

Peripheral blood MФ–cancer cell fusion hybrids identified in cancer patients correlate with disease stage and overall survival.
Abstract
High lethality rates associated with metastatic cancer highlight an urgent medical need for improved understanding of biologic mechanisms driving metastatic spread and identification of biomarkers predicting late-stage progression. Numerous neoplastic cell intrinsic and extrinsic mechanisms fuel tumor progression; however, mechanisms driving heterogeneity of neoplastic cells in solid tumors remain obscure. Increased mutational rates of neoplastic cells in stressed environments are implicated but cannot explain all aspects of tumor heterogeneity. We present evidence that fusion of neoplastic cells with leukocytes (for example, macrophages) contributes to tumor heterogeneity, resulting in cells exhibiting increased metastatic behavior. Fusion hybrids (cells harboring hematopoietic and epithelial properties) are readily detectible in cell culture and tumor-bearing mice. Further, hybrids enumerated in peripheral blood of human cancer patients correlate with disease stage and predict overall survival. This unique population of neoplastic cells provides a novel biomarker for tumor staging, as well as a potential therapeutic target for intervention.
INTRODUCTION
Historic dogma describing tumor evolution is based on outgrowth and expansion of clonal tumor populations; however, it is now appreciated that both genetic and nongenetic mechanisms drive tumor evolution fostering phenotypic variability of neoplastic cells and their clones. These changes underlie aggressive tumor growth, metastatic spread, acquisition of tumor heterogeneity, and therapeutic response or resistance (1, 2). While our understanding of the molecular and cellular mechanisms contributing to intratumoral heterogeneity has significantly expanded, there are no effective therapies quelling heterogeneity to improve patient stratification or response to anticancer therapies, highlighting the need for advances in this area.
Heterotypic cell fusion is a fundamental developmental mechanism serving to enhance cellular diversity; the most notable and best-studied example is fusion of sperm and egg. In adult murine intestines, we previously reported that fusion between hematopoietic and epithelial cells is readily detected in response to injury (3, 4); similar findings have been reported with various cells, including hepatocytes, cardiomyocytes, and skeletal muscles (5–8). In cancer, however, despite a century-old hypothesis that cell fusion contributes to tumor initiation (9–11) and acquisition of metastatic behaviors (10, 12, 13), few experimental studies have mechanistically addressed the functional underpinnings or consequences of cell fusion in the etiology of malignant progression. Reports using various approaches identify human tumor cells with immune and malignant characteristics (13–19); the etiologic mechanism for these cells is attributed to cell fusion, developmental mimicry, transdifferentiation, or other unidentified mechanisms. These previous studies do not address the biologic significance of the hybrid tumor cells or present evidence from experimental models to support the mechanism. Since underlying mechanisms for these cells cannot easily be determined in human subjects, murine models and in vitro studies provide a more appropriate and tractable platform for investigation.
In our previous studies, we reported that in vivo fusion between intestinal epithelial cells and macrophages (MФs) yields hybrid offspring retaining epithelial characteristics defined by their gene expression profile (12). On the basis of this report, and recognizing that MФs are inherently migratory, we sought to determine physiologic relevance of cell fusion to tumor heterogeneity by enhanced somatic diversity of neoplastic hybrids, through increased migratory or invasive properties, and by instilling a selective metastatic advantage. Herein, we present a systematic analysis of MФ–neoplastic cell fusion (referred to as MФ–cancer cell fusion or fusion hybrid) using ex vivo and in vivo murine cancer models to provide evidence that hybrids acquire functional MФ-associated phenotypes that enhance tumor progression. Analyses of human tumor biopsies and peripheral blood reveal a novel circulating hybrid cell (CHC) population (defined as cells harboring hematopoietic and epithelial/tumor properties), whose numbers correlate with disease stage and predict overall outcome, thereby representing a biomarker for patient stratification.
RESULTS
In vitro–derived MФ–neoplastic cell fusion hybrids display biparental lineage
On the basis of our previous findings that MФs are the prominent fusogenic bone marrow–derived cell partner for epithelial cells (12), we used in vitro validation and analyses of MФ–cancer cell fusion hybrids to examine contributions of the neoplastic cell and MФ to the identity of the hybrid cells. To generate in vitro–derived hybrids, we engineered two murine cancer cell lines, colon adenocarcinoma (MC38) and melanoma (B16F10), to stably express Cre recombinase and histone 2B fused to red fluorescent protein (H2B-RFP). In cocultures, engineered MC38 and B16F10 cancer cells spontaneously fused with bone marrow–derived MФs isolated from transgenic mice expressing either actin–green fluorescent protein (GFP) (20) or a yellow fluorescent protein (YFP) Cre reporter (21). This resulted in MФ–cancer cell fusion hybrids identified by coexpression of nuclear RFP and cytoplasmic GFP or YFP (Fig. 1, A and B; figs. S1A and S2; and movie S1). YFP expression enabled subsequent fluorescence-activated cell sorting (FACS) isolation of hybrid cells and downstream validation of their identity using immunoblot analyses and YFP expression (Fig. 1C). Notably, in control experiments where conditioned media from GFP-expressing MФs incubated on MC38s or conditioned media from Cre-expressing MC38s incubated on YFP Cre reporter MФs, fusion hybrids were not detected.
Fig. 1. In vitro–derived MФ–cancer cell fusion hybrids.
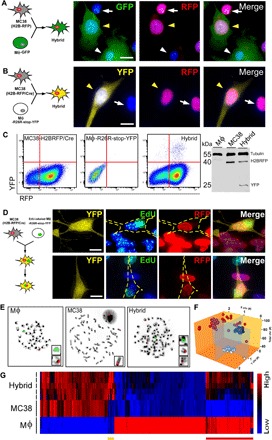
(A) MC38 (H2B-RFP) cancer cells and GFP-expressing MФs cocultured in a ratio of 1:2 result in hybrid cells with RFP nuclei and GFP-expressing cytoplasm (yellow arrowhead) among unfused cancer cells (white arrow) and MФs (white arrowhead). (B) MC38 (H2B-RFP/Cre) cancer cells cocultured with MФs expressing the Cre reporter, R26R-stop-YFP results in YFP-expressing hybrid cells (yellow arrowhead). (C) YFP-expressing hybrids can be FACS-isolated to purify YFP-expressing hybrid cells confirmed by immunoblot. A representative FACS plot is shown. (D) Cocultured MФs labeled with EdU (green) and MC38 (H2B-RFP/Cre) cancer cells produce YFP-expressing hybrids that initially harbor two nuclei—one from each parent; upon mitotic division, these undergo nuclear fusion, resulting in a single nucleus with EdU-labeled and RFP-expressing DNA. Hybrid cell outlined in yellow. Scale bar, 10 μm. (E) Karyotype and X (red) and Y chromosome (green) fluorescence in situ hybridization (FISH) analyses of parental MФs, unfused MC38 cancer cells, and fusion hybrids. (F) Fusion hybrids (red sphere, n = 45) cluster as a unique population based on their chromosome number and sex chromosomes, relative to MФs (white sphere, n = 27) and MC38s (black sphere, n = 28). (G) Microarray analyses of n = 5 independent hybrid isolates and n = 3 each for MC38 and MФ populations. The yellow bar denotes hybrid gene expression unique from MC38s and MФs. The red bar marks hybrid gene expression that is similar to that in MФs.
To demonstrate the biparental lineage of hybrid cells, we used three discrete approaches. First, MΦs labeled with 5-ethynyl-2′-deoxyuridine (EdU) before coculture with H2B-RFP–expressing neoplastic cells produced MФ–cancer cell fusion hybrids that initially harbored two nuclei, one labeled with EdU (MФ origin) and the other expressing H2B-RFP (neoplastic cell origin; Fig. 1D). Upon the first mitotic division, binucleated hybrids underwent nuclear fusion, yielding a single nucleus containing both EdU- and H2B-RFP–labeled DNA (Fig. 1D).
A second approach, karyotype analyses of sex chromosomes, demonstrated that male MФs (XY) fused to neoplastic cells (XO) generated hybrids containing three sex chromosomes (XXY; Fig. 1E), consistent with a fusion event. Chromosome enumeration revealed that hybrids existed as a unique cell population defined by their sex chromosome and total chromosome content when compared to parental MФs or cancer cells (Fig. 1F, red spheres are hybrids, black spheres are MФs, and white spheres are MC38s). Loss of chromosomes observed in hybrid clones occurred with temporal in vitro passage (fig. S3A); karyotype analyses of single hybrid cells revealed variable chromosome numbers (Fig. 1F), indicating that cell fusion contributes to tumor cell heterogeneity.
Finally, transcriptome analyses revealed that MФ–cancer cell hybrids predominantly exhibited neoplastic cell transcriptional identity, while notably, retained MФ gene expression signatures (Fig. 1G, red bar, and table S1) that clustered into gene ontology (GO) biologic functions attributed to MФ behavior (table S2). Of the five independently analyzed hybrid clones, each displayed a high degree of heterogeneity with respect to their MФ gene expression. Together, these findings support the tenet that cell fusion between MФs and neoplastic cells produces heterogeneous hybrid cells sharing characteristics of both parental predecessors but possessing their own characteristics.
Fusion hybrids acquire differential response to the microenvironment
Despite acquiring MФ gene expression profiles, MФ–cancer cell fusion hybrids initially retained in vitro proliferative capacity similar to unfused neoplastic cells, as opposed to MФs (fig. S3B). However, with prolonged culture, that is, past confluence, unfused neoplastic cells pile on themselves, forming cellular aggregates, whereas MФ–cancer cell fusion hybrids remained sheet-like with mesenchymal histologic features, indicating an acquired contact inhibition (fig. S3B and movie S2). These data indicate that, although hybrids have similar division rates, they gain differential growth properties as compared to unfused cancer cells. To determine whether these in vitro differences were recapitulated when cells were grown in vivo, in vitro–derived hybrids from MC38 or B16F10 cells were respectively injected subcutaneously into the flank or intradermally into syngeneic immunocompetent mice. Hybrids retained tumorigenic potential, with MC38 hybrids displaying shorter doubling times as compared to unfused parental cancer cells [data from two hybrid clones and two unfused cancer cell clones (Fig. 2A); B16F10 not shown], indicating that hybrids gain growth advantage in an in vivo microenvironment.
Fig. 2. In vitro–derived fusion hybrid characterization.
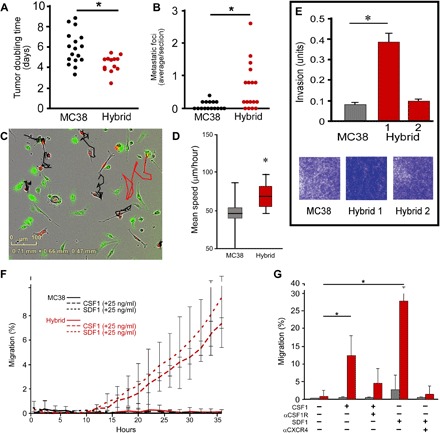
(A) Proliferation analysis of MC38 cells and MC38-derived hybrids injected into flanks of immunocompetent syngeneic mice (n = 13 mice, each from two different hybrid isolates). Each data point reflects tumor growth in a single mouse. (B) Analysis of metastatic seeding potential of hybrids and MC38 cells injected into spleens and area analyzed in H&E-stained tissue sections of the liver [n = 15 mice injected with MC38 cells (three different hybrid clones), n = 17 mice injected with hybrids, with each data point reflecting metastatic tumors analyzed in the liver]. (C) Static portrayal of migration tracks from unfused MC38s (black) and a MC38-derived fusion hybrid (red) generated from live-imaged cocultures. Images reflect representative images. (D) The mean speed of hybrids (red bar) relative to MC38s (gray bar) is statistically significant (*P < 1.1 × 10−9). (E) In vitro invasion assay of MC38 cells and MC38-derived hybrids in Matrigel invasion chambers, stained with crystal violet after 15 hours. Data reflect the average of triplicate samples in biologic replicates. (F) A representative data set evaluating chemotaxis toward CSF1 and SDF1 ligands. Hybrid cell chemotaxis toward CSF1 and SDF1 is statistically significant relative to unfused MC38 cells after 24 hours (P < 0.05). Three independent experiments of triplicates or quadruplicates were conducted for each ligand. Multiple hybrid clones were assessed. (G) Incubation of cells with blocking antibodies to CSF1R and CXCR4 reduces migration of hybrids toward ligands. P < 0.05 and P < 0.01, respectively (hybrid, red bar; MC38, gray bar).
To determine whether hybrid cells acquired enhanced ability to seed and/or proliferate in ectopic microenvironments, we conducted experimental metastases assays. MC38-derived hybrids injected into spleens readily trafficked to the liver and resulted in increased metastatic foci per area compared to unfused parental cancer cells (Fig. 2B), indicating that cancer fusion hybrids gained enhanced properties required for trafficking to metastatic sites, seeding, and/or growing in a new microenvironment. Likewise, B16F10-derived fusions injected intravenously resulted in greater metastatic lung tumor area, relative to control, unfused B16F10 (Fig. 3G), indicating that they trafficked, adhered, or proliferated more efficiently within the lung. These acquired phenotypic behaviors aligned with data from gene expression analyses that identified increased fusion-associated expression of GO pathway genes implicated in metastatic spread (table S2). In particular, those pathways contributing to tumor invasion—attachment, matrix dissolution, and migration—as well as pathways involving response to specific microenvironmental cues (22–24) were up-regulated in hybrids relative to unfused tumor cells.
Fig. 3. B16F10 in vivo–derived fusion hybrids.
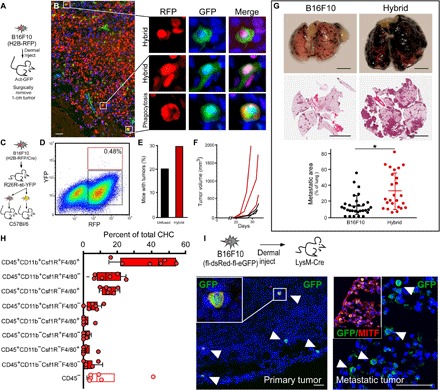
(A) B16F10 (H2B-RFP) cells (5 × 104 cells) intradermally injected into GFP-expressing mice (n = 12, two hybrid clones) were harvested at ~1.0 cm at study end point. (B) Fluorescence analyses of tumor sections for RFP (red) and GFP (green) reveal double-positive hybrids and phagocytosed cancer cells with different nuclear morphology. Scale bar, 25 μm. (C) B16F10 (H2B-RFP/Cre) cells injected (5 × 104 cells) into R26R-stop-YFP transgenic mice (n = 8). (D) Representative FACS plot of hybrid and unfused cancer cells from a dissociated tumor, for example, hybrids (red box) and unfused (black box) cancer cells (n = 6 single tumor analyses, n = 2 pooled tumor analyses, n = 13 mice). (E) Three hundred FACS-isolated cells were injected into wild-type secondary recipient mice (n = 19 unfused, n = 19 hybrids) analyzed for tumor growth at 40 days, and (F) 3000 FACS-isolated cells were injected into syngeneic recipient mice (n = 3 MC38 injected mice, black lines; n = 3 hybrid injected mice, red lines) and temporally monitored for growth. (G) B16F10 (H2B-RFP) or MФ–B16F10-derived hybrid cells tail vein–injected into wild-type mice (n = 12 mice). Macroscopic view of lungs and H&E of a tissue section. Quantification of tumor area. (H) Flow analyses of in vivo–derived B16F10 fusion hybrids from a primary tumor. RFP/GFP coexpressing cells analyzed for cell surface MФ identity. All boxes represent hybrid populations. Open box denote hybrids that have lost CD45 expression (n = 6 mice each). (I) B16F10 (fl-dsRed-fl-eGFP) cells intradermally injected into LysM-Cre mice (n = 4) were harvested at ~1 cm. Primary tumor or metastatic lung tumors stained with antibodies to GFP (green) and the tumor protein microphtalmia-associated transcription factor (MITF, red). Scale bar, 25 μm.
These results led us to ask whether tumor hybrids gain selective advantages in different microenvironments that may reflect primary tumor or metastatic sites. To directly test distinct microenvironmental interactions, we evaluated adhesion phenotypes and cytokine-dependent growth responsiveness of MC38-derived fusion hybrids versus unfused tumor cells using a microenvironment microarray (MEMA) platform (25). This high-throughput assay specifically measures cellular behavior in distinct engineered microenvironments containing variable extracellular matrix (ECM) molecules, growth factors, and chemokines, spotted combinatorially in rows and columns, thus permitting comparison of adhesion phenotypes among unfused cancer cells, MФs, and hybrids. Analysis of microenvironment-specific adhesion revealed that MC38 cells harbor distinct growth factor–independent adhesive preferences for select ECM molecules, most notably, fibronectin (fig. S3C). MФs, by contrast, exhibited enhanced adhesion to collagen XXIII, vitronection (the ECM component), and more uniform adhesion across all MEMA conditions relative to unfused cancer cells (fig. S3C). Fusion hybrids exhibited a combination of adhesion biases, reflecting properties of both parental cells, potentially providing a broader adhesive affinity in different microenvironments. Further analysis, using hierarchical clustering, distinguished hybrids from unfused cancer cells with respect to adhesion on independent microenvironments (fig. S3C).
To extend these observations and determine whether MФ fusion provided cancer cells with a selective proliferative or survival advantage, we directly analyzed effects of >90 different cytokines, chemokines, and soluble factors on unfused MC38s and hybrids (not shown). A number of growth factors induced differential influence on MC38 cells as compared to hybrids, including transforming growth factor (TGFβ1-3), which induced dose-dependent suppression of MC38 proliferation but showed no effect on hybrids (fig. S4, A, C, and D). Likewise, a moderate, dose-dependent growth-suppressing effect of hepatocyte growth factor (HGF) was apparent on MC38 cells but not on hybrids (fig. S4B). More strikingly, hybrids were resistant to tumor necrosis factor–α (TNF-α) that profoundly inhibited proliferation of MC38 cells (fig. S4E). Resistance of hybrids to cytokine concentrations that suppressed MC38 growth indicates that MФ fusion influences selective cellular phenotypes and altered cancer cell responses to microenvironmental factors to yield adhesive, proliferative, and potentially survival advantages.
Fusion hybrids acquire MΦ-associated phenotypes
To determine whether cell fusion provides a mechanism by which neoplastic cells acquire MΦ phenotypes consistent with tumor promotion, we evaluated MФ attributes up-regulated in hybrids identified by GO pathway analysis (table S2); behaviors shared by both MФ and fusion hybrids included migration, invasion, and response to paracrine stimuli (table S3). To determine whether hybrids harbored enhanced functional motility, we evaluated in vitro–derived MC38-derived fusion hybrids and unfused MC38 cells for migratory capacity in live-imaged MФ and neoplastic cell cocultures. Using TrackMate analysis to calculate the mean speed of cells, hybrid clones exhibited increased motility compared to nearby unfused MC38 cells (Fig. 2, C and D). Moreover, when evaluated for invasive properties using a Boyden invasion assay, MC38-derived hybrids exhibited enhanced migration and invasion activity, relative to unfused cancer cells (Fig. 2E); these results were consistent with invasive properties displayed by B16F10-derived hybrids (fig. S1B). Notably, we evaluated two independent hybrid isolates for each cell type; these displayed varying degrees of invasion, supporting our hypothesis that cell fusion can yield heterogeneous clonal outgrowths (Fig. 2E and fig. S1B).
GO genes involved in “response to stimulus” expressed at high levels in MФs were also up-regulated in MФ–cancer cell fusion hybrids. In particular, fusion hybrids harbored elevated expression of the MФ-associated gene colony-stimulating factor 1 receptor (Csf1R), a significant recruitment, differentiation, and survival molecule for MФs (26) implicated in regulating prometastatic MФ effector functions (27). To determine whether acquisition of MФ-associated receptor gene expression translated to functional ligand-mediated migration, we analyzed hybrids and unfused cancer cells in Transwell chemotaxis assays coupled to live imaging (IncuCyte Chemotaxis, Essen BioScience). Under these conditions, fusion hybrids migrated toward the ligand CSF1or stromal cell-derived factor-1 (SDF1) at multiple concentrations (shown at 25 ng/ml), whereas unfused MC38 cancer cells were incapable of responding to either chemoattractant; in contrast, B16F10 cancer cell hybrids exhibited decreased migratory responses (Fig. 2, F and G, and fig. S1C). Notably, the presence of a ligand did not change proliferative dynamics of either fusion hybrids or unfused cancer cells (not shown); however, incubation with blocking antibodies to CSF1R or CXCR4 reduced chemotactic responses of fusion hybrids (Fig. 2G). Some hybrid clones expressed both CSF1R and its main ligand, CSF1.
Tumor cell fusion hybrids are generated in vivo
In vitro–derived fusion hybrids allowed for in-depth functional analyses of acquired MФ behaviors, and FISH analyses of human tumors indicated that cell fusion occurs in vivo. These studies provided only partial insight into the physiologic relevance of fusion hybrids in human cancer. Therefore, to extend the relevance and functional significance of fusion in enhanced tumor heterogeneity and acquired behaviors for tumor progression, we investigated cell fusion in mouse models of cancer. MC38 cancer cells were injected into the flanks of R26R-YFP Cre reporter mice; fusion hybrids were identified as RFP+YFP+ cells detected among unfused tumor cells (RFP+) using immunohistochemical analyses of primary tumors. Orthotopic injection of MC38 cancer cells into the cecum resulted in pervasive peritoneal seeding, limiting the utility of this model. B16F10 melanoma cells injected intradermally into recipient mice readily developed 1.0-cm tumors (Fig. 3A), and subsequent analysis of primary B16F10 tumors revealed presence of RFP+/GFP+ fusion hybrids in actin-GFP recipient mice (Fig. 3B) and RFP+/YFP+ fusion hybrids in R26R-YFP Cre reporter mice (fig. S5, A and B). Using the latter system and FACS analyses, primary tumors were dissociated to single cells to determine the extent of cell fusion (YFP+/RFP+) in primary tumors. Hybrids represented a rare neoplastic cell population within the primary tumor (representative experiment: hybrids, 0.48% of RFP+ cells; but overall hybrids range from 0.03 to 0.69% of RFP+ cells from n = 6 individually analyzed tumors; Fig. 3D and fig. S6). To determine whether hybrids retained tumorigenicity, 300 FACS-isolated in vivo–derived hybrid cells were injected intradermally into secondary recipient mice (n = 19; Fig. 3C) and resulted in tumor growth, demonstrating that hybrid cells retained tumorigenicity (Fig. 3E). To assess tumor heterogeneity, we subsequently isolated and injected 3000 in vivo–derived fusion hybrids to facilitate temporal analyses of robust tumor growth properties in n = 3 mice. In vivo–derived fusion hybrids displayed different rates of tumor growth (Fig. 3F), indicating that hybrid cells have heterogeneous growth capacity and resulted in different rates of tumor growth.
Hybrid cell metastatic potential was evaluated using an experimental metastases model. In vitro–derived hybrids were introduced into circulation by tail vein injection. Tumor cells that trafficked to the lungs and grew as metastatic foci were identified macroscopically by their pigmented appearance and microscopically on tissue section by hematoxylin and eosin (H&E) staining. Metastatic tumor area was quantified; hybrid cells showed markedly greater metastatic burden than injected unfused tumor cells (Fig. 3G).
To determine whether in vivo–derived fusion was generated from MФ fusion partners, primary tumors were dissociated into single cells, and fusion hybrids coexpressing RFP and GFP were analyzed for cell surface MФ antigen expression (Fig. 3H and fig. S7). Identification of discrete populations of hybrids with MФ-associated surface identity points to a MФ fusion partner. Further, to establish the MФ as a fusion partner, B16F10 cells harboring an fl-dsRed-fl-eGFP allele were orthotopically injected into LysM-Cre transgenic mice (Fig. 3I). Analyses of primary tumor and lung metastases revealed MITF-expressing tumor cells harboring Cre-mediated GFP expression (Fig. 3I). Collectively, these data indicate that hybrid cells are of MФ-tumor fusion origin, develop spontaneously in vivo, retain tumorigenic capacity, and exhibit accelerated tumor progression properties.
MФ–tumor cell fusion hybrids are enriched in circulation
Detectible fusion hybrids in both primary and metastatic sites supported the possibility that fused neoplastic cells readily disseminate from primary to distant sites. To explore this, we collected blood from GFP+ mice with established isogenic RFP+ B16F10 tumors (Fig. 4A). Peripheral blood was subjected to flow cytometry for quantification of circulating tumor cells (CTCs) as defined by their RFP expression. RFP+/GFP+ fusion hybrids (or CHC) were easily detectible within the total RFP+ population, representing 90.1% of the tumor cells in circulation, markedly outnumbering unfused RFP+/GFP−ve CTCs (Fig. 4A). Imaging of individual CHCs confirmed their fusion identity and morphologically distinguished them from MФs that had phagocytosed or adhered to cancer cells (Fig. 4A).
Fig. 4. Murine CTCs.
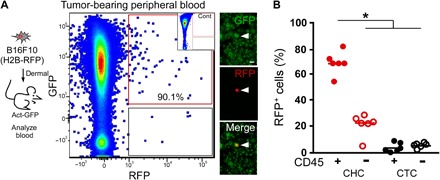
(A) B16F10 (H2B-RFP) cells (5 × 104 cells) intradermally injected into a syngeneic GFP-expressing recipient mouse. Blood collected at time of tumor resection and analyzed by flow cytometry for GFP and RFP expression. RFP+GFP+ cells were detectible in presorted cell preparations by immunofluorescence. Scale bar, 50 μm. We analyzed GFP-expressing blood by flow cytometry as a negative control for (A) inset. (B) Percentages of fusion hybrids (RFP+/GFP+) and unfused CTCs (RFP+/GFP−) expressing the leukocyte antigen CD45 (*P < 0.000002).
The classical definition of CTCs in human cancer is a circulating cell expressing a tumor antigen [typically epithelial cell adhesion molecule (EPCAM) or cytokeratin for epithelial cancers] and not expressing the pan-leukocyte antigen CD45 (28, 29). MФs normally express CD45; therefore, we reasoned that MФ–cancer cell fusion hybrids would also express this cell surface epitope. The majority of RFP+/GFP+ fusion hybrids expressed CD45, while unfused RFP+ cancer cells largely did not (Fig. 4B and fig. S8). The classical isolation approaches for CTCs exclude any CD45-expressing cells and therefore exclude the novel CHC population from routine analyses. The presence of both classical CTCs and novel CHCs highlights heterogeneity of tumor cells in circulation destined to seed metastatic tumors. Moreover, presence of CD45-expressing CHCs in our mouse models prompted us to investigate the presence of this unique hybrid population in human cancer patients.
Hybrids in humans correlate with disease stage and patient survival
To evaluate the biological significance of hybrids in humans, we first determined whether hybrids between blood cells and epithelial-derived cancer cells were detectible. To accomplish this, we exploited a disease scenario that supports identification of hybrids harboring properties of peripheral mononuclear blood cells and epithelial cells (30)—specifically, the analysis of tumor biopsies from female cancer patients who had previously received a sex-mismatched bone marrow transplant and subsequently developed a secondary solid tumor. In these patients, only the male donor–transplanted hematopoietic cells should contain a Y chromosome; therefore, identification of Y chromosome–positive nuclei in cytokeratin-positive cells within the tumor biopsy could indicate fusion between a peripheral mononuclear blood cell and an epithelial tumor cell. We identified tumor epithelia by a pathologic review of H&E-stained tumor biopsies, and then we probed tissue sections with pan-cytokeratin antibodies and interrogated with Y chromosome FISH probes to identify cellular products consistent with fusion between neoplastic cells and transplanted male hematopoietic cells (Fig. 5 and fig. S9). In a biopsy from a female patient with pancreatic ductal adenocarcinoma (PDAC), neoplastic cell nuclei containing a Y chromosome were detectible throughout regions of the tumor (Fig. 5, A to E, and fig. S9, A and B), as well as in premalignant regions of pancreatic intraepithelial neoplasia (PanIN; fig. S9C). Confocal microscopy confirmed that Y chromosomes were located in nuclei of cytokeratin-positive epithelial tumor cells (see higher magnifications in Fig. 5). For these studies, tumor specimens from seven patients were examined, and all contained evidence of fusion by these criteria. Y chromosome–positive epithelial tumor cells were not unique to PDAC, as fusion hybrids were detected in other solid tumors from female recipients of sex-mismatched transplantation, including renal cell carcinoma, head and neck squamous cell carcinoma, and lung adenocarcinoma (fig. S9, D to F). Control tissue staining from female and male tissue samples were carried as controls for Y chromosome detection (fig. S10, A and B). These observations were consistent with previous case reports of cell fusion in human cancer using a variety of other detection methods (11, 17, 31, 32).
Fig. 5. Cell fusion in human tumors.
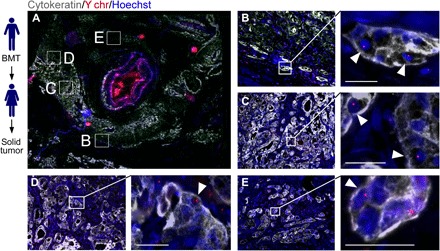
Solid tumors from women (n = 7) with previous sex-mismatched bone marrow transplantation (BMT) permits analysis of cell fusion. (A) PDAC tumor section with cytokeratin (gray), the Y chromosome (Y chr, red), and Hoechst (blue) detection revealed areas of cytokeratin-positive cells with Y chr–positive nuclei (white arrowheads). Boxed representative areas are enlarged in (B) to (E). Scale bars, 25 μm.
To examine hybrid cells in circulation from human patients, we analyzed the peripheral blood from a sex-mismatched bone marrow–transplanted female cancer patient. CHCs that coexpressed CD45, a pan-leukocyte marker, and EPCAM, an epithelial marker, were detected (Fig. 6A). Both of these CHCs and leukocytes expressed the Y chromosome (Fig. 6A and fig. S10B). To determine whether CHCs expressed MФ markers, analogous to fusion hybrids found in the murine tumor model, we identified MФ epitope expression by immunohistochemical (Fig. 6B) and by flow cytometric analyses (Fig. 6C and fig. S11) was performed. A variety of MФ epitopes were expressed on CK+/CD45+ CHCs, including CD163, CD68, CSFR1, and CD66b (Fig. 6B). Similarly, flow cytometric analyses revealed that CHCs from three different PDAC patients expressed MФ epitopes, including CD14, CD16, CD11c, and CD163 (Fig. 6C). CTCs analyzed from the same patients had low expression levels of CD16. These results indicate that MФ–tumor cell hybrids are the predominant tumor cell in circulation, although other leukocyte-tumor cell hybrids with discrete MФ surface antigen expression most likely exist.
Fig. 6. Human CTCs.
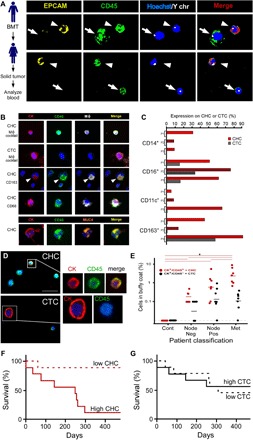
(A) Sex-mismatched bone marrow–transplanted (BMT) patient who acquired a solid tumor (PDAC). Peripheral blood was analyzed for the presence of cell fusion. Two panels displaying cell fusion hybrids (arrowheads) that costain for EPCAM (yellow) and CD45 (green) and have a Y chromosome (white dot) in their nuclei (blue). Arrows denote leukocytes. (B) CHCs and CTCs analyzed from n = 4 patients with PDAC. CHCs (CK+/CD45+) also express MФ proteins (cocktail: CD68, CD163, CD66b, and CSF1R), while CTCs (CK+/CD45−ve) do not. CHCs also express the tumor-specific protein MUC4. (C) CHCs and CTCs analyzed by flow cytometry for CD14, CD16, CD11c, and CD163 expression or the cancer-specific protein MUC4 (n = 4 patients). (D) Human pancreatic cancer patient peripheral blood analyzed for cytokeratin+ (red) and CD45+ (green) expression using in situ analyses and digital scanning. (E) CK+/CD45+ and CK+/CD45− cells quantified in patient blood across cancer stages [analysis of variance (ANOVA), *P < 0.023]. (F and G) Kaplan-Meier curve of dichotomized biomarkers based on median value (CHC and CTC) was associated with statistically significant increased risk of death for CHCs (P = 0.0029) but not for CTCs (P = 0.95).
To explore the presence of CHCs in pancreatic cancer patients diagnosed at various tumor stages, node-negative, node-positive, or metastatic, we collected peripheral blood and performed in situ antibody staining (CD45, and CK) on isolated cells. Digital image analyses allowed validation of double-positive expression of CD45 and CK on CHCs (Fig. 6D) while excluding doublets or clusters of cells that could register as double-positive cells by flow cytometry. We determined that the number of CHCs expressing CD45+/CK+ significantly correlated with advanced disease (Fig. 6E). Notably, CHC enumeration revealing high expression in the blood provided a prognostic indicator of overall survival, regardless of disease stage (Fig. 6F). Conventionally defined CTCs (CD45−ve/CK+) did not correlate with stage or survival (Fig. 6, E and G) and were detected at quantities an order of magnitude lower than CHCs in metastatic disease. These findings identify a novel population of tumor cells in circulation, a population previously overlooked and excluded from routine analyses, which has a biologic function and correlation to clinically relevant disease status in human cancer patients.
DISCUSSION
Cell fusion between immune and neoplastic cells initiating tumorigenesis and affecting progression is an untested, century-old hypothesis (9, 10) that has been only circumstantially examined (10, 12, 17, 31, 33–37). Reports of cells located in tumors containing components of both immune and neoplastic cells are increasingly frequent (1, 10–13, 16, 17, 19, 35, 38–44), although without strong evidence of etiologic mechanism or physiologic relevance. Early in vitro studies revealed that cell fusion hybrids displayed reduced cell doubling times relative to their genetic burden (45). More recently, it was suggested that tumor cell fusions gain shorter cell cycling times compared to either of their parental cells (46, 47). These divergent views add to the controversy of whether cell fusion provides a selective advantage to evolving tumors. Moreover, description of MФ gene expression in metastatic cancer cells as evidence that fusion propagates aggressive metastatic spread of cancer (31, 42) is presented without substantial proof that these cells arise from a fusion event, further diminishing enthusiasm for this mechanism. Despite this, MФ–neoplastic cell fusion does provide an intriguing mechanism for how neoplastic cells rapidly gain discrete cellular behaviors to facilitate metastases and to propagate intratumoral heterogeneity. Before this study, experimental results demonstrating in vivo tumor cell fusion with MФs or investigating a function role for cell fusion in tumor progression were undetermined. Here, we demonstrate that cell fusion occurs spontaneously in a number of systems. Cell fusion can contribute to the generation of diverse neoplastic clones with altered phenotypes, implicating it as a mechanism for gain of intratumoral heterogeneity. This finding may reveal insight into diverse tumor cell pathophysiology that underlies treatment resistance, progression, and posttreatment tumor recurrence in human cancer.
We present a systematic analysis of MФ–cancer cell fusion and provide evidence that hybrids impart physiologically relevant and functionally significant aspects contributing to tumor evolution. Together, our in vitro and in vivo murine data indicate that neoplastic cells fuse spontaneously with leukocytes and myeloid cells (that is, MФs, neutrophils, or dendritic cells) and produce heterogeneous cancer hybrid clones. Despite their diversity, hybrid clones retain MФ genotypes with functional phenotypes, thereby bestowing MФ-like behaviors on neoplastic cells. Fusion hybrids express functional levels of CSF1R, which is relevant to cancer progression exemplified by the association of CSF1 overexpression in lung cancer with increased tumor cell proliferation and invasion (48), by the inhibition of CSF1R with decreased tumor metastasis (49) and by late-stage metastatic breast carcinomas frequently acquiring CSF1R expression (38). In human tumors, the mechanism by which tumor cells gain chemotactic responsive receptor expression remains unclear, but multiple mechanisms likely underlie transcriptional changes. Our data indicate that cell fusion could play a role in the acquisition of migratory/chemotactic functional behavior and may have important clinical implications considering hybrids’ potential response to clinically relevant CSF1R inhibitors (27, 50, 51).
Data presented here indicate that MФ–cancer cell hybrids are differentially responsive to microenvironment-derived regulatory forces. Specific extracellular conditions provide a selective adhesive and/or growth advantage to hybrids but not to unfused neoplastic cells. Given that genotypic and phenotypic diversity provides selective advantages to the fittest neoplastic clones, continued cell fusion may underlie adaptation, survival, and growth of dominant neoplastic clones within the evolving tumor microenvironment during tumor progression. Considering this possibility, cell fusion provides a previously underappreciated mechanism by which neoplastic cells gain phenotypic diversity, increasing opportunities for highly fit subclones to overcome selection pressure and drive tumor progression.
These data indicate that tumor-initiating hybrid populations can acquire behaviors allowing for navigation of the metastatic cascade—from the primary tumor, to survival in circulation, to seeding of ectopic sites, and to propagation of metastatic foci. Further, in vivo–derived hybrid cells were readily detected in peripheral blood. CHCs outnumbered conventionally isolated CTCs in both mice and humans. Moreover, in patients with pancreatic cancer, CHCs directly correlated with tumor stage and inversely correlated with overall survival—highlighting an exciting prognostic opportunity for development of this novel cell population as a liquid biomarker for disease status in discrete cancers where conventional biomarkers (for example, CTCs, cell-free DNA, exosomes, and proteins) have not demonstrated efficacy.
Unlike in murine models, the etiology of human CHCs, while consistent with cell fusion, cannot be conclusively determined. It is possible that tumor cells can gain expression of leukocyte- and MФ-associated proteins by an undetermined mechanism or that CD45-expressing blood cells transdifferentiate into epithelial cancer cells. Despite these unexplored caveats, the CHC is a population of tumor cells previously overlooked and understudied. Our initial investigations indicate that this novel cell population has exciting potential.
Identification of functionally significant properties of this unique tumor population, a chimera of MФs and neoplastic cells, offers opportunities for understanding the dynamic interaction between neoplastic cells and diverse infiltrating immune cell populations. Elevated CHCs relative to CTCs in peripheral blood might suggest that hybrids are immune privileged—a trait bestowed by their leukocyte identity. This scenario could have implications on immune-mediated therapeutic strategies for cancer treatment. Therefore, understanding how hybrids respond to immune therapies, such as inhibitors or agonists to costimulatory and/or coinhibitory receptors, offers an important area of future investigation. Acquisition of functional myelomonocytic receptors on hybrids indicates that they may be vulnerable to targeted therapies such as CSF1/CSF1R blockade, now being investigated in clinical trials (51). Alternatively, these therapies may inhibit MФ–neoplastic cell fusion. The presence of tumor cells with acquired MФ phenotypes supports a cell fusion mechanism in the propagation of intratumoral heterogeneity, introduces a functionally significant aspect of tumor progression and evolution, identifies an unappreciated CHC population, and uncovers a new area of tumor cell biology.
METHODS
Human samples and ethics statement
All human blood and tissue samples were collected and analyzed with approved protocols in accordance with the ethical requirements and regulations of the Oregon Health & Science University (OHSU) institutional review board. Informed consent was obtained from all subjects. Peripheral blood was obtained from cancer patients diagnosed with PDAC at various stages and treated at OHSU, as well as from healthy controls. Identification and acquisition of solid tumor or peripheral blood biopsies from female patients who previously received a gender-mismatched bone marrow transplantation was conducted by screening of the Center for International Bone Marrow Transplant Registry.
FISH and immunohistochemical analyses of human solid tumors and peripheral blood cells
Analyses of human solid tumors
The presence of cell fusion between Y chromosome–containing blood cells and host tumor epithelium was evaluated by dual FISH and immunohistochemical analyses. X and Y chromosome FISH probes were hybridized to 5-μm formalin-fixed paraffin-embedded primary human tumor sections using CEP X (DXZ1 locus) and CEP Y (DYZ1 locus) probes (Abbott Molecular) following the manufacturer’s protocols. Briefly, the tissue was treated with Retrievagen A solutions (BD Biosciences) and Tissue Digestion Kit II reagents (Kreatech) and then hybridized with a probe at 80°C for 5 min and 37°C for 12 hours. Tissue sections were permeabilized with graded detergent washes at 24°C and then processed for immunohistochemical staining. Tissue was incubated with antibodies to pan-cytokeratin (Fitzgerald) and counterstained with Hoechst dye (1 μg/ml). Two slides were analyzed for each tumor section. Slides were digitally scanned and quantified by two independent investigators. Areas with Y chromosome positivity were analyzed by confocal microscopy. H&E staining was conducted on adjacent sections.
In situ analyses of human peripheral blood analyses
Patient peripheral blood was collected in heparinized Vacutainer tubes (BD Biosciences), and then, lymphocytes and peripheral mononuclear cells were isolated using density centrifugation and LeucoSep Centrifuge Tubes (Greiner Bio-One) according to the manufacturer’s protocol. Cells were then prepared for antibody staining. Briefly, cells were adhered to poly-d-lysine–coated slides, fixed, and permeabilized before staining for CD45 and cytokeratin expression using antibodies to CD45 (eBioscience) and human pan-cytokeratin (Fitzgerald). Phenotypes of CHCs were evaluated with additional antibody staining, including to CD66b (BD Pharmingen), CD68 (Abcam), CD163 (Neomarkers), CSF1R (Abcam), and EPCAM (1:200; US Biological). Tissue was developed with appropriate fluorescent-conjugated secondary antibodies [anti-mouse Cy3 (Jackson ImmunoResearch), goat anti-guinea pig 488 (Invitrogen), goat anti-guinea pig 555 (Invitrogen), anti-rabbit A647 (1:500; Thermo Fisher Scientific), and anti-mouse Cy5 (1:500; Jackson ImmunoResearch)] and then was stained with Hoechst (1 μg/ml). Slides were digitally scanned with a Leica DM6000 B microscope or a Zeiss AxioObserverZ1 microscope and analyzed using Ariol or Zeiss Zenblue software.
To determine whether circulating CD45+/CK+ or CD45+/EPCAM+ cells were cell fusion products, patient peripheral blood was subjected to FISH/immunohistochemical analyses as described for solid tissues (see above). Processed peripheral blood was interrogated with Y chromosome FISH (DYZ1 locus) probes (Abbott Molecular) following the manufacturer’s protocols. Briefly, cells were hybridized with a probe at 42°C for 16 to 20 hours and then subjected to graded detergent washes. Cells were then subjected to antibody staining with anti-CD45 conjugated to fluorescein isothiocyanate (FITC) (1:100; BioLegend) and EPCAM (1:100, US Biological) and processed with anti-rabbit AF647 secondary antibodies (1:250; JacksonImmuno Research). Cells were imaged on a Zeiss AxioOberverZ1 microscope. Images were postprocessed to rule out nonspecific staining. Briefly, CZI files were opened using ZEN 2.3 Lite (Blue Edition) and saved as single-channel TIF files [four channels per CZI: EPCAM (white), Y chromosome (red), CD45 (green), and 4′,6-diamidino-2-phenylindole (DAPI; blue)]. Single-channel TIF files were loaded into MATLAB as UINT8 matrices containing RGB information at each pixel. To create binary images, pixel intensity thresholds were set for each channel image separately: Any pixel with a value above the threshold was turned ON (that is, maximum intensity), and the remaining pixels were turned OFF (that is, zero intensity). Two binary channel images were reassigned colors (EPCAM: white → yellow, Y chromosome: red → white); all binary channel images were then overlaid.
Quantification of CHCs in patient blood
Manual quantification by three independent investigators of randomly selected regions containing 2000 cells evaluated CD45 and cytokeratin status of Hoescht+ cells. Percentages of CHCs in the buffy coat correlate with disease stage with significance determined by overall ANOVA post-test [P < 6.3 × 10−8; P values for no nodal-met (0.00035), nodal-met (0.05), and no nodal-nodal (0.15)], while none of the conventional CTCs (that is, CD45−ve) comparisons across stage were statically significant [P values for no nodal-met (0.31) and nodal-met (0.9)]. Survival analysis was conducted on 18 of 20 pancreatic patients (2 patients were lost to follow-up, 9 patients have high levels, and 9 patients have low levels) to correlate CHCs or CTCs with time to death using Kaplan-Meier curve and log-rank test using dichotomized biomarkers based on a median value. High CK+/CD45+ (median, >0.808) was associated with a statistically significant increased risk of death (log-rank test, P = 0.0029) with a hazard ratio of 8.31, but high CK+/CD45−ve (median, >0.101) did not have a statistically significant effect on time to death (log-rank test, P = 0.95).
Flow cytometric analyses of fusion hybrids in peripheral blood
For flow cytometric analysis, patient blood was collected, as described above. Red blood cell (RBC) lysis was performed by a 1-min incubation in 0.2% NaCl, followed by addition of the equivalent volume of 1.6% NaCl. Cells were washed and resuspended in FACS buffer [phosphate-buffered saline (PBS), 1.0 mM EDTA, and 5% fetal bovine serum (FBS)]. Cells were incubated in PBS containing LIVE/DEAD Fixable Aqua (1:500; Invitrogen) with Fc Receptor Binding Inhibitor (1:200; eBioscience). Cells were then incubated in FACS buffer for 30 min with CD45-APC (1:25; Thermo Fisher Scientific), CD11c-APCeF780 (1:100; Thermo Fisher Scientific), CD14-BV785 (1:100; BioLegend), CD163-PECy7 (1:100; BioLegend), EPCAM-FITC (1:100; Abcam), or cytokeratin-PE (1:500; Abcam). A BD LSRFortessa cell analyzer was used for analyses. A gating scheme established with single-color controls is provided in fig. S10. Data reflect analyses from n = 3 patients with PDAC.
Mice
All mouse experiments were performed in accordance to the guidelines issued by the Animal Care and Use Committee at OHSU or the Fred Hutchinson Cancer Center, using approved protocols. Mice were housed in a specific pathogen–free environment under strictly controlled light cycle conditions, fed a standard Rodent Laboratory Chow (#5001 PMI Nutrition International), and provided with water ad libitum. The following strains were used in the described studies: C57BL/6J (JAX #000664), Gt(ROSA)26Sortm(EYFP)Cos/J (R26R-stop-YFP; JAX#006148) (21), Tg(act-EGFP)Y01Osb (Act-GFP; JAX #006567) (20), and B6.129P2-Lyz2tm1(cre)Lfo/J (LysM-Cre; JAX#004781) (52). Mice of both genders were randomized and analyzed at 8 to 10 weeks of age. When possible, controls were littermates housed in the same cage as experimental animals.
Cell culture
MC38 mouse intestinal epithelial cancer cells were provided by J. Schlom [National Cancer Institute (NCI)], and B16F10 mouse melanoma cells were obtained from the American Type Culture Collection. Validation of cell lines was confirmed by polymerase chain reaction and by functional metastasis assay for the latter. Cell lines, both derived from C57BL/6J mice, were cultured in Dulbecco’s modified Eagle’s medium (DMEM) + 10% serum (Life Technologies). Stable cancer cell lines, MC38 (H2B-RFP), MC38 (H2B-RFP/Cre, B16F10 (H2B-RFP), and B16F10 (H2B-RFP/Cre), were generated by retroviral transduction using pBABE-based retroviruses, and polyclonal populations were selected by antibiotic resistance and flow-sorted for bright fluorescence as appropriate. B16F10 (fl-dsRed-fl-eGFP) cells were generated by stably expressing a pMSCV-LoxP-dsRed-LoxP-eGFP-PURO construct (Addgene #32702) into the parental B16F10 cells. Primary MФ derivation was conducted from the bone marrow of R26R-stop-YFP or Act-GFP mice. To elicit MФs, cells were cultured for 6 days in DMEM + 15% serum supplemented with sodium pyruvate, nonessential amino acids (Life Technologies), and CSF1 (25 ng/ml; PeproTech).
Cell fusion hybrid generating cocultures were established in MФ-derivation media without CSF1 for 4 days. MC38 or B16F10 cells and MФs were coseeded at a 1:2 ratio at low density. Hybrid cells were FACS-isolated for appropriate fusion markers on a Becton Dickinson InFlux or FACSVantage SE cell sorters (BD Biosciences). FACS plots are representative of at least 20 independent MC38 or B16F10 hybrid isolates (technical replicates). Low-passage hybrid isolates were established; functional experiments were conducted on passage 8 to 20 hybrid isolates. Live imaging of cocultured cells was performed using an IncuCyte ZOOM automated microscope system and associated software (Essen BioScience). Technical triplicates generated 36 movies that covered 77.4 mm2 and were screened for hybrid generation and division. Movie contains a fusion event; a total of 21 video clips were captured containing fusion events.
EdU-labeling and karyotype analysis
During hybrid generation
Cultured cells were fixed in 4% formaldehyde in PBS and processed for immunohistochemical analyses with antibodies against GFP (1:500; Life Technologies) or RFP (1:1000; Allele Biotechnology). EdU labeling and detection were performed according to the manufacturer’s directions (Life Technologies). Briefly, MФ DNA was labeled with 10 μM EdU supplemented in media for 24 hours before hybrid generation coculture. EdU (10 μM) was used for the determination of S-phase indices as well. Biologic and technical replicates (n = 6) were conducted and screened for biparental hybrids.
For karyotype analyses
Chromosome spreads from cells in S phase were prepared using standard protocols, from cells treated for >12 hours with Colcemid (100 ng/ml; Life Technologies) to induce mitotic arrest. DNA was visualized by staining with DAPI; X and Y chromosomes were identified using fluorescently labeled nucleotide probes (ID Labs) as directed by the manufacturer. Images of stained fixed cells and chromosome spreads were acquired using a 40× 1.35 UApo oil objective on a DeltaVision-modified inverted microscope (IX70; Olympus) using SoftWorx software (Applied Precision) and represented maximum intensity projections of deconvolved z-stacks unless otherwise indicated. Experiments were replicated eight times. Each biologic replicate was analyzed in an independent experiment. A minimum of n = 20 cells were analyzed in each experiment. Chromosomes were counted manually by two independent investigators.
Gene expression analysis
Microarray analysis was performed with Mouse 430.2 gene chips (Affymetrix) at the OHSU Gene Profiling Shared Resource, and data were analyzed using GeneSifter software (Geospiza) to identify relative expression differences between cell types (replicates: MФ, n = 3; MC38, n = 3; hybrids, n = 5 independent isolates) and produce GO analyses. GO category enrichment was calculated using the GOstats R package (53) and visualized using functions from the GOplot R package (54).
Code availability
The source code used to generate figures and corresponding tables is available for download from our public repository (55).
Polymerase chain reaction
DNA was extracted from frozen formalin-fixed melanoma primary tumor and lymph node sections by 40 min of incubation in lysis buffer [25 mM NaOH and 0.2 mM EDTA (pH 12)] at 95°C, followed by neutralization with equal volumes of neutralization buffer [40 mM tris-HCl (pH 5)]. RFP primers were as follows: 5′-CAGTTCCAGTACGGCTCCAAG-3′ (forward) and 5′- CCTCGGGGTACATCCGCTC-3′ (reverse). Actin primers were as follows: 5′-GAAGTACCCCATTGAACATGGC-3′ (forward) and 5′-GACACCGTCCC-CAGAATCC-3′ (reverse). Reactions were run with a 60°C annealing temperature.
Microenvironment arrays
Recombinant proteins (R&D Systems and Millipore) were diluted to desired concentrations in print buffer (ArrayIt), and pairwise combinations of ECM proteins and growth factors or cytokines were made in a 384-well plate. A Q-Array Mini microarray printer (Genetix) was used to draw from the 384-well plate and print protein combinations onto Nunc 8-well chambered cell culture plates (Thermo Fisher Scientific). Each combination was printed in quintuplicate in each array, and arrays were dried at room temperature. Printed MEMAs were blocked for 5 min using 0.25% (w/v) F108 copolymer (Sigma-Aldrich) in PBS and then rinsed with PBS and media before plating cells. Cells were trypsinized, filtered to exclude cell clumps, and counted; 105 cells were plated on each array in 2 ml of DMEM + 2.5% serum and incubated for 30 min in a humidified tissue culture incubator. Unbound cells were gently removed, and fresh media were added. After 12 hours, the arrays were fixed with 4% formaldehyde in PBS for 10 min and stained with DAPI. Adhesion was measured as relative cellular preference: the number of cells occupying a given microenvironment condition relative to the average cell number over all occupied microenvironmental spots across the entire MEMA for each sample. Five replicate samples each for MC38 cells and MФ and five independent MC38-derived hybrid isolates were analyzed. Standard two-tailed t tests were performed with P < 0.05 reported as significant. Error bars represent SEM.
In vitro–derived hybrid proliferation
For phenotypic profiling growth responsiveness to cytokines and soluble factors, 95 different cytokines or soluble signaling molecules were distributed at high, medium, and low concentrations in 384-well plates, in 25 μl of RPMI 1640 (Life Technologies) supplemented with 1% FBS, and 25 μl of a suspension of hybrid or MC38 cells (1.2 × 104 cells/ml) in DMEM + 4% FBS was added to each well. Ninety-nine wells of each plate were left cytokine-free, and no cells were added to 2 of these wells, which served to provide measurements of background signal. Plates were cultured in a humidified incubator for 72 hours, after which 5 μl of MTS reagent was added to each well. Two hours later, absorbance at 490 nm was read with a 384-well plate reader. For each plate, absorbance values for each cytokine-treated well were normalized to the mean absorbance of the cytokine-free wells on that plate and expressed in terms of SDs from the cytokine-free mean. Three independent hybrid isolates and three MC38 replicates were analyzed. Cytokines or factors that showed a potential differential effect on growth of MC38 and hybrid cells were retested in 96-well plates. In these experiments, 2.5 × 104 hybrid or MC38 cells were plated in the presence of three different concentrations for each soluble factor or in media alone (DMEM + 2.5% FBS), in triplicate for each condition. Plates were imaged every 2 hours for 90 hours, and then, cell viability was assessed.
Chemotaxis assay
Chemotaxis assays were performed using IncuCyte Chemotaxis Cell Migration Assay (Essen BioScience) with at least three technical replicates of triplicate samples. Briefly, 1000 cancer cells were plated media for 20 hours. CSF1 or SDF1 ligand (25 ng/ml) was added to the bottom well, and cells were incubated at 37°C for at least 36 hours with live imaging. The neutralizing antibodies to the CSF1R (eBioscience), CXCR4 (BioLegend), and isotype control antibody were added to the top and bottom wells (2.5 ng/μl). Migration was quantified by measuring the phase-contrast area of the top and bottom wells for each time point using IncuCyte ZOOM software. Triplicates of each condition were performed, and the means and SDs were calculated. P < 0.02 for hybrids treated with CSF1 or SDF1 relative to hybrids without CSF1 or SDF1 by unpaired t test. Two independent hybrid isolates were analyzed. Technical octupulicates (MC38) or sextuplicates (B16F10) with biologic quadruplicates or triplicates were analyzed. For inhibitor studies, technical duplicates with biologic triplicates were analyzed.
Migration analysis
From IncuCyte live imaging of cocultured MФs and cancer cells, 24- to 48-hour image series containing a cancer-MФ fusion event was cropped and exported as two separate uncompressed audio video interleave (AVI) files: one containing only the red channel for TrackMate analysis and another containing both red and green channels with a sizing legend. Red-channel AVI files were imported into FIJI and converted to 8-bit image series with a mean filter of 1.5 pixels applied. TrackMate analysis was then performed on nuclei with an estimated diameter of 10 pixels and a tolerance of 17.5. Using the Linear Assignment Problem Tracker, settings for tracking nuclei were as follows: 75.0-pixel frame-to-frame linking and 25.0-pixel and two-frame gap track segment gap closing. Tracks segments were not allowed to split or merge. Using the analysis function in TrackMate, track statistics were exported to an Excel file, and tracks containing 11 or fewer frames were excluded from the analysis. A total of 9 hybrid cells and 536 unfused cells were analyzed with a P < 1.1 × 10−9 by unpaired t test. Error bars represent SD.
Boyden chamber invasion assay
In vitro invasion assay was performed, as described previously (56). Briefly, cellular invasion was measured in a growth factor–reduced Matrigel invasion chamber with 8-μm pores (#354483, Corning). Cells (3 × 105) in a medium containing 0.1% FBS were placed into each Boyden chamber. The medium containing 10% FBS was placed in the lower chamber to facilitate chemotaxis. Invasion assays were run for 15 hours, and then cells that passed through the Matrigel membrane were stained with 0.09% crystal violet/10% ethanol. After extraction by elution buffer [1:2:1 acetate buffer (pH 4.5)/ethanol/deionized water], the stain was measured at 560 nm. Representative images of invaded cells were taken by an Axio Zoom.V16 microscope (Zeiss). The assay was run in triplicate, in biologic replicate.
In vivo analyses of in vitro–derived cell fusion hybrids
For tumor growth, 8-to 12-week-old C57BL/6J mice (Jackson Laboratories) were injected with 5 × 104 cells (MC38 and MC38-derived hybrids) or 5 × 105 cells (B16F10 and B16F10-derived hybrids) subcutaneously or intradermally, respectively. Length (L) and width (W) of palpable tumors were measured three times weekly with calipers until tumors reached a maximum diameter of 2 cm. Tumors were surgically removed in survival surgery, or animals were sacrificed during tumor removal in accordance with OHSU Institutional Animal Care and Use Committee guidelines. Animals were observed for at least 6 months for detection of tumor growth. For each tumor, volume (V) was calculated by the formula V = 1/2 (L × W2); volume doubling time for each tumor was extracted from a curve fit to a plot of log tumor volume over time. Curves with R2 values of less than 0.8 were excluded from analysis, as were tumors with six or fewer dimension measurements; these exclusion criteria were established in response to the unanticipated early ulceration of some tumors, which precluded accurate measurements of length and width (P < 0.05, by Mann-Whitney U test). For growth of tumor at metastatic sites, 1 × 106 MC38 cells were injected into the spleen. Livers were analyzed 3 weeks later for tumor burden by H&E stain. Hybrids formed metastatic foci more readily with a P < 0.008 by Mann-Whitney U test. MC38 (n = 17) and MC38-derived hybrids (n = 13) were injected in four different technical replicate experiments. For B16F10 cells, 2.5 × 105 cells were retro-orbitally injected, and lungs were analyzed 16 days after injection. Melanin-marked tumor metastasis was visualized. Tumor burden was analyzed on paraffin-embedded tissue sections located every 100 μm apart through the entire lung (n = 5 tissue sections per lung). Metastatic foci areas were measured using an Aperio ImageScope V12.3.0.5056 to outline metastatic tumors and quantify area. A nonparametric t test was performed. Duplicate studies of B16F10 and B16F10-derived hybrids (n = 12 mice) were analyzed.
In vivo–derived cell fusion hybrids
For isolation of in vivo–derived hybrids or assessment of CTCs, 5 × 105 B16F10 (H2B-RFP with or without Cre) cells were injected intradermally into R26R-YFP or actin-GFP mice, respectively. Once tumors reached 1 to 2 cm3 in diameter, they were surgically removed for immunohistochemical analyses or for FACS/flow analyses.
For demonstration that tumor cells can fuse with myeloid cells, 5 × 105 B16F10 (fl-dsRed-fl-eGFP) cells were injected intradermally into 6- to 8-week-old LysM-Cre transgenic mice. When tumors reached 1 cm3, primary tumors and lungs were removed for immunohistochemical analyses.
Immunohistochemical analysis of in vivo–derived tumors
B16F10 (H2B-RFP, Cre) primary tumors in Act-GFP or R26R-stop-YFP mice were fixed in 10% buffered formalin, frozen in optimum cutting temperature (OCT), and 5-μm sections were obtained. Tumors from R26R-stop-YFP mice were incubated with antibodies for GFP (1:500; Life Technologies) followed by detection with fluorescent secondary antibody (1:500, Alexa Fluor 488; Jackson ImmunoResearch). Nuclei were counterstained with Hoechst (1 μg/ml). Slides were digitally scanned with a Leica DM6000 B microscope and analyzed using Ariol software. Confocal images were acquired with a FluoView FV1000 confocal microscope (Olympus).
B16F10 (fl-dsRed-fl-eGFP) primary tumors and lungs from LysM-Cre mice were fixed in 4% paraformaldehyde for 2 hours at 20°C, washed, and cryopreserved in 30% sucrose for 16 hours at 4°C and then embedded in OCT. Primary tumors were stained as described in the paragraph above. Lung sections were cut to 8-μm thickness, baked for 30 min at 37°C, then subjected to antigen retrieval under standard conditions (R&D Systems, CTS016), blocked with DAKO Protein Block Serum-Free (Agilent, X090930-2), and incubated for 16 hours at 4°C with primary antibodies [anti-MITF (1:500; Abcam, ab12039), anti-dsRed (1:250; Clontech, 632496), and anti-GFP (1:1000; Abcam, ab13970)] in background-reducing antibody diluent (Agilent, S302281-2). Fluorescent-tagged secondary antibodies were applied, and then sections were mounted in a ProLong Gold antifade reagent (Molecular Probes, P36934). Antibody specificity was determined by immunostaining healthy lungs of nontumor-bearing mice and performing secondary antibody only controls.
FACS isolation and flow cytometric analyses of fusion hybrids
Tumors were diced and digested for 30 min at 37°C in DMEM + Collagenase A (2 mg/ml; Roche) + DNase (Roche) under stirring conditions. Digested tumor cells were filtered through a 40-μm filter and washed with PBS. For FACS isolation, hybrid and unfused cells were isolated by direct fluorescence on a Becton Dickinson InFlux sorter. For flow cytometric analysis, blood was collected retro-orbitally using heparinized microhematocrit capillary tubes (Fisher) into K2EDTA-coated tubes (BD Biosciences). RBC lysis was performed as described above. Cells were washed and resuspended in FACS buffer (PBS, 1.0 mM EDTA, and 5% FBS). Cells were incubated in PBS containing LIVE/DEAD Fixable Aqua (1:500; Invitrogen) with Fc Receptor Binding Inhibitor (1:200; eBioscience). Cells were then incubated in FACS buffer for 30 min with CD45-PeCy7 (1:8000; BioLegend), CSF1R-BV711 (1:200; BioLegend), F4/80-APC (1:400; BioLegend), and CD11b-AF700 (1:200; eBioscience). A BD LSRFortessa FACS machine was used for analyses. A statistical significance of P < 2.2 × 10−6 by unpaired t test was determined for CD45+ hybrid CTCs relative to CD45− hybrid, CD45+ unfused, and CD45− unfused CTCs. Technical duplicates of n = 5 or 6 mice were analyzed.
Tumorigenic analyses of FACS-isolated in vivo–derived hybrids
A total of 100 or 3000 FACS-isolated hybrids and unfused B16F10 cells were injected intradermally into C57BL/6J mice. For experiments with 100 cells, technical octuplicates with biologic duplicates, triplicates, or quadruplicates were performed, depending on the number of hybrids isolated form the primary tumor, for a total of n = 16 mice analyzed. For experiments with 3000 cells injected, technical triplicates were performed.
Statistical analyses and graphical displays
Dot plots, bar charts, and line charts were generated in GraphPad Prism or Excel. GraphPad Prism and Excel were also used for statistical analyses of these data, including ensuring that data met assumptions of the tests used and comparisons of variance between groups when appropriate. Microsoft Excel was used to perform two-tailed t tests. A three-dimensional scatterplot was generated in R using the rgl package. Flow cytometry data were prepared for display using FlowJo software. Microarray gene expression data were displayed as a heatmap prepared using Genesifter software. Heatmap of MEMA data was generated in R using the standard heatmap function and default parameters.
Supplementary Material
Acknowledgments
We acknowledge members of the OHSU Flow Cytometry Shared Resource and Advanced Light Microscopy Core at the Jungers Center for technical assistance, M. Troxell for pathology support, and A. Wong for technical assistance. Funding: Instrumentation used in this work was supported by grant number S10-RR023432 from the National Center for Research Resources, a component of the NIH, to the Advanced Light Microscopy Core at the OHSU Jungers Center. Research reported in this publication was supported by the NCI of the NIH under award numbers U54CA112970 and P30 CA069533 and by the National Heart Lung and Blood Institute of the NIH under award number T32HL007781. Additional support are as follows: NIH/T32GM071388, NCI/T32CA106195, and the American Skin Association (to C.E.G.); the Knight Cancer Institute Development Award/Veterans of Foreign Wars Ladies Auxiliary Group, the Phileo Foundation, and the Collins Medical Trust (to A.D.S.); and the Achievement to College Students Foundation Oregon (to K.T.G.). J.G.B. acknowledges support from NLM/5T15M007088-27. M.H.W. acknowledges support from the OHSU Center for Women’s Health Circle of Giving Grant, the Newton Foundation, the Prospect Creek Foundation, the Brenden-Colson Center for Pancreatic Health, the Crohn’s and Colitis Foundation, and NIH/NCI grants R01CA118235 and R21CA172334. L.M.C. acknowledges support from the NIH/NCI, Department of Defense Breast Cancer Research Program Era of Hope Scholar Expansion Award, Breast Cancer Research Foundation, the Susan G. Komen Foundation, and the Brenden-Colson Center for Pancreatic Health. Author contributions: Conception and design: C.E.G., A.D.S., and M.H.W. Development of methodology: C.E.G., A.D.S., J.K., K.T.G., and J.G. Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): C.E.G., A.D.S., L.R., L.Z., J.R.G., B.O., M.S.P., V.S., M.R.-J., M.S., J.R.S., P.S.D., S.I., P.F., J.J., S.W., J.K., C.D.L., K.G.B., B.C.S., and M.H.W. Analysis and interpretation of data (for example, statistical analysis, biostatistics, and computational analysis): C.E.G., J.B., J.G.B., J.M.F., and M.H.W. Writing, review, and/or revision of the manuscript: C.E.G., A.D.S., J.G.B., K.G.B., S.A.C., L.M.C., and M.H.W. Administrative, technical, or material support (that is, reporting or organizing data and constructing databases): J.G.B. Study supervision: C.E.G., A.D.S., L.Z., and M.H.W. Competing interests: M.H.W. and C.E.G. are inventors on a patent application filed by the OHSU. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/9/eaat7828/DC1
Fig. S1. Characterization of B16F10-derived fusion hybrids.
Fig. S2. Still images from cell fusion movie.
Fig. S3. Differential growth, adhesion, and cytokine response in hybrids.
Fig. S4. Differential growth and cytokine response in hybrids.
Fig. S5. Characterization of in vivo–derived B16F10 fusion hybrids.
Fig. S6. Flow cytometry gating scheme for B16F10-derived cell fusion analyses.
Fig. S7. Gating scheme for flow cytometry of in vivo–derived hybrids from primary tumor.
Fig. S8. Flow cytometry gating scheme for analyses of murine CHCs.
Fig. S9. Cell fusion in PanIN and tumors from other organ sites.
Fig. S10. Control blood samples for immunohistochemical and FISH analyses.
Fig. S11. Flow cytometry gating scheme for analyses of human CHCs.
Table S1. GO terms derived from differentially expressed genes between MC38 and hybrid cells.
Table S2. GO category gene table.
Table S3. MФ-unique or MФ-enriched genes.
Movie S1. Live imaging of MФ–cancer cell fusion.
Movie S2. Live imaging of cultured hybrid cells past confluence.
REFERENCES AND NOTES
- 1.Heppner G. H., Tumor heterogeneity. Cancer Res. 44, 2259–2265 (1984). [PubMed] [Google Scholar]
- 2.Marusyk A., Polyak K., Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 1805, 105–117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rizvi A. Z., Swain J. R., Davies P. S., Bailey A. S., Decker A. D., Willenbring H., Grompe M., Fleming W. H., Wong M. H., Bone marrow-derived cells fuse with normal and transformed intestinal stem cells. Proc. Natl. Acad. Sci. U.S.A. 103, 6321–6325 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies P. S., Powell A. E., Swain J. R., Wong M. H., Inflammation and proliferation act together to mediate intestinal cell fusion. PLOS ONE 4, e6530 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvarez-Dolado M., Pardal R., Garcia-Verdugo J. M., Fike J. R., Lee H. O., Pfeffer K., Lois C., Morrison S. J., Alvarez-Buylla A., Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature 425, 968–973 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Camargo F. D., Green R., Capetanaki Y., Jackson K. A., Goodell M. A., Single hematopoietic stem cells generate skeletal muscle through myeloid intermediates. Nat. Med. 9, 1520–1527 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Terada N., Hamazaki T., Oka M., Hoki M., Mastalerz D. M., Nakano Y., Meyer E. M., Morel L., Petersen B. E., Scott E. W., Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 416, 542–545 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Wang X., Willenbring H., Akkari Y., Torimaru Y., Foster M., Al-Dhalimy M., Lagasse E., Finegold M., Olson S., Grompe M., Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 422, 897–901 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Carter A., Cell fusion theory: Can it explain what triggers metastasis? J. Natl. Cancer Inst. 100, 1279–1281 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Pawelek J. M., Tumour-cell fusion as a source of myeloid traits in cancer. Lancet Oncol. 6, 988–993 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Dittmar T., Zänker K. S., Tissue regeneration in the chronically inflamed tumor environment: Implications for cell fusion driven tumor progression and therapy resistant tumor hybrid cells. Int. J. Mol. Sci. 16, 30362–30381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powell A. E., Anderson E. C., Davies P. S., Silk A. D., Pelz C., Impey S., Wong M. H., Fusion between intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 71, 1497–1505 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lazova R., LaBerge G. S., Duvall E., Spoelstra N., Klump V., Sznol M., Cooper D., Spritz R. A., Chang J. T., Pawelek J. M., A melanoma brain metastasis with a donor-patient hybrid genome following bone marrow transplantation: First evidence for fusion in human cancer. PLOS ONE 8, e66731 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams D. L., Martin S. S., Alpaugh R. K., Charpentier M., Tsai S., Bergan R. C., Ogden I. M., Catalona W., Chumsri S., Tang C.-M., Cristofanilli M., Circulating giant macrophages as a potential biomarker of solid tumors. Proc. Natl. Acad. Sci. U.S.A. 111, 3514–3519 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cogle C. R., Theise N. D., Fu D., Ucar D., Lee S., Guthrie S. M., Lonergan J., Rybka W., Krause D. S., Scott E. W., Bone marrow contributes to epithelial cancers in mice and humans as developmental mimicry. Stem Cells 25, 1881–1887 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Grimm M., Feyen O., Coy J. F., Hofmann H., Teriete P., Reinert S., Analysis of circulating CD14+/CD16+ monocyte-derived macrophages (MDMs) in the peripheral blood of patients with oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 121, 301–306 (2016). [DOI] [PubMed] [Google Scholar]
- 17.LaBerge G. S., Duvall E., Grasmick Z., Haedicke K., Pawelek J., A melanoma lymph node metastasis with a donor-patient hybrid genome following bone marrow transplantation: A second case of leucocyte-tumor cell hybridization in cancer metastasis. PLOS ONE 12, e0168581 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shabo I., Olsson H., Sun X. F., Svanvik J., Expression of the macrophage antigen CD163 in rectal cancer cells is associated with early local recurrence and reduced survival time. Int. J. Cancer 125, 1826–1831 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Yilmaz Y., Lazova R., Qumsiyeh M., Cooper D., Pawelek J., Donor Y chromosome in renal carcinoma cells of a female BMT recipient: Visualization of putative BMT-tumor hybrids by FISH. Bone Marrow Transplant. 35, 1021–1024 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Okabe M., Ikawa M., Kominami K., Nakanishi T., Nishimune Y., ’Green mice’ as a source of ubiquitous green cells. FEBS Lett. 407, 313–319 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Srinivas S., Watanabe T., Lin C.-S., William C. M., Tanabe Y., Jessell T. M., Costantini F., Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bissell M. J., Hines W. C., Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 17, 320–329 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoshino D., Branch K. M., Weaver A. M., Signaling inputs to invadopodia and podosomes. J. Cell Sci. 126 (Pt. 14), 2979–2989 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massagué J., TGFβ in cancer. Cell 134, 215–230 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin C.-H., Lee J. K., LaBarge M. A., Fabrication and use of MicroEnvironment microArrays (MEArrays). J. Vis. Exp., 4152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherr C. J., Rettenmier C. W., The fms gene and the CSF-1 receptor. Cancer Surv. 5, 221–232 (1986). [PubMed] [Google Scholar]
- 27.DeNardo D. G., Brennan D. J., Rexhepaj E., Ruffell B., Shiao S. L., Madden S. F., Gallagher W. M., Wadhwani N., Keil S. D., Junaid S. A., Rugo H. S., Hwang E. S., Jirström K., West B. L., Coussens L. M., Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 1, 54–67 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fehm T., Sagalowsky A., Clifford E., Beitsch P., Saboorian H., Euhus D., Meng S., Morrison L., Tucker T., Lane N., Ghadimi B. M., Heselmeyer-Haddad K., Ried T., Rao C., Uhr J., Cytogenetic evidence that circulating epithelial cells in patients with carcinoma are malignant. Clin. Cancer Res. 8, 2073–2084 (2002). [PubMed] [Google Scholar]
- 29.Racila E., Euhus D., Weiss A. J., Rao C., McConnell J., Terstappen L. W. M. M., Uhr J. W., Detection and characterization of carcinoma cells in the blood. Proc. Natl. Acad. Sci. U.S.A. 95, 4589–4594 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silk A. D., Gast C. E., Davies P. S., Fakhari F. D., Vanderbeek G. E., Mori M., Wong M. H., Fusion between hematopoietic and epithelial cells in adult human intestine. PLOS ONE 8, e55572 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lorico A., Corbeil D., Pawelek J. M., Alessandro R., Transmission of information in neoplasia by extracellular vesicles. Biomed. Res. Int. 2015, 289567 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazova R., Chakraborty A., Pawelek J. M., Leukocyte-cancer cell fusion: Initiator of the warburg effect in malignancy? Adv. Exp. Med. Biol. 714, 151–172 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Halaban R., Nordlund J., Francke U., Moellmann G., Eisenstadt J. M., Supermelanotic hybrids derived from mouse melanomas and normal mouse cells. Somatic Cell Genet. 6, 29–44 (1980). [DOI] [PubMed] [Google Scholar]
- 34.Kerbel R. S., Lagarde A. E., Dennis J. W., Donaghue T. P., Spontaneous fusion in vivo between normal host and tumor cells: Possible contribution to tumor progression and metastasis studied with a lectin-resistant mutant tumor. Mol. Cell. Biol. 3, 523–538 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lizier M., Anselmo A., Mantero S., Ficara F., Paulis M., Vezzoni P., Lucchini F., Pacchiana G., Fusion between cancer cells and macrophages occurs in a murine model of spontaneous neu+ breast cancer without increasing its metastatic potential. Oncotarget 7, 60793–60806 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu X., Kang Y., Cell fusion as a hidden force in tumor progression. Cancer Res. 69, 8536–8539 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shabo I., Midtbö K., Andersson H., Åkerlund E., Olsson H., Wegman P., Gunnarsson C., Lindström A., Macrophage traits in cancer cells are induced by macrophage-cancer cell fusion and cannot be explained by cellular interaction. BMC Cancer 15, 922 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patsialou A., Wang Y., Pignatelli J., Chen X., Entenberg D., Oktay M., Condeelis J. S., Autocrine CSF1R signaling mediates switching between invasion and proliferation downstream of TGFβ in claudin-low breast tumor cells. Oncogene 34, 2721–2731 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mo Y., Giricz O., Hu C., Dahlman K., Bhattacharyya S., Nguyen H., Matusow B., Bhagat T., Yu Y., Shellooe R., Burton E., Habets G., Greally J., Kenny P., Sosman J., Bollag G., West B. L., Verma A., Integrated epigenomic profiling reveals widespread demethylation in melanoma, and reveals aberrant CSF-1 receptor expression as a regulator of malignant growth and invasion inhibited by PLX3397. Cancer Res. 73, Abstract A26 (2013). [Google Scholar]
- 40.Sheng W., Ogunwobi O. O., Chen T., Zhang J., George T. J., Liu C., Fan Z. H., Capture, release and culture of circulating tumor cells from pancreatic cancer patients using an enhanced mixing chip. Lab Chip 14, 89–98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clawson G. A., Kimchi E., Patrick S. D., Xin P., Harouaka R., Zheng S., Berg A., Schell T., Staveley-O’Carroll K. F., Neves R. I., Mosca P. J., Thiboutot D., Circulating tumor cells in melanoma patients. PLOS ONE 7, e41052 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clawson G. A., Matters G. L., Xin P., McGovern C., Wafula E., dePamphilis C., Meckley M., Wong J., Stewart L., D’Jamoos C., Altman N., Imamura Kawasawa Y., Du Z., Honaas L., Abraham T., "Stealth dissemination" of macrophage-tumor cell fusions cultured from blood of patients with pancreatic ductal adenocarcinoma. PLOS ONE 12, e0184451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rappa G., Mercapide J., Lorico A., Spontaneous formation of tumorigenic hybrids between breast cancer and multipotent stromal cells is a source of tumor heterogeneity. Am. J. Pathol. 180, 2504–2515 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu M.-H., Gao X., Luo D., Zhou X.-D., Xiong W., Liu G.-X., EMT and acquisition of stem cell-like properties are involved in spontaneous formation of tumorigenic hybrids between lung cancer and bone marrow-derived mesenchymal stem cells. PLOS ONE 9, e87893 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGill J. R., Lalley P. J., Leach R. J., Johnson T. J., Von Hoff D. D., Chromosomal influence on hybrid cell proliferation. Cell Prolif. 25, 345–355 (1992). [DOI] [PubMed] [Google Scholar]
- 46.Islam M. Q., da S. Meirelles L., Nardi N. B., Magnusson P., Islam K., Polyethylene glycol-mediated fusion between primary mouse mesenchymal stem cells and mouse fibroblasts generates hybrid cells with increased proliferation and altered differentiation. Stem Cells Dev. 15, 905–919 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Xue J., Zhu Y., Sun Z., Ji R., Zhang X., Xu W., Yuan X., Zhang B., Yan Y., Yin L., Xu H., Zhang L., Zhu W., Qian H., Tumorigenic hybrids between mesenchymal stem cells and gastric cancer cells enhanced cancer proliferation, migration and stemness. BMC Cancer 15, 793 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hung J. Y., Horn D., Woodruff K., Prihoda T., LeSaux C., Peters J., Tio F., Abboud-Werner S. L., Colony-stimulating factor 1 potentiates lung cancer bone metastasis. Lab. Invest. 94, 371–381 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Mitchem J. B., Brennan D. J., Knolhoff B. L., Belt B. A., Zhu Y., Sanford D. E., Belaygorod L., Carpenter D., Collins L., Piwnica-Worms D., Hewitt S., Udupi G. M., Gallagher W. M., Wegner C., West B. L., Wang-Gillam A., Goedegebuure P., Linehan D. C., DeNardo D. G., Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 73, 1128–1141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ngiow S. F., Meeth K. M., Stannard K., Barkauskas D. S., Bollag G., Bosenberg M., Smyth M. J., Co-inhibition of colony stimulating factor-1 receptor and BRAF oncogene in mouse models of BRAFV600E melanoma. Oncoimmunology 5, e1089381 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruffell B., Coussens L. M., Macrophages and therapeutic resistance in cancer. Cancer Cell 27, 462–472 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clausen B. E., Burkhardt C., Reith W., Renkawitz R., Förster I., Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 (1999). [DOI] [PubMed] [Google Scholar]
- 53.Falcon S., Gentleman R., Using GOstats to test gene lists for GO term association. Bioinformatics 23, 257–258 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Walter W., Sánchez-Cabo F., Ricote M., GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914 (2015). [DOI] [PubMed] [Google Scholar]
- 55.J. Burkhart, Cell Fusion Potentiated Acquisition of Macrophage-like Behavior in Cancer cells and Its Contribution (Oregon Health & Science University, 2016). [Google Scholar]
- 56.Seals D. F., Azucena E. F. Jr, Pass I., Tesfay L., Gordon R., Woodrow M., Resau J. H., Courtneidge S. A., The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell 7, 155–165 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/9/eaat7828/DC1
Fig. S1. Characterization of B16F10-derived fusion hybrids.
Fig. S2. Still images from cell fusion movie.
Fig. S3. Differential growth, adhesion, and cytokine response in hybrids.
Fig. S4. Differential growth and cytokine response in hybrids.
Fig. S5. Characterization of in vivo–derived B16F10 fusion hybrids.
Fig. S6. Flow cytometry gating scheme for B16F10-derived cell fusion analyses.
Fig. S7. Gating scheme for flow cytometry of in vivo–derived hybrids from primary tumor.
Fig. S8. Flow cytometry gating scheme for analyses of murine CHCs.
Fig. S9. Cell fusion in PanIN and tumors from other organ sites.
Fig. S10. Control blood samples for immunohistochemical and FISH analyses.
Fig. S11. Flow cytometry gating scheme for analyses of human CHCs.
Table S1. GO terms derived from differentially expressed genes between MC38 and hybrid cells.
Table S2. GO category gene table.
Table S3. MФ-unique or MФ-enriched genes.
Movie S1. Live imaging of MФ–cancer cell fusion.
Movie S2. Live imaging of cultured hybrid cells past confluence.