

ABSTRACT
Ankylosing spondylitis (AS) is a chronic autoimmune inflammatory disease with severe inflammatory symptoms in the axial skeleton. The cause of ankylosing spondylitis is unknown. TNFAIP3, also named A20, uses ubiquitin-related functions to regulate immune activation, deficiency of which is highly related to autoimmune disease. However, the role of TNFAIP3 in human AS has not been reported. Our objective was to study the role and mechanism of TNFAIP3 in ankylosing spondylitis. TNFAIP3 expression on different types of immunocytes from AS peripheral blood was measured by flow cytometry. In vitro, monocytes were transfected with a TNFAIP3 shRNA lentivirus, and IL6 and IL1B activation was tested using real-time PCR and ELISA. The novel interaction complex TNFAIP3-DEPTOR was determined through GST pull-down, yeast two-hybrid system, confocal microscopy, and co-immunoprecipitation. Transmission electron microscopy, the RFP-GFP-LC3 adenovirus, and LC3 expression were used for autophagy detection. Here, we show that TNFAIP3 expression in AS peripheral blood non-classical monocytes was decreased. In normal monocytes, TNFAIP3 induced autophagy, which restricted inflammasome activation to the early stage of LPS stimulation. Zinc-finger domains of TNFAIP3 were able to interact and stabilize DEPTOR. TNFAIP3 and DEPTOR together rapidly promoted autophagy after LPS treatment to prevent NLRP3 inflammasome formation. Finally, TNFAIP3 and DEPTOR deficiency in AS non-classical monocytes facilitated inflammasome activation. Our study indicates that TNFAIP3-DEPTOR complex-induced early-onset autophagy is vital for immune inhibition in autoimmune disease.
KEYWORDS: Ankylosing spondylitis, autophagy, DEPTOR, inflammasome, monocytes, NLRP3, TNFAIP3
Introduction
Ankylosing spondylitis (AS) is a chronic inflammatory arthritis that causes inflammation and new bone formation involving the sacroiliac and spinal joints. The strongest known contributing factor to AS is HLA-B27 (major histocompatibility complex, class I, B), a major histocompatibility complex (MHC) class I molecule, and over 90% of white AS patients are HLA-B27-positive [1]. In addition to HLA-B27, other inflammatory cytokine pathways, such as TNF (tumor necrosis factor), IL6 (interleukin 6), IL23 (interleukin 23), and IL17 (interleukin 17), are involved in AS pathogenesis [2]. However, IL1B (interleukin 1 beta) secretion in AS peripheral blood is controversial [3,4].
IL1B is a central mediator of the initiation and amplification of immune responses and is produced by hematopoietic cells such as blood monocytes (especially in CD14+ FCGR3/CD16++ non-classical monocytes) and tissue macrophages [5,6]. In response to toll-like receptor 4(TLR4), activated complement components, and cytokines (such as TNF and IL1B), hematopoietic cells express an IL1B precursor, which is then processed into its mature form by an inflammasome, which secretes the active cytokine into the extracellular space [7,8]. Inflammasomes are assembled with nucleotide-binding domain and leucine-rich repeat containing (NLR) proteins and the ALR member AIM2 (absent in melanoma 2), promote CASP1 (caspase 1) activation, and trigger the maturation of proinflammatory cytokines [9].
As a protective mechanism in cells, autophagy interplays with the inflammasome. For one thing, autophagy inhibits inflammasome activation and secretion. Autophagy regulator ATG16L1 deficiency enables LPS-dependent inflammasome activation [10], and TRIM11 (tripartite motif containing 11) recruits AIM2 in SQSTM1/p62 (sequestosome 1)-related autophagy to inhibit inflammasome formation [11]. Accordingly, proper autophagy in uveitis and liver injury contributes to the inhibition of IL1B secretion and immune activation [12,13]. Additionally, autophagosomes derived from tumor cells induce IL1B release in peripheral blood mononuclear cells (PBMCs), which requires NLRP3 (NLR family pyrin domain containing 3) inflammasome activation [14]. What is more, some microbes balance the levels of inflammasomes and autophagy to avoid further immune supervision [15].
TNFAIP3/A20 (TNF alpha induced protein 3), is a deubiquitinating enzyme that inhibits immune activation [16,17]. Through its interaction and ubiquitination of substrates, such as RIPK1 (receptor interacting serine/threonine kinase 1), TRAF6 (TNF receptor associated factor 6), and UBE2N (ubiquitin conjugating enzyme E2 N), TNFAIP3 negatively regulates NFKB (nuclear factor kappa B) activity [18–20]. Previous works have proven that TNFAIP3 inhibits NLRP3-mediated inflammasome activation without affecting NLRP4 and AIM2 inflammasomes [21]. The promotion of inflammasomes in TNFAIP3-deficient macrophages is also related to pro-IL1B-associated ubiquitination [22]. In mice, TNFAIP3 overexpression inhibits the inflammasome to prevent lupus inflammation and renal injury [23]. Furthermore, TNFAIP3 plays a dual role in autophagy. Shi et al. demonstrated that TNFAIP3 reduced the K63-linked ubiquitination of BECN1 (beclin 1), thereby limiting TLR4 (toll like receptor 4)-related autophagy [24]. However, others have proven that a reduction in TNFAIP3 was responsible for autophagy inhibition through MTOR (mechanistic target of rapamycin kinase) in dendritic cells [25] and CD4 T cells [26].
Different cell type-specific TNFAIP3 deficiencies are linked to multiple autoimmune diseases [27,28]. However, there has been little research revealing the TNFAIP3 level and related function in AS. In this study, TNFAIP3 expression was evaluated in AS peripheral blood and was demonstrated to act on autophagy-associated inflammasome inhibition.
Results
TNFAIP3 expression is decreased in AS monocytes but unchanged in AS T/B lymphocytes
In the GEO databases, GSE25101 and GSE11886, TNFAIP3 mRNA expression was unchanged in AS peripheral blood mononuclear cells (PBMCs) but was reduced in AS monocytes (Figure 1(a)). We used the FSC (forward scatter) and SSC (side scatter) to approximately set the cell groups: group one for lymphocytes; group two for lymphocytes and monocytes; and group three for lymphocytes, monocytes and granulocytes (Figure 1(b)). The TNFAIP3 level was determined by the TNFAIP3 mean fluorescence intensity (MFI) (Figure 1(c)). Whole-blood PBMC TNFAIP3 levels were the same in the AS and healthy control (HC) groups (Figure 1(d)). Then, in group one, CD3E-positive cells were regarded as T lymphocytes and CD19-positive cells as B lymphocytes; ITGAM/CD11b and CD14 double-positive cells in group two were considered monocytes; and CD15-positive cells in group three were considered granulocytes. There was no significant difference in the proportion of cell subtypes between the AS and HC groups (Fig. S1A). Along with the GEO data, TNFAIP3 expression in the lymphocytes and granulocytes of AS patients was the same as that in the HC group, but the TNFAIP3 levels were relatively low in the AS monocytes (Figure 1(e–h)).
Figure 1.
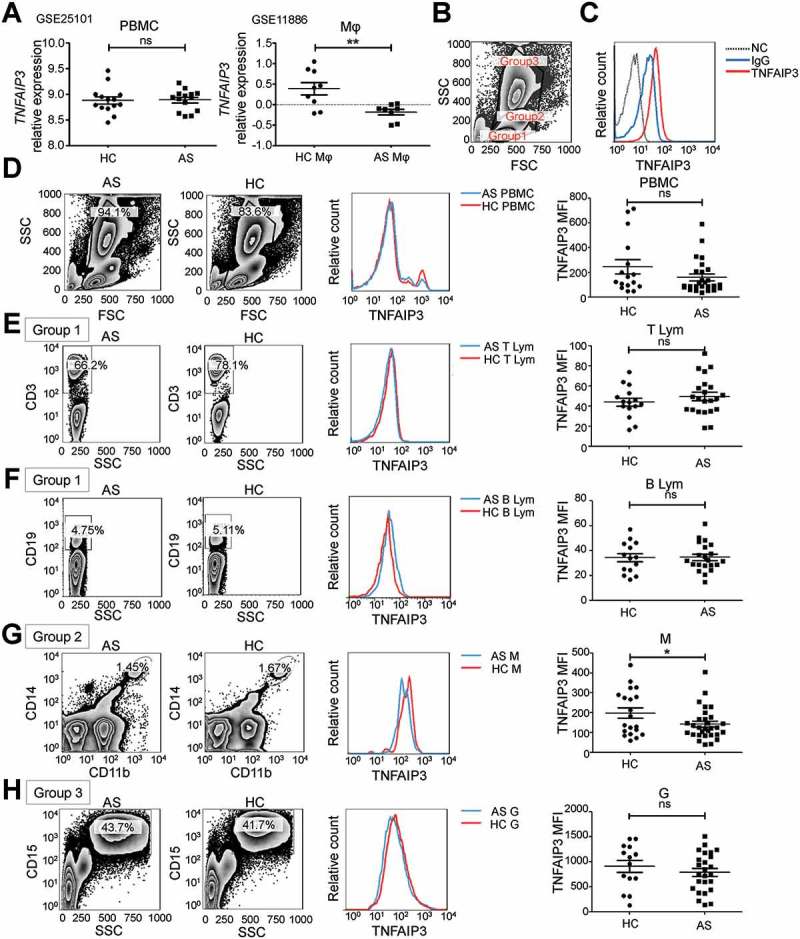
TNFAIP3 expression in AS PBMCs. (a) TNFAIP3 expression in AS PBMCs and macrophages in the GEO datasets GSE25101 and GSE11886. (b) FSC-SSC group settings. (c) TNFAIP3 flow cytometry stain with blank control (NC) and isotype control (IgG). (d–h) Representative results (left) and statistical analysis (right) of TNFAIP3 expression in HC (n = 16) and AS (n = 26) PBMCs (d), in HC (n = 15) and AS (n = 22) T lymphocytes (CD3E-positive in group 1) (e), in HC (n = 14) and AS (n = 21) B lymphocytes (CD19-positive in group 1) (f), in HC (n = 20) and AS (n = 28) monocytes (ITGAM/CD11b and CD14 double-positive in group 2) (g), and in HC (n = 14) and AS (n = 25) granulocytes (CD15-positive in group 3) (h). **, p < 0.01; ns, not significant by unpaired t-test.
For further investigation, we analyzed CD4 and CD8 together with CD3E expression to determine the TNFAIP3 level in CD4 and CD8 T lymphocytes and determined that there was no significant difference in TNFAIP3 expression between AS and HC patients (Fig. S1B). PTPRC (CD45RA and CD45RO) expression was used to determine TNFAIP3 expression in CD4+ or CD8+ naive/memory T lymphocytes, and the TNFAIP3 level was the same in both the AS and HC groups (Fig. S1C). In addition, TNFAIP3 was unchanged in Th17 cells and Treg cells (Fig. S1D).
Furthermore, we detected the TNFAIP3 level in different subtypes of monocytes. CD14 and FCGR3/CD16 were used as markers of classical monocytes (CD14++ FCGR3/CD16−), non-classical monocytes (CD14+ FCGR3/CD16++) and intermediate monocytes (CD14++ FCGR3/CD16+), and the proportions of these 3 groups between the AS and HC groups were not different (Figure 2(a,b)). In AS patients, TNFAIP3 expression in the patrolling CD14+ FCGR3/CD16++ monocytes was lower than in the HC group, while its expression in classical CD14++ FCGR3/CD16− monocytes was unchanged (Figure 2(c,d)),
Figure 2.
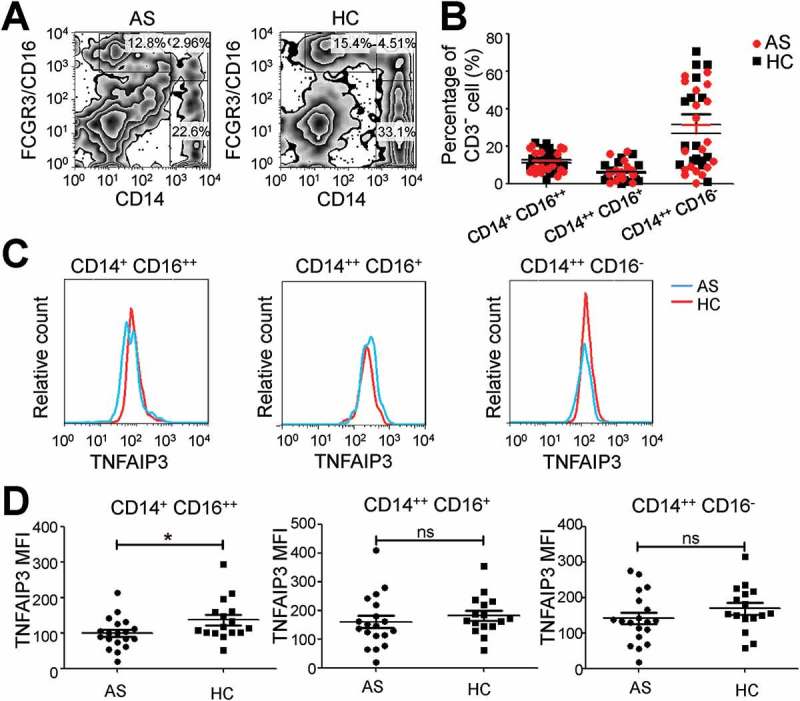
TNFAIP3 expression is reduced in AS CD14+ FCGR3/CD16++ monocytes. (a) CD14 and FCGR3/CD16 group settings. (b) The proportion of classical (CD14++ FCGR3/CD16−), non-classical (CD14+ FCGR3/CD16++) and intermediate (CD14++ FCGR3/CD16+) monocytes. (c, d) Representative results (c) and statistical analysis (d) of TNFAIP3 expression in subtypes of monocytes in AS (n = 19) and HC (n = 16). *, p < 0.05; ns, not significant by ANOVA (b) and unpaired t-test (d). MFI, mean fluorescence intensity.
TNFAIP3 reduces IL1B secretion in monocyte partially depending on the NFKB pathway
To investigate the function of TNFAIP3 in monocytes, we blocked TNFAIP3 expression with TNFAIP3 siRNA and a lentivirus carrying TNFAIP3 shRNA (Figure 3(a)). Under phorbol 12-myristate 13-acetate (PMA, 100 ng/ml) activation, THP-1 cells were activated with morphology changes and CD14 upregulation (Fig. S2A and B). In accordance with previous studies, TNFAIP3 downregulation further increased IL6 and IL1B mRNA expression in PMA-activated THP-1 cells (Fig. S2C). With lipopolysaccharide (LPS, 200 ng/ml) stimulation, IL6 and IL1B mRNA were increased even more significantly in the TNFAIP3 downregulated group (Fig. S2D). Interestingly, when the NFKB inhibitor tosyl phenylalanyl chloromethyl ketone (TPCK) was added along with LPS, IL6 secretion was blocked, while IL1B secretion was partially inhibited (Figure 3(b,c)), which indicated that the TNFAIP3-induced reduction in IL1B secretion was partially depended on the NFKB pathway.
Figure 3.

TNFAIP3 induces autophagy to reduce IL1B secretion in the early stage of LPS stimulation. (a) Downregulation of TNFAIP3 in monocytes with lentivirus carrying TNFAIP3 shRNA or siRNA. NC, negative control. GAPDH mRNA was used to normalize the target mRNA and the relative expression was automatically calculated using the comparative quantification mode of MxPro qPCR system software. (b, c) ELISA assay for IL6 (b) and IL1B (c) secretion in cells treated with LPS and/or the NFKB inhibitor TPCK. (d, e) 3-MA treatment (d) and ATG7 interference (e) of THP-1 cells increased NLRP3 expression during LPS activation. (f) ELISA assay for IL1B secretion after LPS and 3-MA treatment. (g) IL1B secretion after LPS and ATG7 interference. (h) Luciferase reporter assay for NFKB transcription status.The ratio here refers to the ratio of the Firefly luciferase signals to the Renilla luciferase signals. (i) The secretion level of IL1B in control (pcDNA 3.1) and TNFAIP3 overexpression cells after LPS and 3-MA treatment. (j) TNFAIP3 downregulation causes more NLRP3 production under 24-h LPS stimulation. During the early stage of LPS activation, NLRP3 expression is increased when TNFAIP3 expression is knocked down. (k) NLRP3 mRNA expression was not changed in TNFAIP3 downregulated cells. (l) CASP1 expression under LPS and ATP activation in NC vs. TNFAIP3-knockdown cells. (m) LPS activation-induced autophagy is increased in the early stage in NC-treated cells, while in the TNFAIP3 interference group, there was no such change. (n) Overexpression of TNFAIP3 causes less NLRP3 expression and more LC3-II expression under 24-h LPS stimulation. (o) At the early time points (0, 1, 4, 8 h) of LPS treatment, His, LC3 and NLRP3 expression with or without overexpression of TNFAIP3-His. (p) Confocal microscopy assay for cells transfected with mCherry-LC3 and GFP-TNFAIP3 plasmids. Scale bar: 10 μm (q) TEM for autophagosome and autolysosome formation with 1 h LPS activation. More than 40 cells were analyzed. Black arrow indicates an autolysosome, and white arrow indicates an autophagosome. Scale bar: 2 μm (q). The bars represent each sample performed in triplicate. ***, p < 0.001**, p < 0.01 by unpaired t-test (a) and ANOVA (b, c, f–i, k, o, q).
IL1B secretion not only requires NFKB activation but also needs inflammasome-dependent processing. With the autophagy inhibitor 3-methyladenine (3-MA) and downregulation of ATG7, NLRP3 inflammasomes were upregulated (Figure 3(d,e)). Although the IL1B level in cell lysates was not changed after 3-MA inhibition and ATG7 interference, the levels of secreted IL1B were increased (Figure 3(f,g)). Furthermore, we performed luciferase reporter assays to determine if the NFKB transcription status changed under 3-MA treatment. Luciferase reporter readings showed that 3-MA had no impact on the NFKB transcription status (Figure 3(h)), indicating the importance of autophagy regulation in IL1B secretion. Considering the possible involvement of TNFAIP3 in inflammasome regulation through autophagy, 3-MA was used in both control cells and TNFAIP3 overexpression cells. The ELISA results illustrated that TNFAIP3 was able to inhibit IL1B secretion, but when autophagy was blocked, the negative role of TNFAIP3 in immune regulation was attenuated (Figure 3(i)).
TNFAIP3 induces autophagy in the early stage of LPS stimulation
To investigate the role of TNFAIP3 in LPS-related inflammasome activation, NLPR3 mRNA and protein levels were measured with or without TNFAIP3 expression (Figure 3(j,k)). Alteration of TNFAIP3 expression had no impact on the NLRP3 mRNA level but changed NLRP3 protein expression. After 24-h LPS stimulation, NLRP3 expression was significantly increased with siTNFAIP3. In the early stage of LPS stimulation, NLRP3 expression was rapidly and continuously increased without TNFAIP3 expression. Along with NLRP3, active-CASP1 expression (p10 and p12 forms) was also increased with siRNA of TNFAIP3 (Figure 3(l)). In contrast to NLRP3 expression, when TNFAIP3 was downregulated, expression of the marker of autophagy MAP1LC3B/LC3B-II (microtubule associated protein 1 light chain 3 beta) was decreased. LPS activation induced autophagy, while in the TNFAIP3 interference group, there was no such change. (Figure 3(m)), indicating that TNFAIP3 might be responsible for autophagy induction under LPS stimulation. When cells were transfected with adenovirus carrying RFP-GFP-LC3 (Fig. S3A), TNFAIP3 was able to increase autophagy flux, especially autolysosome formation, during the early stage of LPS stimulation (Fig. S3B and C). Overexpression of TNFAIP3 with the His tag in HEK293T (293T) cells (original TNFAIP3 expression was relatively low), upregulated the LPS-induced early autophagy and inhibited NLRP3-related inflammasome formation (Figure 3(n,o)). When cells were transfected with mCherry-LC3 and TNFAIP3-GFPSpark plasmids, the colocalization of TNFAIP3 and LC3-positive autophagic structures was significantly increased (Figure 3(p)). Transmission Electron Microscopy (TEM) results showed that the overexpression of TNFAIP3 was responsible for autolysosome formation during the early stage of LPS activation (Figure 3(q)).
TNFAIP3 interacts with and stabilizes DEPTOR
Next, we investigated the mechanism of TNFAIP3-related autophagy. Previous studies have shown that DEPTOR (DEP domain containing MTOR interacting protein) is capable of inhibiting the MTOR pathway and promotes autophagy [29]. According to the confocal microscopy and co-immunoprecipitation results, TNFAIP3 was able to colocalize and interact with DEPTOR (Figure 4(a,b)). Furthermore, TNFAIP3 and DEPTOR were shown to interact with one another with yeast two-hybrid system analysis and GST affinity-isolation assay (Figure 4(c–e)). The Hitrap purified His-TNFAIP3 had a 50 kD band, which was known as the MALT1 (MALT1 paracaspase)-cleavage product. Others have shown that TNFAIP3 can interact with MTOR [26]. Consistent with that, we found that TNFAIP3 interacted with MTOR and their interaction was enhanced by DEPTOR (Figure 4(f,g)). Downregulation of DEPTOR with siRNA reduced the interaction between TNFAIP3 and MTOR. In the early stage of LPS stimulation (1 and 4 h), the colocalization and interaction of TNFAIP3 and DEPTOR were increased (Figure 4(h,i)), demonstrating that they both play vital roles in the response to LPS.
Figure 4.
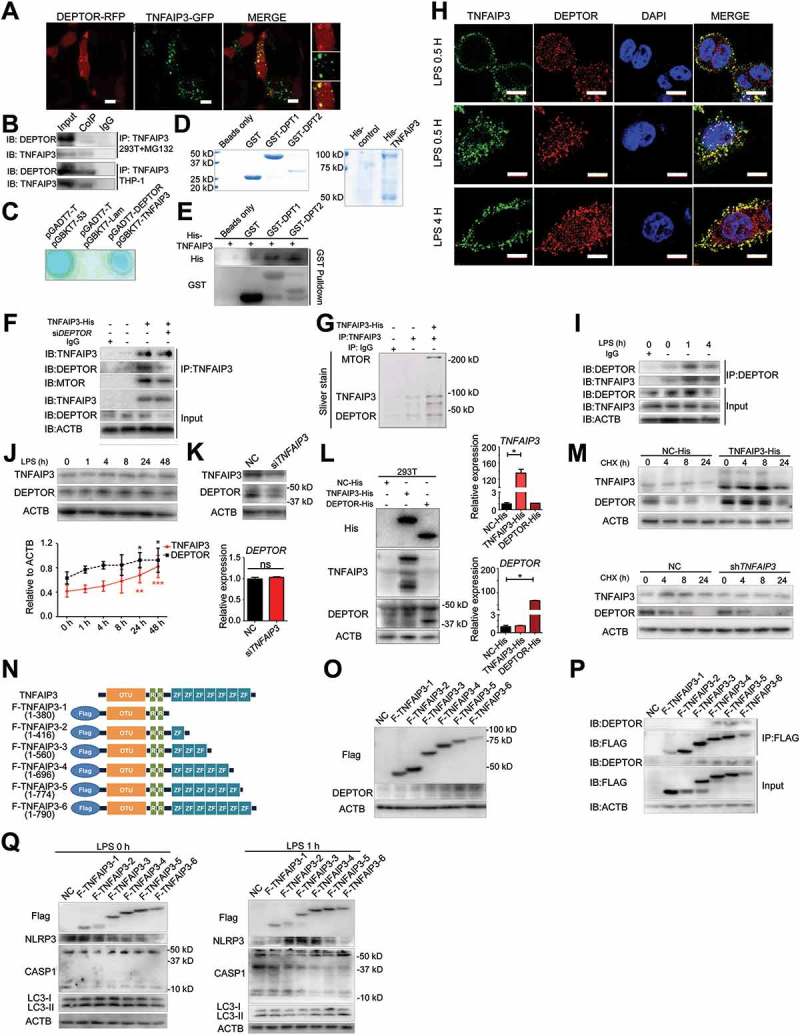
TNFAIP3 interacts with and stabilizes DEPTOR. (a) The confocal microscopy assay indicated that TNFAIP3 and DEPTOR were colocalized in 293T cells. Scale bar: 5 μm. (b) After treatment with MG-132 (0.5 μM) for 12 h, co-immunoprecipitation showed that TNFAIP3 interacts with DEPTOR. (c) The yeast two-hybrid system analysis results suggest that TNFAIP3 (bait) and DEPTOR (prey) interact directly. pGBKT7-53 and pGADT7-T were considered to be the positive controls, while pGBKT7-Lam and pGADT7-T were the negative controls. (d, e) GST pull-down results for TNFAIP3 and DEPTOR stained with either Coomassie Brilliant Blue (d) or antibodies towards His (e). GST-DPT1 (29–219) is GST tag with DEP1 and DEP2 domain of DEPTOR, and GST-DPT2 (328–404) is GST tag with PDZ domain of DEPTOR. (f, g) The co-immunoprecipitation (f) and silver stain results (g) suggest that there is an interaction among TNFAIP3, DEPTOR and MTOR. After downregulation of DEPTOR with siRNA, the interaction between TNFAIP3 and MTOR was reduced. Antibodies of TNFAIP3, DEPTOR, MTOR and ACTB were used in part (f). (h) At early time points (0.5, 1, 4 h) of LPS treatment, the colocalization level of TNFAIP3 and DEPTOR was increased. Red: DEPTOR; Green: TNFAIP3; Blue: DAPI. Scale bar: 10 μm. [Please make the scale bar white for increased visibility.] (i) In THP-1 cells, after LPS treatment for 1 h and 4 h, the interaction between TNFAIP3 and DEPTOR was increased. Antibodies of TNFAIP3, DEPTOR and ACTB were used in here. (j) Western blot and statistic results of TNFAIP3 and DEPTOR expression after LPS treatment. (k) After downregulation of TNFAIP3, the protein level of DEPTOR was reduced; however, the mRNA level was unchanged. (l) Overexpressing TNFAIP3-His in 293T cells resulted in increased expression of the DEPTOR protein, while DEPTOR-His overexpression does not cause a change in TNFAIP3 expression. The mRNA levels of TNFAIP3 and DEPTOR had no effect on one another. (m) CHX treatments (2 μg/ml, 0, 4, 8, 24 h) were used to test the DEPTOR degradation rate. TNFAIP3 overexpression in 293T cells slowed down the degradation of DEPTOR. TNFAIP3 downregulation in monocytes promoted DEPTOR degradation. (n) Diagram of different TNFAIP3 fragments with FLAG tag. (o) Western blot assay for expression of various FLAG-TNFAIP3 fusion proteins and DEPTOR. (p) Co-immunoprecipitation results for interactions of TNFAIP3 fragments and DEPTOR. (q) Western blot assay for FLAG, NLRP3, CASP1 and LC3B in 293T cells expressing different TNFAIP3 fragments following 0 h (left panels) or 1 h (right panels) of LPS stimulation. The bars represent each sample performed in triplicate. *, p < 0.05 by unpaired t-test (k) and ANOVA (j and l) .
The DEPTOR expression levels under LPS stimulation (0, 1, 4, 8, 24, 48 h) were increased with TNFAIP3 (Figure 4(j)). When TNFAIP3 was downregulated in monocytes, the DEPTOR protein level was reduced, while the mRNA level of DEPTOR was unchanged (Figure 4(k)). In HEK293T cells expressing TNFAIP3-His or DEPTOR-His, DEPTOR expression was increased in the TNFAIP3 overexpression cells, but TNFAIP3 was unchanged in the DEPTOR overexpression cells (Figure 4(l)). Then, we investigated the protein degradation rate of DEPTOR. TNFAIP3 overexpression could slow down the degradation of DEPTOR, whereas TNFAIP3 downregulation could promote DEPTOR degradation (Figure 4(m)). Thus, TNFAIP3 could interact with and stabilize DEPTOR, which might be important in TNFAIP3-related autophagy.
Further we established different TNFAIP3 fragment vectors with FLAG tag (Figure 4(n)). Western blot results indicated successful expression of FLAG-TNFAIP3 fragment proteins, and F-TNFAIP3-4, F-TNFAIP3-5 and F-TNFAIP3-6 were able to enhance DEPTOR expression (Figure 4(o)). In addition, through co-immunoprecipitation of FLAG-labeled protein and interaction proteins, F-TNFAIP3-4, F-TNFAIP3-5 and F-TNFAIP3-6 were shown to be responsible for TNFAIP3 and DEPTOR interaction (Figure 4(p)). What is more, with transfection of different TNFAIP3 fragments, TNFAIP3 zinc-finger 4–7 (F-TNFAIP3-4, F-TNFAIP3-5 and F-TNFAIP3-6) was able to induces autophagy and inhibit CASP1 activity in the early stage of LPS stimulation (Figure 4(q)).
TNFAIP3-DEPTOR promotes autophagy and inhibits inflammasome secretion
DEPTOR downregulation increased IL1B secretion, which could not be totally blocked by the NFKB inhibitor TPCK (Figure 5(a,b)). TNFAIP3 deficiency caused a reduction in DEPTOR expression and inhibition in autophagy (Figure 5(c)). With DEPTOR siRNA, NLRP3 expression was increased, but LC3B-II expression was decreased under LPS stimulation (Figure 5(d)). In the early stage of LPS activation, monocytes with DEPTOR knockdown expressed more NLPR3 and cleaved CASP1, but less LC3B-II, especially during LPS stimulation for 1–4 h (Figure 5(e,f)). Similar results were observed in HEK293T DEPTOR overexpression cells. Overexpression of DEPTOR increased autophagy (LC3B-II), while it inhibited NLRP3 expression, effects that were more significant in the early stage of LPS activation (Figure 5(g)). Downregulation of TNFAIP3 and DEPTOR reduced ATG3, ATG5, ATG7, and ATG12 expression (Figure 5(h)).
Figure 5.
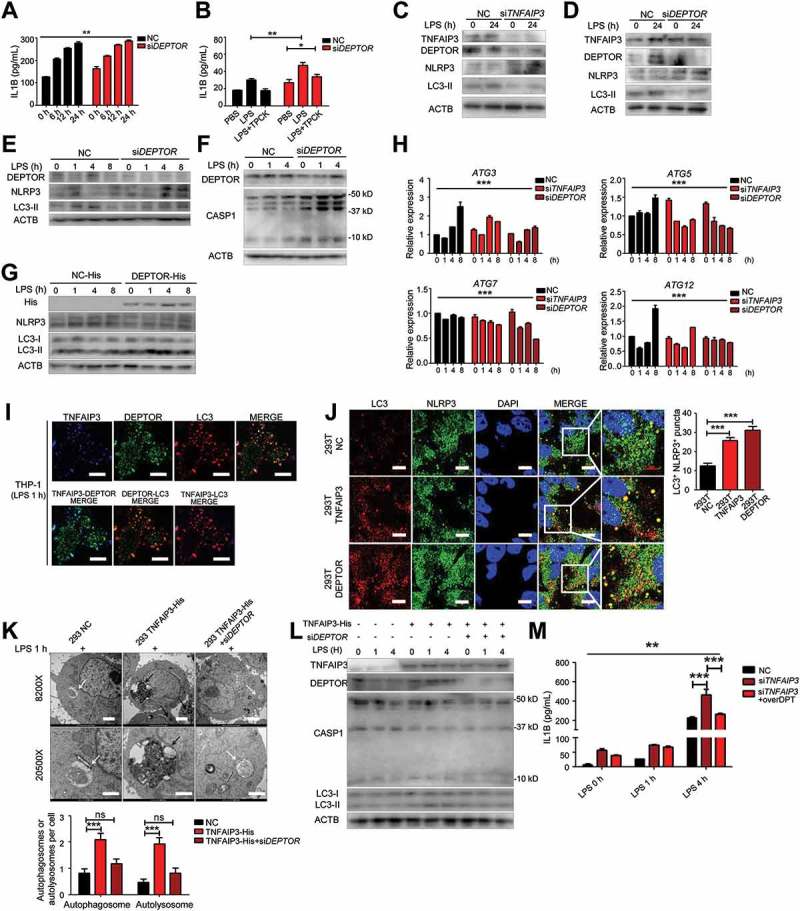
TNFAIP3 and DEPTOR are required for autophagy induction and inflammasome restriction. (a) After LPS stimulation (0, 6, 12, 24 h) and DEPTOR expression blocked with siRNA, IL1B secretion was detected by ELISA. (b) ELISA assay for IL1B secretion with LPS and NFKB inhibitor TPCK treatment, with or without DEPTOR interference. (c) Downregulation of TNFAIP3 caused a reduction in DEPTOR expression, leading to NLRP3 augmentation and LC3-II deficiency in response to LPS (24 h). (d) After blocking DEPTOR expression using siRNA, the expression of TNFAIP3, NLRP3 and LC3-II was tested by western blot (LPS for 24 h). (e) In the DEPTOR interference group, NLRP3 increased more significantly while LC3-II enhanced slower under early stage of LPS activation (0, 1, 4, 8 h). (f) The activity of CASP1 increased more significantly in DEPTOR interference group with LPS treatment. (g) At the early time points (0, 1, 4, 8 h) of LPS treatment, His, LC3, and NLRP3 expression in cells overexpressing DEPTOR-His. Overexpression of DEPTOR causes less NLRP3 activation and more LC3-II expression under LPS stimulation. (h) With downregulation of TNFAIP3 and DEPTOR, the mRNA level of several genes of the ATG family was decreased after LPS treatment for 0, 1, 4 and 8 h. (i) At the early time points (1 h) of LPS treatment, the TNFAIP3, DEPTOR and LC3 were colocalized. Red: LC3; Green: DEPTOR; Blue: TNFAIP3. Scale bar = 10 μm. (j) LC3 and NLRP3 colocalization with 8 h of LPS and chloroquine (CQ) treatment. Scale bar: 10 μm. (k) TEM for autophagosome and autolysosome formation with 1 h LPS activation. Left panel shows control 293T cells, middle panel shows 293T cells with TNFAIP3-His overexpression, and the right panel shows TNFAIP3-His overexpression 293T cells with DEPTOR interference. More than 40 cells were analyzed. Black arrow indicates the autolysosome, and white arrow indicates the autophagosome. Scale bar for the upper row is 2 μm and for the lower row is 1 μm. (l) TNFAIP3, DEPTOR, CASP1, and LC3B expression are determined by western blot with TNFAIP3 overexpression and DEPTOR interference. (m) ELISA assay shows the IL1B levels changed after LPS treatment with TNFAIP3 interference and DEPTOR upregulation. The bars represent each sample performed in triplicate. *, p < 0.05, **, p < 0.01, ***, p < 0.001 by ANOVA (a, b, h, j, k, m).
To further study the role of the TNFAIP3 and DEPTOR complex in autophagy, we used confocal microscopy to localize the TNFAIP3 and DEPTOR complex. Interestingly, the complex was also colocalized with LC3, and the colocalization was increased during the early stage of LPS stimulation (Figure 5(i)), which suggests that TNFAIP3 and DEPTOR participate in LPS-related autophagy activation together. When cells were treated with chloroquine (CQ) and LPS for 8 h, TNFAIP3 or DEPTOR overexpression increased the colocalization of LC3 and NLRP3 in 293T cells (Figure 5(j)). TEM results showed that overexpression of TNFAIP3 was responsible for autolysosome formation during the early stage of LPS activation, which was dependent on DEPTOR, indicating that DEPTOR, together with TNFAIP3, was responsible for early autophagy under LPS stimulation. (Figure 5(k)). The autophagy induction and inflammasome inhibition caused by TNFAIP3 were reversed by DEPTOR interferences (Figure 5(l)). The extra IL1B secretion induced by TNFAIP3 deficiency was partially inhibited by DEPTOR overexpression (Figure 5(m)). Thus, we determined that TNFAIP3 and DEPTOR worked together as a whole to promote autophagy and inhibit inflammasome secretion.
Decrease in DEPTOR expression facilities IL1B expression in AS peripheral blood
To further verify the results of the in vitro study, we measured the IL6, IL1B, and IL18 levels in AS and HC serum. The IL6, IL1B and IL18 levels were higher in the AS than the HC serum (Figure 6(a–c)). The TNFAIP3 expression level in CD14+ FCGR3/CD16++ non-classical monocytes was negatively correlated with the levels of IL1B and IL18, indicating that the loss of TNFAIP3 in AS non-classical monocytes resulted in increased IL1B and IL18 secretion, while IL6 and TNFAIP3 expression were not associated (Figure 6(d–f)). AS IL1B-positive rate and the average IL1B level of the positive samples in AS were higher than HC (Figure 6(g)). We sorted out CD14+ FCGR3/CD16++ non-classical monocytes to test whether there were changes in the TNFAIP3-DEPTOR complex in AS peripheral blood (Figure 6(h)). Consistently, DEPTOR and TNFAIP3 expression was decreased in AS monocytes, resulting in a reduction in autophagy and NLRP3 inflammasome activation (Figure 6(i,j)). In the GSE11886 dataset, DEPTOR mRNA levels were unchanged in AS non-classical monocytes compared with that in HC, which confirmed that TNFAIP3 stabilized DEPTOR at the protein level (Fig. S4A). The levels of some of the autophagy mRNAs were significantly decreased in AS patients, while there were no obvious changes in the inflammasome-related mRNA (Fig. S4B-D).
Figure 6.
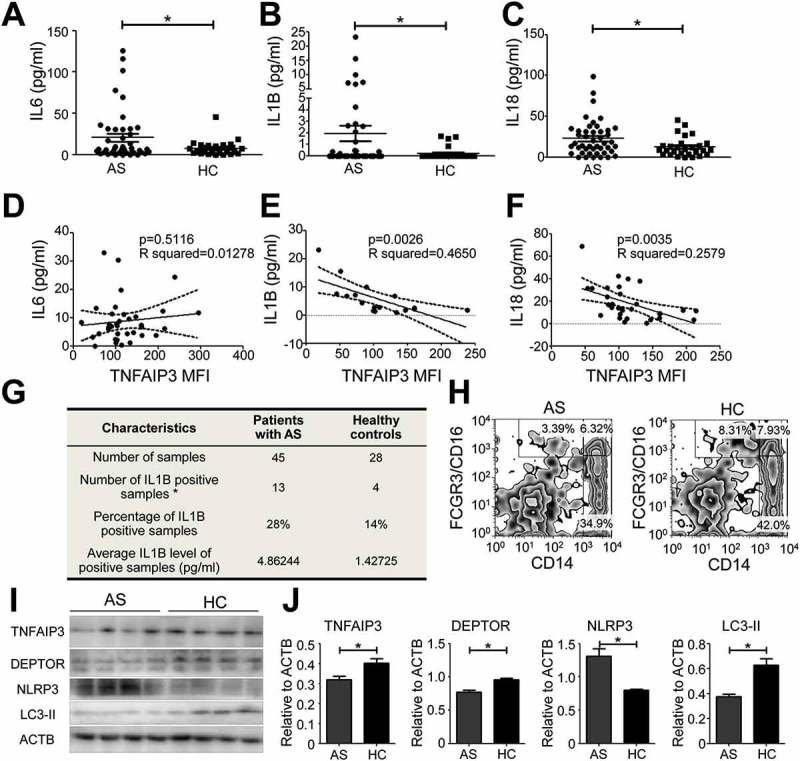
TNFAIP3-DEPTOR deficiency in AS non-classical monocytes increases the serum IL1B level. (a-c) ELISA assay for IL6 (a), IL1B (b), and IL18 (c) in the serum of AS (n = 45) and HC (n = 28) subjects. (d–f) Correlation between non-classical monocytes TNFAIP3 expression and IL6 (d, n = 36, IL6 positive with TNFAIP3 expression data), IL1B (e, n = 17, IL1B positive with TNFAIP3 expression data) and IL18 (f, n = 31, IL18 positive with TNFAIP3 expression data). Each pairing (TNFAIP3 and IL6, TNFAIP3 and IL1B, and TNFAIP3 and IL18) was from the same individual. (g) Characteristics of IL1B secretion in AS and HC subjects. (h) Sorting methods for AS and HC non-classical monocytes. CD14 and FCGR3/CD16 were used as sorting markers, and the CD14+ FCGR3/CD16++ cells were collected for further investigation using a western blot assay. (i, j) Western blot assay (i) and quantification (j) of TNFAIP3, DEPTOR, NLRP3 and LC3-II expression in AS (n = 4) and HC (n = 4) non-classical monocytes. In western blot (i), the four lanes for AS and four for HC represent different individuals. *, p < 0.05 by unpaired t test (a–c, j), Spearman’s correlation test and paired t test (d–f). MFI, mean fluorescence intensity.
Discussion
Our study reveals that in monocytes, TNFAIP3 reduces IL1B secretion, which is partially dependent on the NFKB pathway. Together with DEPTOR, TNFAIP3 induces autophagy in the early stage of LPS stimulation so that the inflammasome level is decreased. Such inflammasome inhibition is absent in AS peripheral non-classical monocytes, causing IL1B secretion and persistent inflammation (Figure 7).
Figure 7.
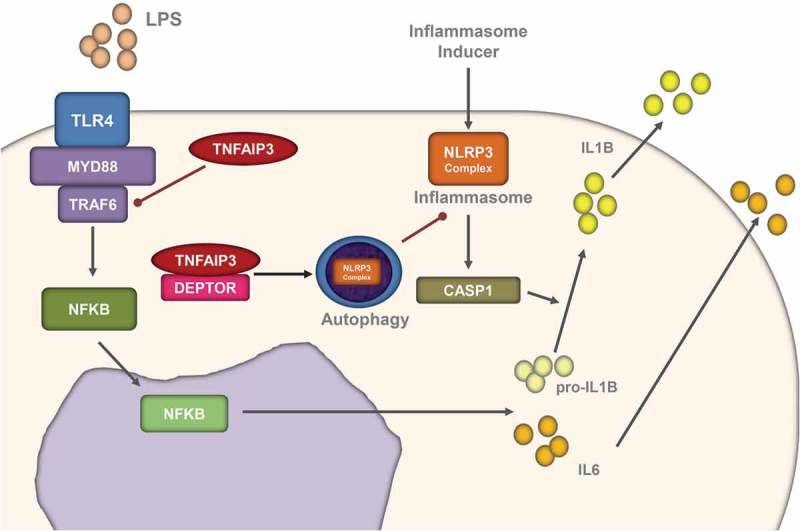
The TNFAIP3-DEPTOR complex inhibits the inflammasome through autophagy. Schematic representation of the TNFAIP3-DEPTOR complex in LPS-related autophagy and inflammasome induction. Together with DEPTOR, TNFAIP3 induces autophagy in the early stage of LPS stimulation so that the inflammasome level is decreased.
In humans, monocytes have been divided into 3 subtypes based on the relative surface expression of CD14 and FCGR3/CD16: classical monocytes (CD14++ FCGR3/CD16−) with strong phagocytic capacity; non-classical monocytes (CD14+ FCGR3/CD16++), also called patrolling monocytes, that secrete TNF and IL1B; and intermediate monocytes (CD14++ FCGR3/CD16+). Previous works have shown that monocytes perform abnormally in AS. For one thing, dysfunction of AS monocytes causes abnormal mesenchymal stem cell osteogenic differentiation and inflammation [30,31]. For another, the blood monocyte population is related to ankylosing spondylitis [32]. What is more, infliximab treatment leads to an increase in non-classical monocytes and a decrease in classical monocytes [33]. As a matter of fact, non-classical monocytes are the main source of IL1B, and the dysfunction of macrophages is likely to affect the processing and secretion of IL1B.
In other studies, the IL1B level in AS peripheral blood has been controversial. In contrast to IL6, which combines with IL6R (interleukin 6 receptor) to induce immunocyte proliferation and differentiation, secreted IL1B is able to further activate the NFKB pathway with its interaction with the IL1B receptor. In addition, activated macrophages are primarily located in tissue, and only the non-classical and intermediate monocytes are responsible for peripheral IL1B secretion. More importantly, as shown by our study, not every AS patient has IL1B-positive blood serum. However, the AS IL1B-positive rate is 28% (14% for HC), and the average IL1B level of the positive samples in AS is 4.86 pg/mL (1.47 pg/mL for HC). These data may explain why anti-IL1 treatment is not ideal [34]. However, considering that IL1B is the central mediator of the initiation and amplification of the immune response, the importance of IL1B should not be neglected.
Autophagy, as a protective mechanism of cells, interacts with the inflammasome in different ways and with different purposes. With LPS stimulation, early-onset autophagy inhibits inflammasome secretion. Pre-existing autophagy-related complexes automatically and immediately activate autophagy under inflammatory stimulation. Without such protection, monocytes secrete more inflammatory cytokines such as IL1B, which later re-stimulate the immunocytes and cause further immune activation. TNFAIP3 is thought to be more responsible for NFKB inhibition, so that TNFAIP3 could reduce inflammasome activation and IL1B secretion through NFKB pathway, which might be irrelevant to the role of TNFAIP3 in autophagy. However, when the inflammasome is activated, autophagy levels should be increased to inhibit the inflammasome [35,36]. In that way, compared to control group, TNFAIP3 overexpression reduces inflammasome activation, which probably leads to a relatively low level of autophagy. In fact, in our experiments, we observed autophagy enhancement during early inflammasome activation in TNFAIP3 overexpression group. Thus, we propose that there is a direct relationship between TNFAIP3 and autophagy.
DEPTOR was first identified as an MTOR-interacting protein that maintained class III phosphatidylinositol 3-kinase and AKT activation and reduced apoptosis [29]. DEPTOR regulates glucose deprivation-induced autophagy, and downregulation of DEPTOR suppresses autophagy [37–39]. However, the mechanism of DEPTOR-induced autophagy is not fully understood. In multiple myeloma and T-cell leukemia, DEPTOR is frequently overexpressed, impacting cell survival and proliferation, as well as endoplasmic reticulum homeostasis [40,41]. There have been a few studies relating DEPTOR to immunocyte activation. Zhong J. et al. demonstrated that DEPTOR accumulation suppressed dendritic cell function through the MTOR pathway [42].
Interestingly, in our study, TNFAIP3 and DEPTOR protein levels were decreased in the AS patients. TNFAIP3 is known to be upregulated by NFKB activation. However, in AS monocytes, where the NFKB pathway is relatively activated, TNFAIP3 expression is decreased. The reason why there is the TNFAIP3 loss in AS monocytes is not clear, but some reasonable assumptions could be made. One possible explanation could be TNFAIP3 protein processing progress is inhibited in AS monocytes. As shown by others, about 90% of AS patients are HLA-B27-positive [1]. The HLA-B27 heavy chain has a tendency to misfold in the endoplasmic reticulum (ER), generates ER stress, and leads to activation of the unfolded protein response [43]. The ER stress causes abnormal protein processing – some proteins including IL23 [44] are dramatically enhanced; but some other proteins, probably TNFAIP3, are inhibited. The other possible reason may be TNFAIP3 is exhausted under HLA-B27 related unfolded protein response. TNFAIP3 is highly related to protein ubiquitination – one of the ways to deal with misfolded and unfolded protein. In order to respond to misfolded HLA-B27, TNFAIP3 could be used to activate ubiquitin-related protein degradation and finally exhausted. Considering that TNFAIP3 is able to stabilize DEPTOR, the loss of TNFAIP3 in AS monocytes could contribute to DEPTOR decrease.
In our study, TNFAIP3 and DEPTOR interact with each other without any stimulation. The original interaction is a kind of pre-existing autophagy inducer. If there is minor inflammasome activation, the autophagy inducer complex is able to eliminate it immediately. We used TPCK to block the NFKB pathway, which inhibited the activation of IL6 by LPS; however, with TNFAIP3 downregulation, IL1B secretion was still present even with the same amount of TPCK. As a matter of fact, in opposition to the effects of TPCK, TNFAIP3 knockdown results in the activation of the NFKB pathway, which indicates that NFKB activation cannot be shut down completely. Thus, the small amount of inflammasome formation is blocked by the pre-existing autophagy inducer; however, without TNFAIP3 or DEPTOR expression, such inhibition is demolished. Interestingly, we found that in monocytes (sorted primary monocytes or THP-1 cells), only LC3-II but not LC3-I was expressed, which emphasized the importance of autophagy in monocytes even without LPS stimulation.
TNFAIP3 is widely known as a ubiquitination-related protein, which, in most cases, recognizes and removes a K63Ub or adds a K48Ub on its substrates [18–20]. However, the interaction between TNFAIP3 and DEPTOR is a direct protein interaction. Shi et al. have shown that TNFAIP3 was able to remove a K63Ub from BECN1 to limit TLR4-related autophagy [24]. TNFAIP3 regulates BECN1 in a ubiquitin-dependent manner, which also requires TLR4 activation. Thus, we consider the interaction of TNFAIP3 and BECN1 to be a type of inflammation-related autophagy inhibitor. During the early stage of LPS stimulation, the pre-existing autophagy inducer (e.g., TNFAIP3-DEPTOR complex) is dominant, triggering autophagy to reduce inflammasome formation. Several hours later, the inflammation-related autophagy inhibitor (e.g. TNFAIP3-BECN1 interaction) becomes more important as inflammation increases. If monocytes lack TNFAIP3, despite the increase in inflammation-related autophagy, inflammation is already at a very high level, and cytokines (such as IL1B) have already been secreted and are activating the corresponding receptors. Therefore, the presence of the pre-existing autophagy inducer is vital to the control of inflammation at the proper level.
The reason why TNFAIP3-DEPTOR complex acts as the pre-existing autophagy inducer is somehow dependent on the characteristics of their interaction. DEPTOR was able to bind to TNFAIP3 zinc-finger domain, especially zinc-finger 4–7, which has been shown to be required for TNFAIP3 lysosomal localization [45]. When there is the signal for autophagy, TNFAIP3-DEPTOR complex moves to the autophagosome and facilitates fusion with lysosomes to form autolysosomes, so that the autophagy flux is highly enhanced and the LPS-related inflammasome is eliminated.
In general, monocytes with a TNFAIP3-DEPTOR deficiency are in danger of over-activation of inflammasome formation. In AS, the TNFAIP3-DEPTOR complex modulates autophagy and reduces inflammasome activation, providing a new theory to better understand the disease.
Materials and methods
Plasmids, virus, and reagents
The plasmids, virus and reagents are used as follows: pCMV3-TNFAIP3-His (Sino Biological, HG12089-CH), pCMV3-DEPTOR-His (Sino Biological, HG20040-CH), pCMV3-TNFAIP3-GFPSpark (Sino Biological, HG12089-ACG), pCMV3-DEPTOR-OFPSpark (Sino Biological, HG20040-ACR), NFKB reporter luciferase (Genomeditech, GM-021001), pmCherry-LC3 (Youbio, VT8111), pCMV3 control vector (Sino Biological, CV015), pCMV-FLAG (Beyotime, D2722), pCMV-FLAG-TNFAIP3 (the different fragment vectors were designed and constructed by AuGCT), the Matchmaker Gold Yeast Two-Hybrid System (Clontech, 630489) containing the pGBKT7 DNA-BD Cloning Vector, pGADT7 AD Cloning Vector, pGBKT7-53 Control Vector, pGADT7-T Control Vector, and pGBKT7-Lam Control Vector, RFP-GFP-LC3 adenovirus (HANBIO, HB-LP210 0001), TNFAIP3 shRNA (h) Lentiviral Particles (Santa Cruz Biotechnology, sc-37655-V), negative control shRNA lentiviral particles (Santa Cruz Biotechnology, sc-108080), phorbol 12-myristate 13-acetate (PMA; Cayman Chemical, 10008014), lipopolysaccharide (Sigma-Aldrich, L2630), chloroquine (Sigma-Aldrich, C6628), cycloheximide (CHX; Cayman Chemical, 17343), MG-132 (Cell Signaling Technology, 2194).
Patients and primary cell preparation
All of the clinical samples were obtained from the Department of Clinical Immunology, Xijing Hospital, Fourth Military Medical University (Xi’an, China). In total, 86 ankylosing spondylitis patients were defined according to the modified New York criteria and participated in the experiments; the patient characteristics in this study are shown in Table S1. In addition, 31 healthy individuals with no symptoms of ankylosing spondylitis were enrolled in the study as the healthy controls (HCs). Informed written consent was obtained from all patients/patients’ families and healthy individuals prior to participation, and the ethics approval was granted by Ethical Committee of Fourth Military Medical University. Primary peripheral blood mononuclear cells (PBMCs) were obtained and enriched by lymphocyte separation medium (MP Biomedicals, 0850494) gradient centrifugation.
Cell culture, transfection, and treatment
THP-1 (TIB-202™) and HEK 293T (293T; CRL-11268™) cells were purchased from the ATCC. Both THP-1 and 293T cells were cultured in RPMI-1640 medium (Thermo Scientific, 11875–093) supplemented with 10% fetal bovine serum (Thermo Scientific, 10100–147 – FBS), 1% penicillin/streptomycin (Thermo Scientific, 15240062), and 2% L-glutamine at 37°C in a humidified atmosphere composed of 5% CO2. The THP-1 cells were stably transfected with TNFAIP3 shRNA (h) lentiviral particles, and puromycin (Thermo Scientific, A1113803) was added for further selection. The vectors pCMV3-TNFAIP3-His, pCMV3-DEPTOR-His and pCMV-FLAG-TNFAIP3 (1–6) were transfected into HEK 293T cells with hygromycin for further stable selection. siRNAs targeting TNFAIP3 and DEPTOR, and negative control siRNA were designed and synthesized by Shanghai GenePharma Company. The negative control shRNA lentiviral particles, pCMV3 vector, and silencer control siRNA served as the negative control (NC). PMA (100 ng/mL) was used to activate THP-1 cells. After 12 h of PMA activation, THP-1 cells were further treated with lipopolysaccharide (LPS, 200 ng/ml; Sigma-Aldrich, L2630) for different time courses.
Flow cytometry and cell sorting
PBMCs from AS and HC subjects were collected for flow cytometry. PBMCs were first stained with antibodies recognizing various surface markers: CD3E-PE (Biolegend, 300308), CD4-PerCP (Biolegend, 317432), CD8A-APC (Biolegend, 301014), ITGAM/CD11b-APC (Biolegend, 301309), CD14-PE/Cy7 (Biolegend, 301,814), CD15-PE (Biolegend, 301905), CD19-PerCP (Biolegend, 302228), IL2RA/CD25-APC (Biolegend, 356109), PTPRC/CD45RA-PE (Biolegend, 304,108), PTPRC/CD45RO-PE/Cy7 (Biolegend, 304229), FCGR3/CD16-APC (Biolegend, 302011). After the surface stain, PBMCs were fixed in 4% formaldehyde in phosphate-buffered saline (PBS; Beyotime, ST476) for 20 min, permeabilized with 0.1% Triton X-100 (20 min; Beyotime, ST795), blocked with goat serum (30 min; Thermo Scientific, 16210072), and then processed for anti-TNFAIP3 (Alexa Fluor® 488; Abcam, ab197541) and intracellular staining of IL-17A-APC (Biolegend, 512333), and FOXP3-PE (Biolegend, 320107). The cells were analyzed by FACSCalibur flow cytometry (BD Pharmingen, San Diego, USA), and the data were processed using FlowJo software. FCGR3/CD16-APC and CD14-PE/Cy7 were used for non-classical monocyte sorting. The primary monocytes were sorted by FACSAria III (BD Pharmingen, San Diego, USA) and processed for western blot assays.
Western blotting
Treated cells were harvested in radioimmunoprecipitation assay buffer (RIPA; Beyotime, P0013B) with protease inhibitor (Roche, 04693159001) and phenylmethylsulfonyl fluoride (PMSF; Beyotime, ST506) on ice, and the protein quantification was determined by using a BCA Protein Assay Kit (Thermo Scientific, 23250). Cellular proteins were subjected to 8–12% SDS-PAGE separation and transferred to polyvinylidene fluoride (PVDF) microporous membranes (EMD Millipore, IPVH00010). Blots were probed for 12 h at 4°C with primary antibodies, and secondary antibodies were incubated with the PVDF membrane for 1 h at room temperature. Visualization was performed with a Molecular Imaging System (CARESTREAM HEALTH, INC. NY, USA). Antibodies used were: anti-TNFAIP3 mouse antibody (Santa Cruz Biotechnology, sc-166692), anti-TNFAIP3 goat antibody (Santa Cruz Biotechnology, sc-32525; this antibody is no longer available from this source), anti-DEPTOR mouse antibody (Santa Cruz Biotechnology, sc-398169), anti-DEPTOR rabbit antibody (Proteintech, 20985–1-AP), anti-MTOR rabbit antibody (Santa Cruz Biotechnology, sc-1550-R), anti-LC3B rabbit antibody (Cell Signaling Technology, 3868), anti-NLRP3 rat antibody (R&D Systems, MAB7578), goat anti-mouse antibody (Thermo Scientific, 31430), goat anti-rabbit antibody (Thermo Scientific, 32460), goat anti-rat antibody (Thermo Scientific, 31470), rabbit anti-goat antibody (Thermo Scientific, 31402), anti-ACTB mouse antibody (HuaBio, M1210-2).
Co-immunoprecipitation (co-IP) and silver stain
The lysates of transfected THP-1 and 293T cells were prepared, and 10 μg of TNFAIP3, DEPTOR or Flag antibodies and 300 μg of each protein sample were used for co-IP assays according to the manufacturer’s protocol (Co-Immunoprecipitation Kit; Thermo Scientific, 26,149). The eluted protein samples were subjected to SDS-PAGE separation and western blot assays. A silver stain was performed with a Protein Stains K Kit (Sangon Biotech, C500021) and visualized with a Gel Doc EZ Imager (BIO-RAD, CA, USA).
Immunostaining and confocal microscopy
Transfected and pretreated cells in dishes were washed twice with PBS. The cells were fixed in 4% paraformaldehyde (Boster Biological Technology, AR1069) and permeabilized with 0.1% Triton X-100 for 20 min separately. Then, the cells were blocked with goat serum in PBS for 1 h. The dishes were first incubated for 12 h at 4°C with primary antibodies, and after the cells were washed 5 times with PBS, they were incubated with the secondary antibodies, donkey anti-mouse antibody, Alexa Fluor 555 conjugate (Thermo Scientific, A-31570), donkey anti-mouse antibody, Alexa Fluor 488 (Thermo Scientific, R37114), donkey anti-rabbit antibody, Alexa Fluor 488 (Thermo Scientific, A-21206), donkey anti-rabbit antibody, Alexa Fluor 555 (Thermo Scientific, A-31572), donkey anti-rat antibody, Alexa Fluor 488 (Thermo Scientific, A-21208), and goat anti-rabbit antibody, DyLight 650 (Thermo Scientific, 84546), for 1 h at room temperature. Cell nuclei were dyed with DAPI (Vector Laboratories, H-1200). The cells were visualized using an IX73 microscope system (Olympus, Tokyo, Japan) and A1R-A1 confocal laser microscope system (Nikon, Tokyo, Japan).
Transmission electron microscopy (TEM)
For transmission electron microscopy, cells were fixed in 4% glutaraldehyde over 24 h. Thin sections (60–80 nm) were cut, mounted on copper grids, and poststained with uranyl acetate and lead citrate. Micrographs were taken using the TECNAI G2 Spirit Biotwin system (FEI company, OR, USA). The autophagy levels were analyzed through the percentage of autophagy area in whole cell area and more than 40 cells in each group were analyzed.
Yeast two-hybrid system
The yeast two-hybrid system analysis was carried out according to the manufacturer’s protocol (Clontech, 630489; Matchmaker Gold Two-Hybrid System). The bait construct on the pGBKT7 DNA-BD Cloning Vector contained the TNFAIP3 mRNA sequence, and DEPTOR was constructed on the pGADT7 AD Cloning Vector. The pGBKT7-53 Control Vector and pGADT7-T Control Vector were used as the positive control, while the pGBKT7-Lam Control Vector and pGADT7-T Control Vector were the negative controls. Both the bait and prey vectors were transfected into yeast AH109. After mating and cloning, the presence of blue positive clones indicated that the two proteins interacted.
GST affinity-isolation assay
The GST-fused DEPTOR and His-tagged proteins were purified as previously described [46]. GST-fused TNFAIP3 incubated with prepared glutathione sepharose beads (BBI Life Sciences, BSP031) on the rotating incubator at 4°C for 1 h, and then the beads were collected and washed by wash/binding buffer 3 times. Then, input His-tagged proteins were incubated with the beads at 4°C for 2 h. After removing the supernatant, the beads were washed with the wash/binding buffer 4 times. The target proteins were washed down with 10% SDS. These elutes were then analyzed and detected by western blotting.
RNA isolation, cDNA production, and real-time PCR
After different treatments, mRNAs were isolated with the E.Z.N.A. Total RNA Kit II (OMEGA BioTek, R6934-02) in RNase-free conditions. Then, reverse transcription of the mRNA was performed using the PrimeScript® RT reagent kit (TaKaRa, DRR037A) following the manufacturer’s instructions. SYBR-Green (TaKaRa, DRR041A) real-time RT-PCR was performed using the Stratagene Mx3005P sequence detection system (Agilent Technologies, CA, USA). GAPDH was used to normalize the target mRNA. All primers were synthesized by Sangon Company. The relative quantity in each experiment was automatically calculated with the comparative quantitation mode of the MxPro qPCR system software.
Luciferase reporter assays
293T cells were transfected with a human TNFAIP3 or control vector using Lipofectamine 2000 (Invitrogen, 11668019) and co-transfected with plasmids encoding the NFKB reporter luciferase (firefly) and Renilla luciferase. After 48 h, the cells were treated with LPS and/or 3-MA. Cells were harvested later, and luciferase reporter assays were performed using the Dual-luciferase reporter assay (Promega, E1960). Firefly luciferase signals were normalized to the Renilla luciferase signals.
Cytokine ELISA
Serum samples were obtained and separated from the AS and HC peripheral blood. Treated cell medium was harvested and centrifuged at 10,000 x g for 5 min to remove cell debris and stored at −80°C until use. IL6, IL1B, and IL18 in serum and culture medium was directly measured with a Human IL6 ELISA Kit (DAKEWE, DKW12-1060–096), Human IL1B ELISA Kit (DAKEWE, DKW12-1012–096), LEGEND MAX Human IL1B ELISA Kit (Biolegend, 437007) and Human IL18 ELISA Kit (Neobioscience, EHC127.96), respectively, according to the manufacturer’s instructions. The optical density was determined with a BIO-RAD Microplate reader (CA, USA), and absorption was measured at 450 nm. A standard curve for each measurement was established using a cytokine standard provided by the ELISA Kit.
GEO data analysis
All of the datasets used in this study were downloaded from the gene expression omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database. The accession codes of the array data were GSE25101 and GSE1188, performed on GPL6947 and GPL570, respectively. The microarray data analysis was done using R and several packages available from CRAN53 and Bioconductor. The raw data (CEL files) were normalized and summarized using the Robust MultiArray Average method.
Statistical analysis
All experiments were performed at least 3 times, and the results were expressed as the mean ± SD. Statistical significance was determined using GraphPad Prism V5.0 software (GraphPad Software, CA, U). Differences were deemed significant if p < 0.05. Two-way ANOVA or one-way ANOVA followed by Dunnett’s post-test (for subgroup analyses) was performed for multiple comparisons; a two-tailed t test was performed for other experiments to compare the mean values; and Spearman’s correlation test was used to analyze the correlation of two molecules. *** indicates p < 0.001, ** indicates p < 0.01, * indicates p < 0.05, and not significant (ns) indicates p > 0.05. Error bars indicate ± SD.
Biography
Design of the study: PZ, Z-nC, F-yL, Y Zhai, JC, and H-yL. Acquisition of data: Y Zhai, PZ, PL, ZF, H-yL, JC, Q Han, Y Zhang, Q He, GN, XL, BW and FF. Interpretation of data: Y Zhai, PZ, Z-nC, H-yL, and F-yL. Manuscript preparation: Y Zhai, PZ, Z-nC, and F-yL.
Funding Statement
This work was supported by the NATIONAL BASIC RESEARCH PROGRAM OF CHINA [2015CB553704].
Abbreviations
- ACTB
actin, beta;
- AIM2
absent in melanoma 2;
- AS
ankylosing spondylitis;
- ATG
autophagy related;
- BECN1
beclin 1;
- CASP1
caspase 1;
- CHX
cycloheximide;
- CQ
chloroquine;
- DEPTOR
DEP domain containing MTOR interacting protein;
- DMSO
dimethyl sulfoxide;
- FSC
forward scatter;
- HLA-B
major histocompatibility complex, class I, B;
- IL
interleukin;
- LPS
lipopolysaccharide;
- MAP1LC3/LC3
microtubule associated protein 1 light chain 3;
- MTOR
mechanistic target of rapamycin kinase;
- MALT1
MALT1 paracaspase;
- MFI
mean fluorescence intensity;
- NFKB
nuclear factor kappa B;
- NLRP3
NLR family pyrin domain containing 3;
- PBS
phosphate-buffered saline;
- PMA
phorbol 12-myristate 13-acetate;
- PMSF
phenylmethylsulfonyl fluoride;
- RIPK1
receptor interacting serine/threonine kinase 1;
- SSC
side scatter;
- TLR4
toll like receptor 4;
- TNFAIP3/A20
TNF alpha induced protein 3;
- TRAF6
TNF receptor associated factor 6;
- TRIM11
tripartite motif containing 11;
- UBE2N
ubiquitin conjugating enzyme E2 N.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental Data
Supplemental data for this article can be accessed here.
References
- [1].Brown MA, Pile KD, Kennedy LG, et al. HLA class I associations of ankylosing spondylitis in the white population in the United Kingdom. Ann Rheum Dis. 1996. April;55(4):268–270. PubMed PMID: 8733445; PubMed Central PMCID: PMC1010149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hreggvidsdottir HS, Noordenbos T, Baeten DL.. Inflammatory pathways in spondyloarthritis. Mol Immunol. 2014. January;57(1):28–37. PubMed PMID: 23969080. [DOI] [PubMed] [Google Scholar]
- [3].Vazquez-Del Mercado M, Garcia-Gonzalez A, Munoz-Valle JF, et al. Interleukin 1beta (IL-1beta), IL-10, tumor necrosis factor-alpha, and cellular proliferation index in peripheral blood mononuclear cells in patients with ankylosing spondylitis. J Rheumatol. 2002. March;29(3):522–526. PubMed PMID: 11908566. [PubMed] [Google Scholar]
- [4].Keystone EC, Jaglal S, Shore A. Interleukin 1 and interleukin 2 generation by peripheral blood cells from patients with ankylosing spondylitis. J Rheumatol. 1986. October;13(5):944–947. PubMed PMID: 3493347. [PubMed] [Google Scholar]
- [5].Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. PubMed PMID: 19302047. [DOI] [PubMed] [Google Scholar]
- [6].Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011. April 7;117(14):3720–3732. PubMed PMID: 21304099; PubMed Central PMCID: PMC3083294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002. August;10(2):417–426. PubMed PMID: 12191486. [DOI] [PubMed] [Google Scholar]
- [8].Afonina IS, Muller C, Martin SJ, et al. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. 2015. June 16;42(6):991–1004. PubMed PMID: 26084020. [DOI] [PubMed] [Google Scholar]
- [9].Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010. March 19;140(6):805–820. PubMed PMID: 20303872. [DOI] [PubMed] [Google Scholar]
- [10].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008. November 13;456(7219):264–268. PubMed PMID: 18849965. [DOI] [PubMed] [Google Scholar]
- [11].Liu T, Tang Q, Liu K, et al. TRIM11 suppresses AIM2 inflammasome by degrading AIM2 via p62-dependent selective autophagy. Cell Rep. 2016. August 16;16(7):1988–2002. DOI: 10.1016/j.celrep.2016.07.019 PubMed PMID: 27498865. [DOI] [PubMed] [Google Scholar]
- [12].Santeford A, Wiley LA, Park S, et al. Impaired autophagy in macrophages promotes inflammatory eye disease. Autophagy. 2016. October 2;12(10):1876–1885. PubMed PMID: 27463423; PubMed Central PMCID: PMC5066937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ding WX, Jaeschke H. Autophagy in macrophages regulates the inflammasome and protects against liver injury. J Hepatol. 2016. January;64(1):16–18. PubMed PMID: 26456339; PubMed Central PMCID: PMC4888871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xing Y, Cao R, Hu HM. TLR and NLRP3 inflammasome-dependent innate immune responses to tumor-derived autophagosomes (DRibbles). Cell Death Dis. 2016. August 04;7(8):e2322 PubMed PMID: 27490927; PubMed Central PMCID: PMC5108312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu C, Yue R, Yang Y, et al. AIM2 inhibits autophagy and IFN-beta production during M. bovis infection. Oncotarget. 2016. July 9 DOI: 10.18632/oncotarget.10503 PubMed PMID: 27409673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Opipari AW Jr., Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem. 1990. September 5;265(25):14705–14708. PubMed PMID: 2118515. [PubMed] [Google Scholar]
- [17].Lee EG, Boone DL, Chai S, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000. September 29;289(5488):2350–2354. PubMed PMID: 11009421; PubMed Central PMCID: PMC3582399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wertz IE, O’Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004. August 05;430(7000):694–699. PubMed PMID: 15258597. [DOI] [PubMed] [Google Scholar]
- [19].Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010. February 26;327(5969):1135–1139. PubMed PMID: 20185725; PubMed Central PMCID: PMC3025292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004. October;5(10):1052–1060. PubMed PMID: 15334086. [DOI] [PubMed] [Google Scholar]
- [21].Duong BH, Onizawa M, Oses-Prieto JA, et al. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity. 2015. January 20;42(1):55–67. PubMed PMID: 25607459; PubMed Central PMCID: PMC4302274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vande Walle L, Van Opdenbosch N, Jacques P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature. 2014. August 7;512(7512):69–73. PubMed PMID: 25043000; PubMed Central PMCID: PMC4126806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li M, Shi X, Qian T, et al. A20 overexpression alleviates pristine-induced lupus nephritis by inhibiting the NF-kappaB and NLRP3 inflammasome activation in macrophages of mice. Int J Clin Exp Med. 2015;8(10):17430–17440. PubMed PMID: 26770333; PubMed Central PMCID: PMC4694233. [PMC free article] [PubMed] [Google Scholar]
- [24].Shi CS, Kehrl JH. Traf6 and A20 differentially regulate TLR4-induced autophagy by affecting the ubiquitination of Beclin 1. Autophagy. 2010. October;6(7):986–987. PubMed PMID: 20798608; PubMed Central PMCID: PMC3039745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hubbard-Lucey VM, Shono Y, Maurer K, et al. Autophagy gene Atg16L1 prevents lethal T cell alloreactivity mediated by dendritic cells. Immunity. 2014. October 16;41(4):579–591. PubMed PMID: 25308334; PubMed Central PMCID: PMC4237219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Matsuzawa Y, Oshima S, Takahara M, et al. TNFAIP3 promotes survival of CD4 T cells by restricting MTOR and promoting autophagy. Autophagy. 2015;11(7):1052–1062. PubMed PMID: 26043155; PubMed Central PMCID: PMC4590588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol. 2012. November;12(11):774–785. PubMed PMID: 23059429; PubMed Central PMCID: PMC3582397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Catrysse L, Vereecke L, Beyaert R, et al. A20 in inflammation and autoimmunity. Trends Immunol. 2014. January;35(1):22–31. PubMed PMID: 24246475. [DOI] [PubMed] [Google Scholar]
- [29].Peterson TR, Laplante M, Thoreen CC, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009. May 29;137(5):873–886. PubMed PMID: 19446321; PubMed Central PMCID: PMC2758791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xie Z, Wang P, Li J, et al. MCP1 triggers monocyte dysfunctions during abnormal osteogenic differentiation of mesenchymal stem cells in ankylosing spondylitis. J Mol Med. 2017. February;95(2):143–154. PubMed PMID: 27921117. [DOI] [PubMed] [Google Scholar]
- [31].Wright C, Edelmann M, diGleria K, et al. Ankylosing spondylitis monocytes show upregulation of proteins involved in inflammation and the ubiquitin proteasome pathway. Ann Rheum Dis. 2009. October;68(10):1626–1632. PubMed PMID: 18952638. [DOI] [PubMed] [Google Scholar]
- [32].Surdacki A, Sulicka J, Korkosz M, et al. Blood monocyte heterogeneity and markers of endothelial activation in ankylosing spondylitis. J Rheumatol. 2014. March;41(3):481–489. PubMed PMID: 24488416. [DOI] [PubMed] [Google Scholar]
- [33].Aeberli D, Kamgang R, Balani D, et al. Regulation of peripheral classical and non-classical monocytes on infliximab treatment in patients with rheumatoid arthritis and ankylosing spondylitis. RMD Open. 2016;2(1):e000079 PubMed PMID: 26819749; PubMed Central PMCID: PMC4716562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Toussirot E. Biologics in spondyloarthritis: tNFalpha inhibitors and other agents. Immunotherapy. 2015;7(6):669–681. PubMed PMID: 26058432. [DOI] [PubMed] [Google Scholar]
- [35].Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell. 2016. July 14;166(2):288–298. PubMed PMID: 27419869; PubMed Central PMCID: PMC4947210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chiu HW, Chen CH, Chang JN, et al. Far-infrared promotes burn wound healing by suppressing NLRP3 inflammasome caused by enhanced autophagy. J Mol Med. 2016. July;94(7):809–819. PubMed PMID: 26864306. [DOI] [PubMed] [Google Scholar]
- [37].Zhang H, Chen J, Zeng Z, et al. Knockdown of DEPTOR induces apoptosis, increases chemosensitivity to doxorubicin and suppresses autophagy in RPMI-8226 human multiple myeloma cells in vitro. Int J Mol Med. 2013. May;31(5):1127–1134. PubMed PMID: 23503641. [DOI] [PubMed] [Google Scholar]
- [38].Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell. 2011. October 21;44(2):304–316. PubMed PMID: 22017876; PubMed Central PMCID: PMC3216641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yang D, Zhao Y, Liu J, et al. Protective autophagy induced by RBX1/ROC1 knockdown or CRL inactivation via modulating the DEPTOR-MTOR axis. Autophagy. 2012. December;8(12):1856–1858. PubMed PMID: 22965024; PubMed Central PMCID: PMC3541304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Catena V, Bruno T, De Nicola F, et al. Deptor transcriptionally regulates endoplasmic reticulum homeostasis in multiple myeloma cells. Oncotarget. 2016. September 16 DOI: 10.18632/oncotarget.12060 PubMed PMID: 27655709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hu Y, Su H, Liu C, et al. DEPTOR is a direct NOTCH1 target that promotes cell proliferation and survival in T-cell leukemia. Oncogene. 2016. September 5 DOI: 10.1038/onc.2016.275 PubMed PMID: 27593934. [DOI] [PubMed] [Google Scholar]
- [42].Cheng M, Hu S, Wang Z, et al. Inhibition of neddylation regulates dendritic cell functions via Deptor accumulation driven mTOR inactivation. Oncotarget. 2016. June 14;7(24):35643–35654. PubMed PMID: 27224922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mear JP, Schreiber KL, Munz C, et al. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunology. 1999. December 15;163(12):6665–6670. PubMed PMID: 10586062. [PubMed] [Google Scholar]
- [44].DeLay ML, Turner MJ, Klenk EI, et al. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. 2009. September;60(9):2633–2643. PubMed PMID: 19714651; PubMed Central PMCID: PMC2893331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li L, Hailey DW, Soetandyo N, et al. Localization of A20 to a lysosome-associated compartment and its role in NFkappaB signaling. Biochimica Et Biophysica Acta. 2008. June;1783(6):1140–1149. PubMed PMID: 18329387; PubMed Central PMCID: PMC2587335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang WJ, He YX, Yang Z, et al. Crystal structure of glutathione-dependent phospholipid peroxidase Hyr1 from the yeast Saccharomyces cerevisiae. Proteins. 2008. December;73(4):1058–1062. PubMed PMID: 18767166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.