

Abstract
Introduction
Despite extensive multidisciplinary team (MDT) assessment, some patients have interstitial lung disease (ILD) that is considered unclassifiable (uILD), for which there are currently no approved treatments. This study will assess the efficacy and safety of the antifibrotic pirfenidone in treating uILD.
Methods and analysis
This double-blind, randomised, placebo-controlled phase II trial is enrolling adults with fibrosing ILD, including uILD that fulfils proposed research criteria for interstitial pneumonia with autoimmune features (IPAF), that cannot be classified with moderate or high confidence to any category of ILD following MDT discussion. Study participants must have >10% fibrosis on high-resolution CT scan within the previous 12 months, forced vital capacity (FVC) ≥45% and diffusing capacity of the lung for carbon monoxide ≥30% of predicted values. Study participants will be randomised to receive 801 mg pirfenidone or placebo three times daily for 24 weeks. The efficacy of pirfenidone vs placebo will be assessed by daily measurement of FVC using a handheld spirometer over the treatment period. Other functional parameters, patient-reported outcomes, samples for biomarker analysis and safety endpoints will be collected. Additionally, the study will assess the efficacy and safety of pirfenidone with and without concomitant mycophenolate mofetil treatment and in study participants with or without IPAF.
Ethics and dissemination
This trial is being conducted in accordance with the International Conference on Harmonisation E6 guideline for Good Clinical Practice, Declaration of Helsinki and local laws for countries in which the research is conducted.
Trial registration number
Keywords: unclassifiable interstitial lung disease, interstitial pneumonia with autoimmune features, antifibrotic, pirfenidone
Key messages.
This trial, assessing the efficacy and safety of pirfenidone in patients with fibrosing uILD, will be the first controlled study assessing a potential treatment option solely in this population.
The study design has considered the definition of uILD, current treatment practice in uILD and the most appropriate measurements of treatment efficacy in this population.
It is hoped that these methodological considerations will allow meaningful clinical data to be generated that would inform treatment strategies and improve outcomes for patients with fibrotic uILD.
Introduction
Interstitial lung disease (ILD) comprises a large group of diseases characterised by inflammation or fibrosis of pulmonary tissue.1 2 ILDs can be caused by environmental exposures or may be secondary to another condition, such as connective tissue disease (CTD) or sarcoidosis; alternatively, ILDs may not have a clear predisposing factor (idiopathic interstitial pneumonias (IIPs)).1 2 Idiopathic pulmonary fibrosis (IPF) is the most common IIP.3 Other IIPs include idiopathic non-specific interstitial pneumonia, respiratory bronchiolitis-ILD, desquamative interstitial pneumonia, cryptogenic organising pneumonia, acute interstitial pneumonia and some rare forms of IIP.2 4
However, 4%–24% of patients with ILD cannot be given a specific diagnosis, even after thorough investigation by a multidisciplinary team (MDT)5–11; in these cases, the disease is considered an ‘unclassifiable ILD’ (uILD).1–4 12 An ILD might be labelled unclassifiable for various reasons, including a lack of biopsy, an inadequate or non-diagnostic biopsy and/or major discrepancies between clinical, radiological and pathological findings.1 3 Therefore, uILD represents a heterogeneous collection of undiagnosed ILDs with patients displaying clinical features of IPF and other non-IPF ILDs.5 Diagnosis of uILD is associated with survival rates and disease progression that are either intermediate between those associated with IPF and non-IPF ILD or similar to those of non-IPF ILD.5 11
Interstitial pneumonia with autoimmune features (IPAF) is a specific research classification scheme that clinically remains within the category of uILD because, although a variety of clinical, serological or pulmonary morphological autoimmune features may be identified, individuals with IPAF do not have a characterisable CTD.13 An estimated 7% of patients with ILD have IPAF14; these patients cannot be diagnosed with a specific type of IIP or CTD-ILD13 and seem to have similar or marginally better survival rates than patients with IPF.14 15
Determining treatment strategies for patients with features of multiple diagnoses, such as those with IPF or chronic hypersensitivity pneumonitis,1 13 can be particularly challenging, as immunosuppressive or antifibrotic approaches are both possible options.1 Although there have been no controlled clinical trials in patients with uILD and no pharmacological treatments are approved for the treatment of these populations, data have been published on the treatment of specific patients with uILD with immunosuppressive therapies and/or corticosteroids, which were generally associated with good initial responses to treatment.16–18 Clinical trials have demonstrated that the antifibrotic drugs pirfenidone and nintedanib reduce disease progression in patients with IPF, and both are approved for the treatment of IPF.19–21
Given the overlap between clinical, radiological and histopathological features of IPF and uILD, antifibrotic therapy may be beneficial in patients with uILD, particularly in cases characterised by considerable fibrosis. The current double-blind, randomised, placebo-controlled phase II trial therefore aims to evaluate the efficacy and safety of pirfenidone in patients with fibrosing uILD, including those who meet proposed research criteria for IPAF.
Methods and analysis
Study design
This is a multicentre, international, double-blind, two-arm, randomised, placebo-controlled phase II trial (Clinicaltrials.gov: NCT03099187) that plans to enrol approximately 250 patients across 60 clinical centres in Australia, Canada, Europe and Israel (for details of study sites visit: https://clinicaltrials.gov/ct2/show/study/NCT03099187). Patient enrolment began in April 2017. The trial will evaluate the efficacy and safety of pirfenidone in study participants over 24 weeks (figure 1). Study data will be managed according to the procedures described in online supplementary appendix 1.
Figure 1.

Study design. *Washout period for study participants taking prohibited medications prior to screening; patients not taking a prohibited medication will forgo the washout period and directly enter screening. †Informed consent may be obtained either at the washout (if applicable) or screening visits and must be obtained before any trial-specific screening procedure is performed. ‡After completion of the treatment period and follow-up visit, study participants will be given the opportunity to take part in an open-label extension of pirfenidone for up to 12 months; a final follow-up visit will be performed 4 weeks after the last open-label dose of pirfenidone. ICF, informed consent form.
bmjresp-2018-000289supp001.pdf (89.7KB, pdf)
Study participants
Patients aged ≥18–85 years with fibrosing uILD will be eligible for recruitment into this study. This trial is being conducted in accordance with the International Conference on Harmonisation E6 guideline for Good Clinical Practice and the principles of the Declaration of Helsinki, or the laws and regulations of the countries in which the research is conducted, whichever affords the greater protection to the individual. Informed consent will be obtained from each patient by the study investigator before any trial-specific screening procedure is performed. Fibrosing uILD will be defined as cases of fibrosing ILD that cannot be classified with moderate or high confidence to any category of ILD after MDT discussion. Levels of confidence in a diagnosis will be defined as follows: (1) high: a specific diagnosis is highly likely; (2) moderate: the MDT arrives at a working diagnosis of a particular ILD that is sufficient to lead to a specific therapeutic strategy; (3) low: the MDT may have a suspicion of a particular ILD but considers the available evidence insufficient (ie, <50% likelihood22) to inform therapeutic strategy. The MDT at each centre will use all available clinical (comprising medical history including prior exposures, physical examination, pulmonary function and exercise capacity tests and so on), radiological and pathological evidence to attempt to diagnose study participants with a category of fibrosing ILD and may involve rheumatology expertise at their discretion. If a surgical lung biopsy or transbronchial lung cryobiopsy was not undertaken to provide evidence for a specific diagnosis, the investigator should state the reason why this was the case. Study participants with uILD who fulfil proposed research classification criteria for IPAF13 will be eligible for inclusion. Study participants will be excluded from this study if IPF is the main differential or working diagnosis, irrespective of confidence level in the diagnosis. Examples of the types of patients who could be enrolled in this study can be found in the patient case studies (figures 2–4).
Figure 2.

Patient case study 1—chronic fibrosing ILD in the setting of IPAF. CCP, cyclic citrullinated peptide; DLco, diffusing capacity of the lung for carbon monoxide; FVC, forced vital capacity; HRCT, high-resolution CT; IIP, interstitial pneumonia; ILD, interstitial lung disease; IPAF, interstitial pneumonia with autoimmune features; MDT, multidisciplinary team; UIP, usual interstitial pneumonia.
Figure 3.
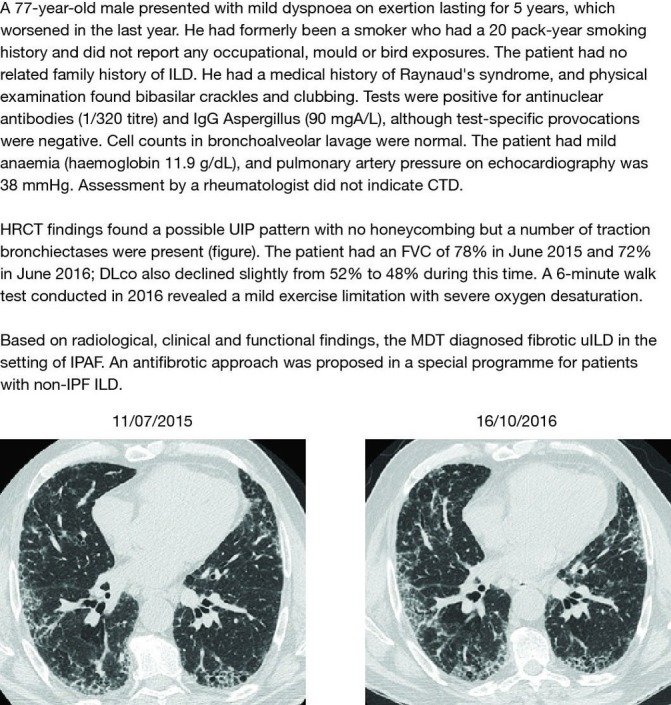
Patient case study 2—fibrotic uILD in the setting of IPAF. CTD, connective tissue disease; DLco, diffusing capacity of the lung for carbon monoxide; FVC, forced vital capacity; HRCT, high-resolution CT; ILD, interstitial lung disease; IPAF, interstitial pneumonia with autoimmune features; IPF, idiopathic pulmonary fibrosis; MDT, multidisciplinary team; uILD, unclassifiable interstitial lung disease; UIP, usual interstitial pneumonia.
Figure 4.

Patient case study 3—low-confidence chronic hypersensitivity pneumonitis. DLco, diffusing capacity of the lung for carbon monoxide; FVC, forced vital capacity; HRCT, high-resolution CT; MDT, multidisciplinary team; uILD, unclassifiable interstitial lung disease.
Eligible participants must have >10% extent of fibrosis (eg, reticulation) on visual scoring of a high-resolution CT (HRCT) scan, as assessed by the thoracic radiologist at each centre, within the previous 12 months to be considered for the trial, with forced vital capacity (FVC) ≥45% and diffusing capacity of the lung for carbon monoxide (DLco) ≥30% of predicted values. Study participants must also have progressive disease defined as either absolute decline in per cent predicted FVC >5%23 or significant symptomatic worsening not due to cardiac, pulmonary, vascular or other causes (as determined by the investigator) within the previous 6 months. Other inclusion and exclusion criteria for this study are listed in box 1.
Box 1. Entry criteria.
Description of inclusion criteria
Extent of fibrosis >10% on HRCT (visual scoring) within the last 12 months.
Spirometry criteria: FVC ≥45% predicted, DLco ≥30% predicted, FEV1/FVC ratio ≥0.7.
6MWD ≥150 m.
Progressive disease: absolute decline in per cent predicted FVC >5% or significant symptomatic worsening not due to cardiac, pulmonary, vascular or other causes in the previous 6 months.
For women of childbearing potential: agreement to remain abstinent (refrain from heterosexual intercourse) or use a non-hormonal or hormonal contraceptive method with a failure rate of <1% per year during the treatment period and for at least 58 days after the last dose of trial treatment.
For men: agreement to remain abstinent (refrain from heterosexual intercourse) or use contraceptive measures and agreement to refrain from donating sperm.
Signed informed consent form and able to comply with the study protocol, according to the investigator’s judgement.
Description of exclusion criteria
Study participants previously treated with pirfenidone or nintedanib.
Participation in a trial of an investigational medicinal product within the last 4 weeks.
Drug treatment for any type of pulmonary hypertension (eg, sildenafil, ERA).
Concomitant use of fluvoxamine.
Significant coexistent emphysema (extent greater than extent of fibrosis on HRCT within the last 12 months).
Clinical evidence of any active infection which, according to the investigator’s judgement, may interfere with trial conduct or measurement of pulmonary function or impact the course of the ILD.
History of unstable angina or myocardial infarction during the previous 6 months.
An ECG with a heart-rate-corrected QT interval (corrected using QTcF) ≥500 ms at screening or a family or personal history of long QT syndrome.
-
Any history of hepatic impairment, elevation of transaminase enzymes or the confirmation of any of the following liver-function test criteria above the specified limits:
Total bilirubin above the ULN.
AST or ALT >1.5 × ULN.
Alkaline phosphatase >2.0 × ULN.
Creatinine clearance <30 mL/min, calculated using the Cockcroft-Gault formula.
Significant other-organ comorbidity, including hepatic or renal impairment.
Any serious medical condition, clinically significant abnormality on an ECG at screening or laboratory test results (haematology, serum chemistry and urinalysis) which, in the opinion of the investigator, may pose an additional risk to the study participant following the administration of trial treatment.
Planned major surgery during the trial.
Predicted life expectancy <12 months or on an active transplant waiting list.
Use of any tobacco product in the 12 weeks prior to the start of screening or any unwillingness to abstain from their use through to the follow-up visit.
Illicit drug or alcohol abuse within 12 months prior to screening, according to the investigator’s judgement.
Pregnant or lactating or intending to become pregnant during the trial.
A positive urine pregnancy test, which was confirmed with a positive serum pregnancy test.
Women of childbearing potential not using a reliable contraceptive method.
Previous intolerance or allergy to the trial treatment.
Hypersensitivity to the active substance or to any of the excipients of pirfenidone.
History of angioedema.
6MWD, 6 min walk distance; ALT, alanine aminotransferase; AST, aspartate aminotransferase; DLco, diffusing capacity of the lung for carbon monoxide; ERA, endothelin receptor antagonist; FVC, forced vital capacity; FEV1, forced expiratory volume in 1 second; HRCT, high-resolution CT; ILD, interstitial lung disease; QTcF, Fridericia’s correction formula; QT, interval time between Q and T waves; ULN, upper limit of normal.
Study drug administration and blinding
Enrolled patients will be randomised on Day 1 of the study in a 1:1 ratio to receive 2403 mg/day pirfenidone or placebo. The randomisation process will be conducted using a validated interactive voice or web-based response system (IxRS). To prevent selection bias and allow subgroup comparison, randomisation will be stratified by use of concomitant mycophenolate mofetil (MMF; including mycophenolate sodium and mycophenolic acid) and presence or absence of IPAF.
Pirfenidone will be administered orally in 267 mg capsules taken with food at the same times each day. The dose of pirfenidone will be titrated over 2 weeks from an initial dose of 801 mg/day during Week 1 (one capsule three times a day) to 1602 mg/day during Week 2 (two capsules three times a day) to a maintenance dose of 2403 mg/day from Week 3 onwards (three capsules three times a day). The dose of pirfenidone used for each individual study participant will be at the discretion of the investigator. Temporary dose reduction, treatment interruption or discontinuation will be considered to manage treatment-emergent adverse events. Treatment will be discontinued in patients who become pregnant, experience hepatic or renal impairment or angioedema or have substantial liver-function test elevations. Patients who are non-adherent with their dosing regimen or titration schedule will be withdrawn from the study.
To ensure blinding of the treatment group to investigators, study participants and the sponsor, study participants randomised to the placebo group will be administered placebo capsules with identical appearance, size and taste to pirfenidone capsules. If unblinding is necessary due to, for example, a serious adverse event, the study investigator will be able to reveal the patient’s allocated intervention by contacting the IxRS.
Concomitant therapies
Study participants who are receiving a stable dose of MMF for >3 months before screening and who are expected to remain on a stable dose of MMF, are eligible for inclusion in the trial. MMF has been used to successfully manage patients with a diverse range of ILDs (see Discussion section) and recruitment into the trial would be limited if concomitant MMF treatment was not permitted for patients receiving therapeutic benefit from this treatment. Use of other immunosuppressants is not permitted during the trial. Study participants may not receive treatment with high-dose systemic corticosteroids (ie, >15 mg/day prednisolone or equivalent) for >4 weeks during the trial. N-acetyl cysteine (NAC) is not permitted for the treatment of fibrotic lung disease within 4 weeks of the screening period; intermittent use of NAC for other conditions is permitted. Inhibitors or inducers of cytochrome P450 1A2 are not permitted during the trial due to the potential for drug interactions altering the exposure to pirfenidone. Study participants receiving prohibited medications will need to taper or discontinue these during the 4 weeks before screening (washout period).
Study objectives and endpoints
The primary objective of this study is to evaluate the efficacy of pirfenidone vs placebo on FVC decline. This will be assessed by daily measurement of FVC using a handheld spirometer over the 24-week double-blind treatment period. Study participants will perform a single spirometry reading using a portable handheld Micro spirometer (CareFusion, Kent, England) at approximately the same time each day. Study participants will be given 60 min dedicated instruction on how to undertake spirometry at the screening visit. They will be asked to use the device during the screening period and will be subsequently retrained at the baseline visit, with additional refresher training provided after Month 1, between Months 2 and 3 and between Months 4 and 5. Measurements will be downloaded at each visit and patients with multiple missing values will be retrained either by the investigator at the centre or by a home nursing staff member on an additional visit.
The secondary objective is to evaluate the efficacy of pirfenidone vs placebo on other functional parameters and patient-reported outcomes (box 2 24). Clinic-based spirometry will be conducted at baseline and then every 4 weeks until Week 24 (or the early treatment discontinuation visit). All other assessments (DLco, 6 min walk distance, University of California–San Diego Shortness of Breath Questionnaire, St. George’s Respiratory Questionnaire and cough scores) will be conducted at baseline, Week 12 and Week 24 (or the early treatment discontinuation visit).
Box 2. Study endpoints.
Primary endpoint
Change in FVC (in mL) measured by daily home spirometry.
Secondary endpoints
Change in FVC (in mL and per cent predicted) measured by clinic-based spirometry.
Categorical change in FVC of >5% or >10% (absolute change in per cent predicted and relative change in mL), measured by both daily spirometry and spirometry during clinic visits.
Change in per cent predicted DLco.
Change in 6MWD.
Change in UCSD-SOBQ score.
Change in score in Leicester Cough Questionnaire.
Change in cough visual analogue scale.
Change in total and subscores of the SGRQ.
Non-elective hospitalisation, both respiratory and all cause.
Incidence of, and time to first, investigator-reported acute exacerbations (analogous to the criteria for acute exacerbation of IPF proposed by Collard et al 24).
PFS, defined as the time to the first occurrence of a >10% absolute decline in per cent predicted FVC (measured during a clinic visit), a >50 m decline of 6MWD or death.
PFS, alternatively defined as the time to the first occurrence of a >10% relative decline in FVC (measured during a clinic visit), non-elective respiratory hospitalisation or death.
Time to death from any cause.
Time to death from respiratory diseases.
Safety endpoints
Nature, frequency, severity and timing of treatment-emergent adverse events.
Dose reductions and treatment interruptions.
Clinical laboratory test results.
12-lead ECGs.
Withdrawals from trial treatment or trial discontinuations.
6MWD, 6 min walk distance; DLco, diffusing capacity of the lung for carbon monoxide; FVC, forced vital capacity; IPF, idiopathic pulmonary fibrosis; PFS, progression-free survival; SGRQ, St. George’s Respiratory Questionnaire; UCSD-SOBQ, University of California–San Diego Shortness of Breath Questionnaire.
The safety and tolerability of pirfenidone in this population will be assessed by collection of the nature, frequency, severity and timing of treatment-emergent adverse events. Information on dose reductions, treatment interruptions and premature discontinuation of treatment will also be collected.
Biomarker collection
To evaluate the impact of pirfenidone on potential inflammatory and fibrotic biomarkers associated with fibrosis and ILD, serum, plasma and whole blood will be collected from study participants at baseline, Week 4, Week 12 and Week 24 (or the early treatment discontinuation visit). Transcriptomic and proteomic profiling of markers associated with molecular pathways and cellular processes of lung injury and fibrosis will be measured.
Research Biosample Repository (RBR) whole-blood samples for DNA extraction will also be collected at baseline to examine genetic polymorphisms and their potential role in the pathogenesis and associated clinical outcomes of uILD. Specimens for the RBR will be collected from study participants who give specific consent to participate in this optional research. RBR specimens will be stored until they are no longer needed or until they are exhausted. The RBR storage period will be in accordance with informed consent and applicable laws, such as health authority requirements.
Statistical analysis
The planned sample size of 250 patients is based on the statistical hypothesis of the primary endpoint and assumes 80% power and a two-sided α of 5% using a student’s t-test. After inspection of historical data, it is assumed that, over the course of the trial, FVC decline in the placebo arm will be 85 mL with a common SD of 70 mL, which can be reduced to 60 mL with a common SD of 70 mL in the pirfenidone arm. In this scenario, 125 patients per treatment arm are needed to detect this treatment effect with 80% power.
The primary endpoint will be analysed in a two-step approach: first, individual FVC decline for each patient will be estimated by applying a linear regression model to daily home spirometry measurements during the 24-week treatment period. Second, mean FVC decline in each treatment arm, calculated using estimated FVC decline for each individual patient, will be compared using a student’s t-test with a two-sided significance level α=0.05. The primary analysis will be based on the intent-to-treat population; patients who discontinue treatment prematurely will be analysed based on the available data and no imputation method will be applied for missing data. A linear mixed-effects model will be included as a sensitivity analysis to account for codependency of daily FVC measurements.
For the secondary endpoints, all data from baseline to Week 24 will be used without imputation, and p values will be reported in a descriptive fashion with no adjustment for multiplicity (further details of statistical analysis of the secondary endpoints can be found in online supplementary appendix 2).
bmjresp-2018-000289supp002.pdf (73.6KB, pdf)
The safety analysis will include investigation of the nature, frequency, severity and timing of treatment-emergent adverse events. The incidence, type and severity of adverse events will be summarised according to primary System Organ Class and subcategorised by Medical Dictionary for Regulatory Activities version 19.1 preferred term. Adverse events Grade ≥3 according to National Cancer Institute Common Terminology Criteria for Adverse Events V.4.0, adverse events of special interest (cases of potential drug-induced liver injury) and serious adverse events will be analysed in a similar way to all adverse events. Descriptive statistics will be presented for dose reductions and treatment interruptions, with adverse events leading to these occurrences summarised.
Data Monitoring Committee
There are no planned efficacy interim analyses for this trial; however, an independent Data Monitoring Committee (iDMC) will perform interim safety analyses and advise on trial conduct at least three times during the trial (6, 12 and 18 months after the start of recruitment). Additional ad hoc meetings can be requested at any time by the iDMC or sponsor if necessary. The iDMC will be an independent body, unblinded to treatment allocation, who will recommend that the study should be continued, modified or stopped during each meeting.
Open-label extension study
All study participants will be offered the opportunity to receive open-label pirfenidone within the trial protocol during the follow-up visit at Week 28. In order to maintain blinding of the controlled period of the study, all study participants will discontinue treatment by Week 24 and return for a follow-up visit 4 weeks later; study participants eligible to participate in the 12-month extension will be initiated on open-label pirfenidone during this visit (restarting the dose titration from one capsule three times a day). During the long-term extension period, study participants will be evaluated for safety endpoints only. Study participants should be evaluated by the investigator approximately every 3 months during the 12-month safety follow-up (liver function tests will be measured monthly during the first 6 months); a final follow-up visit will take place 4 weeks after the last dose of pirfenidone is taken.
Discussion
This study will be the first randomised controlled trial of an antifibrotic agent specifically in patients with uILD and aims to establish whether pirfenidone reduces the rate of FVC decline in this population. A study of nintedanib is underway in patients with progressive, ‘physician-diagnosed fibrosing ILD’ including unclassifiable IIP25; however, the study of pirfenidone is unique in that only patients with uILD will be included. A number of important methodological questions have been considered in the study design in order to generate meaningful clinical data and provide important insights into treatment strategies for this patient population.
There is currently a lack of consensus on the definition of uILD, so the first key question in designing this trial was how best to properly identify patients with the greatest unmet need for inclusion in the study. In order to enrol patients for whom there is currently insufficient evidence available to inform a therapeutic strategy, uILD will be defined as fibrosing ILD (>10% fibrosis on HRCT) that cannot be classified with moderate or high confidence to any category of ILD and that cannot be classified as IPF at any level of confidence. In patients with IPF, a decline in FVC of ≥10% over 12 months is generally taken to indicate progressive disease; however, FVC decline in patients with uILD is not well understood and therefore a threshold of 5% absolute decline (or significant symptomatic worsening, which would indicate later FVC decline) over 6 months was selected, the aim being to avoid restricting the eligible population of patients and to only exclude patients with very stable disease. Nevertheless, variability between FVC measurements is a potential limitation with a 5% threshold.
Importantly, diagnosis of uILD must be made by consensus following an MDT discussion. The use of MDTs in the diagnosis of IPF has improved the confidence in diagnosis,26 and it is expected that making MDT discussion mandatory before the qualifying diagnosis of uILD can be made will ensure that study participants who can be diagnosed with a specific ILD are not included. Although the requirement for MDT discussion should reduce the chance that patients with a classifiable ILD are included in the study, it should be acknowledged that there may be differences in interpretation between centres, particularly in those centres with a lower ILD case load. In an attempt to minimise this, training and case studies were provided at an investigator meeting. In addition, it could be argued that practice may vary between expert and non-expert centres with regard to assigning a diagnosis of IPF or other ILDs and the number of patients who could be classified as having uILD. Future diagnosis of uILD should ideally be made at an expert centre to avoid misclassification and allow for close monitoring of these patients.
One particular area of contention in defining uILD is whether a surgical lung biopsy should be mandatory before designating ILD as unclassifiable.1 12 22 It can be argued that uILD in patients who have not undergone a biopsy should be labelled ‘unclassified’ rather than ‘unclassifiable’, as a biopsy may have provided the necessary information for a diagnosis.1 Although a biopsy may provide critical diagnostic information in patients where a diagnosis cannot be made from HRCT and other clinical tests, this is not always possible due to medical risk or patient choice.1 12 Both surgical lung biopsy and transbronchial lung cryobiopsy were included as options in this study. Cryobiopsy has been shown to provide meaningful data to aid MDT diagnosis of ILD and is generally considered to be less invasive than surgical lung biopsy.27 A diagnosis of uILD can be assigned regardless of whether a surgical lung biopsy has been performed, and the current study protocol does not specify which diagnostic tests should be conducted to make a diagnosis of uILD. The number of patients who undergo each procedure will be documented, and if a biopsy (either surgical biopsy or cryobiopsy) has not been conducted, then the protocol requests a reason to be given. Although inclusion of patients without biopsy data is a potential limitation, it does reflect clinical reality where many patients will not or cannot have a biopsy.
Another question relates to the inclusion of patients with uILD who fulfil research criteria for IPAF.13 From a clinical diagnostic perspective, these patients remain unclassifiable and will therefore be included in the trial. However, assessment of treatment effect in patients with IPAF may be limited by heterogeneity. Therefore, randomisation will be stratified at baseline depending on whether a study participant meets IPAF research criteria, so that the effect of pirfenidone in these participants can be analysed separately.
Current treatment practice for patients with uILD is another important factor considered in the design of this trial. Although no therapies have been recommended for treatment of patients with uILD, MMF has been used to successfully manage patients with a diverse range of ILDs in controlled clinical trials (in systemic sclerosis-related (SSc)-ILD)28 and observational studies (in CTD-ILD and IPAF)18 29 and is therefore a likely concomitant medication in patients screened for entry into this trial. The combination of pirfenidone and MMF has previously been studied in the phase II LOTUSS study of the safety and tolerability of pirfenidone in patients with SSc-ILD.30 A total of 63.5% of patients in the LOTUSS trial received MMF and there were no clinically significant effects on overall tolerability compared with patients who did not receive MMF.30 Therefore, this study allows participants to receive concomitant MMF therapy during the treatment period and, as in the LOTUSS trial, the effect of MMF on outcomes will be investigated by stratifying the population at randomisation based on whether or not they are receiving MMF at baseline. However, use of inhibitors or inducers of cytochrome P450 1A2, NAC for the treatment of fibrotic lung disease or other immunosuppressants besides MMF (eg, azathioprine or cyclophosphamide) will not be permitted in this study due to potential drug interactions or negative effects in combination with pirfenidone.28 29 31–33 The dose of pirfenidone (2403 mg/day) and initial dose titration used in this trial will be the same as the clinically recommended dose and titration used in patients with IPF.32 33 In the absence of any clinically approved treatment for uILD, study participants in the control group will receive placebo during the 24-week double-blind treatment period.
Methods of assessing efficacy in the study population are another important consideration to ensure that meaningful clinical data are collected. Change in FVC is a generally accepted measure of disease course and a predictor of mortality risk in patients with ILD and has been examined in clinical studies that enrolled patients with uILD.5 34 The primary endpoint in this trial will therefore assess rate of FVC decline. In order to ensure the 24-week treatment period is sufficient to observe any clinically significant differences in FVC decline, FVC will be measured daily by study participants using a handheld spirometer. Recent data collected in patients with IPF demonstrate the feasibility of asking patients to perform daily or weekly FVC measurements.35 36 Results from Russell et al suggest that daily FVC measurements correlated well with measurements recorded during clinic visits, and daily readings correlated with mortality over 3, 6 and 12 months.35 Weekly FVC measurements over 24 weeks have been found to result in enhanced precision and power compared with FVC measurements taken in clinic on Weeks 1 and 24.36 Furthermore, previous studies of pirfenidone in IPF have demonstrated a treatment effect within a 24-week timeframe.37
The analysis of the primary endpoint in this study will be via a linear regression model of individual FVC measurements made each day over the 24-week treatment period. It is acknowledged that daily measurements of FVC can be considered as dependent measurements and that a mixed-effects model may be more appropriate. Indeed, a mixed-effects model will be conducted as a sensitivity analysis. However, the appropriate methods available to calculate sample size and the power for such a model require assumptions on the correlation structure across all patients and observations a priori and this information is not available. Therefore, a simple linear regression model without assumptions on the aforementioned correlation structure was considered more appropriate for the purpose of sample size and power calculation. Furthermore, FVC decline will not be linear in every patient, as demonstrated by Russell et al.35 However, in the absence of any specific non-linear models for FVC decline over time, it was determined that a linear model was appropriate for this analysis.
Certain biomarkers, such as chemokine ligand 18 (CCL18), matrix-metalloproteinase 7 (MMP7), chemokine ligand 13 (CXCL13), cartilage oligomeric matrix protein (COMP), plasma surfactant protein-D and osteopontin, are differentially expressed in other fibrotic lung diseases38–42 and thus could be used to potentially distinguish subtypes of uILD. Therefore, samples will be collected during this study to allow an exploratory assessment of biomarkers in uILD. The exploratory biomarker analysis will aim to identify any biomarkers that change as a result of pirfenidone therapy or could be used to predict response to pirfenidone. In addition, the analysis will aim to identify protein or RNA biomarkers which are related to disease progression. Study participants will also be given the option to provide samples for DNA extraction at baseline to enable investigation of genetic polymorphisms and their potential role in the pathogenesis and associated clinical outcomes of uILD.
Summary
This randomised, controlled phase II trial of the efficacy and safety of pirfenidone in patients with fibrosing uILD, including those who meet proposed research criteria for IPAF, will be the first controlled study assessing a potential treatment option in this population. The study design has considered a number of methodological factors, including the definition of uILD, current treatment practice in uILD and the most appropriate measurements of treatment efficacy in this population. It is hoped that these methodological considerations will allow meaningful clinical data to be generated that would inform treatment strategies for fibrotic uILD and thereby improve patient outcomes.
Acknowledgments
Medical writing support was provided by Rebekah Waters of CMC AFFINITY, a division of Complete Medical Communications, Ltd., Manchester, UK, funded by F. Hoffmann-La Roche, Ltd. TMM is supported by an NIHR-funded Clinician Scientist Fellowship (NIHR Ref: CS-2013-13-017) and British Lung Foundation Chair in Respiratory Research (C17-3).
Footnotes
Contributors: All authors provided substantial contributions to the conception and/or design of the work, revised the manuscript critically for important intellectual content, approved the version to be published and agree to be accountable for all aspects of the work.
Funding: This study is sponsored by F. Hoffmann-La Roche, Ltd., Basel, Switzerland, which was involved in study design and will be involved in the collection, analysis and interpretation of the data. This trial was initially developed as part of an NIHR-funded Clinician Scientist Fellowship awarded to Toby M Maher (NIHR Ref: CS-2013-13-017) and subsequently adapted following discussions with F. Hoffmann-La Roche, Ltd.
Competing interests: TMM has received industry-academic research funding from GlaxoSmithKline R&D and UCB, has served on a clinical trial advisory board for GlaxoSmithKline R&D, has received stock options from Apellis and has received consultancy or speaker fees from AstraZeneca, Bayer, Biogen Idec, Boehringer Ingelheim, Cipla, GlaxoSmithKline R&D, ProMetic, Roche, Sanumed and UCB. TJC has received unrestricted educational grants, travel assistance and speaker fees from and served on an advisory board for Boehringer Ingelheim, has received unrestricted educational grants and speaker fees from and served on an advisory board for Roche, has received unrestricted educational grants from Actelion Ltd., Bayer, BMS and Sanofi and has served on an advisory board for AstraZeneca. AF serves as a consultant and steering committee member for Boehringer Ingelheim and Roche. MK, or his institution, has received unrestricted educational grants, speaker fees and research grants from Boehringer Ingelheim and Roche and has served on advisory boards for Boehringer Ingelheim and Roche. DJL is a steering committee member for this study and has received consulting fees from and has served on advisory boards for Genentech-Roche and Veracyte, has served on advisory boards for Boehringer Ingelheim, has served on an adjudication committee for Degge Group, has received lecture fees and fees for generation of educational content from and served on a steering committee for the France Foundation and has received consulting fees from FibroGen, Global Blood Therapeutics, ImmuneWorks, Patara Pharmaceuticals, Philips Respironics and Sanofi Genzyme. DJL’s institution, Columbia University, has received funding for clinical trials in IPF from Boehringer Ingelheim, FibroGen and Global Blood Therapeutics and has received consulting fees from the Pulmonary Fibrosis Foundation. MM-M, or her institution, has received grants from AstraZeneca, Boehringer Ingelheim, BRN, Chiesi, GlaxoSmithKline, InterMune and Roche. JA is an employee of F. Hoffmann-La Roche, Ltd. K-UK is an employee and shareholder of F. Hoffmann-La Roche, Ltd. VC receives consultancy and lecture fees and support for travel to medical meetings from Actelion and Roche, receives consultancy and lecture fees, support for travel to medical meetings and fees for development of educational presentations from Boehringer Ingelheim, receives consultancy and lecture fees from Novartis and Sanofi, receives consultancy fees from Bayer, Galapagos, GlaxoSmithKline and MSD, is a member of an adjudication committee for Gilead, is a chair of the DSMB for Promedior, is a member of the DSMB for Celgene and receives grants to his institution from Boehringer Ingelheim and Roche.
Ethics: The Ethics Committees of the local study centres: Australia: Bellberry Human Research Ethics Committee; Belgium: UZ Leuven campus Gasthuisberg Ethische commimissie onderzoek; Canada: UBC-Providence Health Care Research Institut; Czech Republic: Etická komise Všeobecné fakultní nemocnice v Praze; Denmark: Regionshuset, Viborg, Regionssekretariatet, Juridisk kontor De Videnskabsetiske Komiteer, For Region Midtjylland; Germany: Justus-Liebig-Universität Gießen, Ethik-Kommission des FB Medizin; Greece: National Ethics Committee; Ireland: Clinical Research Ethics Committee of the Cork Teaching Hospitals; Israel: Meir Medical Center, Helsinki Committee; Italy: Comitato Etico IRST IRCCS; Poland: Bioethics Committee of Medical University in Łódź; Portugal: CEIC-Comissão de Ética para Investigação Clínica; Spain: CEIC Hospital de Bellvitge; UK: North East-Newcastle & North Tyneside 1 Research Ethics Committee.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data statement: Qualified researchers may request access to individualpatient level data through the clinical study data request platform (www.clinicalstudydatarequest.com). Further details on Roche’s criteria for eligible studies are available here (https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx). For further details on Roche’s Global Policy on the Sharing of ClinicalInformation and how to request access to related clinical study documents, seehere (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
References
- 1. Skolnik K, Ryerson CJ. Unclassifiable interstitial lung disease: A review. Respirology 2016;21:51–6. 10.1111/resp.12568 [DOI] [PubMed] [Google Scholar]
- 2. Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013;19:453–9. 10.1097/MCP.0b013e328363f48d [DOI] [PubMed] [Google Scholar]
- 3. American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med 2002;165:277–304. 10.1164/ajrccm.165.2.ats01 [DOI] [PubMed] [Google Scholar]
- 4. Travis WD, Costabel U, Hansell DM, et al. . An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013;188:733–48. 10.1164/rccm.201308-1483ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ryerson CJ, Urbania TH, Richeldi L, et al. . Prevalence and prognosis of unclassifiable interstitial lung disease. Eur Respir J 2013;42:750–7. 10.1183/09031936.00131912 [DOI] [PubMed] [Google Scholar]
- 6. Hyldgaard C, Hilberg O, Muller A, et al. . A cohort study of interstitial lung diseases in central Denmark. Respir Med 2014;108:793–9. 10.1016/j.rmed.2013.09.002 [DOI] [PubMed] [Google Scholar]
- 7. Karakatsani A, Papakosta D, Rapti A, et al. . Epidemiology of interstitial lung diseases in Greece. Respir Med 2009;103:1122–9. 10.1016/j.rmed.2009.03.001 [DOI] [PubMed] [Google Scholar]
- 8. Troy L, Glaspole I, Goh N, et al. . Prevalence and prognosis of unclassifiable interstitial lung disease. Eur Respir J 2014;43:1529–30. 10.1183/09031936.00003414 [DOI] [PubMed] [Google Scholar]
- 9. Zhang D, Liu Y. Surgical lung biopsies in 418 patients with suspected interstitial lung disease in China. Intern Med 2010;49:1097–102. 10.2169/internalmedicine.49.3225 [DOI] [PubMed] [Google Scholar]
- 10. Musellim B, Okumus G, Uzaslan E, et al. . Epidemiology and distribution of interstitial lung diseases in Turkey. Clin Respir J 2014;8:55–62. 10.1111/crj.12035 [DOI] [PubMed] [Google Scholar]
- 11. Hyldgaard C, Bendstrup E, Wells AU, et al. . Unclassifiable interstitial lung diseases: Clinical characteristics and survival. Respirology 2017;22:494–500. 10.1111/resp.12931 [DOI] [PubMed] [Google Scholar]
- 12. Cottin V, Wells A. Unclassified or unclassifiable interstitial lung disease: confusing or helpful disease category? Eur Respir J 2013;42:576–9. 10.1183/09031936.00107713 [DOI] [PubMed] [Google Scholar]
- 13. Fischer A, Antoniou KM, Brown KK, et al. . An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 2015;46:976–87. 10.1183/13993003.00150-2015 [DOI] [PubMed] [Google Scholar]
- 14. Ahmad K, Barba T, Gamondes D, et al. . Interstitial pneumonia with autoimmune features: Clinical, radiologic, and histological characteristics and outcome in a series of 57 patients. Respir Med 2017;123:56–62. 10.1016/j.rmed.2016.10.017 [DOI] [PubMed] [Google Scholar]
- 15. Oldham JM, Adegunsoye A, Valenzi E, et al. . Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J 2016;47:1767–75. 10.1183/13993003.01565-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Leung SC, Churg AM, Leipsic JA, et al. . Unclassifiable interstitial lung disease: an unresolved diagnostic dilemma. Respirol Case Rep 2015;3:85–8. 10.1002/rcr2.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sasano H, Hagiwara E, Kitamura H, et al. . Long-term clinical course of anti-glycyl tRNA synthetase (anti-EJ) antibody-related interstitial lung disease pathologically proven by surgical lung biopsy. BMC Pulm Med 2016;16:168 10.1186/s12890-016-0325-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chartrand S, Swigris JJ, Stanchev L, et al. . Clinical features and natural history of interstitial pneumonia with autoimmune features: A single center experience. Respir Med 2016;119:150–4. 10.1016/j.rmed.2016.09.002 [DOI] [PubMed] [Google Scholar]
- 19. King TE Jr, Bradford WZ, Castro-Bernardini S, et al. . A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2083–92. 10.1056/NEJMoa1402582 [DOI] [PubMed] [Google Scholar]
- 20. Noble PW, Albera C, Bradford WZ, et al. . Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011;377:1760–9. 10.1016/S0140-6736(11)60405-4 [DOI] [PubMed] [Google Scholar]
- 21. Richeldi L, du Bois RM, Raghu G, et al. . Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2071–82. 10.1056/NEJMoa1402584 [DOI] [PubMed] [Google Scholar]
- 22. Ryerson CJ, Corte TJ, Lee JS, et al. . A standardized diagnostic ontology for fibrotic interstitial lung disease. An international working group perspective. Am J Respir Crit Care Med 2017;196:1249–54. 10.1164/rccm.201702-0400PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Richeldi L, Ryerson CJ, Lee JS, et al. . Relative versus absolute change in forced vital capacity in idiopathic pulmonary fibrosis. Thorax 2012;67:407–11. 10.1136/thoraxjnl-2011-201184 [DOI] [PubMed] [Google Scholar]
- 24. Collard HR, Ryerson CJ, Corte TJ, et al. . Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med 2016;194:265–75. 10.1164/rccm.201604-0801CI [DOI] [PubMed] [Google Scholar]
- 25. Flaherty KR, Brown KK, Wells AU, et al. . Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir Res 2017;4:e000212 10.1136/bmjresp-2017-000212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walsh SLF, Wells AU, Desai SR, et al. . Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016;4:557–65. 10.1016/S2213-2600(16)30033-9 [DOI] [PubMed] [Google Scholar]
- 27. Tomassetti S, Wells AU, Costabel U, et al. . Bronchoscopic lung cryobiopsy increases diagnostic confidence in the multidisciplinary diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2016;193:745–52. 10.1164/rccm.201504-0711OC [DOI] [PubMed] [Google Scholar]
- 28. Tashkin DP, Roth MD, Clements PJ, et al. . Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med 2016;4:708–19. 10.1016/S2213-2600(16)30152-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fischer A, Brown KK, Du Bois RM, et al. . Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. J Rheumatol 2013;40:640–6. 10.3899/jrheum.121043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khanna D, Albera C, Fischer A, et al. . An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: the LOTUSS trial. J Rheumatol 2016;43:1672–9. 10.3899/jrheum.151322 [DOI] [PubMed] [Google Scholar]
- 31. Behr J, Bendstrup E, Crestani B, et al. . Safety and tolerability of acetylcysteine and pirfenidone combination therapy in idiopathic pulmonary fibrosis: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir Med 2016;4:445–53. 10.1016/S2213-2600(16)30044-3 [DOI] [PubMed] [Google Scholar]
- 32. Genentech. Product label: Full prescribing information - Esbriet® 2017. http://www.gene.com/download/pdf/esbriet_prescribing.pdf (accessed 19 Jun 2018).
- 33. European Medicines Agency. Summary of Product Characteristics - Esbriet (pirfenidone). 2018. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002154/human_med_001417.jsp&mid=WC0b01ac058001d124 (accessed 19 Jun 2018).
- 34. Solomon JJ, Ryu JH, Tazelaar HD, et al. . Fibrosing interstitial pneumonia predicts survival in patients with rheumatoid arthritis-associated interstitial lung disease (RA-ILD). Respir Med 2013;107:1247–52. 10.1016/j.rmed.2013.05.002 [DOI] [PubMed] [Google Scholar]
- 35. Russell AM, Adamali H, Molyneaux PL, et al. . Daily home spirometry: an effective tool for detecting progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2016;194:989–97. 10.1164/rccm.201511-2152OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johannson KA, Vittinghoff E, Morisset J, et al. . Home monitoring improves endpoint efficiency in idiopathic pulmonary fibrosis. Eur Respir J 2017;50:1602406 10.1183/13993003.02406-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Noble PW, Albera C, Bradford WZ, et al. . Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016;47:243–53. 10.1183/13993003.00026-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vuga LJ, Milosevic J, Pandit K, et al. . Cartilage oligomeric matrix protein in idiopathic pulmonary fibrosis. PLoS One 2013;8:e83120 10.1371/journal.pone.0083120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vuga LJ, Tedrow JR, Pandit KV, et al. . C-X-C motif chemokine 13 (CXCL13) is a prognostic biomarker of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2014;189:966–74. 10.1164/rccm.201309-1592OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rosas IO, Richards TJ, Konishi K, et al. . MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med 2008;5:e93 10.1371/journal.pmed.0050093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Prasse A, Pechkovsky DV, Toews GB, et al. . CCL18 as an indicator of pulmonary fibrotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheum 2007;56:1685–93. 10.1002/art.22559 [DOI] [PubMed] [Google Scholar]
- 42. White ES, Xia M, Murray S, et al. . Plasma surfactant protein-D, matrix metalloproteinase-7, and osteopontin index distinguishes idiopathic pulmonary fibrosis from other idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2016;194:1242–51. 10.1164/rccm.201505-0862OC [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjresp-2018-000289supp001.pdf (89.7KB, pdf)
bmjresp-2018-000289supp002.pdf (73.6KB, pdf)