

Abstract
Cobalt oxide Co3O4 has recently emerged as promising, noble metal-free catalyst for oxidation reactions but a better understanding of the active catalyst under working conditions is required for further development and potential commercialization. An operando approach has been applied, combining near ambient (atmospheric) pressure X-ray photoelectron spectroscopy (NAP-XPS), Fourier transform infrared spectroscopy (FTIR), or X-ray diffraction (XRD) with simultaneous catalytic tests of CO oxidation on Co3O4, enabling one to monitor surface and bulk states under various reaction conditions (steady-state and dynamic conditions switching between CO and O2). On the basis of the surface-specific chemical information a complex network of different reaction pathways unfolded: Mars-van-Krevelen (MvK), CO dissociation followed by carbon oxidation, and formation of carbonates. A possible Langmuir–Hinshelwood (LH) pathway cannot be excluded because of the good activity when no oxygen vacancies were detected. The combined NAP-XPS/FTIR results are in line with a MvK mechanism above 100 °C, involving the Co3+/Co2+ redox couple and oxygen vacancy formation. Under steady state, the Co3O4 surface appeared oxidized and the amount of reduced Co2+ species at/near the surface remained low up to 200 °C. Only in pure CO, about 15% of surface reduction were detected, suggesting that the active sites are a minority species. The operando spectroscopic studies also revealed additional reaction pathways: CO dissociation followed by carbon reoxidation and carbonate formation and its decomposition. However, due to their thermal stability in various atmospheres, the carbonates are rather spectators and also CO dissociation seems a minor route. This study thus highlights the benefits of combining operando surface sensitive techniques to gain insight into catalytically active surfaces.
Keywords: CO oxidation, Co3O4, operando, NAP-XPS, FTIR, XRD, surface spectroscopy
1. Introduction
Controlling automotive emissions during an engine’s cold start is an unresolved issue, resulting from the strong CO adsorption on noble metals, poisoning the catalyst at low temperature (<200 °C).1,2 Due to their low-temperature CO oxidation ability, transition metal oxides,3 especially cobalt oxide, are promising.4,5 Despite intensive studies, the nature of the active sites of cobalt oxide catalysts as well as exact reaction mechanisms are still debated and contradicting results concerning the role of Co3+/Co2+, of different oxygen species, oxygen vacancies, and carbonates were reported.
The active sites are often attributed to the abundance of specific cobalt cations. In 2009, Xie et al. reported that Co3O4 nanorods, predominately exposing (110) planes and enriched in Co3+ cations, exhibit very high activity for low-temperature CO oxidation. The high catalytic activity was ascribed to Co3+ cations,5 but no spectroscopic evidence was given. Two years later the same group reported that Co3O4 nanosheets with (111) planes, enriched in Co2+ cations, were the most active among nanorods, nanocubes, and nanoparticles,6 contrasting with the previous report. Jia et al. observed that a Co3O4–SiO2 nanocomposite catalyst, without ordered planes, but enriched in Co2+, exhibited very high activity at low temperature.4 Recently, it was reported that CoO with octahedrally coordinated Co2+ had unexpectedly high activity due to the ease of surface oxidation of Co2+ to Co3+.7 Nevertheless, all conclusions were primarily based on catalytic studies and no direct spectroscopic evidence of the active surface oxidation state was provided.
A recent study by Iablokov et al. using different particle sizes (2 to 10 nm) showed a maximum rate between 5 and 8 nm. This was explained by a higher concentration of Co3+ 3d states, as revealed by XPS.8 Ding et al. reported an enhanced activity of flower-like Co3O4 with an increased number of surface Co3+.9 All findings illustrate that there are still open questions concerning the nature of active sites, i.e., the abundance of Co3+/Co2+ and Co3O4/CoO.
With respect to the reaction mechanism, it was suggested that CO adsorbs to Co3+ cations, followed by abstracting surface lattice oxygen coordinated to three Co3+ cations, with the oxygen vacancy later replenished by gas phase oxygen (Mars-van-Krevelen mechanism).5 Studying the effect of pretreatment conditions, Yu et al. proposed a reaction between a molecularly adsorbed CO and molecularly adsorbed O–O peroxo species, but without spectroscopic evidence.10 However, Jia et al. did not detect peroxo O–O species by in situ Raman spectroscopy.4 In an in situ IR study, Pollard et al.11 suggested that CO adsorbed on Co2+ sites reacted with an oxygen atom bonded to a neighboring Co3+ cation, and the oxygen vacancy was replenished by oxygen from the gas phase. In his early work on the CO oxidation mechanism, Jansson et al. had already proven lattice oxygen extraction using isotopes, i.e., the redox Mars-van-Krevelen mechanism.12−14 He also found that CO disproportionation takes place during CO oxidation. Mechanistic insights were also gained from reaction orders. The study of Perti et al. on CO oxidation kinetics on Co3O4–Al2O3 revealed reaction orders of CO and O2 being 0 and 0.42, respectively15−17 and their study suggested two competitive mechanisms: reaction of CO with adsorbed oxygen (Langmuir–Hinshelwood LH) and with lattice oxygen (MvK).15−17
The CO oxidation mechanism on cobalt oxide has also been examined by theoretical work,18−21 also showing divergences. For example, Jiang et al. suggested a MvK mechanism for Co3O4 (110).21 Pang et al. proposed two scenarios: direct reaction of CO with surface lattice oxygen atoms (MvK) but also direct reaction of preadsorbed molecular O2 with molecular CO from the gas phase.20,22
A better understanding of CO oxidation on cobalt oxide would clearly benefit from an evaluation of the surface oxidation state of the active catalyst, as well as of the surface species present. Applying operando (in situ) techniques for studying the catalyst surface under reaction conditions may enable to reveal the reaction network, which is required for setting up a mikrokinetic model that could eventually explain the macroscopic kinetics.23−25 The importance of surface composition changes of a cobalt oxide model catalyst during methanol oxidation was, e.g., revealed by operando near atmospheric pressure X-ray photoelectron spectroscopy (NAP-XPS).26 The goal of the current study was to improve the understanding of the reaction pathways occurring during CO oxidation on Co3O4. Thus, NAP-XPS and Fourier transform infrared (FTIR) spectroscopy monitoring the surface changes and surface species were applied with simultaneous catalytic activity tests by gas chromatography (GC) or mass spectroscopy (MS). Experiments were performed under steady-state conditions (CO + O2), in CO atmosphere, and upon switching between CO and O2. In addition, the effect of pretreatment conditions on catalyst structure and activity was examined by operando XRD/MS. Altogether, the operando studies provided new insight into the active catalyst surface, several possible reaction pathways, and the presence and stability of surface species.
2. Experimental Section
2.1. Catalyst
Co3O4 was used as received from Fluka (purity 99.5%). The average crystallite size of Co3O4 determined by X-ray diffraction was 28 nm, the BET specific surface area was 38 m2g–1. According to TEM measurements, the material was composed of nanospheres of 20 to 50 nm in size. A detailed catalyst characterization was reported previously.27
2.2. Catalytic CO Oxidation
CO oxidation was performed in a continuous-flow fixed-bed quartz reactor under atmospheric pressure. The sample (ca. 20 mg, mixed with 100 mg quartz powder to avoid mass and heat transfer limitations) was loaded into the reactor and pretreated with synthetic air at 400 °C for 30 min (50 mL min–1, heating rate of 10 °C min–1, standard pretreatment procedure). For studying the effect of pretreatment conditions, oxidation at 400 °C in synthetic air was followed by reduction at 100 °C, 200 °C, 300 °C, or 400 °C in 5 vol % H2 in N2. After cooling to 30 °C, temperature-dependent CO oxidation was performed either with 5 vol % CO and 10 vol % O2 in He or 5 vol % CO and 2.5 vol % O2 in He. The total flow rate was 50 mL min–1, and the heating rate was 2 °C min–1. The catalysts rapidly deactivate at room temperature (RT), why rather higher reaction temperatures were examined. The concentrations of CO and CO2 in the outlet stream were monitored by gas chromatography (GC) using a HP-PLOT Q column and a flame-ionization detector (FID) with a methanizer.
2.3. Operando XRD Measurements
Operando XRD experiments were performed on a laboratory diffractometer (XPERT III: PANalytical XPert Pro MPD) using Cu–Kα radiation (1.54 Å) operating in the Bragg–Brentano reflection geometry. The diffractometer is equipped with an Anton Paar XRK 900 high-temperature gas cell. In situ diffraction patterns were recorded in the scanning range from 25° to 70° (2θ) using a step scan mode with steps of 0.05° (2θ) and a time per step of 2 s. XRD data were analyzed with the HighScore Plus program.
The inlet of the Anton Paar XRK 900 high-temperature gas cell is connected to a gas manifold system with calibrated mass flow controllers; the outlet of the cell is connected to a quadrupole mass spectrometer (QMS) (PrismaPlus QMG 220, Pfeiffer Vacuum, SEM detector). The Co3O4 (ca. 20 mg) was placed into a sample holder, inserted into the cell and pretreated in synthetic air at 400 °C (30 min). Afterward, the reaction mixture of 5 vol % CO, 10 vol % O2, and 85 vol % He was introduced and XRD patterns were recorded at room temperature (RT), 100 °C, 150 °C, 200 °C, and 250 °C. The same experiment was done after pretreatment in synthetic air at 400 °C followed by reduction in 5 vol % H2 in He at 400 °C (30 min). For in situ H2-temperature-programmed reduction, the samples were pretreated in synthetic air at 400 °C (30 min), then cooled to RT, purged with He, and then 5 vol % H2 in He was introduced. XRD patterns were recorded at RT, 100 °C, 200 °C, 300 °C, and 400 °C. All experiments were performed at atmospheric pressure with a total flow of 50 mL min–1.
2.4. Operando NAP-XPS Measurements
Operando near ambient (atmospheric) pressure X-ray photoelectron spectroscopy (NAP-XPS) was performed at the ISISS beamline at the synchrotron radiation facility BESSY II of the Helmholtz-Zentrum Berlin, Germany. The setup consisted of a reaction cell attached to a set of differentially pumped electrostatic lenses and a separately pumped analyzer (Phoibos 150 Plus, SPECS GmbH), as described elsewhere.28 Typically, the powder sample (ca. 30 mg) was pressed into a tantalum grid (to minimize potential charging effects) together with a K-type thermocouple, fixed to a sapphire sample holder and mounted inside the XPS reaction cell in front of the first aperture of the differentially pumped electrostatic lens system. The heating of the samples was done from the back using an infrared laser. Before the catalytic experiments, the sample was pretreated in the XPS reaction cell by oxidation (0.5 mbar O2 at 400 °C) until all residual surface carbon and carbonates disappeared. After cooling the sample to RT, the CO + O2 reaction mixture (1:2 ratio at 0.5 mbar) was introduced with the partial pressure of the gases controlled by calibrated mass flow controllers, and the photoemission spectra were recorded. Then, the sample was heated to 100 °C, 150 °C, and 200 °C with a heating rate of 5 °C min–1, and photoemission spectra were again acquired at these temperatures. For CO vs O2 switching experiments, CO (0.15 mbar) was introduced at RT and photoemission spectra were recorded for ∼30 min, then the atmosphere was changed to O2 (0.15 mbar), again collecting spectra for ∼30 min. Then, Co3O4 was heated in O2 to the next temperature and the procedure was repeated. Such experiments were performed at RT, 100, 150, and 200 °C. To ensure surface sensitivity, the Co 2p and C 1s core-level regions were recorded using selected photon energies that resulted in photoelectrons with 200 eV kinetic energy and a ∼0.6 nm inelastic mean free path. The gas phase composition was monitored online by an electron impact quadrupole mass spectrometer, which was connected to the XPS cell via a gas dosing valve.
XPS spectra were analyzed using the CasaXPS package. All binding energies (BE) were calibrated using the second-order O 1s peak. The accuracy of the BE calibration was estimated to be around 0.05 eV. A Shirley-type function was used to remove the background arising from energy loss for Co 2p, and either a Shirley- or a linear-type function was used in the case of C 1s. The extracted spectra were then fitted with a combined Gaussian and Lorentzian line profile. The peak positions of Co 2p and C 1s, full width at half-maximum (fwhm), and constraints are presented in Tables S1 and S2 (Supporting Information, SI).
2.5. Operando FTIR Measurements
Operando FTIR studies were carried out in transmission mode using a Bruker Vertex 70 spectrometer (liquid N2-cooled MCT detector, resolution of 4 cm–1) in a stainless steel transmission flow cell equipped with CaF2 windows. The inlet of the cell was connected to a gas manifold system with calibrated mass flow controllers. The sample (ca. 4–5 mg) was pressed into a pellet, which consisted of a thin catalyst layer supported on a KBr pellet, and placed in a small cylindrical stainless steel sample holder equipped with a ring-shaped furnace and a type-K thermocouple. All infrared spectra were collected in the 4000–900 cm–1 range by averaging 256 scans to achieve good signal-to-noise ratio. A spectrum of the empty sample holder recorded in He was used for background subtraction for all spectra, which were evaluated with the OPUS 4.0 software. During operando FTIR measurements catalytic CO oxidation was simultaneously monitored by gas chromatography (Figure S5).
Before each experiment, the catalyst was pretreated in synthetic air (50 mL min–1) at 400 °C for 30 min (heating rate 10 °C min–1), cooled to 30 °C under a flow of synthetic air, and purged with He for 10 min.
CO Oxidation
the reaction mixtures, (i) 5 vol % CO, 10 vol % O2 in He; (ii) 5 vol % CO, 5 vol % O2 in He; or (iii) 5 vol % CO, 2.5 vol % O2 in He were passed through the cell at 25 mL min–1, and temperature-dependent spectra were recorded while heating to 250 °C with a heating rate of 2 °C min–1.
CO-Temperature-Programmed Reduction Followed by Heating in O2 or He
Five vol % CO in He (25 mL min–1) was continuously introduced to the cell and temperature-dependent spectra were recorded while heating to 250 °C. Then, the sample was cooled to room temperature and temperature-dependent spectra were recorded during heating to 200 °C in 5 vol % O2 in He or in pure He.
CO vs O2 Switching Experiments
CO (5 vol % CO in He) was introduced and five IR spectra were consecutively recorded within 10 min, then the atmosphere was changed to O2 (5 vol % O2 in He) again collecting five IR spectra within 10 min. Then, Co3O4 was heated in O2 to the next temperature and the procedure was repeated. Such experiments were performed at RT, 100 °C, 150 °C, 200 °C, and 250 °C.
CO2 Adsorption
Five vol % CO2 in He (25 mL min–1) was continuously passed through the cell and temperature-dependent spectra were recorded while heating to 200 °C.
3. Results
3.1. Operando XRD under Steady State Conditions: Influence of Pretreatment on CO Oxidation Activity
The CO oxidation activity strongly depended on the CO/O2 ratio (Figure S1; as reported previously), why a 1:2 ratio was used for most experiments. To examine the influence of the (bulk) cobalt oxidation state, the Co3O4 catalyst was pretreated in different ways: by oxidation in synthetic air at 400 °C; or by oxidation at 400 °C followed by reduction in 5 vol % H2 in N2 at 100 °C, 200 °C, 300 °C, and 400 °C. After pretreatment, CO oxidation was carried out in 5 vol % CO and 10 vol % O2 in He. The temperature-dependent CO conversion for differently pretreated Co3O4 is presented in Figure 1a and summarized in Table 1. Co3O4 pretreated in synthetic air or in synthetic air/H2 (100 °C or 200 °C) was active already at ∼80 °C, reaching 100% CO conversion at ∼110–120 °C (apparent activation energy around 70 kJ/mol). In contrast, for preoxidized Co3O4 reduced in H2 at 300 °C or 400 °C, CO conversion started at higher temperature (∼120 °C) and reached 100% at 160 °C and 170 °C, respectively. This is accompanied by an increase in apparent activation energy to around 110 kJ/mol, suggesting a different rate-determining step and different active sites (Figure S2).
Figure 1.

(a) Temperature-dependence of the CO oxidation activity for a reaction mixture of 5 vol % CO, 10 vol % O2, and 85 vol % He (total flow 50 mL min–1) for differently pretreated Co3O4; (b) operando XRD during CO oxidation at 200 °C after oxidation pretreatment of Co3O4 at 400 °C in synthetic air and after reduction of preoxidized Co3O4 in 5 vol % H2 at 400 °C. The XRD patterns before starting the reaction are included for comparison.
Table 1. CO Conversion for a Reaction Mixture of 5 vol.% CO, 10 vol.% O2, and 85 vol.% He (total flow 50 mL min–1, 20 mg Catalyst) for Co3O4 Pretreated in Synthetic Air (400 °C) or in Synthetic Air (400 °C) Followed by Hydrogen at the Indicated Temperatures.
| pretreatment | T10% (°C)a | T50% (°C)b | T90% (°C)c | r90 °C (mol/s·g)d | TOF90 °C (s–1)e |
|---|---|---|---|---|---|
| Syn_air_400 °C | 79 | 104 | 115 | 1.64 × 10–5 | 1.9–3.8 × 10–2 |
| +H2_100 °C | 76 | 101 | 108 | 2.18 × 10–5 | 2.6–5.2 × 10–2 |
| +H2_200°C | 88 | 105 | 114 | 9.37 × 10–6 | 1.1–2.2 × 10–2 |
| +H2_300°C | 132 | 146 | 157 | n.a. | n.a. |
| +H2_400°C | 130 | 145 | 164 | n.a. | n.a. |
Reaction temperature for 10% CO conversion.
Reaction temperature for 50% CO conversion;
Reaction temperature for 90% CO conversion.
Reaction rate of CO oxidation at 90 °C per gram of a catalyst.
Turnover frequency of the Co3+ sites at 90 °C. This estimation is based on the procedure suggested by Xie et al. for nanoparticles of similar mean size and shape, assuming 5–10% of Co3+ in surface defects as active sites.5
Operando XRD upon CO oxidation on Co3O4 pretreated in synthetic air and in synthetic air followed by 5 vol % H2 at 400 °C are shown in Figure 1b for 200 °C, with more details in Figure S2. After standard preoxidation, Co3O4 remained Co3O4 at all reaction temperatures (Figure S2a), in agreement with the in situ XRD study of Jansson et al.14 Although the bulk structure of Co3O4 does not undergo structural changes during CO oxidation, the topmost surface layers could still be affected, as discussed below.
The low catalytic activity after reduction at 300 °C or 400 °C is due to the reduction of Co3O4 to metallic cobalt, as revealed by in situ XRD (Figures 1b and S3) and in situ XAS at the Co K edge reported in our previous study.27 Upon heating in the 1:2 CO/O2 mixture the metallic cobalt is gradually reoxidized and CO conversion increases at 150–200 °C. This is evident from operando XRD (Figure 1b) indicating metallic cobalt, CoO and Co3O4 at 200 °C. At 250 °C the oxidation was still incomplete (Figure S2b). As mentioned, XRD is a bulk characterization technique, and the surface composition of the catalyst might be different, calling for surface sensitive techniques, such as X-ray photoelectron spectroscopy and infrared spectroscopy.
3.2. Operando Spectroscopy under Steady State Conditions
3.2.1. Operando NAP-XPS
To gain spectroscopic insight into CO oxidation, we have utilized operando NAP-XPS, which provides surface-specific information on the cobalt oxidation state and formation of oxygen vacancies (by probing the Co 2p region), as well as on adsorbate species (by probing the C 1s region). Within our previous study of preferential CO oxidation (PROX),27 CO-temperature-programmed reduction (CO-TPR) NAP-XPS indicated that CO reduced the top surface layers of Co3O4, forming CoO, Co, and oxygen vacancies, whereas elementary carbon, carbonates, and CO-Co3+/CO-Co2+ were observed in the C 1s region. Similarly, herein operando NAP-XPS (KE = 200 eV, probing depth ∼0.6 nm) was carried out during CO oxidation (0.15 mbar CO and 0.3 mbar O2), as a function of reaction temperature (Figure 2). The assignment of Co 2p peaks was based on literature:29 the peak at 779.4 eV corresponds to Co3+, the peak at 780.9 eV to Co2+ in CoO, and that at 782.4 eV to Co2+ in Co(OH)2.
Figure 2.

Operando NAP-XPS during CO oxidation on Co3O4 from RT to 200 °C (0.15 mbar CO and 0.3 mbar O2): (a) the Co 2p region (hν = 1015 eV, KE = 200 eV); (b) MS catalytic data recorded during NAP-XPS; (c) the C 1s region (hν = 465 eV, KE = 200 eV); (d) the absolute amount of carbon species; and (e) the relative ratio between carbon species, both calculated from a linear peak fit.
At first, Co 2p XPS did not indicate any surface reduction of Co3O4 in CO+O2, neither at RT nor at higher temperature (i.e., there were no shakeup satellites)29,30 (Figure 2a). To better reveal changes, all spectra were normalized, plotted together (Figure S4a) and a difference spectrum of spectra at 200 °C and RT was calculated (Figure S4b), which point to minor surface reduction at the detection level at 200 °C. Note that (pure) CO induced surface reduction and formation of oxygen vacancies particularly above 100 °C.27 Thus, during CO oxidation (i.e., in the presence of an excess of O2) only minute surface reduction occurred. This indicates a rapid dynamic reduction/reoxidation of the cobalt oxide surface in the CO+O2 mixture under steady state. Catalytic data recorded in parallel to NAP-XPS (Figure 2b) agreed with those from the fixed-bed flow reactor.
The C 1s region revealed three peaks during CO oxidation (Figure 2c), characterizing carbonates (288.2 eV),31−33 elementary carbon (284.7 eV),34 and CO adsorbed to cobalt (i.e., CO-Co3+/CO-Co2+) (286.1 eV).35 The peak positions and fwhm are listed in Table S1. The absolute amount of elementary carbon, carbonates, and weakly adsorbed CO increased from RT to 100 °C, but decreased upon further heating (Figure 2d). The relative amount of C was 32% at RT and 44–47% at 100–200 °C (Figure 2e), assuming identical sensitivity for all species.
In contrast, during CO-TPR (O2 absent) the intensity of carbon species increased with temperature.27 The ratio of elementary carbon to carbonates and adsorbed CO was larger: the fraction of C was 52% at RT and 78% at 200 °C.27 Also, increasing temperature in CO led to Co3O4 reduction to CoOx and formation of oxygen vacancies. Since the C concentration was much higher in pure CO, the oxygen vacancies are likely the reaction sites of CO dissociation (cooperating with neighboring Co cations). In the presence of oxygen, vacancies are refilled and less carbon is produced. Thus, O2 of the reaction mixture may hinder the growth of C by keeping the surface oxidized and thus preventing CO dissociation, or O2 just reoxidizes C deposited by CO dissociation.
3.2.2. Operando FTIR Spectroscopy
Operando FTIR spectroscopy enables studying adsorbed species, thus contributing in identifying reaction pathways. A number of IR studies have reported carbonates,4,13,14,32 but their exact role, whether being reaction intermediates, spectators, or poisons remained unclear. Carbonates have also frequently been observed for CO-containing reactions on oxide supported metals.36,37 Systematic operando FTIR studies were performed to unravel their role, applying three feed compositions (Figure 3): CO/O2 = 1:2; 1:1; 2:1.
Figure 3.
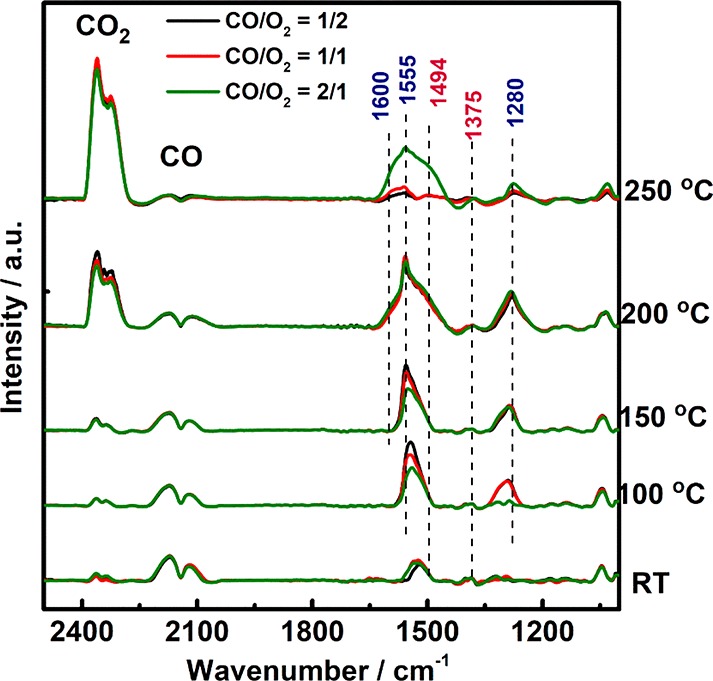
Operando FTIR spectra recorded during CO oxidation on Co3O4 in flow mode (25 mL min–1) from RT to 250 °C in different gas mixtures: 50 mbar CO, 100 mbar O2 (black); 50 mbar CO, 50 mbar O2 (red); and 50 mbar CO, 25 mbar O2 (green).
All operando FTIR spectra exhibited the characteristic gas-phase bands of CO (2110 and 2170 cm–1) as well as bands in the 1000–1650 cm–1 region, attributed to various carbonate vibrations. In contrast to NAP-XPS, operando FTIR did not show any bands of (linearly adsorbed) CO on Co2+ or Co3+. The CO stretching frequencies of cobalt carbonyls were reported at 2023–2025 cm–1 for metallic cobalt, 2070–2110 cm–1 for Co+, 2120–2170 cm–1 for Co2+, and 2178–2180 cm–1 for Co3+.14 Thus, the 2178–2180 cm–1 on Co3+ band could be masked by gas-phase CO.
In the FTIR cell, carbonate formation started at RT whereas CO2 production began around 150 °C, increasing with temperature. The assignment of surface carbonates is based on literature38,39 and IR stretching vibrations are presented in Table S3. The carbonates at RT were mainly monodentates (1494, 1375, 1320 cm–1). Upon temperature increase, bidentates evolved (additional bands at 1620, 1555, and 1280 cm–1). It is important to note that all FTIR spectra were recorded on the same pellet of Co3O4, allowing to quantitatively compare intensities and the amount of adsorbed species.
FTIR spectra of the three gas mixtures mainly differed in the stability of the carbonates. Under O2-rich conditions (CO/O2 = 1 to 2) the signal intensity increased from RT to 170 °C, between 170 °C and 200 °C additional bidentate carbonates were formed, but at 220 °C carbonates started to disappear and no carbonates were present above 220 °C (Figure 3). The appearance of bidentate carbonates was accompanied by an increase in CO conversion (CO2 gas-phase peak; Figure 3). The disappearance of carbonate peaks was accompanied by a further increase of the CO2 gas-phase peak and an overall increase of CO conversion. The decreasing carbonate intensity above 220 °C is likely due to lower stability of carbonates in O2 excess.
In the reaction mixture with an initial CO/O2 ratio of 1:1, a relatively high amount of carbonates was still present at 220 °C and higher temperatures were required to make them disappear. When the CO to O2 ratio was stoichiometric (CO/O2 = 2:1), the amount of carbonates was even higher and the concentration of carbonates increased continuously up to 250 °C.
In summary, the more reducing (CO rich) the reaction atmosphere was, the higher the amount and stability of surface carbonates became. This suggests that upon temperature increase more sites were formed (likely by reduction of the Co3O4 surface), that were active for CO adsorption and carbonate formation. Whether excess O2 reduces the amount of carbonates formed or just facilitates carbonate decomposition/desorption to CO2 cannot be answered at this point. On the basis of static IR experiments, the potential contribution of carbonates to the overall activity cannot be assessed. Thus, gas switching experiments were performed both for NAP-XPS and FTIR.
3.3. Operando Spectroscopy upon Switching between CO and O2
3.3.1. NAP-XPS
To lift the limitations of steady state spectroscopy, switching experiments were monitored by NAP-XPS (Figures 4, 5, S6, S7, and S8). CO/O2 switching (each 0.15 mbar for ∼50 min) was carried out at RT, 100 °C, 150 °C, and 200 °C, simultaneously recording Co 2p and C 1s spectra (KE = 200 eV; Figures 4, 5, and S7) and MS of reactants and CO2 (Figure S6). Taking into account the attenuation of photoelectrons in the gas phase, a CO/O2 ratio of 1 was used (0.15 mbar CO vs 0.15 mbar O2; similar scattering was confirmed by comparing Co 2p peak intensities).
Figure 4.

Operando NAP-XPS on Co3O4 during CO/O2 switching (0.15 mbar CO vs 0.15 mbar O2), Co 2p3/2 region (hν = 1015 eV; KE = 200 eV): comparison in CO and O2 at (a) RT and (b) 200 °C; the Co 2p3/2 region at various temperatures in (c) CO and (d) O2; (e) difference spectrum (Co 2p3/2 200 °C in CO-Co 2p3/2 RT in O2).
Figure 5.

Operando NAP-XPS C 1s region (hν = 465 eV; KE = 200 eV) on Co3O4 during CO/O2 switching (0.15 mbar CO vs 0.15 mbar O2). The absolute amount of carbon species was calculated from a linear peak fit.
Figure S6b demonstrates that changing the atmosphere from O2 to CO produced CO2 already at RT, but increased with temperature. Note that before introducing the reacting gas (e.g., CO) into the NAP-XPS chamber, the other reacting gas (e.g., O2) was evacuated from the chamber, in order to keep the Co3O4 catalyst free from physisorbed molecules.
A significant increase in CO2 evolution at 200 °C and continuous CO2 production for ∼30 min upon switching to CO indicates that not only surface lattice oxygen may take part in CO2 production, but also oxygen from the bulk. The shape of the MS curve, a sharp peak followed by a slow decrease, points to fast reaction with surface oxygen (decaying fast upon consumption) and a slower reaction with bulk oxygen, which needs to diffuse to the surface. If only surface oxygen would react, then CO2 production should have dropped quickly. A gradual (slow) decrease in CO2 evolution is observed at 200 °C, which may be caused by catalyst deactivation by carbon (C 1s region, Figure 5) or by slow diffusion of oxygen from deeper (bulk) layers.
CO disproportionation to C and CO2 may also contribute to the slower pathway. The amount of CO2 produced at 200 °C during steady state CO oxidation (0.15 mbar CO and 0.3 mbar O2; Figure 2b) was 1.34 × 10–10 a.u. The amount of CO2 produced at 200 °C in the presence of pure CO (0.15 mbar CO; Figure S6b) was 6.10 × 10–12 a.u. (after ∼5 min) and 3.61 × 10–12 a.u. (after ∼30 min) that is 22–37 times lower than the catalytic activity in CO + O2.
In the opposite case, when O2 was introduced after CO, there was only minor CO2 production, likely due to surface carbonate decomposition (and then O2 reoxidized the surface). The MS data were confirmed by analogous experiments in the catalytic flow reactor.
Figures 4 and 5 display the corresponding Co 2p3/2 and C 1s spectra. Upon switching to CO, the Co3O4 surface oxidation state did not change at RT (no satellites appeared in Co 2p), whereas more pronounced changes of Co3+/Co2 were observed at higher temperature, especially at 200 °C (Figures 4a,b, and S7). In CO at 200 °C, the Co3+ concentration decreased by 15%, as compared to O2. The amount of reduced sites upon switching was quantified via the spectral intensities (i.e., the ratio between Co3O4 and CoO). This indicates that CO reduces part of the surface, while O2 reoxidizes it, as expected for the Mars-van-Krevelen mechanism (which has been proven by numerous experimental and theoretical studies.5,12,13,18−21) Figure 4d suggests that reoxidation (0.15 mbar O2) was not complete though, but a tiny amount of reduced Co2+ species persisted.
With respect to the C 1s region, elementary carbon increased with temperature in CO, whereas switching to O2 strongly decreased the (amorphous) carbon (interestingly, O2 partially oxidizes carbon even at RT) (Figures 5 and S8). Thus, it is possible that a (minor) reaction pathway toward CO2 proceeds via CO dissociation followed by carbon oxidation to CO/CO2. In contrast, the carbonates decreased to a considerably lower extent at higher temperature, indicating that carbonate decomposition was more difficult than the reoxidation of carbon.
Carbonate formation has been previously reported for Co3O4,4,13,14 but the observed carbon deposition has been scarcely discussed in literature (indirect investigations by Jansson12,13 in the course of isotope studies). The current C 1s NAP-XPS data directly indicate carbon formation on Co3O4 during CO exposure and CO oxidation.
The simultaneous NAP-XPS/MS observations are in line with the redox Mars-van-Krevelen mechanism, but also point to a possible contribution of CO dissociation, elementary carbon deposition, and carbon reoxidation, the extent of which is however unknown. The role of the various carbonates remains ambiguous, why “switching” operando FTIR spectroscopy was employed.
3.3.2. FTIR Spectroscopy
Figure 6 shows operando FTIR spectra upon CO/O2 switching (50 mbar CO vs 50 mbar O2) on Co3O4. CO was introduced first and consecutive IR spectra were acquired for 10 min (each spectrum taking about 2 min), then the atmosphere was changed to O2 and IR spectra were acquired after 2 and 10 min. Then, Co3O4 was heated in O2 to the next temperature before introducing CO again (performed at RT, 100 °C, 150 °C, 200 °C, and 250 °C). FTIR shows that neither at RT nor at 100 °C carbonate decomposition by O2 took place, not even during 10 min O2 exposure. Mainly monodentate carbonates, apparently being quite stable, were present. At 150 °C, the carbonate peak area decreased during the first 2 min, and higher temperature decreased both mono- and bidentate carbonates. At 200 °C and 250 °C, the carbonates strongly decreased after 2 min and after 10 min no carbonates were present anymore (Figure 6). Overall, the surface carbonates seemed rather stable spectators, with their intensity decreasing rather slowly and mostly at high temperature, which rather excludes carbonates as reaction intermediates of fast CO oxidation. Thus, carbonate formation/decomposition plays rather a minor role beside fast CO oxidation (MvK). Deactivation/site blocking by carbonates can also rather be excluded.
Figure 6.
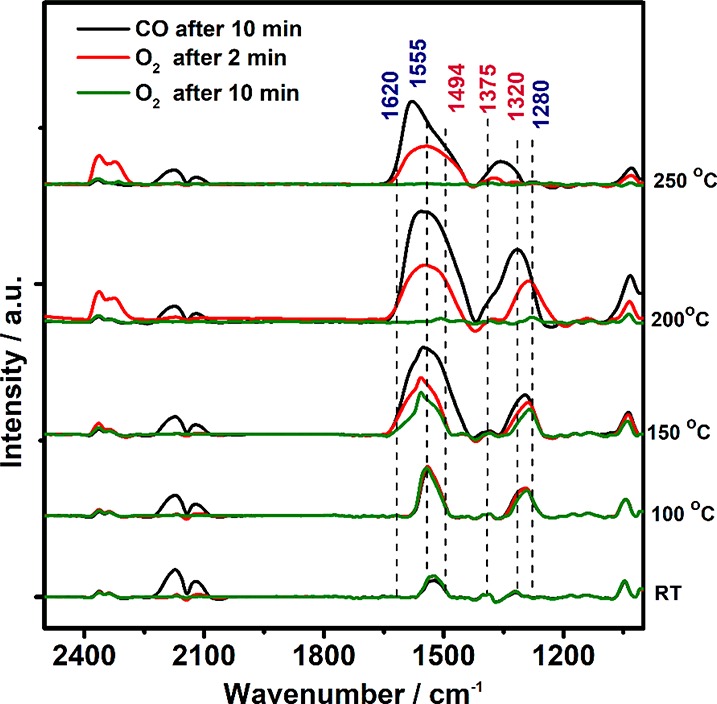
Operando FTIR on Co3O4 upon CO/O2 switching experiments (50 mbar CO vs 50 mbar O2): spectra recorded during the 10th minute of CO exposure (black), during the 2nd minute of O2 exposure (red) and during the 10th minute of O2 exposure (green).
3.4. Carbonate Formation and Stability: FTIR Spectroscopy
3.4.1. Interaction of Co3O4 with Carbon Monoxide
To further examine carbonate formation, reactivity and stability, the interaction of CO with Co3O4 was studied by FTIR from RT to 350 °C (Figure 7a). Similar to CO+O2, no adsorbed CO but only carbonates were observed. Mainly monodentate carbonates were formed at RT, bidentate carbonates emerged at 100 °C, and the overall carbonate intensity increased with temperature. This suggests that at higher temperature more reactive sites are formed that enable CO adsorption as a carbonate. Recall that NAP-XPS revealed partial reduction of the Co3O4 surface and surface oxygen vacancy formation in CO, especially from 100 °C to 200 °C.27 When more vacancies were created, CO could adsorb as carbonate. Accordingly, heating in CO favors lattice oxygen extraction, increasing the concentration of carbonates. Note that in CO the amount of carbonates was significantly higher than that during CO oxidation (even in a stoichiometric mixture at low temperature) (Figure S9). The higher concentration of carbonates in CO as compared to O2-containing feed suggests that partial Co3O4 surface reduction is beneficial for carbonate formation. This will be discussed in the following.
Figure 7.

FTIR spectra on Co3O4 recorded during: (a) adsorption of CO (50 mbar) in flow mode (total flow 25 mL min–1) from RT to 350 °C; (b) pretreatment with CO at 200 °C, cooling in CO to RT and heating in O2 (100 mbar O2 bar in He, total flow 25 mL min–1); and (c) pretreatment with CO at 200 °C, cooling in CO to RT and heating in He (total flow 25 mL min–1).
3.4.2. Investigation of Carbonate Stability in O2 and He Atmosphere
The thermal stability of carbonates was examined in O2 and He atmospheres. FTIR spectra were recorded after exposing Co3O4 to CO at 200 °C and cooling in CO to RT (i.e., initial spectrum) and during heating in 50 mbar O2 in He or in pure He. As evident from Figure 7b, introducing O2 at RT already changed the carbonates, forming additional monodentate carbonates appearing as a shoulder at ∼1530–1470 cm–1. Heating in O2 to 100 °C decomposed both mono- and bidentate carbonates. At 130 °C, the intensity of carbonate bands decreased to 1/3 of its initial value. At 150 °C, only a small amounts of monodentate carbonates were left on the cobalt oxide surface, while at 200 °C it was free of carbonates.
The thermal stability of carbonates was also studied in He (Figure 7c). Again, FTIR spectra were recorded after exposing Co3O4 to CO at 200 °C, cooling down in CO to RT and heating in He. Up to 100 °C, no significant carbonate decomposition was observed. At 130 °C, carbonates started to decompose. However, the decrease was faster for the 1380–1280 cm–1 peaks than for the 1600–1430 cm–1 peaks, indicating different thermal stability and reactivity of surface carbonates. Importantly, at 150 °C carbonates were still detected in He, whereas in O2 only a small amount was present. The observation of carbonate peaks disappearing at about 200 °C is in agreement with CO-TPD reported in our previous study (CO exposure at RT for 30 min followed by heating in He to 700 °C).27 Only CO2 evolved (and no CO) suggesting lattice oxygen extraction from cobalt oxide.
3.4.3. Interaction of Co3O4 with Carbon Dioxide
The results presented so far (CO vs CO/O2) suggest that carbonate formation requires, or is at least facilitated, by partial surface reduction by CO. However, it is still unclear whether carbonates may also form by readsorption of the reaction product CO2. Thus, preoxidized Co3O4 was exposed to CO2 at RT and heated to 200 °C (Figure S10a). Interestingly, neither at RT nor during heating any CO2 adsorption/carbonate was observed; CO2 remained in the gas phase (2336 and 2360 cm–1). This is in contrast to CO2 adsorption on many other oxides such as ZrO238,40 or Al2O341 when CO2 easily forms carbonates. The different affinity to CO2 may be due to specific active sites or functional groups (i.e., OH, basic surface oxygen) that are missing on Co3O4.
In another series of experiments, the interaction of CO2 with Co3O4 prereduced by CO or H2 (intended to create oxygen vacancies that seem required for CO2 adsorption) was examined. (Figures S10b, S11, and S12). However, there was no CO2 adsorption or carbonate formation under all conditions. Thus, it can be ruled out that the observed carbonates originate from CO2 readsorption. Rather, they are only formed by direct interaction of Co3O4 with CO that partly reduces the Co3O4 surface. The higher concentration of surface carbonates in CO as compared to O2-containing feed also suggests this picture.
3.5. Discussion
Using operando NAP-XPS, FTIR, and XRD we have examined the (surface) oxidation state of Co3O4 and adsorbates present during CO oxidation, and compared them with those in pure CO and upon switching between CO and O2. In particular, the comparison of static (steady-state) and switching experiments provided valuable information.
Combining information from NAP-XPS/FTIR and activity tests revealed a complex network of different reaction pathways contributing to CO oxidation on Co3O4. In the following Osurf, O#, and Oads refer to surface (lattice) oxygen, oxygen (surface) vacancies, and adsorbed atomic oxygen. Some pathways are reversible, others have been shown to be irreversible.
-
(1)
Direct MvK pathway:
-
(2)
via CO dissociation and carbon oxidation:
-
(3)
via carbonates as intermediates:
Operando NAP-XPS and FTIR spectroscopy—in particular based on switching experiments—were in accordance with a fast CO oxidation route via a Mars-van-Krevelen mechanism (1), i.e., alternating reduction–oxidation of the Co3O4 surface by CO and O2. Carbon monoxide adsorbed to cobalt cations reacts with lattice surface oxygen, and the created oxygen vacancies (O#) are replenished by O2 from the gas phase, further creating active Oads species.
In addition, adsorbed CO (likely near O#) may undergo dissociation, filling an oxygen vacancy and depositing elementary carbon on the surface, which can be reoxidized by O2 (once more near O#) (2). CO dissociation likely occurs at/near oxygen vacancies, as indicated by the larger amount of carbon present under more reducing conditions when more oxygen vacancies are formed. The carbon is quite reactive and is oxidized even at RT. Overall, CO dissociation and carbon reoxidation seems an (unavoidable) additional pathway of CO oxidation on Co3O4.
The pronounced decline of surface carbonates appearing at higher temperature (at 200–250 °C the surface of Co3O4 is almost free of carbonates) suggests that the stable carbonates play a minor role, being rather spectators (3). Decomposition of carbonates to CO2 is, however, facilitated by (excess) O2.
Below 100–150 °C, oxygen vacancies cannot be detected but Co3O4 exhibits considerable activity. A contribution via a Langmuir–Hinshelwood (LH) reaction can thus not be excluded. For methane oxidation on cobalt oxide spinel nanocubes Zasada et al.42 reported that at 300–450 °C the LH mechanism was dominant, whereas above 450 °C, LH and MvK coexisted.
The spectroscopically examined possible reaction pathways/elementary steps of CO oxidation on Co3O4 are summarized in Figure 8. Clearly, surface reduction by CO, reoxidation by O2, CO dissociation/reoxidation presumably at vacancies, and (to a lesser extent) carbonate formation/decomposition are key processes, demonstrating the complexity of the seemingly simple CO oxidation.
Figure 8.

Schematic representation of CO oxidation on Co3O4.
4. Conclusions
Combining surface-specific chemical information from operando NAP-XPS and FTIR spectroscopy with activity tests revealed a complex network of four different reaction pathways of CO oxidation on Co3O4: redox Mars-van-Krevelen, CO dissociation followed by carbon oxidation, formation of carbonate spectators, and possibly Langmuir–Hinshelwood at low temperature (which can currently not be excluded). Experiments performed under steady-state and dynamic conditions were in accordance with a MvK mechanism above 100 °C, involving the Co3+/Co2+ redox couple and oxygen vacancy formation. Under steady state, the Co3O4 surface appeared oxidized and only in pure CO about 15% Co2+ species at/near the surface were detected, suggesting the active sites being minority species. CO dissociation followed by carbon reoxidation and carbonate formation/decomposition are additional reaction pathways. Nevertheless, stable carbonates are rather spectators and also CO dissociation may be a minor route.
This work demonstrates the benefits of combining several operando techniques for studying catalysts under steady state and dynamic conditions, revealing a complex network of reaction pathways. Nevertheless, to assess the relative contributions of all pathways and to determine the rate limiting step, further studies would be required (e.g., by transient/concentration modulation IR spectroscopy43 and detailed (micro)kinetic modeling).23−25
Acknowledgments
We acknowledge the Helmholtz-Zentrum Berlin for synchrotron radiation beamtime at ISISS beamline of BESSY II. The research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 312284. We are grateful to Dr. Klaudia Hradil and DI Werner Artner for assistance with the in situ XRD measurements. L.L. is grateful to Dr. Leon van de Water from Johnson Matthey for useful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b01237.
CO conversion at different CO/O2 ratios, in situ XRD during CO oxidation, temperature-programmed reduction in H2 followed by XRD, NAP-XPS during CO oxidation at different (additional) temperatures, MS data and Co 2p XP spectra obtained in the switching experiments, supplementary FTIR spectra of CO adsorption and CO oxidation, FTIR spectra of CO2 adsorption, C 1s spectral fitting parameters, spectral ranges, and assignment of surface carbonates (PDF)
Author Present Address
⊥ Johnson Matthey, PO Box 1, Belasis Avenue, Billingham, Cleveland, TS23 1LB, U.K.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the Austrian Science Fund (FWF) in the framework of the Doctoral School “Building Solids for Function (“Solids4Fun”) [project W1243] and the ComCat Project [I 1041-N28].
The authors declare no competing financial interest.
Supplementary Material
References
- Ertl G. Reactions at Surfaces: From Atoms to Complexity (Nobel Lecture). Angew. Chem., Int. Ed. 2008, 47, 3524–3535. 10.1002/anie.200800480. [DOI] [PubMed] [Google Scholar]
- Zorn K.; Giorgio S.; Halwax E.; Henry C. R.; Gronbeck H.; Rupprechter G. CO Oxidation on Technological Pd-Al2O3 Catalysts: Oxidation State and Activity. J. Phys. Chem. C 2011, 115, 1103–1111. 10.1021/jp106235x. [DOI] [Google Scholar]
- Royer S.; Duprez D. Catalytic Oxidation of Carbon Monoxide over Transition Metal Oxides. ChemCatChem 2011, 3, 24–65. 10.1002/cctc.201000378. [DOI] [Google Scholar]
- Jia C.-J.; Schwickardi M.; Weidenthaler C.; Schmidt W.; Korhonen S.; Weckhuysen B. M.; Schüth F. Co3O4–SiO2 Nanocomposite: A Very Active Catalyst for CO Oxidation with Unusual Catalytic Behavior. J. Am. Chem. Soc. 2011, 133, 11279–11288. 10.1021/ja2028926. [DOI] [PubMed] [Google Scholar]
- Xie X.; Li Y.; Liu Z.-Q.; Haruta M.; Shen W. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 2009, 458, 746–749. 10.1038/nature07877. [DOI] [PubMed] [Google Scholar]
- Teng Y.; Kusano Y.; Azuma M.; Haruta M.; Shimakawa Y. Morphology effects of Co3O4 nanocrystals catalyzing CO oxidation in a dry reactant gas stream. Catal. Sci. Technol. 2011, 1, 920–922. 10.1039/c1cy00113b. [DOI] [Google Scholar]
- Gu D.; Jia C.-J.; Weidenthaler C.; Bongard H.-J.; Spliethoff B.; Schmidt W.; Schüth F. Highly Ordered Mesoporous Cobalt-Containing Oxides: Structure, Catalytic Properties, and Active Sites in Oxidation of Carbon Monoxide. J. Am. Chem. Soc. 2015, 137, 11407–11418. 10.1021/jacs.5b06336. [DOI] [PubMed] [Google Scholar]
- Iablokov V.; Barbosa R.; Pollefeyt G.; Van Driessche I.; Chenakin S.; Kruse N. Catalytic CO Oxidation over Well-Defined Cobalt Oxide Nanoparticles: Size-Reactivity Correlation. ACS Catal. 2015, 5, 5714–5718. 10.1021/acscatal.5b01452. [DOI] [Google Scholar]
- Ding K.; Wang D.; Yang P.; Hou P.; Cheng X. Enhanced CO catalytic oxidation of flower-like Co3O4 composed of small nanoparticles. RSC Adv. 2016, 6, 16208–16214. 10.1039/C6RA01092J. [DOI] [Google Scholar]
- Yu Y.; Takei T.; Ohashi H.; He H.; Zhang X.; Haruta M. Pretreatments of Co3O4 at moderate temperature for CO oxidation at – 80 °C. J. Catal. 2009, 267, 121–128. 10.1016/j.jcat.2009.08.003. [DOI] [Google Scholar]
- Pollard M. J.; Weinstock B. A.; Bitterwolf T. E.; Griffiths P. R.; Piers Newbery A.; Paine Iii J. B. A mechanistic study of the low-temperature conversion of carbon monoxide to carbon dioxide over a cobalt oxide catalyst. J. Catal. 2008, 254, 218–225. 10.1016/j.jcat.2008.01.001. [DOI] [Google Scholar]
- Jansson J. Low-Temperature CO Oxidation over Co3O4/Al2O3. J. Catal. 2000, 194, 55–60. 10.1006/jcat.2000.2924. [DOI] [Google Scholar]
- Jansson J.; Skoglundh M.; Fridell E.; Thormählen P. A Mechanistic Study of Low Temperature CO Oxidation over Cobalt Oxide. Top. Catal. 2001, 16, 385–389. 10.1023/A:1016681620216. [DOI] [Google Scholar]
- Jansson J.; Palmqvist A. E. C.; Fridell E.; Skoglundh M.; Österlund L.; Thormählen P.; Langer V. On the Catalytic Activity of Co3O4 in Low-Temperature CO Oxidation. J. Catal. 2002, 211, 387–397. 10.1006/jcat.2002.3738. [DOI] [Google Scholar]
- Perti D.; Kabel R. L. Kinetics of CO oxidation over Co3O4/γ-Al2O3. Part I: Steady state. AIChE J. 1985, 31, 1420–1426. 10.1002/aic.690310903. [DOI] [Google Scholar]
- Perti D.; Kabel R. L. Kinetics of CO oxidation over Co3O4/γ-Al2O3. Part II: Reactor dynamics. AIChE J. 1985, 31, 1427–1434. 10.1002/aic.690310904. [DOI] [Google Scholar]
- Perti D.; Kabel R. L.; McCarthy G. J. Kinetics of CO oxidation over Co3O4/γ-Al2O3. AIChE J. 1985, 31, 1435–1440. 10.1002/aic.690310905. [DOI] [Google Scholar]
- Broqvist P.; Panas I.; Persson H. A DFT Study on CO Oxidation over Co3O4. J. Catal. 2002, 210, 198–206. 10.1006/jcat.2002.3678. [DOI] [Google Scholar]
- Wang H.-F.; Kavanagh R.; Guo Y.-L.; Guo Y.; Lu G.; Hu P. Origin of extraordinarily high catalytic activity of Co3O4 and its morphological chemistry for CO oxidation at low temperature. J. Catal. 2012, 296, 110–119. 10.1016/j.jcat.2012.09.005. [DOI] [Google Scholar]
- Pang X.-Y.; Liu C.; Li D.-C.; Lv C.-Q.; Wang G.-C. Structure Sensitivity of CO Oxidation on Co3O4: A DFT Study. ChemPhysChem 2013, 14, 204–212. 10.1002/cphc.201200807. [DOI] [PubMed] [Google Scholar]
- Jiang D.-e.; Dai S. The role of low-coordinate oxygen on Co3O4(110) in catalyticCO oxidation. Phys. Chem. Chem. Phys. 2011, 13, 978–984. 10.1039/C0CP01138J. [DOI] [PubMed] [Google Scholar]
- Luo J.-Y.; Meng M.; Li X.; Li X.-G.; Zha Y.-Q.; Hu T.-D.; Xie Y.-N.; Zhang J. Mesoporous Co3O4–CeO2 and Pd/Co3O4–CeO2 catalysts: Synthesis, characterization and mechanistic study of their catalytic properties for low-temperature CO oxidation. J. Catal. 2008, 254, 310–324. 10.1016/j.jcat.2008.01.007. [DOI] [Google Scholar]
- Vogel D.; Spiel C.; Suchorski Y.; Trinchero A.; Schlögl R.; Grönbeck H.; Rupprechter G. Local Catalytic Ignition during CO Oxidation on Low-Index Pt and Pd Surfaces: A Combined PEEM, MS, and DFT Study. Angew. Chem., Int. Ed. 2012, 51, 10041–10044. 10.1002/anie.201204031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markova V. K.; Philbin J. P.; Zhao W.; Genest A.; Silvestre-Albero J.; Rupprechter G.; Rösch N. Catalytic Transformations of 1-Butene over Palladium. A Combined Experimental and Theoretical Study. ACS Catal. 2018, 8, 5675–5685. 10.1021/acscatal.8b01013. [DOI] [Google Scholar]
- Suchorski Y.; Datler M.; Bespalov I.; Zeininger J.; Stöger-Pollach M.; Bernardi J.; Grönbeck H.; Rupprechter G. Visualizing catalyst heterogeneity by a multifrequential oscillating reaction. Nat. Commun. 2018, 9, 600. 10.1038/s41467-018-03007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafeiratos S.; Dintzer T.; Teschner D.; Blume R.; Hävecker M.; Knop-Gericke A.; Schlögl R. Methanol oxidation over model cobalt catalysts: Influence of the cobalt oxidation state on the reactivity. J. Catal. 2010, 269, 309–317. 10.1016/j.jcat.2009.11.013. [DOI] [Google Scholar]
- Lukashuk L.; Föttinger K.; Kolar E.; Rameshan C.; Teschner D.; Hävecker M.; Knop-Gericke A.; Yigit N.; Li H.; McDermott E.; Stöger-Pollach M.; Rupprechter G. Operando XAS and NAP-XPS studies of preferential CO oxidation on Co3O4 and CeO2-Co3O4 catalysts. J. Catal. 2016, 344, 1–15. 10.1016/j.jcat.2016.09.002. [DOI] [Google Scholar]
- Knop-Gericke A.; Kleimenov E.; Hävecker M.; Blume R.; Teschner D.; Zafeiratos S.; Schlögl R.; Bukhtiyarov V. I.; Kaichev V. V.; Prosvirin I. P.; Nizovskii A. I.; Bluhm H.; Barinov A.; Dudin P.; Kiskinova M.. X-Ray Photoelectron Spectroscopy for Investigation of Heterogeneous Catalytic Processes. In Advances in Catalysis; Academic Press, 2009; Vol. 52; pp 213–272. [Google Scholar]
- Biesinger M. C.; Payne B. P.; Grosvenor A. P.; Lau L. W. M.; Gerson A. R.; Smart R. S. C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. 10.1016/j.apsusc.2010.10.051. [DOI] [Google Scholar]
- Grosvenor A. P.; Wik S. D.; Cavell R. G.; Mar A. Examination of the Bonding in Binary Transition-Metal Monophosphides MP (M = Cr, Mn, Fe, Co) by X-Ray Photoelectron Spectroscopy. Inorg. Chem. 2005, 44, 8988–8998. 10.1021/ic051004d. [DOI] [PubMed] [Google Scholar]
- Feng Z. A.; Machala M. L.; Chueh W. C. Surface electrochemistry of CO2 reduction and CO oxidation on Sm-doped CeO2-x: coupling between Ce3+ and carbonate adsorbates. Phys. Chem. Chem. Phys. 2015, 17, 12273–12281. 10.1039/C5CP00114E. [DOI] [PubMed] [Google Scholar]
- Ferstl P.; Mehl S.; Arman M. A.; Schuler M.; Toghan A.; Laszlo B.; Lykhach Y.; Brummel O.; Lundgren E.; Knudsen J.; Hammer L.; Schneider M. A.; Libuda J. Adsorption and Activation of CO on Co3O4(111) Thin Films. J. Phys. Chem. C 2015, 119, 16688–16699. 10.1021/acs.jpcc.5b04145. [DOI] [Google Scholar]
- Shchukarev A. V.; Korolkov D. V. XPS Study of group IA carbonates. cent.eur.j.chem. 2004, 2, 347–362. 10.2478/BF02475578. [DOI] [Google Scholar]
- Wolfbeisser A.; Klotzer B.; Mayr L.; Rameshan R.; Zemlyanov D.; Bernardi J.; Fottinger K.; Rupprechter G. Surface modification processes during methane decomposition on Cu-promoted Ni-ZrO2 catalysts. Catal. Sci. Technol. 2015, 5, 967–978. 10.1039/C4CY00988F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsvik T.; Borg A.; Kildemo M.; Raaen S.; Matsuura A.; Jaworowski A. J.; Worren T.; Leandersson M. Molecular vibrations in core-ionised CO adsorbed on Co(0 0 0 1) and Rh(1 0 0). Surf. Sci. 2001, 492, 152–160. 10.1016/S0039-6028(01)01446-7. [DOI] [Google Scholar]
- Diemant T.; Bansmann J.; Behm R. J. CO oxidation on planar Au/TiO2 model catalysts: Deactivation and the influence of water. Vacuum 2009, 84, 193–196. 10.1016/j.vacuum.2009.04.004. [DOI] [Google Scholar]
- Denkwitz Y.; Zhao Z.; Hörmann U.; Kaiser U.; Plzak V.; Behm R. J. Stability and deactivation of unconditioned Au/TiO2 catalysts during CO oxidation in a near-stoichiometric and O2-rich reaction atmosphere. J. Catal. 2007, 251, 363–373. 10.1016/j.jcat.2007.07.029. [DOI] [Google Scholar]
- Köck E.-M.; Kogler M.; Bielz T.; Klötzer B.; Penner S. In Situ FT-IR Spectroscopic Study of CO2 and CO Adsorption on Y2O3, ZrO2, and Yttria-Stabilized ZrO2. J. Phys. Chem. C 2013, 117, 17666–17673. 10.1021/jp405625x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov A.: Molecular Spectroscopy of Oxide Catalyst Surfaces; John Wiley & Sons Ltd: The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, England, 2003; pp 95–136. [Google Scholar]
- Pokrovski K.; Jung K. T.; Bell A. T. Investigation of CO and CO2 Adsorption on Tetragonal and Monoclinic Zirconia. Langmuir 2001, 17, 4297–4303. 10.1021/la001723z. [DOI] [Google Scholar]
- Fottinger K.; Schlogl R.; Rupprechter G. The mechanism of carbonate formation on Pd-Al2O3 catalysts. Chem. Commun. 2008, 320–322. 10.1039/B713161E. [DOI] [PubMed] [Google Scholar]
- Zasada F.; Janas J.; Piskorz W.; Gorczyńska M.; Sojka Z. Total Oxidation of Lean Methane over Cobalt Spinel Nanocubes Controlled by the Self-Adjusted Redox State of the Catalyst: Experimental and Theoretical Account for Interplay between the Langmuir–Hinshelwood and Mars-Van-Krevelen Mechanisms. ACS Catal. 2017, 7, 2853–2867. 10.1021/acscatal.6b03139. [DOI] [Google Scholar]
- Haghofer A.; Ferri D.; Föttinger K.; Rupprechter G. Who Is Doing the Job? Unraveling the Role of Ga2O3 in Methanol Steam Reforming on Pd2Ga/Ga2O3. ACS Catal. 2012, 2, 2305–2315. 10.1021/cs300480c. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.