

Abstract
Circulating naive T cells exist in a quiescent state. After TCR contact with the cognate peptide presented by APCs in secondary lymphoid structures, T cells undergo a period of rapid transcriptional changes that set the stage for fate-determining effector or memory programming. We describe a novel method to analyze TCR signaling pathway activation in nuclei isolated from primary mouse naive T cells after stimulation with natural peptide Ags. We prelabeled cells with cell tracking dye to easily distinguish CD8+ T cell nuclei from APC nuclei by conventional flow cytometry. Using this approach, we observed clear digital activation of NFAT1 transcription factor in OT-I T cells stimulated with OVA peptide presented by bulk splenocytes. OVA concentration had discrete control over the fraction of the cells that translocated NFAT1, indicating that a distinct threshold amount of TCR signaling is required to switch on NFAT1 in naive T cells. This behavior was cell contact dependent and qualitatively more exact than the NFAT1 response in ionomycin-stimulated naive T cells. These data contribute to our understanding of the digital behavior of TCR signaling components documented in other studies and indicate how T cells might discriminate log-fold changes in Ag availability during an actual infection. Overall, these results highlight the potential of this coculture nuclei isolation protocol to address stimulation-dependent translocation of proteins in primary lymphocytes.
INTRODUCTION
Following a virus infection, naive CD8+ T lymphocytes have the ability to carefully discern differences in cognate Ag quality and concentration and to use this information to generate the appropriate antiviral response. The molecular details of the interactions between peptide/MHC (pMHC) molecules and TCRs control the initiation of multiple signaling pathways that induce activation-dependent transcriptional programs. The strength of the TCR stimulus variesbecauseof differences incognate pMHC density on APCs and in the affinity of each TCR for this pMHC complex. In turn, this variability affects the amount of T effector or memory gene products made in activated cells, but the mechanistic details linking TCR signal strength to differential gene expression and downstream cell fate decisions are not well understood (1–3). Close examination of signaling pathway dynamics downstream of the TCR in single cells will provide important information to improve our understanding of how individual CD8+ T cellscommit to effector or memory cell fates (1, 3, 4).
NFAT is a well-characterized family of factors known to be critical for optimal T cell activation–associated transcription (5). In naive resting T cells, NFAT1 is hyperphosphorylated and sequestered in the cytoplasm. TCR stimulation induces strong intracellular cytoplasmic calcium (Ca2+) elevation and activates calcium-sensitive calcineurin, which dephosphorylates NFAT1 and permits translocation to the nucleus to bind target genes (6). Therefore, methods that measure NFAT1 nuclear translocation reveal immediate effects of TCR stimulation.
In this study, we aimed to characterize the TCR-dependent changes in NFAT1 localization in primary mouse naive CD8+ T cells responding to varying concentrations of pMHC. Because the strength of TCR stimulation has been shown to tune the expression of effector-associated transcription factors such as IRF4, we predicted that TCR-proximal signaling pathways would also produce graded responses to variable TCR stimulation (7). To measure NFAT translocation in human T cell lines, others have used imaging flow cytometry (8). However, the small size of primary naive mouse lymphocytes restricts accurate measurements using these methods. In another study, flow cytometry of isolated human memory T cell nuclei displayed an “all-or-none” (digital) NFAT1 response (9). During stimulation with phorbol ester (PMA) and the calcium ionophore ionomycin, addition of a calcineurin inhibitor reduced the fraction of the population that had NFAT1 present in their nuclei at a given time. These findings raised questions about the behavior of NFAT1 nuclear localization in naive T cells in response to stimulation with natural pMHC ligands to induce TCR signaling. To address these questions, we developed a conventional flow cytometry–based assay to assess NFAT1 nuclear localization in primary naive CD8+ T cells stimulated with cognate pMHC on splenic APCs.
In this study we outline a high-throughput method to analyze transcription factor localization in isolated splenic OT-I TCR transgenic T cell nuclei after stimulation with APCs pulsed with OVA peptide. To distinguish OT-I nuclei from APC nuclei, we labeled CD8+-enriched OT-I T cells with a fluorescent cell tracking reagent prior to combination with APCs in coculture. After isolation of cell nuclei, fluorescently labeled OT-I nuclei are distinguishable from APC nuclei in flow cytometric analyses. Labeled, isolated cell nuclei are of high purity and able to be stained with other fluorescently conjugated Abs for conventional flow analysis. We report rapid NFAT1 nuclear translocation in CD8+ OT-I cells stimulated with cognate OVA peptide and demonstrate that this response exhibits a strong digital behavior that is modulated by peptide dose. Lower OVA concentration reduces the fraction of cells within the stimulated population that respond and shows slower accumulation of NFAT1-positive nuclei. Peptide control over NFAT1 activation is also much more dynamic and discrete compared with stimulation with different doses of ionomycin. This method could be extended to assess nuclear translocation of other proteins and will be useful to address numerous research questions in the future.
MATERIALS AND METHODS
Mice and cell culture
OT-I TCR transgenic Rag2−/− mice and C57BL/6 mice were purchased from Taconic Biosciences and were housed and bred in a specific pathogen–free environment at the University of Massachusetts Medical School facilities in accordance with Institutional Animal Care and Use Committee guidelines. OT-I splenocytes were harvested and pooled from 8- to 12-wk-old gender-matched littermatesand enriched using a CD8+ magnetic negative selection kit (STEMCELL Technologies). Prior to coculture, OT-I cells were labeled with CellTrace Violet (Thermo Fisher Scientific) reagent for 20 min in 1× PBS according to the manufacturer’s specifications. Splenocytes from 8- to 12-wk-old C57BL/6 mice were pulsed with OVA SIINFEKL peptide (21st Century Biochemicals) for 30 min in RPMI 1640 with 10% FBS. In a 96-well round-bottom plate, bulk splenocytes were mixed with dye-labeled, OT-I CD8+–selected T cells 3:1 on ice (normally ?~1 × 106 OT-I cells plus 3 × 106 bulk splenocytes per well), with media containing additional OVA peptide (during stimulations >1 h only). The 96-well plate containing the cell mixture was briefly centrifuged to produce a loose pellet and was then placed in a 37°C CO2 incubator. After the incubation, the plate was immediately transferred to ice. For timecourse experiments, individual wells were transferred to a new 96-well plate sitting on ice after a specified time.
Nuclei isolation
The base nuclei isolation protocol was adapted from online sources (National Cancer Institute Experimental Transplantation and Immunology Branch Flow Cytometry Core Laboratory; home.ccr.cancer.gov/med/flowcore) and previously published reports (9). In brief, the 96-well plate containing stimulated cells was spun at 300 × g and 4°C, and the pellets were immediately resuspended with 250 μl of ice-cold Buffer A containing 320 mM sucrose, 10 mM HEPES (Life Technologies), 8 mM MgCl2, 1× Roche EDTA-free cOmplete Protease Inhibitor, and 0.1% (v/v) Triton X-100 (Sigma-Aldrich). After 15 min on ice, the plate was spun at 2000 × g and 4°C for 10 min. This was followed by two 250-μl washes with Buffer B (Buffer A without Triton X-100) and spinning at 2000 × g and 4°C. After the final wash, pellets were resuspended with 200 μl Buffer B containing 4% paraformaldehyde (electron microscopy grade; Electron Microscopy Sciences), and nuclei were rested on ice for 30 min for fixation, followed by two washes and resuspension in 1× PBS with 2% FBS, centrifuging at 1000 × g to sufficiently pellet nuclei. Nuclei were kept at 4°C until flow cytometry analysis. A detailed, step-by-step protocol describing both coculture and nuclei isolation is available upon request.
Flow cytometry
Whole cells were fixed with 4% paraformaldehyde in 1× PBS. Both fixed whole cells and fixed nuclei were permeabilized with 0.3% Triton X-100 in1× PBS and 2% FBS and stained with fluorescent Abs for 15 min at room temperature followed by three washes in 1× PBS and 2% FBS. To sufficiently pellet nuclei, centrifugation was performed at 1000 × g. The following Abs were used for staining: β-tubulin-AF647 (9F3, #3624; Cell Signaling Technology), CD3ε-APC (145–2C11; BD Biosciences), NFAT1-AF488 (D43B1, #14324; Cell Signaling Technology), and NF-κB (p65) (D14E12, #8242; Cell Signaling Technology). Propidium iodide was sourced from Thermo Fisher Scientific. Samples were collected on an LSR 11 flow cytometer (BD Biosciences) and analyzed with FlowJo 10.4.2 (BD Biosciences/Tree Star). After gating on singlets, OT-I nuclei were identified as CellTrace Violethi, β-tubulinlo. At least 10,000 OT-I nuclei events were recorded per sample.
Confocal imaging
For imaging, whole cells and nuclei were fixed and stained with fluorescent Abs similarly to flow cytometry preparation. After staining, pellets were resuspended in ~10 μl of PBS, pipetted onto glass slides, and mounted with ProLong Gold Antifade Reagent with DAPI (#8961; Cell Signaling Technology). Images were collected on a Leica SP5 confocal microscope with a 63× oil objective using Leica LAS AF software. Images were processed and merged using ImageJ software (National Institutes of Health).
RESULTS
Nuclear isolation of naive CD8+ T cells
Common methods to measure protein localization such as immunoblotting and confocal imaging do not allow for singlecell analysis or suffer from limited throughput, respectively. To observe single-cell NFAT1 translocation kinetics in a pMHC-stimulated cell population, we modified existing nuclei isolation methods and paired them with flow cytometry (9). In brief, stimulated cells were centrifuged in buffer-containing sucrose and nonionic detergent to disrupt the outer membrane. After centrifugation and washes with buffer without detergent, the resulting nuclei were immediately fixed with paraformaldehyde and stained with fluorescent Abs for analysis via flow cytometry. Mouse naive CD8+ T cell nuclei isolated using this protocol were distinguishable from whole cells in forward and side scatter, appearing slightly smaller in size (Fig. 1A). Also, proteins found in the cytosol and membrane (e.g., β-tubulin and CD3ε) have little fluorescence in isolated nuclei when compared with the fluorescence of permeabilized whole cells, but the nuclei stained highly for the DNA-intercalating dye propidium iodide (Fig. 1B). In restingnaive CD8+ T cells, NFAT1 is sequestered in the cytoplasm, which was observed as a ring of fluorescence using confocal imaging techniques (Fig. 1C). After treatment with Ca2+ ionophore ionomycin, NFAT1 fluorescence shifted predominantly to the nucleus. Isolated nuclei lack the cytoplasmic NFAT1 signal seen in unstimulated whole-cell samples and only exhibited NFAT1 fluorescence in ionomycin-treated samples. Adopting this method for use with flow cytometry easily permits analysis of thousands of single cells for NFAT1 activation and characterization of the responding stimulated population.
FIGURE 1. Isolation of stimulated OT-I nuclei and analysis via conventional flow cytometry.
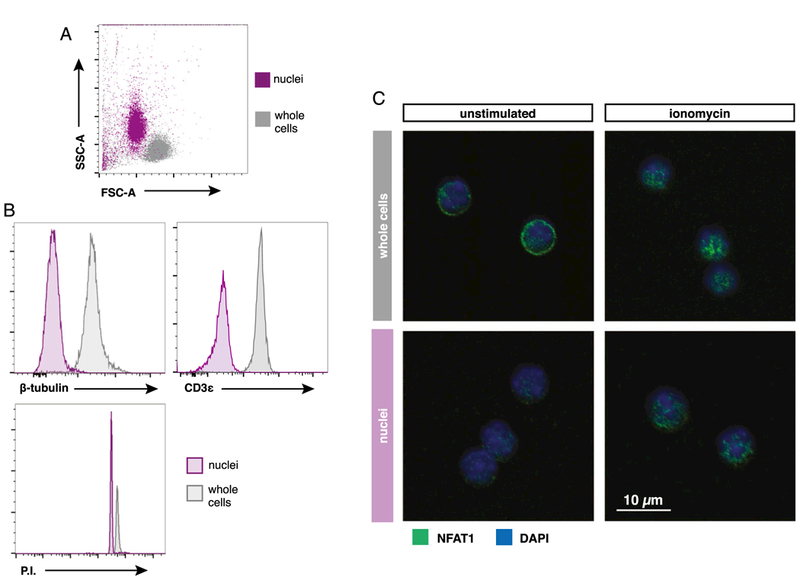
(A) Representative forward scatter (FSC) and side scatter (SSC) plot of OT-I nuclei isolated and fixed in sucrose buffer compared with OT-I whole cells. (B) Flow cytometry histograms comparing nuclei versus whole cells stained with indicated Abs or propidium iodide (P.I.). (C) Confocal images displaying NFAT1 localization in isolated OT-I nuclei compared with whole cells after 30 min of stimulation with or without 1 μg/ml ionomycin.
NFAT1 nuclear translocation in peptide-stimulated naive CD8+ T cells
Similar methods of nuclei isolation and flow cytometry have been used previously in one study to track NFAT1 translocation in primary human CD4+ T cells after stimulation with ionomycin and PMA with or without a calcineurin inhibitor (9). To carefully quantify the NFAT1 activation response in naive CD8+ T cells stimulated by natural peptide ligands, we used an experimental approach that mixes wild-type C57BL/6 splenocytes pulsed with SIINFEKL OVA peptide with splenic OT-I CD8+ T cells, followed by nuclei isolation (Fig. 2A, 2B). Because nuclei have been stripped of cell surface markers, tools such as congenic markers cannot be used to distinguish OT-I CD8+ T cell nuclei from bulk splenocyte nuclei. To resolve this, OT-I cells were labeled with CellTrace Violet proliferation dye prior to stimulation by peptide-pulsed wild-type splenocytes. Isolated OT-I nuclei retained CellTrace fluorescence and were thus distinguishable from wild-type unlabeled cells (Fig. 2C). When mixed with 1nM OVA peptide-pulsed bulk splenocytes for 30 min, OT-I cell nuclei displayed clear increases in NFAT1 fluorescence, whereas flow cytometry of whole cells from the same stimulation conditions exhibited little difference in total NFAT1 fluorescence (Fig. 1D).
FIGURE 2. CellTrace labeling of primary OT-I T cells allows for identification of OT-I nuclei after coculture.
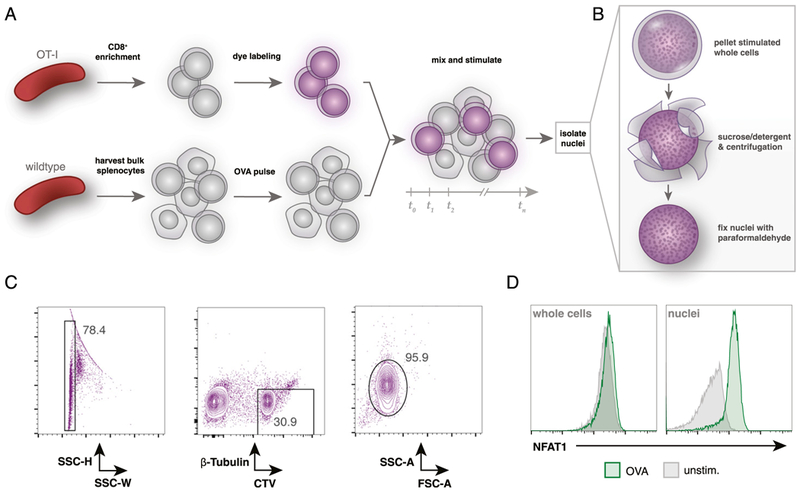
(A) Overview of coculture experimental setup. OT-I splenocytes are enriched for CD8+ T cells by negative selection and then labeled with CellTrace Violet dye. Wild-type splenocytes are harvested separately and pulsed with OVA peptide. Labeled OT-I cells are mixed (~1:3) with bulk splenocytes for desired time. (B) Summary of the nuclei isolation protocol. After coculture stimulation, all mixed OT-I and wild-type cells are lysed in a sucrose and detergent buffer, centrifuged, and washed twice prior to fixation with 4% paraformaldehyde. (C) Flow cytometry gating strategy to identify OT-I nuclei after coculture stimulation. Fixed nuclei are stained with a fluorescent Ab against β-tubulin prior to analysis. OT-I nuclei are identified by gating on singlets by side scatter height (SSC-H) and SSC width (SSC-W) and then on β-tubulinlo and CellTrace Violethi events. (D) Flow cytometry comparison of isolated OT-I nuclei or whole cells (prelabeled with CellTrace Violet) after 30 min of coculture with 1 nM OVA peptide-pulsed bulk C57BL/6 splenocytes. Gated on CellTrace Violethi and β-tubulinlo events.
Peptide concentration controls digital NFAT1 nuclear localization within a population of naive CD8+ T cells
We wished to fully characterize NFAT1 activation in naive OT-I cells presented with natural pMHC ligands. Flow cytometry of nuclei revealed a bimodal NFAT1 response to OVA peptide stimulation within the OT-I population. When analyzed over a 60-min timecourse, stimulation with 10 pM of OVA peptide produced a progressive increase in the fraction of NFAT1-positive nuclei (Fig. 3A). Stimulated populations lacked nuclei with intermediate levels of NFAT1 fluorescence and instead exhibited a digital or all-or-none NFAT1 response to TCR signaling. This finding indicates there may be a threshold of peptide-mediated TCR stimulation that triggers maximal NFAT1 translocation on a per-cell basis. Importantly, we also found that the concentration of peptide controlled the proportion of NFAT1-positive nuclei within the population at a given timepoint (Fig. 3B). Specifically, higher doses of OVA peptide (1nM) induced NFAT1 activation in 87% of the OT-I cells after 45 min, whereas only 41% of nuclei were NFAT1-positive when stimulated with 10 pM OVA for the same duration. Compilation of peptide titration experiments indicated that OVA peptide concentration controlled the maximum percentage of OT-I cells that translocated NFAT1 within 2 h (Fig. 3C). A low dose of OVA (10 pM) produces a maximum of ~55% of OT-I cells responding, whereas 80% of OT-I cells respond to a 100-fold-higher dose of peptide (1000 pM). Broadly, a greater concentration of peptide more rapidly induced a larger proportion of NFAT1 responders.
FIGURE 3. Peptide concentration controls digital NFAT1 nuclear translocation kinetics in a responder fraction of stimulated naive OT-I cells.
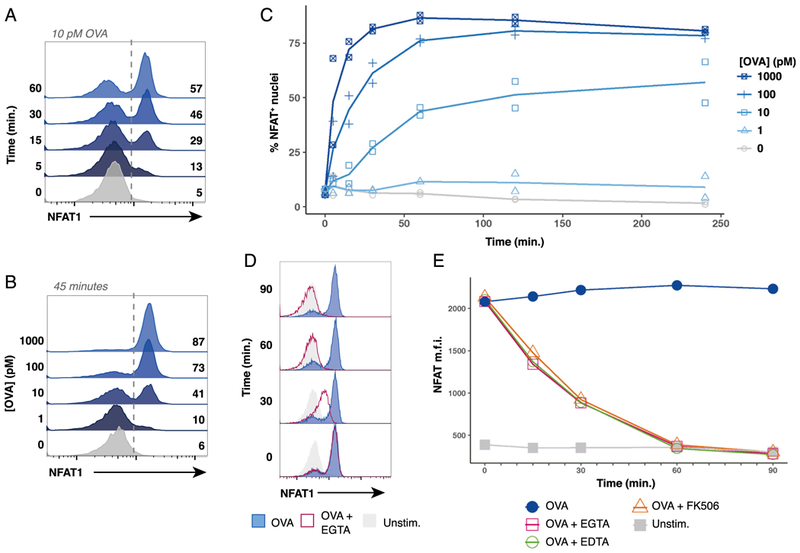
(A) Representative flow cytometry histograms displaying a timecourse of NFAT1 staining in OT-I nuclei after coculture with bulk splenocytes pulsed with 10 pM of OVA peptide. (B) Representative flow cytometry histograms of NFAT1-positive OT-I nuclei after 45 min of coculture with bulk splenocytes pulsed with indicated dose of OVA peptide. (C) Plots depicting compiled timecourse results from two experiments measuring NFAT1 localization in OT-I nuclei after coculture with bulk splenocytes pulsed with the indicated dose of OVA peptide. (D) Representative histograms of three experiments performed show that EGTA addition causes NFAT1 to exit OT-I nuclei. OT-I cells were cocultured with bulk splenocytes pulsed with 100 pM of OVA peptide. After stimulation for 30 min to allow for nuclear translocation of NFAT1, 5 mM EGTA was added to culture, and samples were harvested after 0,30, 60, or 90 min. (E) Line plots depicting median fluorescence intensity (m.f.i.) of NFAT1 in isolated OT-I nuclei stimulated similarly to (D). After 30 min of stimulation with OVA peptide, either 5 mM EDTA, 5 mM EGTA, or 100 nM FK506 was added to culture media where indicated. Samples were collected 0, 15, 30, 60, and 90 min after adding inhibitors. In (A)-(E), OT-I nuclei were identified as CellTrace Violethi β-tubulinlo
Addition of the calcium ion chelating agent EGTA to media of activated T cells causes the immediate cessation of calcium influx into the cells, leading to the rapid loss of calcineurin activity and the return of NFAT1 to the phosphorylated, cytoplasmic inactive state (5,6, 10). When EGTA was added to our OT-I cultures 30 min after stimulation, we observed a gradual loss of NFAT1 fluorescence from nuclei, indicating NFAT1 export to the cytoplasm (Fig. 3D, 3E). Similar NFAT1 export behavior was observed when the divalent ion chelator EDTA or the calcineurin inhibitor FK506 were added to the culture media (Fig. 3E). The rate at which cells lost nuclear NFAT1 are consistent with previous studies performed using direct biochemical measurement of activated NFAT1 or as assessed by confocal microscopy (11). Interestingly, these data revealed the presence of an NFAT1-intermediate population after inhibitor addition, indicating that NFAT1 nuclear export is not regulated digitally as is the process inducing NFAT1 nuclear entry following TCR stimulation. Overall, these findings support the conclusion that the OVA peptide concentration determines the percentage of cells in the population that activate NFAT in response to TCR stimulation as well as the kinetics of this response. In contrast, our data do not support a model in which the responding percentage of cells and the bimodal population response result from a process of rapid nuclear NFAT1 import/export equilibrium in stimulated cells.
Digital NFAT1 activation threshold in naive OT-I cells is a characteristic of TCR signaling
To determine whether the bimodal population response of NFAT1 activation was a specific feature of TCR signaling or a more general feature of NFAT1 activation in T cells, we performed a titration of the calcium ionophore ionomycin. Unlike the response observed with increasing doses of OVA peptide presented on splenic APCs, titration of ionomycin concentration does not produce a clear bimodal response of NFAT1 activation in naive OT-I cells (Fig. 4A, 4B). Instead, lower doses of ionomycin a create less precise, intermediate amount of nuclear NFAT1 in OT-I cells that was more prevalent after longer stimulation. As expected, addition of 10 ng/ml PMA in the culture media did not appear to alter the behavior of NFAT1 translocation because of ionomycin (Supplemental Fig. 1A, 1B). Because ionomycin activation of Ca2+ signaling is TCR independent, these data imply that dynamic control of the NFAT1 activation threshold in naive cells, producing a digital NFAT1 response, is likely to be dependent on pMHC-TCR interactions or the TCR-proximal downstream signaling mechanisms.
FIGURE 4. Titrated doses of ionomycin does not accurately induce a digital NFAT1 response in OT-I cells.
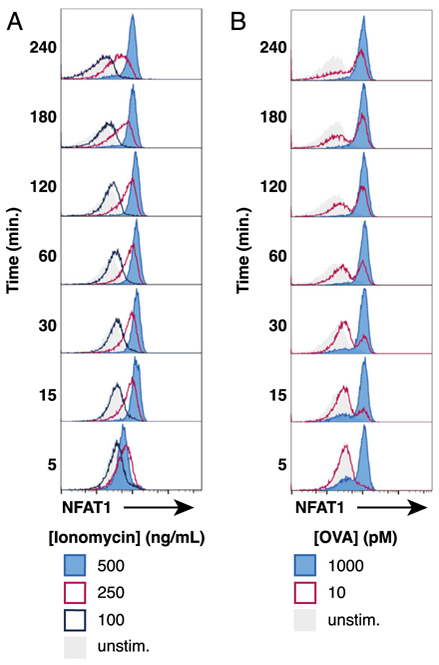
(A) NFAT1 localization in OT-I nuclei after different doses of ionomycin over the course of 240 min. Representative histograms displaying NFAT1-positive nuclei with indicated dose of ionomycin present in the coculture media. Nuclei were identified as CellTrace Violethi β-tubulinlo (B) For comparison, results of NFAT1-positive OT-I nuclei during coculture with bulk splenocytes pulsed with indicated dose of OVA peptide.
DISCUSSION
We present an easily accessible and adaptable method to measure localization of transcription factors in nuclei from stimulated single naive CD8+ T cells. Importantly, our innovations allow for measurement of CD8+ T cell nuclei after stimulation in cocultures with peptide-pulsed APCs. Stimulation of TCR transgenic CD8+ T cells by bulk splenocytes is commonly used to measure gene expression after 24 h (or more) of stimulation (12, 13). Using these established approaches, activated T cells are identified via flow cytometry with Abs against T lineage markers (CD8, CD3), activation markers (CD69, CD25), and congenic markers (e.g., CD45.1/2). However, identification via surface markers is not possible when analyzing isolated nuclei. Thus, we employed CellTrace labeling prior to stimulation, which allows for easy discrimination of OT-I T cell nuclei from nuclei of other cocultured cells. We first validated our assay and then used it to profile the translocation behavior of NFAT1 in OT-I T cells responding to varying concentrations of OVA peptide on APCs. This method is readily adaptable to measurements of additional nuclear proteins in a wide range of cells and coculture conditions.
Alternative methods currently exist to measure nuclear translocation of selected proteins in T cells (8, 9, 14). In comparison, we find our method is more adaptable to larger cell numbers, more amenable to quantification, and better suited for analyses of mouse primary naive T cells. We first attempted to use imaging flow cytometry to measure NFAT1 localization in APC-stimulated T cells, as others have had success with this approach in human Jurkat tumor cells (8). Naive T cells present a more difficult challenge to analyze by imaging because they are substantially smaller than cultured tumor cells (<10 μm) and have a minimal amount of cytoplasm. These properties confounded the analysis, and in our hands, imaging flow cytometry could not discern cytoplasmic versus nuclear signal with sufficient resolution to allow for accurate quantification of the responses to signaling (data not shown). In contrast, our conventional flow cytometry-based method used to analyze isolated nuclei avoided these difficulties.
The analysis of OT-I nuclei revealed clear digital (all-or-none) behavior of NFAT1 activation in single cells responding to pMHC stimulation (Fig. 3A, 3B). These results validate the observations of Podtschaske et al. (9), who demonstrated that digital NFAT1 translocation was regulated in part by calcineurin. We have expanded upon this concept and shown that direct TCR stimulation with cognate peptide Ag modulates a digital NFAT1 response. The concentration of OVA peptide sets the maximum percentage of NFAT1 responders when cells were analyzed for up to 4 h following initial APC contact (Fig. 3C). In naive T cells, NFAT activation is downstream of store-operated calcium entry events (15). Single-cell calcium imaging of OT-I T cells stimulated with varying doses of OVA peptide on B cell APCs show that reducing the peptide dose increases the proportion of nonresponding cells in the population but, importantly, has no effect on the magnitude of the peak calcium signal in the responding subset (16). Bone marrow-derived macrophages pulsed with varying doses of OVA peptide also induce calcium responders and nonresponders within a stimulated naive OT-I cell population (17). These data are consistent with our NFAT1 translocation data and together indicate that the all-or-none calcium response in turn produces an all-or-none response of NFAT1 activation.
Based on our results, we suggest that higher densities of peptide on the APCsurface increase the probability that any individual naive T cell will digitally activate calcium and then NFAT. The single-cell calcium imaging study also reported that lower doses of peptide stimulation produced delayed and less synchronized Ca2+ activation in the population of responding cells (16). These data agree with our observations of NFAT1 translocation, in which within the first 45 min of stimulation, lower concentrations of OVA-APCstimulation slowed the rate at which individual nuclei became NFAT1-positive compared with cells stimulated with a higher dose of APC peptide (Fig. 3C). In the same study, when T cells were restimulated after APC peptide stimulation, individual cells were likely to repeat the same pattern of Ca2+ signaling observed during the primary APC peptide stimulation, indicating a cell-intrinsic heterogeneity among the naive OT-I T cell population. Therefore, assuming all OT-I cells have equal access to Ag, the bimodal NFAT1 response we observe is also likely indicative of some heterogeneity.
Ionomycin titration did not reliably elicit digital NFAT1 activation in naive OT-I cells but instead translocated graded amounts of NFAT1 (Fig. 4). Phosphorylation of kinases in the TCR complex and downstream pathways causes accumulation of the second messenger IP3. Once a threshold level of IP3 is reached, endoplasmic reticulum Ca2+ releases, resulting in digital activation of Ca2+ release-activated channels (CRAC) (15,18,19). Ionomycin induces Ca2+ pathway responses by directly causing Ca2+ influx, bypassing the TCR machinery; thus, it is possible that low levels of ionomycin produce graded Ca2+ influx patterns that are not identical to those induced by the TCR machinery. Our experiments help showcase how T cell populations discern and respond to a wide range of peptide concentrations through digital activation of individual cells.
Most experiments exploring other TCR signaling pathways also observe digital switch-like behavior when naive T cells are activated (20, 21). For example, a minimal number of OVA-bound MHC molecules on an APC line induce a responder fraction of OT-I cells with activated MAPK signaling (22). A later study with human Jurkat T cells discovered that this sharp threshold in TCR-induced MAPK signaling was due to a feedback loop in Son of Sevenless signaling that amplifies Ras activation (23). Our results support the notion that TCR-induced NFAT signaling behaves like a digital switch. However, it is challenging to understand how such behavior in individual naive T cells translates to dynamic and finely tuned gene responses hours after activation.
Unlike our experiments using monoclonal TCR transgenic T cells in culture, naive T cells in vivo face numerous obstacles influencing their fate. Ag may be disproportionally presented within secondary lymphoid structures, naive cells must compete with other clones for limited Ag, and APCs may have varying levels of activation status (20, 21, 24–26). Although digital NFAT signaling does seem to correlate with digital activation of some select genes (IL-2 and IFN-γ) in cells stimulated in vitro (9), current models propose that in vivo, T cells integrate digital signals over longer periods of time after numerous successive contacts with APCs (11, 20, 21, 25, 27). It is unclear whether gene expression 24 h after Ag encounter in vitro is the result of many successive, separate contacts with APCs. However, the NFAT1 translocation we measure is likely cell contact dependent, as NFAT1 exits OT-I nuclei after addition of EGTA or other inhibitors that immediately arrest calcium influx (Fig. 3D, 3E). Interestingly, others have documented that NFAT-dependent transcription for privileged genes (Egr2 but not Ifng) continues after breaking contacts between T cells and APCs (11). This outlines one manner whereby digital signaling events may contribute to differential gene expression programs. Serial, digital activation of factors like NFAT1 may drive the accumulation of target gene products at early times after initial activation. For instance, the amount of the cell cycle regulator c-Myc that accumulates within an early time frame after initial T cell activation dictates the total number of cell divisions effector T cells undergo during clonal expansion (28). TCR signal strength determines the initial frequency of digital c-Myc expression within a stimulated OT-I population, but continued c-Myc expression is dependent on the expression level of IL-2, a well-characterized NFAT target gene (6, 29). Thus, successive digital NFAT responses may be integrated to influence clonal scaling and other graded effector gene expression patterns through this type of mechanism.
In summary, we describe a powerful and easily adaptable conventional flow cytometry approach for examination of the nuclear localization of specific proteins in single cells present in coculture assays. This method is amenable to exploring the dynamics of other T cell transcription factors in response to variable quantity or quality of Ag stimulation, including but not limited to both naive and activated states of CD8+ or CD4+ primary T cells.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Nicholas A. Spidale for initial encouragement with nuclei isolation protocols and Dr. Jeff D. Colbert and Dr. Friedrich M. Cruz for guidance in acquiring confocal images. We thank Regina Whitehead and Sharlene Hubbard for technical assistance. We thank the Department of Animal Medicine at the University of Massachusetts Medical School for maintaining the mouse colonies.
This work was supported by National Institutes of Health Grant AI32419 (to L.J.B.).
Abbreviation used in this article:
- pMHC
peptide/MHC
Footnotes
The online version of this article contains supplemental material.
DISCLOSURES
The authors have no financial conflicts of interest.
REFERENCES
- 1.Zehn D, King C, Bevan MJ, and Palmer E. 2012. TCR signaling requirements for activating T cells and for generating memory. Cell. Mol. Life Sci. 69: 1565–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huber M, and Lohoff M. 2014. IRF4 at the crossroads of effector T-cell fate decision. Eur. J. Immunol. 44: 1886–1895. [DOI] [PubMed] [Google Scholar]
- 3.Kaech SM, and Cui W. 2012. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 12: 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corse E, Gottschalk RA, and Allison JP. 2011. Strength of TCR-peptide/MHC interactions and in vivo T cell responses. J. Immunol. 186: 5039–5045. [DOI] [PubMed] [Google Scholar]
- 5.Hogan PG, Chen L, Nardone J, and Rao A. 2003. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17: 2205–2232. [DOI] [PubMed] [Google Scholar]
- 6.Macian F 2005. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 5: 472–484. [DOI] [PubMed] [Google Scholar]
- 7.Nayar R, Enos M, Prince A, Shin H, Hemmers S, Jiang JK, Klein U, Thomas CJ, and Berg LJ. 2012. TCR signaling via Tec kinase ITK and interferon regulatory factor 4 (IRF4) regulates CD8+ T-cell differentiation. Proc. Natl. Acad. Sci. USA 109: E2794–E2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maguire O, O’Loughlin K, and Minderman H. 2015. Simultaneous assessment of NF-κB/p65 phosphorylation and nuclear localization using imaging flow cytometry. J. Immunol. Methods 423: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Podtschaske M, Benary U, Zwinger S, Höfer T, Radbruch A, and Baumgrass R. 2007. Digital NFATc2 activation per cell transforms graded T cell receptor activation into an all-or-none IL-2 expression. PLoS One 2: e935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dolmetsch RE, Xu K, and Lewis RS. 1998. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392: 933–936. [DOI] [PubMed] [Google Scholar]
- 11.Marangoni F, Murooka TT, Manzo T, Kim EY, Carrizosa E, Elpek NM, and Mempel TR. 2013. The transcription factor NFAT exhibits signal memory during serial T cell interactions with antigen-presenting cells. Immunity 38: 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stoycheva D, Deiser K, Stärck L, Nishanth G, Schlüter D, Uckert W, and Schüler T. 2015. IFN-γ regulates CD8+ memory T cell differentiation and survival in response to weak, but not strong, TCR signals. J. Immunol. 194: 553–559. [DOI] [PubMed] [Google Scholar]
- 13.Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, and Hogquist KA. 2011. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med. 208: 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poglitsch M, Katholnig K, Säemann MD, and Weichhart T. 2011. Rapid isolation of nuclei from living immune cells by a single centrifugation through a multifunctional lysis gradient. J. Immunol. Methods 373: 167–173. [DOI] [PubMed] [Google Scholar]
- 15.Hogan PG, Lewis RS, and Rao A. 2010. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 28: 491–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dura B, Dougan SK, Barisa M, Hoehl MM, Lo CT, Ploegh HL, and Voldman J. 2015. Profiling lymphocyte interactions at the singlecell level by microfluidic cell pairing. Nat. Commun. 6: 5940. [DOI] [PubMed] [Google Scholar]
- 17.Le Borgne M, Raju S, Zinselmeyer BH, Le VT, Li J, Wang Y, Miller MJ, and Shaw AS. 2016. Real-time analysis of calcium signals during the early phase of T cell activation using a genetically encoded calcium biosensor. J. Immunol. 196: 1471–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parekh AB, Fleig A, and Penner R. 1997. The store-operated calcium current I(CRAC): nonlinear activation by InsP3 and dissociation from calcium release. Cell 89: 973–980. [DOI] [PubMed] [Google Scholar]
- 19.Kar P, Nelson C, and Parekh AB. 2012. CRAC channels drive digital activation and provide analog control and synergy to Ca(2+)-dependent gene regulation. Curr. Biol. 22: 242–247. [DOI] [PubMed] [Google Scholar]
- 20.Tkach K, and Altan-Bonnet G. 2013. T cell responses to antigen: hasty proposals resolved through long engagements. Curr. Opin. Immunol. 25: 120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayya V, and Dustin ML. 2016. What scales the T cell response? Trends Immunol. 37: 513–522. [DOI] [PubMed] [Google Scholar]
- 22.Altan-Bonnet G, and Germain RN. 2005. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 3: e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das J, Ho M, Zikherman J, Govern C, Yang M, Weiss A, Chakraborty AK, and Roose JP. 2009. Digital signaling and hysteresis characterize ras activation in lymphoid cells. Cell 136: 337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henrickson SE, Mempel TR, Mazo IB, Liu B, Artyomov MN, Zheng H, Peixoto A, Flynn MP, Senman B, Junt T, et al. 2008. T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for T cell activation. Nat. Immunol. 9: 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henrickson SE, Perro M, Loughhead SM, Senman B, Stutte S, Quigley M, Alexe G, Iannacone M, Flynn MP, Omid S, et al. 2013. Antigen availability determines CD8+ T cell-dendritic cell interaction kinetics and memory fate decisions. Immunity 39: 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia Z, Pradelli E, Celli S, Beuneu H, Simon A, and Bousso P. 2007. Competition for antigen determines the stability of T celldendritic cell interactions during clonal expansion. Proc. Natl. Acad. Sci. USA 104: 4553–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mempel TR, Henrickson SE, and Von Andrian UH. 2004. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 427: 154–159. [DOI] [PubMed] [Google Scholar]
- 28.Heinzel S, Binh Giang T, Kan A, Marchingo JM, Lye BK, Corcoran LM, and Hodgkin PD. 2017. A Myc-dependent division timer complements a cell-death timer to regulate T cell and B cell responses. Nat. Immunol. 18: 96–103. [DOI] [PubMed] [Google Scholar]
- 29.Preston GC, Sinclair LV, Kaskar A, Hukelmann JL, Navarro MN, Ferrero I, MacDonald HR, Cowling VH, and Cantrell DA. 2015. Single cell tuning of Myc expression by antigen receptor signal strength and interleukin-2 in T lymphocytes. EMBO J. 34: 2008–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.