

Abstract
Modifying Pt catalysts using hydrophobic ionic liquids (ILs) has been demonstrated to be a facile approach for boosting the performance of Pt catalysts for the oxygen reduction reaction (ORR). This work aims to deepen the understanding and initiate a rational molecular tuning of ILs for improved activity and stability. To this end, Pt/C catalysts were modified using a variety of 1-methyl-3-alkylimidazolium bis(trifluoromethanesulfonyl)imide ([CnC1im][NTf2], n = 2–10) ILs with varying alkyl chain lengths in imidazolium cations, and the electrocatalytic properties (e.g., electrochemically active surface area, catalytic activity, and stability) of the resultant catalysts were systematically investigated. We found that ILs with long cationic chains (C6, C10) efficiently suppressed the formation of nonreactive oxygenated species on Pt; however, at the same time they blocked active Pt sites and led to a lower electrochemically active surface area. It is also disclosed that the catalytic activity strongly correlates with the alkyl chain length of cations, and a distinct dependence of intrinsic activity on the alkyl chain length was identified, with the maximum activity obtained on Pt/C-[C4C1im][NTf2]. The optimum arises from the counterbalance between more efficient suppression of oxygenated species formation on Pt surfaces and more severe passivation of Pt surfaces with elongation of the alkyl chain length in imidazolium cations. Moreover, the presence of an IL can also improve the electrochemical stability of Pt catalysts by suppressing the Pt dissolution, as revealed by combined identical-location transmission electron microscopy (TEM) and in situ inductively coupled plasma mass spectrometry (ICP-MS) analyses.
Keywords: fuel cell, oxygen reduction, Pt catalyst, ionic liquid, electrocatalysis, Pt dissolution
1. Introduction
Low-temperature fuel cells (LTFCs) featuring high efficiency and low emission to the environment are expected to play an important role in the clean energy scenario of the future.1−3 However, the slow kinetics of the oxygen reduction reaction (ORR) at the cathode poses major barriers to broad-based applications of LTFC technology, particularly in the automotive industry.4,5 Although carbon-supported Pt nanoparticles set the benchmark for the most efficient ORR catalysts, an undesirable overpotential (300–400 mV) is still needed to accelerate the ORR process to proceed at a significant rate.6−8 Researchers have struggled for decades to improve the catalytic performance of cathode catalysts toward the ORR, either by engineering the structure of Pt catalysts through tuning their particle shapes, sizes, and/or surface compositions or by developing alternative non-precious-metal catalysts (NPMCs), which mainly include transition-metal oxides9−11 and carbon-based materials (e.g., pyrolyzed Fe-N-C, metal-free functionalized carbons, heteroatom-doped carbons, and metal–organic framework (MOF)/zeolitic imidazolate framework (ZIF)-derived carbon).12−15 Despite the great achievement in developing highly active NPMCs whose activity is comparable to or even surpasses that of the state of the art Pt catalysts, the stability performance of NPMCs under real fuel cell operation conditions (e.g., 0.6 V) or after frequent cathode potential excursions to >1.5 V (i.e., mimicking the start-up/shutdown operations) is still far from satisfactory or comparable to that of Pt catalysts, which has prohibited NPMCs from penetrating the commercial market in the short term.16 So far, combining Pt with base metals (e.g., Fe, Co, Ni, Cu, Mn) still represents the most successful attempt in the search for high-performing ORR catalysts,17−27 as showcased by the record high specific activity of Pt3Ni(111)28 and practical applications of PtCo catalysts in recently developed fuel cell electric vehicles.29
In addition to direct engineering of the structure of active sites, introduction of a subtle amount of hydrophobic ionic liquids (ILs) into Pt-based catalysts which follows the so-called “solid catalyst with ionic liquid layer (SCILL)” concept30 has also been demonstrated in recent years to be an effective approach to improving Pt activity toward the ORR.31−36 The pioneering work was conducted by Erlebacher et al., who found that the specific activity of IL ([MTBD][beti])-modified porous PtNi films or nanoparticles toward the ORR was enhanced by 2–3 times relative to that of their pristine counterparts.31,33 The enhanced activity was mainly attributed to the high O2 solubility in the IL phase.31,33 Similarly, Zheng et al. reported that IL ([MTBD][NTf2])-impregnated graphene-supported Pt nanoparticles exhibited 2 times higher ORR activity and improved methanol tolerance in comparison to its counterpart without IL.32 We also studied in detail the boosting effect of an IL ([MTBD][NTf2]) on the ORR over Pt/C catalysts, by varying the filling degrees (α) of the IL within the pores of Pt/C catalysts. It was disclosed that both the catalytic activity and stability showed dependence on the α value, and the maximum activity was obtained at α = 50%, as a result of the tradeoff between the activity-boosting effect and the enhanced mass transfer resistance in the presence of the IL.35 More recently, we discovered that modifying a Pt/C catalyst with a conventional and inexpensive IL, i.e., [C4C1im][NTf2], can also bring about remarkable enhancement in the catalytic activity of Pt/C toward the ORR. It is disclosed that the presence of IL can boost the overall reaction rate of the ORR by suppressing the formation of nonreactive species (identified mainly as OHad) on Pt sites.36 The successful application of the SCILL concept in boosting ORR catalysts was also reflected by some more recent studies, where the catalytic activity of both conventional Pt-based catalysts and emerging non-precious-metal catalysts such as Fe-N-C and N-doped graphene was found to be enhanced after IL modifications.37−41 All of these inspiring works imply that the IL modification has the potential to become a generic method to improve the catalytic performance of cathodic ORR catalysts, while a fundamental understanding of the boosting effect from IL modification at a molecular level is still needed.
Intrigued by the great structural flexibility of imidazolium-based ILs,42,43 in the current study we synthesized a series of IL-modified Pt/C catalysts using imidazolium-based ILs with varied cationic alkyl chain lengths ranging from C2 to C10. The structures of these ILs are illustrated in Figure 1. We systematically explore the electrocatalytic and structural properties of these IL-modified Pt/C catalysts, in the hope of gaining some new insight into complex interactions of IL with solid catalysts and the promoting role of IL toward ORR over a Pt/C catalyst. It turns out that both the electrochemically active surface area (ECAS) and catalytic properties of Pt toward the ORR can be readily manipulated by varying the side chain lengths of the imidazolium ions. A distinct dependence of catalytic activity of Pt toward ORR on the side chain length is identified, and the maximum activity is obtained using an IL with a medium chain length (C4), which originates from the counterbalance between more efficiently suppressed formation of nonreactive oxygenated species on Pt and more severe passivation of Pt surfaces with elongation of the cationic chain length. Identical-location TEM and in situ ICP-MS analyses were conducted on the pristine and IL-modified Pt/C samples, and we found that the presence of ILs can effectively suppress the Pt dissolution which is thought to be the major cause for the degradation of the pristine Pt/C catalyst. We believe that these new findings will enrich our knowledge of the SCILL concept and provide some guidelines for designing efficient SCILL systems for other electrocatalytic and/or heterogeneous catalytic applications.
Figure 1.
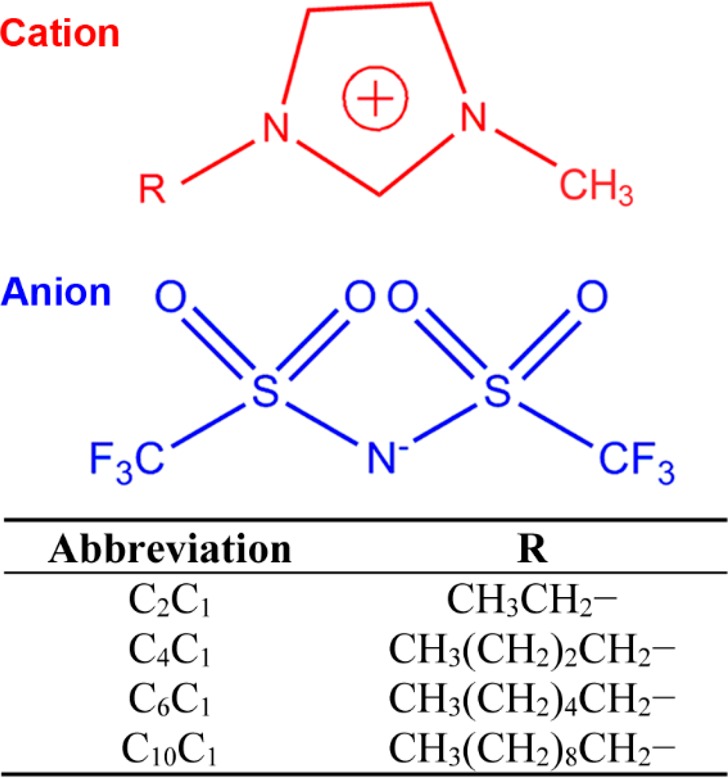
Specifications of the investigated imidazolium-based ILs.
2. Experimental Section
2.1. Materials
Pt/C catalyst (20 wt %, HiSPEC-3000, Johnson Matthey) was purchased from Alfa Aesar. HClO4 solution (70%) was purchased from Sigma-Aldrich. 1-Ethyl-3-methylimidazolium bis(trifluomethanesulfonyl)imide ([C2C1im][NTf2]) and lithium bis(trifluomethanesulfonyl)imide (NTf2) (99%) were purchased from IoLiTec GmbH. 1-Bromobutane (≥98%) and 1-bromodecane (≥98%) were obtained from Merck. 1-Bromohexane (≥99%) and 1-bromooctane (≥99%) were provided by Acros Organics. 1-Methylimidazole (99%) was supplied by Solvent Innovation GmbH. All chemicals were used as received without further purification.
2.2. Synthesis
2.2.1. Synthesis of Ionic Liquids
The imidazolium-based ILs reported in this work were synthesized from 1-methylimidazole, 1-bromobutane, 1-bromohexane, 1-bromodecane, and lithium bis(trifluomethanesulfonyl)imide salt according to a literature protocol,44 and the detailed synthetic procedure for each IL can be found in the Supporting Information. The ILs were obtained as pale yellow or colorless liquids and were characterized by NMR spectroscopy (Figure S1), where peaks marked with X are attributed to the presence of water traces in the DMSO solvent used for analyses.
2.2.2. Synthesis of IL-Modified Pt/C Samples
The IL-modified Pt/C samples were synthesized by impregnating commercial Pt/C catalysts with the as-prepared ILs according to our previously developed protocol.35 A typical procedure is as follows: 90 mg of Pt/C catalyst was first mixed with 10 mL of isopropyl alcohol solution containing a calibrated amount of IL (corresponding to a final pore filling degree of 10%) with intense stirring at room temperature. After a 20 min ultrasonic treatment of this mixture, the isopropyl alcohol was slowly removed from the slurry by rotary evaporation under low-vacuum conditions (137 mbar, 60 °C), followed by further evaporation at 8 mbar vacuum to facilitate complete intrusion of IL into pores. Finally, the sample was dried under high-vacuum conditions (2 × 10–3 mbar, room temperature) overnight in order to ensure complete removal of the solvent.
2.3. Instrumentation
1H NMR spectra of the ILs were recorded using an NMR tube filled with DMSO-d6 on an ECX 400 spectrometer (JEOL). Transmission electron microscopy (TEM) images were captured using a JEOL JEM-2100F microscope operated at 200 kV. The TEM samples were prepared by placing a drop of catalyst powder dispersion in deionized water onto a carbon-film-coated Cu grid, followed by drying under ambient conditions. Identical location TEM characterizations were carried out by using carbon-coated Au finder grids (G200F1, Quantifoil). More details about identical location TEM measurements can be found in Figure S2 in the Supporting Information. The loading amounts of Pt on SCILL samples were determined using inductively coupled plasma atomic emission spectrometry (ICP-AES, PerkinElmer Plasma 400). Electrochemical Pt dissolution tests were carried out on an in situ scanning flow cell coupled to an inductively coupled plasma mass spectrometer (SFC-ICP-MS) (NexION 300X, PerkinElmer) as described in previous works.45,46 The measurement procedures are detailed in the Supporting Information.
2.4. Electrochemical Measurements
Electrochemistry measurements were conducted on a PARSTAT 4000 Potentiostat/Galvanostat (AMETEK). A double-junction Ag/AgCl electrode (Sigma-Aldrich) and a platinum coil (PINE) isolated by a fritted glass tube were used as reference and counter electrodes, respectively. A glassy-carbon rotating disk electrode (RDE, 5 mm, PINE) was used as the working electrode. All potentials reported here were calibrated against the reversible hydrogen electrode (RHE). To determine the electron transfer number of ORR on different samples, the rotating ring disk electrode (RRDE) technique was employed. The RRDE working electrode is made of a glassy-carbon-disk electrode and a Pt-ring electrode with a collection efficiency of 37% (PINE). During the RRDE measurements, two potentiostats (PMC-1000, AMETEK) were used to control the potentials of disk and ring electrodes independently, and the potential of the Pt ring electrode was set to 1.4 V. Prior to each experiment, the RDE was polished to a mirror finish using an alumina suspension (0.05 μm, BUEHLER), followed by cleaning ultrasonically with ethanol, acetone, and then deionized water, respectively. Catalyst ink solutions were prepared by sonicating a suspension of catalyst powder (5 mg) in a mixture of deionized water, isopropyl alcohol, and 5 wt % Nafion solution in a volume ratio of 4:1:0.025. A calibrated amount of catalyst ink was then applied onto the RDE and dried under a gentle argon flow in order to form a uniform catalyst layer. The Pt loading was controlled at 20.4 μg cm–2 on the glassy-carbon electrode.
All electrochemical measurements were carried out in a N2- or O2-saturated 0.1 M HClO4 solution. The catalyst-coated working electrode was first pretreated by using a cyclic voltammetry (CV) technique in the potential range of 0–1.3 V (versus RHE) for 20 cycles at a scan rate of 100 mV s–1. In order to determine the ECAS values of different electrocatalysts, CV measurements were performed at a scan rate of 20 mV s–1, and ECAS values were then evaluated by integrating the H adsorption signals on the CV curves, assuming 210 μC cm–2 for calibrating the charge of a H monolayer per unit Pt surface area.47,48 Before ORR measurements, the electrolyte was first saturated with O2 by bubbling high-purity O2 into the HClO4 solution for at least 30 min, and an O2 flow was then maintained to avoid any disturbance from the ambient atmosphere during measurements. The ORR tests were carried out with a rotating RDE at a rate of 1600 rpm, and polarization curves were recorded at a scan rate of 10 mV s–1. The ohmic drop effect was compensated for all the ORR measurements. The ohmic resistance was determined by conducting electrochemical impedance spectroscopy (EIS) analysis with an ac signal amplitude of 5 mV.
For CO-stripping measurements, the catalysts were first saturated with CO by bubbling high-purity CO through the electrolyte at open circuit potential for 15 min. The electrolyte was then purged with N2 for another 15 min before the stripping measurement. The CO-stripping curves were recorded at a scan rate of 20 mV s–1. In order to better distinguish the CO-stripping signals, the as-measured CO-stripping curves were corrected by subtracting background currents associated with the double-layer charging and surface oxidation, which were recorded in the subsequent anodic-scan voltammograms. The ECASCO value was evaluated from the charge corresponding to the CO-stripping signal after background subtraction, assuming a specific charge of 420 μC cm–2 for a monolayer of CO. To distinguish the individual contributions of prepeak and main peak to ECASCO, the integration was also made by splitting the CO-stripping signal at the valley between two peaks (∼0.68 V) for the pristine Pt/C catalyst.
3. Results
3.1. Structural Analysis
The ILs investigated in this work were synthesized through a metathesis reaction of halide salts of the imidazolium ions with varied alkyl chain lengths and Li[NTf2]. The successful formation and purity of the as-synthesized ILs were verified by using 1H NMR (Figure S1), where the characteristic proton chemical shift peaks can be clearly observed. To explore the possible structural change of Pt/C samples after IL modification, we conducted TEM analyses on the pristine Pt/C and the Pt/C-[C4C1im][NTf2]. As displayed in Figure 2, for both samples Pt nanoparticles are well dispersed on carbon supports without any severe particle aggregation. The statistical analyses based on over 200 randomly selected Pt nanoparticles also confirm that there is negligible change in average particle size/distribution before and after the IL modification.
Figure 2.

Representative TEM images and their size histograms of the pristine (a) and the IL ([C4C1im][NTf2]) modified (b) Pt/C samples. More than 200 Pt nanoparticles were randomly selected in different images to determine the average particle sizes and their distribution.
3.2. Electrochemical Measurements
The electrochemical properties of the pristine and IL-modified Pt/C samples were first probed using CV techniques, and their corresponding CV curves are shown in Figure 3a. The characteristic signals featuring hydrogen adsorption/desorption on Pt can be seen for all the samples in the potential range of 0.05–0.40 V, along with the oxidation signals beyond 0.6 V corresponding to formation of surface oxygenated species on Pt surfaces.47,49,50 The ECAS values of the pristine and IL-modified Pt/C samples derived from the hydrogen adsorption signals (HUPD) are summarized in Figure 3b. The samples prepared by using ILs with relatively short alkyl chains (n < 6) possess ECAS values (96.0, 94.6, and 94.0 m2 gPt–1) quite comparable to that of the pristine Pt/C (99.0 m2 gPt–1), while further elongation of the alkyl chain leads to a dramatic drop in ECAS values: to be specific, from 94.6 m2 gPt–1 on Pt/C-[C4C1im][NTf2], to 61.9 m2 gPt–1 on Pt/C-[C6C1im][NTf2], and then 41.0 m2 gPt–1 on Pt/C-[C10C1im][NTf2]. It is not uncommon to observe the ECAS loss for a Pt/C catalyst after IL modification. For instance, we observed in our previous work that ECAS of Pt/C was reduced by up to 19.2% after surface modification using [MTBD][NTf2] (Scheme S1). Similarly, Huang et al. reported that Pt/C modified with [MTBD][C4F9SO3] (Scheme S1) suffered 28% ECAS loss relative to the pristine Pt/C.37 Nevertheless, our current work confirms that the effect of IL modification on ECAS depends sensitively on the specific structure of ILs, even for the ILs from the same category.
Figure 3.
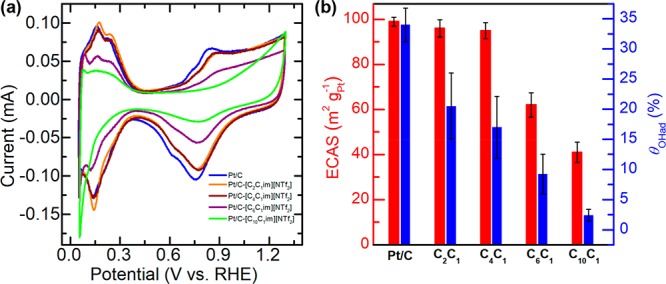
(a) Cyclic voltammetry curves of IL-modified Pt/C catalysts recorded in N2-saturated 0.1 M HClO4 solution at a scan rate of 20 mV s–1. (b) ECAS and surface coverage of oxygenated species calculated from CV curves on the Pt/C catalysts with and without IL modification.
Meanwhile, it can be noticed that the oxidation peaks beyond 0.6 V are attenuated on all the IL-modified samples with respect to that of the Pt/C (Figure 3a). This indicates that the presence of the IL phase can suppress the formation of oxygenated species on Pt, which is consistent with previous reports.31,33,35−37 For the IL-modified Pt/C catalysts, the hydrophobicity of the IL phase at the Pt/C catalyst surfaces is thought to create a hydrophobic microenvironment at the interface between the catalyst and aqueous electrolyte, which can help preserve the active Pt sites from being partially oxidized or forming oxygenated species by repelling water molecules.36 It should be be noted that the surface oxygenated species on Pt is considered to be nonreactive and will lead to blockage of Pt active sites.28 Herein, attempts were also made to quantify the coverage of the nonreactive oxygenated species (θOHad) on Pt. As displayed in Figure 3b, side chain elongation in imidazolium cations leads to a substantial decrease in θOHad values. It can be found that, despite the unwanted passivation of active Pt surfaces, the presence of an IL with a longer cationic chain (n > 6) can indeed make the rest of the active surfaces be less prone to forming nonreactive oxygenated species.
The electrocatalytic properties of these IL-modified samples toward the ORR were investigated and compared with those of the pristine Pt/C using thin-film RDE techniques. Figure 4a compares their ORR polarization curves recorded in O2-saturated 0.1 M HClO4 electrolyte, where it can be seen that the curves of most of the IL-modified Pt/C samples have positively shifted relative to that of the pristine Pt/C. The only exception is Pt/C-[C10C1im][NTf2], whose polarization curve has shifted negatively by 53 mV instead. It is well documented that the ORR can proceed either through the two-electron pathway to produce H2O2 or four-electron pathway to produce water. To clarify whether the cationic chain length of ILs has an influence on the ORR pathways, we have conducted RRDE measurements on Pt/C samples with and without IL. As shown in Figure S3, in which the upper panel displays the mole fraction of H2O2 formation, it can be seen that the H2O2 fraction is lower than 4% for all of the investigated samples, and their plots of mole fraction of the formed H2O2 are comparable. These results indicate that the ORR is proceeding mainly through the preferred four-electron pathway to produce water, and neither the presence of IL nor the cationic chain length has imposed any significant effect on the ORR pathway on these catalysts. To quantify their difference in ORR activity, the specific activity (SA) and mass specific activity (MSA) at 0.9 V were calculated and are compared in Figure 4b. It is notable that Pt activity strongly correlates with the cationic chain length of ILs, and a distinct dependence of SA and MSA on the cationic chain length with the maximum activity obtained on Pt/C-[C4C1im][NTf2] can be identified. The hydrophobicity of the catalyst surface conveyed by hydrophobic ILs is believed to help preserve active sites from being oxidized by water and thereafter results in an enhanced overall reaction rate. Thus, it is expected that the more hydrophobic IL with a longer alkyl chain length would more efficiently boost the ORR rate, and it is not surprising to observe that ORR proceeded more quickly on Pt/C-[C4C1im][NTf2] in comparison to that on Pt/C-[C2C1im][NTf2]. However, further elongation of the alkyl chain length from C4 to C10 leads to an decrease in SA from 1.10 mA cmPt–2 on Pt/C-[C4C1im][NTf2], to 0.90 mA cmPt–2 on Pt/C-[C6C1im][NTf2] and 0.18 mA cm–2Pt on Pt/C-[C10C1im][NTf2]. This trend is quite similar to the ECAS drop in response to the elongation of the alkyl chain from C4 to C10, which hints that the critical changes in structure/property of these IL-modified samples would occur at the transition from Pt/C-[C4C1im][NTf2] to Pt/C-[C6C1im][NTf2].
Figure 4.

(a) ORR polarization curves of IL-modified Pt/C catalysts and the pristine Pt/C recorded in O2-saturated 0.1 M HClO4 electrolyte at a scan rate of 10 mV s–1. (b) Comparisons of mass and specific activity of Pt toward the ORR at 0.9 V on the pristine and IL-modified Pt/C catalysts.
In order to probe the interactions between ILs and Pt nanoparticles, we carried out CO-stripping measurements on the pristine and IL-modified Pt/C samples. The background-subtracted CO-stripping voltammetric profiles are shown in Figure 5a. It can be seen that the CO-stripping signal on Pt/C features a prepeak and a main peak centered at 0.75 and 0.83 V, respectively. Interestingly, the clearly resolved prepeak on Pt/C, which is thought to originate from CO oxidation on low-coordinated Pt sites,51−53 becomes less pronounced on all of the IL-modified Pt/C samples. To better distinguish the influence of chain length on the CO-stripping signals, the ECAS values obtained accordingly to CO stripping charges are compared in Figure 5b. The individual contributions of the prepeak and the main peak to ECASCO for each sample were roughly estimated by splitting the CO-stripping signal as described in the Experimental Section. It can be seen clearly that the share of the prepeak to the total CO-stripping signals decreases with elongation of the cationic chain length from C4 (6.2%) to C6 (3.9%) and almost completely vanishes on C10 (0.7%). Furthermore, the main CO-stripping peaks on all of the IL-modified samples are positively shifted, indicating a stronger bonding of CO molecules on Pt surfaces in the presence of IL, which is consistent with our previous report,36 while the main peaks on the Pt/C samples modified by ILs with relatively shorter cationic side chains (i.e., [C2C1im][NTf2] and [C4C1im][NTf2]) can retain the same peak height as that on the pristine Pt/C. However, further elongation of the cationic side chain from C4 to C10 would lead to a reduction in peak area (ECASCO) contributed by the main peaks (Figure 5b), which is in line with the aforementioned decrease of the ECAS in response to the side-chain elongation. These results imply that the IL molecules would preferentially locate at the low-coordinated edge/corner/defect Pt sites regardless of the cationic chain length, while with the elongation of the side chain (n > 6), the IL molecules will start interacting with the terrace sites probably through the long side chain, leading to blockage of terrace Pt sites.
Figure 5.

(a) Background subtracted CO-stripping curves and (b) summary of ECAS calculated on the basis of CO-stripping peaks on the pristine and IL-modified Pt/C catalysts. The values in (b) indicate the proportion of ECAS contributed by the prepeaks.
3.3. Accelerated Degradation Tests
To assess the electrochemical stability of different catalysts toward the ORR, we adapted a relatively harsh accelerated degradation test (ADT) protocol by conducting repeated CV tests in the potential range of 0.4–1.4 V at a scan rate of 1 V s–1.54 The loading amount of Pt on RDE was fixed (20.4 μg·cm–2) to enable a fair comparison, considering that the exact loading of Pt may influence the stability performance.54,55 The voltammograms and the ORR polarization curves of different catalysts before and after the ADT are shown in Figures S4 and S5, respectively. Figure 6 summarizes the corresponding changes in ECAS and the ORR half-wave potential (E1/2). After 2000 cycles, the ORR polarization curve of the pristine Pt/C was negatively shifted by 30 mV, along with a reduction in ECAS by 65%, demonstrating that the pristine Pt/C sample suffered severe degradation during the ADT. It can be seen in Figure 6 that the IL-modified samples generally exhibit improved electrochemical stability relative to the pristine Pt/C, as reflected by the fact that the ECAS value is reduced by 51% on the Pt/C-[C2C1im][NTf2], 47% on the Pt/C-[C4C1im][NTf2], and 38% on the Pt/C-[C6C1im][NTf2], while the ECAS of Pt/C-[C10C1im][NTf2] is fully retained after the ADT. It is also noteworthy that after the ADT both Pt/C-[C2C1im][NTf2] and Pt/C-[C4C1im][NTf2] still show higher half-wave potentials in comparison to the pristine Pt/C, despite the fact that their polarization curves are negatively shifted by 21 and 18 mV, respectively (Figure 6b). Meanwhile, it can be observed that the stabilizing effect of the IL seems to be insensitive to the nature of the cations, as indicated by the comparable drop in the half-wave potentials for the IL-modified samples with IL cationic chain length ranging from C2 to C6. The only exception is Pt/C-[C10C1im][NTf2], its ORR polarization curve being positively shifted, which might arise from the restructuring of IL molecules that strongly interact with the Pt surfaces. Further experiment and/or simulation work needs to be carried out to verify this hypothesis, while these efforts would be beyond the scope of the current work. In any event, the above results demonstrate that the presence of the IL helps improve the electrochemical stability of a Pt catalyst, which is consistent with the previous reports that the presence of an IL phase can bring enhanced electrochemical stability of various Pt-based electrocatalysts relative to their counterparts without IL.35,41,56 At the same time, it is verified that the boosting effect from IL modification can be maintained even under the harsh ADT conditions, which is of great significance for their practical applications.
Figure 6.

Comparison of the electrochemically active surface area (a) and half-wave potential (b) of the pristine and IL-modified Pt/C catalysts before and after 2000 potential cycles between 0.4 and 1.4 V in O2-saturated 0.1 M HClO4.
In order to unambiguously explore the degradation mechanism of these Pt/C samples toward the ORR, we employed the identical-location TEM technique to probe the structural change of the pristine Pt/C and the most promising Pt/C-[C4C1im][NTf2] before and after ADT tests. As shown in Figure 7a–d, the most striking effect induced by the potential cycling on the structures of the pristine Pt/C sample is severe dissolution of Pt nanoparticles, as evidenced by the fact that the same particles become much smaller after the ADT. Some particles even completely disappear after the ADT, which can be caused by either Pt dissolution or particle detachment from carbon supports. While considering that the carbon support is still intact and no obvious change in shape of the carbon spheres can be observed, we tend to believe that the particle loss would mainly take place following the dissolution mechanism, which is in accordance with previous reports.57,58 A different picture emerges for Pt/C-[C4C1im][NTf2], for which the Pt nanoparticles are well retained without any pronounced change in particle sizes after the ADT, as illustrated in Figure 7e–h. The integrity of the carbon-supported Pt nanoparticles strongly indicates that the presence of an IL can suppress the Pt dissolution, which agrees with the aforementioned suppressed oxidation of Pt on IL-modified Pt/C catalysts. At the same time, it can be seen that particles in close vicinity tend to merge into larger ones, corresponding to “particle coalescence”, another mechanism leading to degradation of Pt catalysts. On the basis of these results, it can be concluded that the presence of an IL can indeed improve the electrochemical stability the Pt/C sample mainly by suppressing the dissolution of Pt nanoparticles; however, the IL seems incapable of fully preventing the particles from coalescing.
Figure 7.

Identical-location TEM images of the pristine Pt/C (a–d) and the Pt/C-[C4C1im][NTf2] (e–h) before and after the stability test.
To further probe the Pt dissolution behavior of the pristine and IL-modified Pt/C catalysts during the ADTs, the concentration of dissolved Pt during the ADT was quantified using an in situ SFC-ICP-MS setup.46,59 As summarized in Figure S6, we can see a trend in stability toward Pt dissolution for IL-modified catalysts. Quantification shows that the Pt mass losses after the ADT are decreased from 46.6 ± 7.8% on the pristine Pt/C to 35.7 ± 1.8% on the Pt/C-[C4C1im][NTf2], providing the second piece of evidence that the presence of an IL can stabilize Pt catalysts during the ADT. At the same time, we can also observe that the IL-modified Pt/C samples exhibit similar dissolution amounts of Pt after ADT regardless of the cationic chain length of the ILs, which is consistent with the above electrochemical measurements. It appears that, although the nature of cations imposes a significant effect on the catalytic activity of these IL-modified Pt/C catalysts, it may play a minor role in affecting the electrochemical stability.
Moreover, to better resolve the Pt dissolution at the pristine and IL-modified Pt/C catalysts, we carried out additional SFC-ICP-MS experiments by monitoring the Pt dissolution profiles during four consecutive CV tests between 0.05 and 1.5 V at a scan rate of 10 mV s–1. As shown in Figure 8, for each CV cycle there are two significant dissolution peaks for both samples, while the more dominant dissolution peak emerges in the reverse scan. It can also be noticed that the dissolution peak intensity decreases with an increase in the cycle number, i.e., the Pt dissolution rate is gradually decreasing, which is in accordance with a previous report.59 At the same time, it is noteworthy that the Pt dissolution peak intensity is significantly lower on the Pt/C-[C4C1im][NTf2] than on the pristine Pt/C. These results confirm again that the presence of an IL plays an important role in stabilizing the Pt catalysts.
Figure 8.

Dissolution profiles of Pt/C and Pt/C-[C4C1im][NTf2] samples during four consecutive CV tests between 0.05 and 1.5 V at a scan rate of 10 mV s–1.
4. Discussion
We observed that the electrochemical properties as ECAS, electrocatalytic properties toward the ORR, and CO-stripping behavior exhibited a unique dependent behavior on the cationic chain length of the ILs used for Pt surface modification. Especially intriguing is that two regimes can be observed, when the cationic chain length of ILs is elongated. The first regime is from C2 to C4, where ECAS values similar to that of the pristine catalyst can be obtained, but the specific activity increases with IL modification and chain length. The second regime is from C6 to C10, where a higher specific activity (C6) can also be obtained, but the ECAS and mass specific activity drop significantly when the chain length is elongated. This implies that cationic chain elongation from C4 to C6 pronouncedly changes the interactions taking place among the IL, aqueous electrolyte, and platinum.
For ORR in aqueous electrolyte, water is believed to play an important role in the whole process. For instance, water molecules are considered to be the major source of the nonreactive oxygenated species,28,60,61 and interface water itself at electrode surfaces may also directly block the Pt sites, as suggested by Jinnouchi et al. on the basis of their combined DFT and Poisson–Boltzmann calculations.62 Moreover, the dissolved water in the IL phase could promote proton transportation from the aqueous electrolyte to the Pt surfaces through the IL phase.33,36 Both theory and experiment indicate that ILs with longer cationic chains possess lower water content,63,64 which can be rationalized by the fact that a longer nonpolar cationic tail of the IL would cause larger structural mismatching on interaction with water molecules, leading to a higher misfit energy and consequently a lower water solubility.64 Therefore, an intuitive rationale for the dependence of electrochemical properties of those IL-modified catalysts on the cationic chain lengths of ILs would lie in their different water contents. If this is the case, we would expect a significant decrease in water contents when the cationic chain is elongated from C4 to C6, to be compliant with the dramatically different structural/catalytic properties of Pt/C-[C4C1im][NTf2] and Pt/C-[C6C1im][NTf2]. Nevertheless, the difference in water content between [C4C1im][NTf2] and [C6C1im][NTf2] is actually quite minor, i.e., 0.25 vs 0.21 in terms of the mole fraction solubility of water in ILs at room temperature,63 which seems unlikely to be fully responsible for the significantly different ECAS and θOHad values between Pt/C-[C4C1im][NTf2] (94.6 m2 gPt–1, 17%), and Pt/C-[C6C1im][NTf2] (61.9 m2 gPt–1, 9%). Actually, the difference in water content between [C2C1im][NTf2] (0.30) and [C4C1im][NTf2] (0.25) is even slightly larger,63 and the comparable ECAS and θOHad values of Pt/C-[C2C1im][NTf2] and Pt/C-[C4C1im][NTf2] further verify that, although the water content of ILs would decrease with the elongation of cationic chains, it might play a minor role in determining the electrocatalytic performance of these IL-modified Pt/C catalysts. Therefore, apart from the differences in water content among various ILs, elongation of the cationic chain must have caused some essential changes in the structures of ILs over Pt surfaces, which will be discussed in the following section.
It is well documented that ILs with long alkyl chains tend to form lipid- or micelle-like aggregates.65−69 For instance, Perkin et al. investigated the layered structures of [C4C1im][NTf2] and [C6C1im][NTf2] at an atomically smooth mica surface by using a surface force balance and revealed that elongation of alkyl chain length from C4 to C6 led to a structure transformation from the alternating cation and anion monolayer structure of [C4C1im][NTf2] to the lipid-like bilayer structure of [C6C1im][NTf2].69 Voth et al. proposed on the basis of their molecular dynamics (MD) simulations that the alkyl chain of imidazolium cations would aggregate and form micelle-like structures when the cationic chain is elongated from C4 to C8.67 Similarly, Klein et al. investigated the effect of alkyl chain length of imidazolium cations on the IL structure in the presence of water and suggested that a micelle-like structure would form for [C6C1im][Br] and [C8C1im][Br], but this is not the case for [C4C1im][Br], the short butyl chain of which is less likely to aggregate.70−72 Venkatnathan et al. also reported that an IL with long alkyl chain length, i.e., [C6C1im][NTf2], tended to form a micelle-like structure in contact with an excess amount of water on the basis of their MD simulations.65 Taking these experiment and simulation findings into account, it seems that the lipid-/micelle-like structure would likely form for ILs with long alkyl chain (n ≥ 6) due to the spontaneous aggregation of their long nonpolar cationic tails.
Here in the current study, we also found that the essential change in structural and catalytic properties of these IL-modified Pt/C catalysts would occur when the cationic side-chain length of ILs increased from C4 to C6. As evidenced by the CO-stripping measurements, the IL molecules would preferentially interact with the low-coordinated Pt sites (e.g., edge, corner) regardless of the alkyl chain length, while for the ILs with relatively long cationic chains (n > 6), the IL cations would block the terrace Pt sites and lead to significant loss in ECSA, which is not the case for the short-chain ILs. These findings lead us to hypothesize that the interactions between IL molecules and Pt surfaces would be dependent on the specific cationic chain length. The ILs with long cationic chains, i.e. [C6C1im][NTf2] and [C10C1im][NTf2], would likely compromise their mobility adapting the aforementioned lipid-/micelle-like structure on solid Pt catalysts due to the spontaneous aggregation of their long nonpolar cation tails.
Nevertheless, in the aforementioned studies, the formation of the lipid-/micelle-like structures of ILs was predicted without considering the Coulomb interactions of IL anions/cations with electrified surfaces or the associated formation of an electrochemical double layer whose structure depends strongly on the applied electrode potentials. For the electrochemical measurements carried out by applying variable potentials, as in the current study, a dynamic structure of ILs in contact with platinum is to be expected, which complicates the picture of the interfacial structure of IL molecules at electrode surfaces.73−76 Unfortunately, the dynamic response of IL structures to the variation of electrode potentials is still poorly understood, while consensus suggests that the ILs would adopt a charge-separated layered structure: i.e., with alternating cation- and anion-rich layers at electrified surfaces.73 Following this line of reasoning, at a positively polarized electrode, which is relevant to the ORR conditions, the [NTf2]− anions are enriched at the innermost layer (Stern) of the electrode to compensate for the positive charge of the electrode, while the second layer (overlayer) will be dominated by the cations. The ion proportions at each layer depend on the magnitude of electrode polarization.75 Taking all this information into account, we can expect that the IL structures at the electrode surfaces are determined by the counterbalance between van der Waals and Coulombic interactions. Accordingly, a simplified picture of IL structures on electrode catalyst is shown schematically in Figure 9, where water and oxygen molecules are not shown for clarity. A positively charged surface was chosen for illustration because it is more relevant to the operation conditions of a cathode (positive electrode) in a fuel cell, and the innermost layer close to the electrode is expected to be enriched by the anions due to electrostatic attraction. For the long-chain ILs, cations would self-assemble into a bilayer structure with polar head groups (imidazolium ring) pointing toward the aqueous electrolyte or interacting with the innermost anion layer as proposed by Kornyshev,74 while this may not be the case for the short-chain cations which should have much higher mobility (Figure 9a). We have to admit that this picture should be viewed with caution, especially considering the poorly understood dynamics of IL structures in response to the varying potentials and the ill-defined nature of interactions between IL cations/anions and metal surfaces. For instance, we cannot rule out the possibility that some cations would still stay at the innermost layer at the positively polarized electrode, which can be a consequence of either the strong interaction between cations and metal surfaces or the tardy dynamic response of cations to the varying potentials. As demonstrated by Nelson et al. on the basis of their neutron reflectometry and electrochemical analyses, a cation ([C4C1pyr]+)-rich interface was found even at a positively charged Au electrode (1.3 V above the potential of zero charge) due to the specific adsorption of [C4C1pyr]+ on Au, while as expected the cations at the innermost layer depopulated at increasing potentials.77
Figure 9.

Schematic illustrations for the likely structures of IL-modified Pt/C samples with short (a) and long (b) alkyl chains in imidazolium cations on positively polarized electrodes. Water and oxygen molecules are not shown in the schemes for clarity. The schemes are not drawn to scale.
Despite this uncertainty, as presented in Figure 9, the most significant feature for the long-chain ILs would be their tendency to form a lipid-like bilayer structure, which turns out to have both positive and negative consequences. On one hand, this structure can more efficiently protect Pt sites from being oxidized or forming nonreactive oxygenated species by repelling water molecules due to its rigid bilayer structure along with the highly hydrophobic microenvironment created at the interface between the solid catalyst and aqueous electrolyte, which can be justified by the lower θOHad values of Pt/C-[C6C1im][NTf2] and Pt/C-[C10C1im][NTf2] in comparison to those of the other samples; on the other hand, this structure causes partial blockage of Pt sites, leading to a much lower ECAS value for the sample with long-chain ILs. The attenuation or vanishment of the prepeaks along with the reduction in the main peak intensity of CO stripping on Pt/C-[C6C1im][NTf2] and Pt/C-[C10C1im][NTf2] also indicates that the long-chain ILs can partially passivate/block the Pt terrace sites. In addition, the much lower specific activity of Pt/C-[C10C1im][NTf2] (0.18 mA cmPt–2) for the ORR implies that the surface blockage of Pt surface sites not only leads to the loss of Pt active site number but may also break the ensemble of Pt active sites required for high turnover rates of the ORR through the so-called third-body effect.78 Moreover, as evidenced by the positively shifted CO-stripping peaks in the CO-stripping measurements, the presence of ILs on Pt catalysts can also strengthen the bonding of CO molecules on Pt. These results indicate that the IL might also play a role in altering the surface electronic structure through a ligand effect, promoting the activation of O2 molecules on Pt, which can also contribute to the accelerated ORR kinetics on the IL-modified Pt catalysts. On the basis of these results, we can get a more comprehensive picture about the role of ILs in affecting the electrocatalytic properties of a solid catalyst. That is, in addition to the well-documented beneficial effect from IL modification including high O2 solubility in IL and suppressed formation of nonreactive oxygenated species, the ligand effect, surface passivation/blockage, and ensemble (third-body) effect originating from IL modification have long been overlooked and have turned out to play important roles in controlling the overall reaction rate, which can be optimized by finely tuning the IL molecular structures.
The above picture also sheds new light on understanding the improved electrochemical stability of the IL-modified Pt/C catalysts. As mentioned above, Pt dissolution is found to be the major reason for the degradation of the pristine Pt/C catalyst, and it is documented that low-coordinated (edge/corner/defect) Pt sites are more prone to dissolution and indeed act as the starting sites for the initial oxidative dissolution of a Pt nanoparticle.79,80 Interestingly, as evidenced by the attenuated CO-stripping prepeaks on all of the IL-modified Pt/C samples, the IL molecules (most likely the [NTf2]− anions for a positively polarized electrode) would preferentially interact with those low-coordinated Pt sites. These results lead us to conclude that the presence of an IL improves the electrochemical stability of Pt/C catalysts through suppression of the formation of chemisorbed oxygenated species and/or the initial oxidation of low-coordinated Pt sites located at the edge, corner, and/or defect Pt sites. Thus, the [NTf2]− anions seem to be mainly responsible for increasing the electrochemical stability, which would explain the observed similarly lowered dissolution rate of Pt irrespective of the cations. This result highlights the importance of understanding the nature of the molecular interactions between IL molecules and electrode surfaces, especially under dynamic potentials. Further extensive spectroscopic (e.g., IRAS, XPS) and microscopic (e.g., STM, AFM) analyses of the IL-modified model/practical catalysts, especially under the electrochemical operation conditions, and also the perspectives of simulation studies (e.g., MD simulations of electrified surfaces in contact with ILs) are still greatly needed before a more fundamental understanding can be obtained about the interactions between varied IL molecules and electrified metal surfaces.
5. Conclusions
In conclusion, the current work verified again the boosting effect of ILs toward the ORR on Pt catalysts by using a wide variety of imidazolium-based ILs. More importantly, we have discovered that the electrocatalytic properties of IL-modified Pt/C catalysts can be regulated by rationally tuning the structures of ILs at a molecular level, and the proper length of the cationic alkyl chain holds the key to obtaining a promising ORR catalyst. Specifically, the IL with an alkyl chain that is too short (e.g., [C2C1im][NTf2]) cannot fully boost the activity of Pt for the ORR because its limited hydrophobicity cannot efficiently suppress the formation of nonreactive oxygenated species on Pt, while an IL with alkyl chain that is too long (e.g., [C6C1im][NTf2], [C10C1im][NTf2]) would lead to a dramatic loss in both active surface area and intrinsic activity of Pt probably due to the formation of rigid lipid-like bilayer structures. Therefore, the chain length of an IL can be used as a regulator to tune the electrocatalytic properties of a Pt-based ORR catalyst, and an optimized IL structure for an active Pt catalyst is thought to have a proper chain length which can provide sufficient hydrophobicity without triggering the formation of a lipid-/micelle-like structure or loss of active sites. While the cationic structure of ILs has a great influence on the intrinsic activity of Pt, the promising improvement in electrocatalytic stability of Pt/C catalysts appears to stem from the interaction/blockage of low-coordinated Pt sites by the anions. As evidenced by the identical-location TEM and in situ SFC-ICP-MS analyses, the presence of ILs could effectively suppress Pt dissolution, which is thought to be the major cause for the degradation of the pristine Pt/C catalyst. We believe that these findings could be exploited to develop active and stable Pt-saving electrocatalysts for fuel cell catalysis and may also have broad implications for constructing other high-performing catalytic systems following the SCILL concept.
Acknowledgments
The authors acknowledge funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No. 681719). The authors are also thankful to Dr. Stefan Lauterbach for his kind help in the identical-location TEM measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b02018.
Additional information about IL syntheses, determination of ORR kinetic current, procedures of identical-location TEM and SFC-ICP-MS measurements, and supplementary figures/schemes (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Jiao Y.; Zheng Y.; Jaroniec M.; Qiao S. Z. Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 2015, 44, 2060–2086. 10.1039/C4CS00470A. [DOI] [PubMed] [Google Scholar]
- Dai L.; Xue Y.; Qu L.; Choi H.-J.; Baek J.-B. Metal-Free Catalysts for Oxygen Reduction Reaction. Chem. Rev. 2015, 115, 4823–4892. 10.1021/cr5003563. [DOI] [PubMed] [Google Scholar]
- Guo S. J.; Zhang S.; Sun S. H. Tuning Nanoparticle Catalysis for the Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2013, 52, 8526–8544. 10.1002/anie.201207186. [DOI] [PubMed] [Google Scholar]
- Wang Y.-J.; Zhao N.; Fang B.; Li H.; Bi X. T.; Wang H. Carbon-Supported Pt-Based Alloy Electrocatalysts for the Oxygen Reduction Reaction in Polymer Electrolyte Membrane Fuel Cells: Particle Size, Shape, and Composition Manipulation and Their Impact to Activity. Chem. Rev. 2015, 115, 3433–3467. 10.1021/cr500519c. [DOI] [PubMed] [Google Scholar]
- Debe M. K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. 10.1038/nature11115. [DOI] [PubMed] [Google Scholar]
- Gasteiger H. A.; Kocha S. S.; Sompalli B.; Wagner F. T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal., B 2005, 56, 9–35. 10.1016/j.apcatb.2004.06.021. [DOI] [Google Scholar]
- Feng Y. Y.; Zhang G.-R.; Ma J. H.; Liu G.; Xu B. Q. Carbon-supported Pt̂Ag nanostructures as cathode catalysts for oxygen reduction reaction. Phys. Chem. Chem. Phys. 2011, 13, 3863–3872. 10.1039/c0cp01612h. [DOI] [PubMed] [Google Scholar]
- Zhang G.-R.; Wöllner S. Hollowed structured PtNi bifunctional electrocatalyst with record low total overpotential for oxygen reduction and oxygen evolution reactions. Appl. Catal., B 2018, 222, 26–34. 10.1016/j.apcatb.2017.09.066. [DOI] [Google Scholar]
- Liang Y.; Li Y.; Wang H.; Zhou J.; Wang J.; Regier T.; Dai H. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 2011, 10, 780–786. 10.1038/nmat3087. [DOI] [PubMed] [Google Scholar]
- Ge X.; Sumboja A.; Wuu D.; An T.; Li B.; Goh F. W. T.; Hor T. S. A.; Zong Y.; Liu Z. Oxygen Reduction in Alkaline Media: From Mechanisms to Recent Advances of Catalysts. ACS Catal. 2015, 5, 4643–4667. 10.1021/acscatal.5b00524. [DOI] [Google Scholar]
- Wang Y.; Li J.; Wei Z. Transition-metal-oxide-based catalysts for the oxygen reduction reaction. J. Mater. Chem. A 2018, 6, 8194–8209. 10.1039/C8TA01321G. [DOI] [Google Scholar]
- Yan X.-H.; Zhang G.-R.; Xu B.-Q. Performance of polyaniline-derived Fe-N-C catalysts for oxygen reduction reaction in alkaline electrolyte. Chin. J. Catal. 2013, 34, 1992–1997. 10.1016/S1872-2067(12)60714-1. [DOI] [Google Scholar]
- Tang C.; Zhang Q. Nanocarbon for Oxygen Reduction Electrocatalysis: Dopants, Edges, and Defects. Adv. Mater. 2017, 29, 1604103. 10.1002/adma.201604103. [DOI] [PubMed] [Google Scholar]
- Shi J.-L.; Tang C.; Huang J.-Q.; Zhu W.; Zhang Q. Effective exposure of nitrogen heteroatoms in 3D porous graphene framework for oxygen reduction reaction and lithium–sulfur batteries. J. Energy Chem. 2018, 27, 167–175. 10.1016/j.jechem.2017.09.014. [DOI] [Google Scholar]
- Wu M.; Wang K.; Yi M.; Tong Y.; Wang Y.; Song S. A Facile Activation Strategy for an MOF-Derived Metal-Free Oxygen Reduction Reaction Catalyst: Direct Access to Optimized Pore Structure and Nitrogen Species. ACS Catal. 2017, 7, 6082–6088. 10.1021/acscatal.7b01649. [DOI] [Google Scholar]
- Banham D.; Ye S.; Pei K.; Ozaki J.-i.; Kishimoto T.; Imashiro Y. A review of the stability and durability of non-precious metal catalysts for the oxygen reduction reaction in proton exchange membrane fuel cells. J. Power Sources 2015, 285, 334–348. 10.1016/j.jpowsour.2015.03.047. [DOI] [Google Scholar]
- Chung D. Y.; Yoo J. M.; Sung Y.-E. Highly Durable and Active Pt-Based Nanoscale Design for Fuel-Cell Oxygen-Reduction Electrocatalysts. Adv. Mater. 2018, 1704123. 10.1002/adma.201704123. [DOI] [PubMed] [Google Scholar]
- Strasser P.; Gliech M.; Kuehl S.; Moeller T. Electrochemical processes on solid shaped nanoparticles with defined facets. Chem. Soc. Rev. 2018, 47, 715–735. 10.1039/C7CS00759K. [DOI] [PubMed] [Google Scholar]
- Wu J. B.; Qi L.; You H. J.; Gross A.; Li J.; Yang H. Icosahedral Platinum Alloy Nanocrystals with Enhanced Electrocatalytic Activities. J. Am. Chem. Soc. 2012, 134, 11880–11883. 10.1021/ja303950v. [DOI] [PubMed] [Google Scholar]
- Choi S. I.; Xie S. F.; Shao M. H.; Odell J. H.; Lu N.; Peng H. C.; Protsailo L.; Guerrero S.; Park J. H.; Xia X. H.; Wang J. G.; Kim M. J.; Xia Y. N. Synthesis and Characterization of 9 nm Pt-Ni Octahedra with a Record High Activity of 3.3 A/mg(Pt) for the Oxygen Reduction Reaction. Nano Lett. 2013, 13, 3420–3425. 10.1021/nl401881z. [DOI] [PubMed] [Google Scholar]
- Guo S. J.; Li D. G.; Zhu H. Y.; Zhang S.; Markovic N. M.; Stamenkovic V. R.; Sun S. H. FePt and CoPt Nanowires as Efficient Catalysts for the Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2013, 52, 3465–3468. 10.1002/anie.201209871. [DOI] [PubMed] [Google Scholar]
- Wang D. L.; Xin H. L. L.; Hovden R.; Wang H. S.; Yu Y. C.; Muller D. A.; DiSalvo F. J.; Abruna H. D. Structurally ordered intermetallic platinum-cobalt core-shell nanoparticles with enhanced activity and stability as oxygen reduction electrocatalysts. Nat. Mater. 2013, 12, 81–87. 10.1038/nmat3458. [DOI] [PubMed] [Google Scholar]
- Strasser P.; Koh S.; Anniyev T.; Greeley J.; More K.; Yu C.; Liu Z.; Kaya S.; Nordlund D.; Ogasawara H.; Toney M. F.; Nilsson A. Lattice-strain control of the activity in dealloyed core–shell fuel cell catalysts. Nat. Chem. 2010, 2, 454. 10.1038/nchem.623. [DOI] [PubMed] [Google Scholar]
- Kang Y.; Murray C. B. Synthesis and Electrocatalytic Properties of Cubic Mn–Pt Nanocrystals (Nanocubes). J. Am. Chem. Soc. 2010, 132, 7568–7569. 10.1021/ja100705j. [DOI] [PubMed] [Google Scholar]
- Shen L.-L.; Zhang G.-R.; Miao S.; Liu J.; Xu B.-Q. Core–Shell Nanostructured Au@NimPt2 Electrocatalysts with Enhanced Activity and Durability for Oxygen Reduction Reaction. ACS Catal. 2016, 6, 1680–1690. 10.1021/acscatal.5b02124. [DOI] [Google Scholar]
- Liu W.; Rodriguez P.; Borchardt L.; Foelske A.; Yuan J. P.; Herrmann A. K.; Geiger D.; Zheng Z. K.; Kaskel S.; Gaponik N.; Kotz R.; Schmidt T. J.; Eychmuller A. Bimetallic Aerogels: High-Performance Electrocatalysts for the Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2013, 52, 9849–9852. 10.1002/anie.201303109. [DOI] [PubMed] [Google Scholar]
- Xue Q.; Xu G.; Mao R.; Liu H.; Zeng J.; Jiang J.; Chen Y. Polyethyleneimine modified AuPd@PdAu alloy nanocrystals as advanced electrocatalysts towards the oxygen reduction reaction. J. Energy Chem. 2017, 26, 1153–1159. 10.1016/j.jechem.2017.06.007. [DOI] [Google Scholar]
- Stamenkovic V. R.; Fowler B.; Mun B. S.; Wang G. F.; Ross P. N.; Lucas C. A.; Markovic N. M. Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability. Science 2007, 315, 493–497. 10.1126/science.1135941. [DOI] [PubMed] [Google Scholar]
- Yoshida T.; Kojima K. Toyota MIRAI fuel cell vehicle and progress toward a future hydrogen society. Electrochem. Soc. Interface 2015, 24, 45–49. 10.1149/2.F03152if. [DOI] [Google Scholar]
- Kernchen U.; Etzold B.; Korth W.; Jess A. Solid catalyst with ionic liquid layer (SCILL) - A new concept to improve selectivity illustrated by hydrogenation of cyclooctadiene. Chem. Eng. Technol. 2007, 30, 985–994. 10.1002/ceat.200700050. [DOI] [Google Scholar]
- Snyder J.; Fujita T.; Chen M. W.; Erlebacher J. Oxygen reduction in nanoporous metal-ionic liquid composite electrocatalysts. Nat. Mater. 2010, 9, 904–907. 10.1038/nmat2878. [DOI] [PubMed] [Google Scholar]
- Tan Y. M.; Xu C. F.; Chen G. X.; Zheng N. F.; Xie Q. J. A graphene-platinum nanoparticles-ionic liquid composite catalyst for methanol-tolerant oxygen reduction reaction. Energy Environ. Sci. 2012, 5, 6923–6927. 10.1039/c2ee21411c. [DOI] [Google Scholar]
- Snyder J.; Livi K.; Erlebacher J. Oxygen Reduction Reaction Performance of [MTBD][beti]-Encapsulated Nanoporous NiPt Alloy Nanoparticles. Adv. Funct. Mater. 2013, 23, 5494–5501. 10.1002/adfm.201301144. [DOI] [Google Scholar]
- Chen C.; Kang Y. J.; Huo Z. Y.; Zhu Z. W.; Huang W. Y.; Xin H. L. L.; Snyder J. D.; Li D. G.; Herron J. A.; Mavrikakis M.; Chi M. F.; More K. L.; Li Y. D.; Markovic N. M.; Somorjai G. A.; Yang P. D.; Stamenkovic V. R. Highly Crystalline Multimetallic Nanoframes with Three-Dimensional Electrocatalytic Surfaces. Science 2014, 343, 1339–1343. 10.1126/science.1249061. [DOI] [PubMed] [Google Scholar]
- Zhang G.-R.; Munoz M.; Etzold B. J. Boosting Performance of Low Temperature Fuel Cell Catalysts by Subtle Ionic Liquid Modification. ACS Appl. Mater. Interfaces 2015, 7, 3562–3570. 10.1021/am5074003. [DOI] [PubMed] [Google Scholar]
- Zhang G.-R.; Munoz M.; Etzold B. J. Accelerating Oxygen-Reduction Catalysts through Preventing Poisoning with Non-Reactive Species by Using Hydrophobic Ionic Liquids. Angew. Chem., Int. Ed. 2016, 55, 2257–2261. 10.1002/anie.201508338. [DOI] [PubMed] [Google Scholar]
- Huang K.; Song T.; Morales-Collazo O.; Jia H.; Brennecke J. F. Enhancing Pt/C Catalysts for the Oxygen Reduction Reaction with Protic Ionic Liquids: The Effect of Anion Structure. J. Electrochem. Soc. 2017, 164, F1448–F1459. 10.1149/2.1071713jes. [DOI] [Google Scholar]
- Martinaiou I.; Wolker T.; Shahraei A.; Zhang G.-R.; Janßen A.; Wagner S.; Weidler N.; Stark R. W.; Etzold B. J. M.; Kramm U. I. Improved electrochemical performance of Fe-N-C catalysts through ionic liquid modification in alkaline media. J. Power Sources 2018, 375, 222–232. 10.1016/j.jpowsour.2017.07.028. [DOI] [Google Scholar]
- Qiao M.; Tang C.; Tanase L. C.; Teodorescu C. M.; Chen C.; Zhang Q.; Titirici M.-M. Oxygenophilic ionic liquids promote the oxygen reduction reaction in Pt-free carbon electrocatalysts. Mater. Horiz. 2017, 4, 895–899. 10.1039/C7MH00298J. [DOI] [Google Scholar]
- Pham Truong T. N.; Randriamahazaka H.; Ghilane J. Polymer Brushes Ionic Liquid as a Catalyst for Oxygen Reduction and Oxygen Evolution Reactions. ACS Catal. 2018, 8, 869–875. 10.1021/acscatal.7b03158. [DOI] [Google Scholar]
- Tran Q. C.; Dao V.-D.; Kim H. Y.; Jung K.-D.; Choi H.-S. Pt-based alloy/carbon black nanohybrid covered with ionic liquid supramolecules as an efficient catalyst for oxygen reduction reactions. Appl. Catal., B 2017, 204, 365–373. 10.1016/j.apcatb.2016.11.051. [DOI] [Google Scholar]
- Zhang G.-R.; Etzold B. J. M. Ionic liquids in electrocatalysis. J. Energy Chem. 2016, 25, 199–207. 10.1016/j.jechem.2016.01.007. [DOI] [Google Scholar]
- Holbrey J. D.; Seddon K. R. The phase behaviour of 1-alkyl-3-methylimidazolium tetrafluoroborates; ionic liquids and ionic liquid crystals. J. Chem. Soc., Dalton Trans. 1999, 2133–2140. 10.1039/a902818h. [DOI] [Google Scholar]
- Huddleston J. G.; Visser A. E.; Reichert W. M.; Willauer H. D.; Broker G. A.; Rogers R. D. Characterization and comparison of hydrophilic and hydrophobic room temperature ionic liquids incorporating the imidazolium cation. Green Chem. 2001, 3, 156–164. 10.1039/b103275p. [DOI] [Google Scholar]
- Schuppert A. K.; Topalov A. A.; Katsounaros I.; Klemm S. O.; Mayrhofer K. J. J. A Scanning Flow Cell System for Fully Automated Screening of Electrocatalyst Materials. J. Electrochem. Soc. 2012, 159, F670–F675. 10.1149/2.009211jes. [DOI] [Google Scholar]
- Cherevko S.; Kulyk N.; Mayrhofer K. J. J. Durability of platinum-based fuel cell electrocatalysts: Dissolution of bulk and nanoscale platinum. Nano Energy 2016, 29, 275–298. 10.1016/j.nanoen.2016.03.005. [DOI] [Google Scholar]
- Zhang G.-R.; Zhao D.; Feng Y. Y.; Zhang B. S.; Su D. S.; Liu G.; Xu B. Q. Catalytic Pt-on-Au Nanostructures: Why Pt Becomes More Active on Smaller Au Particles. ACS Nano 2012, 6, 2226–2236. 10.1021/nn204378t. [DOI] [PubMed] [Google Scholar]
- Zhang G.-R.; Xu B. Q. Surprisingly strong effect of stabilizer on the properties of Au nanoparticles and Pt boolean AND Au nanostructures in electrocatalysis. Nanoscale 2010, 2, 2798–2804. 10.1039/c0nr00295j. [DOI] [PubMed] [Google Scholar]
- Zhang G. R.; Wu J.; Xu B. Q. Syntheses of Sub-30 nm Au@Pd Concave Nanocubes and Pt-on-(Au@Pd) Trimetallic Nanostructures as Highly Efficient Catalysts for Ethanol Oxidation. J. Phys. Chem. C 2012, 116, 20839–20847. 10.1021/jp304570c. [DOI] [Google Scholar]
- Ariyanto T.; Kern A. M.; Etzold B. J. M.; Zhang G.-R. Carbide-derived carbon with hollow core structure and its performance as catalyst support for methanol electro-oxidation. Electrochem. Commun. 2017, 82, 12–15. 10.1016/j.elecom.2017.07.010. [DOI] [Google Scholar]
- Lopez-Cudero A.; Cuesta A.; Gutierrez C. Potential dependence of the saturation CO coverage of Pt electrodes: The origin of the pre-peak in CO-stripping voltammograms. Part 1: Pt(111). J. Electroanal. Chem. 2005, 579, 1–12. 10.1016/j.jelechem.2005.01.018. [DOI] [Google Scholar]
- Urchaga P.; Baranton S.; Coutanceau C.; Jerkiewicz G. Electro-oxidation of COchem on Pt Nanosurfaces: Solution of the Peak Multiplicity Puzzle. Langmuir 2012, 28, 3658–3663. 10.1021/la202913b. [DOI] [PubMed] [Google Scholar]
- Chen Q. S.; Vidal-Iglesias F. J.; Solla-Gullon J.; Sun S. G.; Feliu J. M. Role of surface defect sites: from Pt model surfaces to shape-controlled nanoparticles. Chem. Sci. 2012, 3, 136–147. 10.1039/C1SC00503K. [DOI] [Google Scholar]
- Meier J. C.; Galeano C.; Katsounaros I.; Witte J.; Bongard H. J.; Topalov A. A.; Baldizzone C.; Mezzavilla S.; Schuth F.; Mayrhofer K. J. J. Design criteria for stable Pt/C fuel cell catalysts. Beilstein J. Nanotechnol. 2014, 5, 44–67. 10.3762/bjnano.5.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeley G. P.; Cherevko S.; Mayrhofer K. J. J. The Stability Challenge on the Pathway to Low and Ultra-Low Platinum Loading for Oxygen Reduction in Fuel Cells. ChemElectroChem 2016, 3, 51–54. 10.1002/celc.201500425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W.; He S.; Ding L.; Yao Y.; Wan L.; Miao S.; Xu J. Supported Ionic-Liquid “Semi-Heterogeneous Catalyst”: An Interfacial Chemical Study. J. Phys. Chem. C 2013, 117, 7026–7038. 10.1021/jp310117x. [DOI] [Google Scholar]
- Gilbert J. A.; Kariuki N. N.; Wang X.; Kropf A. J.; Yu K.; Groom D. J.; Ferreira P. J.; Morgan D.; Myers D. J. Pt Catalyst Degradation in Aqueous and Fuel Cell Environments studied via In-Operando Anomalous Small-Angle X-ray Scattering. Electrochim. Acta 2015, 173, 223–234. 10.1016/j.electacta.2015.05.032. [DOI] [Google Scholar]
- Pizzutilo E.; Geiger S.; Grote J.-P.; Mingers A.; Mayrhofer K. J. J.; Arenz M.; Cherevko S. On the Need of Improved Accelerated Degradation Protocols (ADPs): Examination of Platinum Dissolution and Carbon Corrosion in Half-Cell Tests. J. Electrochem. Soc. 2016, 163, F1510–F1514. 10.1149/2.0731614jes. [DOI] [Google Scholar]
- Cherevko S.; Zeradjanin A. R.; Topalov A. A.; Kulyk N.; Katsounaros I.; Mayrhofer K. J. J. Dissolution of Noble Metals during Oxygen Evolution in Acidic Media. ChemCatChem 2014, 6, 2219–2223. 10.1002/cctc.201402194. [DOI] [Google Scholar]
- Holewinski A.; Linic S. Elementary mechanisms in electrocatalysis: revisiting the ORR Tafel slope. J. Electrochem. Soc. 2012, 159, H864–H870. 10.1149/2.022211jes. [DOI] [Google Scholar]
- Subramanian N. P.; Greszler T. A.; Zhang J.; Gu W.; Makharia R. Pt-Oxide Coverage-Dependent Oxygen Reduction Reaction (ORR) Kinetics. J. Electrochem. Soc. 2012, 159, B531–B540. 10.1149/2.088205jes. [DOI] [Google Scholar]
- Jinnouchi R.; Anderson A. B. Electronic structure calculations of liquid-solid interfaces: Combination of density functional theory and modified Poisson-Boltzmann theory. Phys. Rev. B: Condens. Matter Mater. Phys. 2008, 77, 245417. 10.1103/PhysRevB.77.245417. [DOI] [Google Scholar]
- Freire M. G.; Carvalho P. J.; Gardas R. L.; Marrucho I. M.; Santos L. M. N. B. F.; Coutinho J. A. P. Mutual Solubilities of Water and the [Cnmim][Tf2N] Hydrophobic Ionic Liquids. J. Phys. Chem. B 2008, 112, 1604–1610. 10.1021/jp7097203. [DOI] [PubMed] [Google Scholar]
- Zhou T.; Chen L.; Ye Y.; Chen L.; Qi Z.; Freund H.; Sundmacher K. An Overview of Mutual Solubility of Ionic Liquids and Water Predicted by COSMO-RS. Ind. Eng. Chem. Res. 2012, 51, 6256–6264. 10.1021/ie202719z. [DOI] [Google Scholar]
- Ramya K. R.; Kumar P.; Kumar A.; Venkatnathan A. Interplay of Phase Separation, Tail Aggregation, and Micelle Formation in the Nanostructured Organization of Hydrated Imidazolium Ionic Liquid. J. Phys. Chem. B 2014, 118, 8839–8847. 10.1021/jp504270b. [DOI] [PubMed] [Google Scholar]
- Su Y. Z.; Fu Y. C.; Wei Y. M.; Yan J. W.; Mao B. W. The Electrode/Ionic Liquid Interface: Electric Double Layer and Metal Electrodeposition. ChemPhysChem 2010, 11, 2764–2778. 10.1002/cphc.201000278. [DOI] [PubMed] [Google Scholar]
- Feng S.; Voth G. A. Molecular dynamics simulations of imidazolium-based ionic liquid/water mixtures: Alkyl side chain length and anion effects. Fluid Phase Equilib. 2010, 294, 148–156. 10.1016/j.fluid.2010.02.034. [DOI] [Google Scholar]
- Jiang W.; Wang Y.; Voth G. A. Molecular Dynamics Simulation of Nanostructural Organization in Ionic Liquid/Water Mixtures. J. Phys. Chem. B 2007, 111, 4812–4818. 10.1021/jp067142l. [DOI] [PubMed] [Google Scholar]
- Smith A. M.; Lovelock K. R. J.; Perkin S. Monolayer and bilayer structures in ionic liquids and their mixtures confined to nano-films. Faraday Discuss. 2014, 167, 279–292. 10.1039/c3fd00075c. [DOI] [PubMed] [Google Scholar]
- Bhargava B. L.; Klein M. L. Initial Stages of Aggregation in Aqueous Solutions of Ionic Liquids: Molecular Dynamics Studies. J. Phys. Chem. B 2009, 113, 9499–9505. 10.1021/jp903560y. [DOI] [PubMed] [Google Scholar]
- Bhargava B. L.; Klein M. L. Molecular Dynamics Studies of Cation Aggregation in the Room Temperature Ionic Liquid [C10mim][Br] in Aqueous Solution. J. Phys. Chem. A 2009, 113, 1898–1904. 10.1021/jp8068865. [DOI] [PubMed] [Google Scholar]
- Bhargava B. L.; Yasaka Y.; Klein M. L. Computational studies of room temperature ionic liquid-water mixtures. Chem. Commun. 2011, 47, 6228–6241. 10.1039/c1cc10575b. [DOI] [PubMed] [Google Scholar]
- Wen R.; Rahn B.; Magnussen O. M. Potential-Dependent Adlayer Structure and Dynamics at the Ionic Liquid/Au(111) Interface: A Molecular-Scale In Situ Video-STM Study. Angew. Chem., Int. Ed. 2015, 54, 6062–6066. 10.1002/anie.201501715. [DOI] [PubMed] [Google Scholar]
- Kornyshev A. A. Double-Layer in Ionic Liquids: Paradigm Change?. J. Phys. Chem. B 2007, 111, 5545–5557. 10.1021/jp067857o. [DOI] [PubMed] [Google Scholar]
- Uysal A.; Zhou H.; Feng G.; Lee S. S.; Li S.; Cummings P. T.; Fulvio P. F.; Dai S.; McDonough J. K.; Gogotsi Y.; Fenter P. Interfacial ionic ‘liquids’: connecting static and dynamic structures. J. Phys.: Condens. Matter 2015, 27, 032101. 10.1088/0953-8984/27/3/032101. [DOI] [PubMed] [Google Scholar]
- Gebbie M. A.; Valtiner M.; Banquy X.; Fox E. T.; Henderson W. A.; Israelachvili J. N. Ionic liquids behave as dilute electrolyte solutions. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 9674–9679. 10.1073/pnas.1307871110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauw Y.; Horne M. D.; Rodopoulos T.; Lockett V.; Akgun B.; Hamilton W. A.; Nelson A. R. J. Structure of [C4mpyr][NTf2] Room-Temperature Ionic Liquid at Charged Gold Interfaces. Langmuir 2012, 28, 7374–7381. 10.1021/la3005757. [DOI] [PubMed] [Google Scholar]
- Strmcnik D.; Escudero-Escribano M.; Kodama K.; Stamenkovic V. R.; Cuesta A.; Marković N. M. Enhanced electrocatalysis of the oxygen reduction reaction based on patterning of platinum surfaces with cyanide. Nat. Chem. 2010, 2, 880. 10.1038/nchem.771. [DOI] [PubMed] [Google Scholar]
- Cherevko S.; Zeradjanin A. R.; Keeley G. P.; Mayrhofer K. J. J. A Comparative Study on Gold and Platinum Dissolution in Acidic and Alkaline Media. J. Electrochem. Soc. 2014, 161, H822–H830. 10.1149/2.0881412jes. [DOI] [Google Scholar]
- Topalov A. A.; Cherevko S.; Zeradjanin A. R.; Meier J. C.; Katsounaros I.; Mayrhofer K. J. J. Towards a comprehensive understanding of platinum dissolution in acidic media. Chem. Sci. 2014, 5, 631–638. 10.1039/C3SC52411F. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.