AV, Skepper JN, Murphy G, English WR. PLoS One. 2012;7:e30753. doi: 10.1371/journal.pone.0030753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ehsan SM, Welch-Reardon KM, Waterman ML, Hughes CCW, George SC. Integr. Biol. 2014;6:603. doi: 10.1039/c3ib40170g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mason BN, Starchenko A, Williams RM, Bonassar LJ, Reinhart-King Ca. Acta Biomater. 2013;9:4635. doi: 10.1016/j.actbio.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Armstrong SJ, Wiberg M, Terenghi G, Kingham PJ. Tissue Eng. 2007;13:2863. doi: 10.1089/ten.2007.0055. [DOI] [PubMed] [Google Scholar]
- 128.Kraus D, Boyle V, Leibig N, Stark G, Penna V. J. Neurosci. Methods. 2015;246:97. doi: 10.1016/j.jneumeth.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 129.Theiss F, Mirsaidi A, Mhanna R, Kümmerle J, Glanz S, Bahrenberg G, Tiaden AN, Richards PJ. Biomaterials. 2015;69:99. doi: 10.1016/j.biomaterials.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 130.Te Hou Y, Ijima H, Takei T, Kawakami K. J. Biosci. Bioeng. 2011;112:265. doi: 10.1016/j.jbiosc.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 131.Hou Y, Ijima H, Shirakigawa N, Takei T, Kawakami K. Biochem. Eng. J. 2012;69:172. [Google Scholar]
- 132.Murphy KC, Fang SY, Leach JK. Cell Tissue Res. 2014;357:91. doi: 10.1007/s00441-014-1830-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Martinez EC, Vu D-T, Wang J, Lilyanna S, Ling LH, Gan SU, Tan AL, Phan TT, Lee CN, Kofidis T. Stem Cells Dev. 2013;22:3087. doi: 10.1089/scd.2013.0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nishikawa Y, Tokusashi Y, Kadohama T, Nishimori H, Ogawa K. Exp. Cell Res. 1996;223:357. doi: 10.1006/excr.1996.0091. [DOI] [PubMed] [Google Scholar]
- 135.Nishikawa Y, Doi Y, Watanabe H, Tokairin T, Omori Y, Su M, Yoshioka T, Enomoto K. Am. J. Pathol. 2005;166:1077. doi: 10.1016/S0002-9440(10)62328-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nishikawa Y, Sone M, Nagahama Y, Kumagai E, Doi Y, Omori Y, Yoshioka T, Tokairin T, Yoshida M, Yamamoto Y, Ito A, Sugiyama T, Enomoto K. J. Cell. Biochem. 2013;114:831. doi: 10.1002/jcb.24424. [DOI] [PubMed] [Google Scholar]
- 137.Desmaison A, Frongia C, Grenier K, Ducommun B, Lobjois V. PLoS One. 2013;8:e80447. doi: 10.1371/journal.pone.0080447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ho WJ, Pham EA, Kim JW, Ng CW, Kim JH, Kamei DT, Wu BM. Cancer Sci. 2010;101:2637. doi: 10.1111/j.1349-7006.2010.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Härmä V, Schukov H-P, Happonen A, Ahonen I, Virtanen J, Siitari H, Åkerfelt M, Lötjönen J, Nees M. PLoS One. 2014;9:e96426. doi: 10.1371/journal.pone.0096426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Peter HJ, Gerber H, Studer H, Groscurth P, Zakarija M. Endocrinology. 1991;128:211. doi: 10.1210/endo-128-1-211. [DOI] [PubMed] [Google Scholar]
- 141.Li Q, Chen C, Kapadia A, Zhou Q, Harper MK, Schaack J, Labarbera DV. J. Biomol. Screen. 2011;16:141. doi: 10.1177/1087057110392995. [DOI] [PubMed] [Google Scholar]
- 142.Muir CP, Adams Ma, Graham CH. Breast Cancer Res. Treat. 2006;96:169. doi: 10.1007/s10549-005-9076-9. [DOI] [PubMed] [Google Scholar]
- 143.Li L, Fukunaga-kalabis M, Herlyn M. Hum. Cell Cult. Protoc. 2012;806:15. doi: 10.1007/978-1-61779-367-7_2. [DOI] [PubMed] [Google Scholar]